Neutrophil-specific Granule Deficiency Results from a Novel Mutation with Loss of Function of the Transcription Factor CCAAT/Enhancer Binding Protein ε (original) (raw)
Abstract
Neutrophil-specific granule deficiency (SGD) is a rare disorder characterized by recurrent pyogenic infections, defective neutrophil chemotaxis and bactericidal activity, and lack of neutrophil secondary granule proteins. CCAAT/enhancer binding protein (C/EBP)ε, a member of the leucine zipper family of transcription factors, is expressed primarily in myeloid cells, and its knockout mouse model possesses distinctive defects, including a lack of neutrophil secondary granule proteins. Sequence analysis of the genomic DNA of a patient with SGD revealed a five-basepair deletion in the second exon of the C/EBPε locus. The predicted frame shift results in a truncation of the 32-kD major C/EBPε isoform, with loss of the dimerization domain, DNA binding region, and transcriptional activity. The multiple functional defects observed in these early neutrophil progenitor cells, a consequence of C/EBPε deficiency, define SGD as a defect in myelopoiesis and establish the requirement for C/EBPε for the promyelocyte–myelocyte transition in myeloid differentiation.
Keywords: myelopoiesis, lactoferrin, granulocyte, immunodeficiency, neutrophil
Neutrophil-specific granule deficiency (SGD) is a rare congenital disorder marked by frequent and severe bacterial infections. The five reported cases consistently describe pleiotropic characteristics, including lack of secondary granule proteins and defensins, abnormalities in neutrophil migration and disaggregation, atypical nuclear morphology, and impaired bactericidal activity (1–11). More recent work has revealed additional granule abnormalities in the eosinophils of SGD patients, with absence of eosinophil-specific granule contents, including eosinophil cationic protein, eosinophil-derived neurotoxin, and major basic protein (12). Platelet disorders and associated bleeding diatheses, including the neutrophilic phagocytosis of platelets (13) and the absence of platelet–high-molecular-mass von Willebrand factor multimers stored in platelet α granules (14), have also been reported in SGD patients. In contrast to these seemingly genetically unrelated manifestations, these patients express normal levels of salivary lactoferrin (8, 15, 16), a characteristic specific granule marker absent in neutrophils in SGD, suggesting that the responsible defect involves myeloid-specific transcriptional regulation.
CCAAT/enhancer binding proteins (C/EBPs) comprise a family of transcription factors that are key regulators of cellular differentiation and function in a variety of tissues (17). The prototypic C/EBP is a modular protein consisting of one or more activation domains, a dimerization basic zipper domain and a DNA binding region (18). C/EBPs are least conserved in their activation domains and vary from dominant negative repressors to strong activators.
C/EBPε, the newest member of the family, is expressed exclusively in cells of myeloid and T cell lineage (19–22). The human C/EBPε gene encodes four mRNA isoforms with varying splice patterns, driven from two alternative promoters, and from which are translated three protein isoforms (23). Analogous to what has been shown for C/EBPα and C/EBPβ (24, 25), in vitro transfection data suggest that the full length, 32-kD isoform of C/EBPε (C/EBPε32) possesses the fully active transcriptional activation domain, whereas the short, 14.2-kD isoform (C/EBPε14) lacks transcriptional activity (23).
Nearly 60% of C/EBPε knockout mice (26) succumb to low pathogenicity bacterial infections by 4–6 mo of age. Neutrophils from C/EBPε knockout mice have morphological features similar to human SGD neutrophils, including bilobed nuclei, absent specific and tertiary granule contents, and defective chemotaxis and bactericidal activity (27). The striking phenotypic similarities between SGD defects and the C/EBPε knockout model prompted a search for a C/EBPε knockout mutation in an SGD patient's genomic locus.
Materials and Methods
Patient.
Material from a previously described (5, 6) male patient lacking neutrophil-specific granules was studied. Research was conducted with informed consent under the guidelines of a National Institutes of Health (NIH) Internal Review Board– approved protocol, no. 92-I-99. The patient died from complications of pneumonia at age 20.
DNA, RNA, and Protein Extraction.
Peripheral blood neutrophils were isolated as described (28), cryopreserved with dimethylformamide (Sigma Chemical Co.), and maintained at −140°C. Cell proteins were extracted as described (29). DNA extraction from cryopreserved fibroblasts proceeded as described (30). RNA was extracted from patient bone marrow aspirate using RNAzol reagent (Teltest) as per manufacturer's protocol. Normal human bone marrow RNA was purchased from Clontech.
PCR Amplification of Genomic Sequence.
PCR reaction was performed using Platinum taq DNA polymerase (Life Technologies) per manufacturer's instructions and cycled as follows: 96°C for 12 min, followed by a three-step cycle—94°C for 30 s, 60°C for 30 s, and 72°C for 2 min—for 35–40 cycles. PCR products were gel purified and recovered using Gene Clean (Bio101). Products were sequenced with an ABI Prism Dye Terminator Cycle Sequencing Ready Reaction Kit (Perkin-Elmer Corp.). Primers were chosen from a published sequence available from EMBL/GenBank/DDBJ under accession no. U48865. Primer sets (upstream, downstream): B, 5′-AGC GGC CAT GCA AAA GGA AAG ACA, 5′-TCC ACC TAC CCC CAA GAG AAA GTT (bp 667–1186); C, 5′-CCC ACG GGA CCT ACT ACG A, 5′-GGG CTG GCC TGC TCT TAC (bp 1818–2343); F, 5′-CTC CCC GGC TGG CCC CTT ACA C, 5′-GCC AAC AGT CCC AAC ACC CAG TCA (bp 3133–3615); G, 5′-GGA GGT GGG GCT ACA AAA GAA ACT, 5′-TCA GGG AGG GGC AGG ACA (bp 1143–1553); H, 5′-ACA GGA GTG GGT GAC AGA GGA GAC, 5′-GGG CCG AAG GTA TGT GGA GGG TAG (bp 1563–2104); I, 5′-CCA TGC CCC CTC CTC TTG TTT CTC, 5′-ACT GCC TTC TTG CCC TTG TGT AA (bp 2594–3171); K, 5′-AAC TTT CTC TTG GGG GTA GGT GGA, 5′-TCG TAG TAG GTC CCG TGG (bp 1163– 1837). Homozygosity was determined by hybridization of the PCR product fragment C to a [32P]γdATP-endlabeled internal oligonucleotide (H. downstream; sequence above). Labeled oligo was mixed with hybridization buffer (75 mM NaCl, 5 mM EDTA) in a ratio of 1:10, and 10 μl was added to 30 μl PCR product. Hybridization was cycled in a thermal cycler: 95°C for 5 min and 55°C for 10 min. Reactions were immediately placed on ice. Products were resolved on 4–20% Tris/borate/EDTA polyacrylamide gel (Novex) at 250 V.
RNA Blotting Assay.
10 μg of total RNA isolated from the patient's bone marrow and 0.25, 0.5, and 1 μg of control polyadenylated (pA)-mRNA was electrophoretically separated, blotted, hybridized, and washed as described (27). The membrane was stripped by boiling and stored at −20°C.
Immunoblotting.
Protein quantitation was performed using a BCA Protein Assay kit (Pierce Chemical Co.) according to the manufacturer's instructions. 10–100 μg protein extracts were electrophoretically separated, transferred to nitrocellulose, and incubated with primary antibody as described (29). Primary antibody was generated in rabbits by Research Genetics, Inc., using a synthetic peptide encoded in exon 2 of C/EBPε, downstream of the SGD deletion (DPRAVAVKEEPRGPEGSR). The membranes were washed, incubated with anti–rabbit horseradish peroxidase conjugate antibody (ECL Western blot kit; Amersham Pharmacia Biotech, Inc.), and developed according to the manufacturer's instructions. Membranes were stripped and reblotted with anti–mouse human β actin antibody (Boehringer Mannheim) to control for protein loading.
In Vitro Mutagenesis Assay.
The patient's mutation was introduced into the pCMV-C/EBPε32 expression vector using a Stratagene QuikChange site-directed mutagenesis kit per manufacturer's instructions using a complementary oligonucleotide (PAGE purified; purchased from Genosys Biotech) containing the deletion (5′-CCA CTA CTT GCC GCC CTC GGC CCT TTG CCT ACC). Presence of the mutation and maintenance of the vector sequences was verified by sequencing and restriction enzyme digestion, respectively.
Transient Transfections.
HeLa cells were maintained in DMEM (BioWhittaker) supplemented with 10% heat-inactivated FBS (Life Technologies, Inc.) and penicillin/streptomycin at 37°C and 5% CO2. Cells were plated in 6-well plates and transfected within 24 h, at 30–50% confluency. Transfections, using the Mammalian Transfection System (Stratagene), were performed using 5 μg reporter plasmid (G-CSF receptor promoter-luc); 1, 2, or 5 μg inducer plasmid (pCMV-C/EBPα, pCMV-C/EBPε32 isoform, pCMV-C/EBPε14 isoform, or pCMV-C/EBPε32-SGD, described above); and 0.5 μg pCMVβ, as described (23, 31). The DNA content of transfections was normalized, and transfection was performed according to the manufacturer's instructions, with 300 μl transfection solution applied to the cells. Samples were harvested 24 h after transfection.
Luciferase and β-galactosidase activities were measured using a Dual-Light kit (Tropix, Inc.), according to the manufacturer's protocol, on a Turner 20/20 Luminometer. Samples were read for 15 s after a 3-s delay.
Results and Discussion
Sequencing of PCR products from genomic DNA detected a 5-bp deletion, TGACC, in exon 2 of the patient's C/EBPε sequence. Fig. 1 A shows sequence data from one normal control (top sequence) and the SGD patient (bottom sequence). The mutation predicts a frameshift and a premature termination of the encoded C/EBPε32 isoform (Fig. 1 B). The missense code after the frameshift results in the loss of the critical DNA binding domain and leucine zipper region required for C/EBP dimerization and function. C/EBPε transcripts encoding the shorter 27- and 14-kD isoforms are predicted to be unaffected, based upon the splice donor and acceptor and translational start sites (23).
Figure 1.



(A) Sequence data from PCR products of normal control genomic DNA (top) and SGD patient DNA (bottom). Sequencing was performed on three separate PCRs from the SGD patient and three normal controls. Color coding of nucleotides on sequence scan is red, T; green, A; black, G; blue, C. Underlined nucleotides, 5-bp deletion. Arrowhead, location of deletion in the SGD patient sequence. Schematic drawing of C/EBPε locus shows three exons; two alternative promoters, Pα and Pβ; translational start codons; and bZIP region. (B) Schematic drawing of the three human C/EBPε protein isoforms. Second drawing (from top) shows the C/EBPε32-SGD isoform with predicted missense region and premature termination codon occurring after the arrowhead. (C) PCR products after liquid hybridization of DNA region containing 5-bp deletion. Lanes 1 and 2, normal controls. Lane 4, patient DNA. Lane 3, PCR products from normal control DNA mixed equimolar with patient DNA. Arrowheads indicate normal allele (top) and SGD allele (bottom). Over 30 normal controls were tested.
Homozygosity of the deletion was determined by PCR amplification of the affected region and resolution of the DNA fragments on a 4–20% polyacrylamide gel (Fig. 1 C). DNA from one normal control and the SGD patient were mixed before amplification and electrophoresis (lane 3), showing bands from both affected and normal alleles. In comparison, PCR products from the SGD patient (lane 4) and normal controls (lanes 1 and 2) show only one fragment, indicating homozygosity for their respective alleles.
RNA blot analysis of the SGD patient's bone marrow total RNA showed decreased amounts of C/EBPε transcripts in comparison with control human bone marrow pA-RNA (Fig. 2 A). Hybridization with a [32P]dCTP- labeled actin probe (provided by L. Perera, National Cancer Institute, NIH) showed that 10 μg of SGD patient bone marrow total RNA was equivalent to 1 μg of normal bone marrow pA-mRNA and verified the stability and quality of the patient's RNA preparation. Specific loss of C/EBPε transcripts in the SGD patient is likely due to mRNA instability secondary to the frameshift and the premature termination codon, as seen in other similar gene mutations (32, 33). Residual C/EBPε message is likely comprised by C/EBPε14 and C/EBPε27 transcripts, which are unaffected by the 5-bp deletion and similar in size to the C/EBPε32 transcript. Transcripts of C/EBPα were present in normal amounts. C/EBPα has a more proximal role in the myelopoietic pathway and specifically induces expression of C/EBPε (31, 34, 35). As expected, message for lactoferrin was not detected in the SGD patient's bone marrow RNA.
Figure 2.

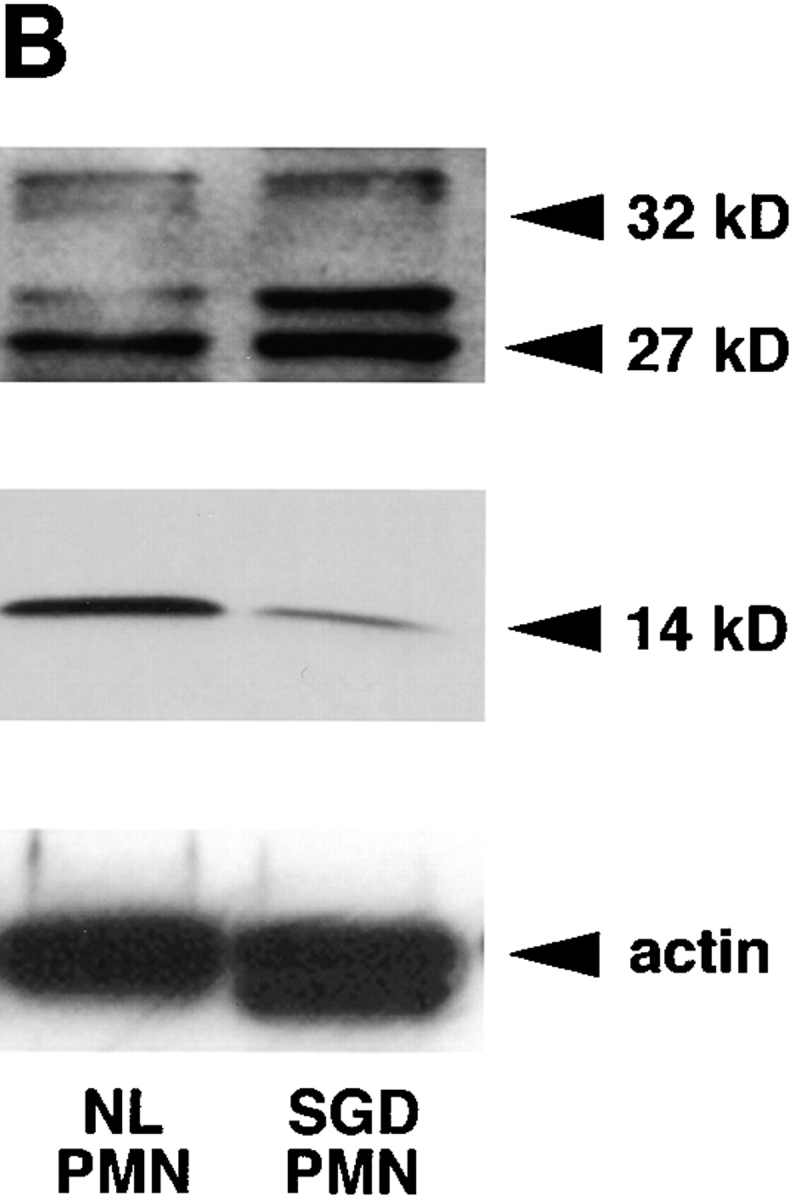
(A) RNA blot of SGD patient bone marrow total RNA and normal human bone marrow pA-RNA. Three concentrations of control RNA, 1, 0.5, and 0.25 μg, and 10 μg of SGD patient RNA were used, as indicated below the lanes. Blot was probed with [32P]dCTP-labeled probes, as indicated, and stripped between hybridizations. (B) Immunoblotting of cellular proteins extracted from peripheral blood neutrophils isolated as described. Top panel was blotted with rabbit anti–human polyclonal C/EBPε antibody. Center panel was blotted with rabbit anti–rat C/EBP–related protein 1 (Santa Cruz Biotechnology). Bottom panel shows immunoblotting with mouse anti–human actin antibody. Arrowheads indicate C/EBPε32, C/EBPε27, and C/EBPε14 isoforms.
As predicted from the C/EBPε transcript maps (Fig. 1 B), immunoblotting detected C/EBPε27 and C/EBPε14 isoforms, but not C/EBPε32, in neutrophils from the SGD patient (Fig. 2 B). All three isoforms were seen in the normal control. The antibody used is specific for a peptide sequence immediately downstream of the 5-bp mutation and should not bind the C/EBPε32-SGD protein.
Transient transfection assays in HeLa cells, using the G-CSF receptor promoter driving the luciferase gene (31), compared the transactivation potentials of the inducer genes C/EBPα, C/EBPε32, C/EBPε14, and C/EBPε32-SGD (Fig. 3). C/EBPε32 has been shown to transactivate the G-CSF receptor promoter, whereas the C/EBPε14 isoform lacks transactivating function (23). Transient transfection of these plasmid constructs showed a significant loss of transactivation with the C/EBPε32-SGD isoform (P = 0.02, Mann-Whitney U test). The demonstrated in vitro data, as well as the in vivo SGD phenotype, mark the full length, 32-kD isoform as the major transactivator encoded in the C/EBPε locus.
Figure 3.

Transcriptional activation of normal C/EBPε and C/EBPε32-SGD. Transient transfections in HeLa cells were performed as described. Concentrations of labeled inducer plasmids (pCMV-C/EBPα, pCMV- C/EBPε32 isoform, pCMV-C/ EBPε14 isoform, and pCMV-C/ EBPε32-SGD) and reporter plasmid, G-CSF receptor promoter-luc, are given (μg). Sample activity was adjusted based on transfection efficiency, measured by β-galactosidase activity. Fold increase in luciferase activity was calculated from reporter-only baseline. Data shown represent the mean (dot) and SE (box) of four independent experiments. Significant decreases in luciferase activity were observed between the pCMV-C/EBPε32 and pCMV-C/EBPε32-SGD isoforms (P = 0.02, Mann-Whitney U test.)
The temporal link between granule protein production and myeloid lineage differentiation is well described: primary granule proteins are synthesized in myeloblasts and promyelocytes, secondary granules are produced in myelocytes and metamyelocytes, and tertiary granule proteins are generated in band and segmented neutrophils (36).
Previous work suggested that C/EBPε functions at the terminal stages of myeloid differentiation (23, 26). However, the total absence of patient neutrophil secondary granules and the selective loss of primary granule defensins marks an early myelopoietic block at the promyelocyte transition (Fig. 4). Further evidence for this conclusion comes from in vitro differentiation experiments using C/EBPε-deficient stem cells, which do not proceed beyond the promyelocyte stage (26). Other functional defects seen in mouse and human C/EBPε-deficient neutrophils, such as loss of tertiary granule gelatinase (27) and abnormalities in chemotaxis and cytokine expression (6, 27), may occur secondary to the block at the promyelocyte or later stage.
Figure 4.

Neutrophil granule expression during myelopoiesis and abnormalities in neutrophil-specific granule deficiency (broken arrows). The contents of primary granules, with the exception of defensins, are present in SGD neutrophils; however, secondary and tertiary granules are absent. G-CSF induces C/EBPε early in myelopoiesis, which initiates transcription of granule components as indicated.
Functional analysis of the previously developed C/EBPε knockout mouse model (26, 27) was critical for the interpretation of the C/EBPε mutation in SGD. The apparent multiplicity of C/EBPε target genes at different cell stages suggests that C/EBPε transactivates a set of early cell stage– specific genes, inducing normal promyelocyte differentiation and granule development. Additional evidence supporting these conclusions comes from recent observations suggesting that C/EBPε is induced by and transduces the G-CSF signal in neutrophils early in myelopoiesis (37). Absence of secondary granules, defensins, eosinophil cationic protein, eosinophil-derived neurotoxin (12), and platelet α granule high-molecular-mass von Willebrand factor (14) in SGD demonstrates a critical role for C/EBPε in the development of granules and their contents in multiple myeloid lineages.
Acknowledgments
We are grateful to Dr. Helene Rosenberg for providing SGD patient bone marrow RNA and Dr. Mitchell Horwitz for providing normal peripheral blood CD34+ selected cells and expertise.
References
- 1.Spitznagel JK, Cooper MR, McCall AE, DeChatelet LR, Welsh IRH. Selective deficiency of granules associated with lysozyme and lactoferrin in human polymorphs with reduced microbicidal capacity. J Clin Invest. 1972;51:93a. . (Abstr.) [Google Scholar]
- 2.Strauss RG, Bove KE, Jones JF, Mauer AM, Fulginiti VA. An anomaly of neutrophil morphology with impaired function. N Engl J Med. 1974;290:478–484. doi: 10.1056/NEJM197402282900903. [DOI] [PubMed] [Google Scholar]
- 3.Parmley RT, Ogawa M, Darby CP, Spicer SS. Congenital neutropenia: neutrophil proliferation with abnormal maturation. Blood. 1975;46:723–734. [PubMed] [Google Scholar]
- 4.Komiyama A, Morosawa H, Nakahata T, Miyagawa Y, Akabane T. Abnormal neutrophil maturation in a neutrophil defect with morphologic abnormality and impaired function. J Pediatr. 1979;94:19–25. doi: 10.1016/s0022-3476(79)80343-1. [DOI] [PubMed] [Google Scholar]
- 5.Breton-Gorius J, Mason DY, Bruiot D, Vilde JL, Griscelli C. Lactoferrin deficiency as a consequence of a lack of specific granule in neutrophils from a patient with recurrent infections. Am J Pathol. 1980;99:413–419. [PMC free article] [PubMed] [Google Scholar]
- 6.Gallin JI, Fletcher MP, Seligmann BE, Hoffstein S, Cehrs K, Mounessa N. Human neutrophil-specific granule deficiency: a model to assess the role of neutrophil-specific granules in the evolution of the inflammatory response. Blood. 1982;59:1317–1329. [PubMed] [Google Scholar]
- 7.Boxer LA, Coates TD, Haak RA, Wolach JB, Hoffstein S, Baehner RL. Lactoferrin deficiency associated with altered granulocyte function. N Engl J Med. 1982;307:404–410. doi: 10.1056/NEJM198208123070704. [DOI] [PubMed] [Google Scholar]
- 8.Ambruso DR, Sasada M, Nishiyama H, Kubo A, Komiyama A, Allen RH. Defective bactericidal activity and absence of specific granules in neutrophils from a patient with recurrent bacterial infections. J Clin Immunol. 1984;4:23–30. doi: 10.1007/BF00915283. [DOI] [PubMed] [Google Scholar]
- 9.Borregaard N, Boxer LA, Smolen JE, Tauber AI. Anomolous neutrophil granule distribution in a patient with lactoferrin deficiency. Am J Hematol. 1985;18:255–260. doi: 10.1002/ajh.2830180306. [DOI] [PubMed] [Google Scholar]
- 10.Ganz T, Metcalf JA, Gallin JI, Boxer LA, Lehrer RI. Microbicidal/cytotoxic proteins of neutrophils are deficient in two disorders: Chediak-Higashi Syndrome and “specific” granule deficiency. J Clin Invest. 1988;82:552–556. doi: 10.1172/JCI113631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamura A, Agematsu K, Mori T, Kawai H, Kuratsuji T, Shimane M, Tani K, Asano S, Komiyama A. A marked decrease in defensin mRNA in the only case of congenital neutrophil-specific granule deficiency reported in Japan. Int J Hematol. 1994;59:137–142. [PubMed] [Google Scholar]
- 12.Rosenberg HF, Gallin JI. Neutrophil-specific granule deficiency includes eosinophils. Blood. 1993;82:268–273. [PubMed] [Google Scholar]
- 13.Sakura T, Murakami H, Matsushima T, Tamura J, Sawamura M, Tsuchiya J. Ultrastructure of neutrophilic phagosome of autologous platelet in vivo in specific granule deficiency. Am J Hematol. 1993;43:149–150. doi: 10.1002/ajh.2830430216. [DOI] [PubMed] [Google Scholar]
- 14.Parker RI, McKeown LP, Gallin JI, Gralnick HR. Absence of the largest platelet-von Willebrand multimers in a patient with lactoferrin deficiency and a bleeding tendency. Thromb Haemost. 1992;67:320–324. [PubMed] [Google Scholar]
- 15.Lomax KJ, Gallin JI, Rotrosen D, Raphael GD, Kaliner MA, Benz EJ, Boxer LA, Malech HL. Selective defect in myeloid cell lactoferrin gene expression in neutrophil-specific granule deficiency. J Clin Invest. 1989;83:514–519. doi: 10.1172/JCI113912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raphael GD, Davis JL, Fox PC, Malech HL, Gallin JI, Baraniuk JN, Kaliner MA. Glandular secretion of lactoferrin in a patient with neutrophil lactoferrin deficiency. J Allergy Clin Immunol. 1989;84:914–919. doi: 10.1016/0091-6749(89)90389-8. [DOI] [PubMed] [Google Scholar]
- 17.Lekstrom-Himes JA, Xanthopoulos KG. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem. 1998;273:28545–28548. doi: 10.1074/jbc.273.44.28545. [DOI] [PubMed] [Google Scholar]
- 18.Williams SC, Cantwell CA, Johnson PF. A family of C/EBP-related proteins capable of forming covalently linked leucine zipper dimers in vitro. Genes Dev. 1991;5:1553–1567. doi: 10.1101/gad.5.9.1553. [DOI] [PubMed] [Google Scholar]
- 19.Antonson P, Stellan B, Yamanaka R, Xanthopoulos KG. A novel human CCAAT/enhancer binding protein gene, C/EBPε, is expressed in cells of lymphoid and myeloid lineages and is localized on chromosome 14q11.2 close to the T-cell receptor α/δ locus. Genomics. 1996;35:30–38. doi: 10.1006/geno.1996.0319. [DOI] [PubMed] [Google Scholar]
- 20.Chumakov AM, Grillier I, Chumakova E, Chih D, Slater J, Koeffler HP. Cloning of the novel human myeloid-cell-specific C/EBPε transcription factor. Mol Cell Biol. 1997;17:1375–1386. doi: 10.1128/mcb.17.3.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koike M, Chumakov AM, Takeuchi S, Tasaka T, Yang R, Nakamaki T, Tsuruoka N, Koeffler HP. C/EBP-ε: chromosomal mapping and mutational analysis of the gene in leukemia and preleukemia. Leuk Res. 1997;21:833–839. doi: 10.1016/s0145-2126(97)00072-6. [DOI] [PubMed] [Google Scholar]
- 22.Williams SC, Du Y, Schwartz RC, Weiler SR, Ortiz M, Keller JR, Johnson PF. C/EBPε is a myeloid-specific activator of cytokine, chemokine and macrophage-colony-stimulating factor receptor genes. J Biol Chem. 1998;22:13493–13501. doi: 10.1074/jbc.273.22.13493. [DOI] [PubMed] [Google Scholar]
- 23.Yamanaka R, Kim G-D, Rodomska HS, Lekstrom-Himes J, Smith LT, Antonson P, Tenen DG, Xanthopoulos KG. CCAAT/enhancer binding protein ε is preferentially up-regulated during granulocytic differentiation and its functional versatility is determined by alternative use of promoters and differential splicing. Proc Natl Acad Sci USA. 1997;94:6462–6467. doi: 10.1073/pnas.94.12.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ossipow V, Descombes P, Schibler U. CCAAT/enhancer-binding protein mRNA is translated into multiple proteins with different transcription activation potentials. Proc Natl Acad Sci USA. 1993;90:8219–8223. doi: 10.1073/pnas.90.17.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 26.Yamanaka R, Barlow C, Lekstrom-Himes J, Castilia L, Liu P, Eckhaus M, Decker T, Wynshaw-Boris A, Xanthopoulos KG. Impaired granulopoiesis, myelodysplasia, and early lethality in C/EBPε deficient mice. Proc Natl Acad Sci USA. 1997;94:13187–13192. doi: 10.1073/pnas.94.24.13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lekstrom-Himes JA, Xanthopoulos KG. C/EBPε is critical for effective neutrophil-mediated response to inflammatory challenge. Blood. 1999;93:3096–3105. [PubMed] [Google Scholar]
- 28.Kuhns DB, Young HA, Gallin EK, Gallin JI. Ca2+-dependent production and release of IL-8 in human neutrophils. J Immunol. 1998;161:4332–4339. [PubMed] [Google Scholar]
- 29.Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon γ receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–2369. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith LT, Hohaus S, Gonzalez DA, Dziennis SE, Tenen DG. PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood. 1996;88:1234–1247. [PubMed] [Google Scholar]
- 32.Hovnanian A, Rochat A, Bodemer C, Petit E, Rivers CA, Prost C, Fraitag S, Christiano AM, Uitto J, Lathrop M, et al. Characterization of 18 new mutations in COL7A1 in recessive dystrophic epidermolysis bullosa provides evidence for distinct molecular mechanisms underlying defective anchoring fibril formation. Am J Hum Genet. 1997;61:599–610. doi: 10.1086/515495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christiano AM, Amano S, Eichenfield LF, Burgeson RE, Uitto J. Premature termination codon mutations in the type VII collagen gene in recessive dystrophic epidermolysis bullosa result in nonsense-mediated mRNA decay and absence of functional protein. J Invest Dermatol. 1997;109:390–394. doi: 10.1111/1523-1747.ep12336276. [DOI] [PubMed] [Google Scholar]
- 34.Zhang D-E, Zhang P, Wang N-D, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–574. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–4314. doi: 10.1128/mcb.18.7.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borregaard N, Sehested M, Nielsen BS, Sengelov H, Kjeldsen L. Biosynthesis of granule proteins in normal human bone marrow cells. Gelatinase is a marker of terminal neutrophil differentiation. Blood. 1995;85:812–817. [PubMed] [Google Scholar]
- 37.Nakajima H, Cleveland JL, Nagata S, Ihle JN. Granulocyte colony-stimulating factor regulates myeloid differentiation through CCAAT enhancer binding protein ε. Blood. 1998;92:712a. doi: 10.1182/blood.v98.4.897. [DOI] [PubMed] [Google Scholar]