Differentiating between Memory and Effector Cd8 T Cells by Altered Expression of Cell Surface O-Glycans (original) (raw)
Abstract
Currently there are few reliable cell surface markers that can clearly discriminate effector from memory T cells. To determine if there are changes in O-glycosylation between these two cell types, we analyzed virus-specific CD8 T cells at various time points after lymphocytic choriomeningitis virus infection of mice. Antigen-specific CD8 T cells were identified using major histocompatibility complex class I tetramers, and glycosylation changes were monitored with a monoclonal antibody (1B11) that recognizes O-glycans on mucin-type glycoproteins. We observed a striking upregulation of a specific cell surface O-glycan epitope on virus-specific CD8 T cells during the effector phase of the primary cytotoxic T lymphocyte (CTL) response. This upregulation showed a strong correlation with the acquisition of effector function and was downregulated on memory CD8 T cells. Upon reinfection, there was again increased expression of this specific O-glycan epitope on secondary CTL effectors, followed once more by decreased expression on memory cells. Thus, this study identifies a new cell surface marker to distinguish between effector and memory CD8 T cells. This marker can be used to isolate pure populations of effector CTLs and also to determine the proportion of memory CD8 T cells that are recruited into the secondary response upon reencounter with antigen. This latter information will be of value in optimizing immunization strategies for boosting CD8 T cell responses.
Keywords: memory T cells, effector T cells, cytotoxic T lymphocytes, viral immunity, O-glycosylation
Introduction
T cells can be classified into three separate populations: naive, effector, and memory 1. There are several cell surface markers that distinguish naive from activated T cells, but there are very few reliable markers that distinguish between effector and memory T cells 2 3. It is possible to differentiate effector and memory T cells by the presence of specific effector molecules, such as perforin or cytokines 3 4. However, direct ex vivo staining of cytokines in effector T cells is difficult, since cytokines are rapidly secreted from these cells in vivo. Moreover, the fixation procedures used for staining intracellular cytokines or perforin kills the cells, and therefore these molecules cannot be used to isolate T cells for subsequent functional studies. For these reasons, cell surface markers that distinguish between memory and effector T cells would be of more value. CD69 and CD25 have been quite useful in this regard and are widely used to identify recently activated T cells. However, expression of both of these molecules is very transient, especially that of CD69, and it is possible to miss effector cells based on CD69 and CD25 staining. Thus, it would be useful to identify cell surface markers to reliably discriminate between effector and memory T cells.
Lymphocytic choriomeningitis virus (LCMV) infection of mice is an excellent model to study T cell immunity, and the use of MHC class I tetramers has allowed us to directly visualize antigen-specific CD8 T cells at various times after LCMV infection 1 4 5. We have previously shown that there are changes in the glycosylation patterns of T cells after viral infection based on binding to peanut agglutinin (PNA; reference 6). PNA binding differentiated between naive and activated T cells but failed to provide a clear distinction between memory and effector CD8 T cells. To further investigate potential glycosylation differences between effector and memory T cells, we chose the 1B11 antibody that recognizes core 2 O-glycans on mucin-type glycoproteins such as CD43 7 8. Using MHC class I tetramers, we examined the expression of the 1B11 epitope on virus-specific CD8 T cells and show that memory T cells can be distinguished from effector T cells based on altered expression of cell surface O-glycans.
Materials and Methods
Mice, Virus, and Adoptive Transfers.
BALB/cByJ, C57BL/6J, and P14 LCMV TCR-transgenic mice 2 were purchased from The Jackson Laboratory. For primary LCMV responses, naive adult mice were infected with 2 × 105 pfu of LCMV-Armstrong intraperitoneally. For secondary responses, LCMV immune mice were rechallenged with 2 × 106 pfu of LCMV-clone 13 or varying doses of LCMV-Armstrong intravenously. In experiments analyzing LCMV-transgenic CD8 T cells, 2 × 106 P14 splenocytes were transferred intravenously into naive nonirradiated B6 recipient mice and infected intraperitoneally with 2 × 105 pfu of LCMV-Armstrong.
Surface/Tetramer Staining and Flow Cytometry.
Production of MHC class I tetramers and cell surface staining/sorting was done as previously described 4. Anti-CD8α–PE or PerCP, 1B11–FITC or PE, and tetramer–APC were used. All antibodies were purchased from PharMingen.
Direct Ex Vivo CTL Assay.
LCMV-specific CTL activity was determined by a 6-h 51Cr-release assay as previously described 4.
Results
Kinetics of Effector CD8 CTL Response during Acute LCMV Infection.
An example of the kinetics of direct ex vivo virus-specific CTL activity during acute LCMV infection and after rechallenge is shown in Fig. 1 A. The obvious conclusion from this data is that effector CD8 T cells are present at days 5, 8, 15, and after rechallenge but not at day 80. However, a problem with such an analysis is that the number of virus-specific CD8 T cells is vastly different in each sample. Thus, it could be argued that the differences seen in the levels of killing do not necessarily represent a difference in the killing function of antigen-specific CD8 T cells present in these samples but are a reflection of the differences in the frequency of virus-specific CD8 T cells. For example, the frequency of LCMV NP118-specific CD8 T cells in the day 8 spleen is ∼1/8 (25% of splenocytes are CD8, and 50% of these are NP118 specific, i.e., 12.5% of spleen cells are virus specific), compared with a frequency of ∼1/100 in LCMV immune mice (10% of spleen cells are CD8, and of these, 10% are NP118 specific, i.e., only 1% are virus specific; reference 4). This means that in the CTL assay shown in Fig. 1 A, although the E/T ratio based on total splenocytes is the same (200:1) for the d8 and d80 samples, the E/T ratio based on the number of virus-specific CD8 T cells is quite different, 25:1 for the d8 sample and 2:1 for the d80 sample. To determine if there is truly a difference in the cytolytic activity between “effector” and “memory” CD8 T cells, we performed a CTL assay in which all samples contained the same number of antigen-specific CD8 T cells (Fig. 1 B). This was done by determining the frequency of NP118-specific CD8 T cells before the CTL assay by staining with Ld(NP118) tetramer and then appropriately diluting (i.e., normalizing) the samples using spleen cells from naive BALB/c mice. As shown in Fig. 1 B, even when the number of LCMV-specific CD8 T cells was the same in all samples, there was still a striking difference in CTL activity. The effector cells (days 5 and 8 after primary infection and day 4 after rechallenge) exhibited high cytolytic activity, whereas the same number of LCMV-specific memory CD8 T cells showed a minimal amount of killing.
Figure 1.
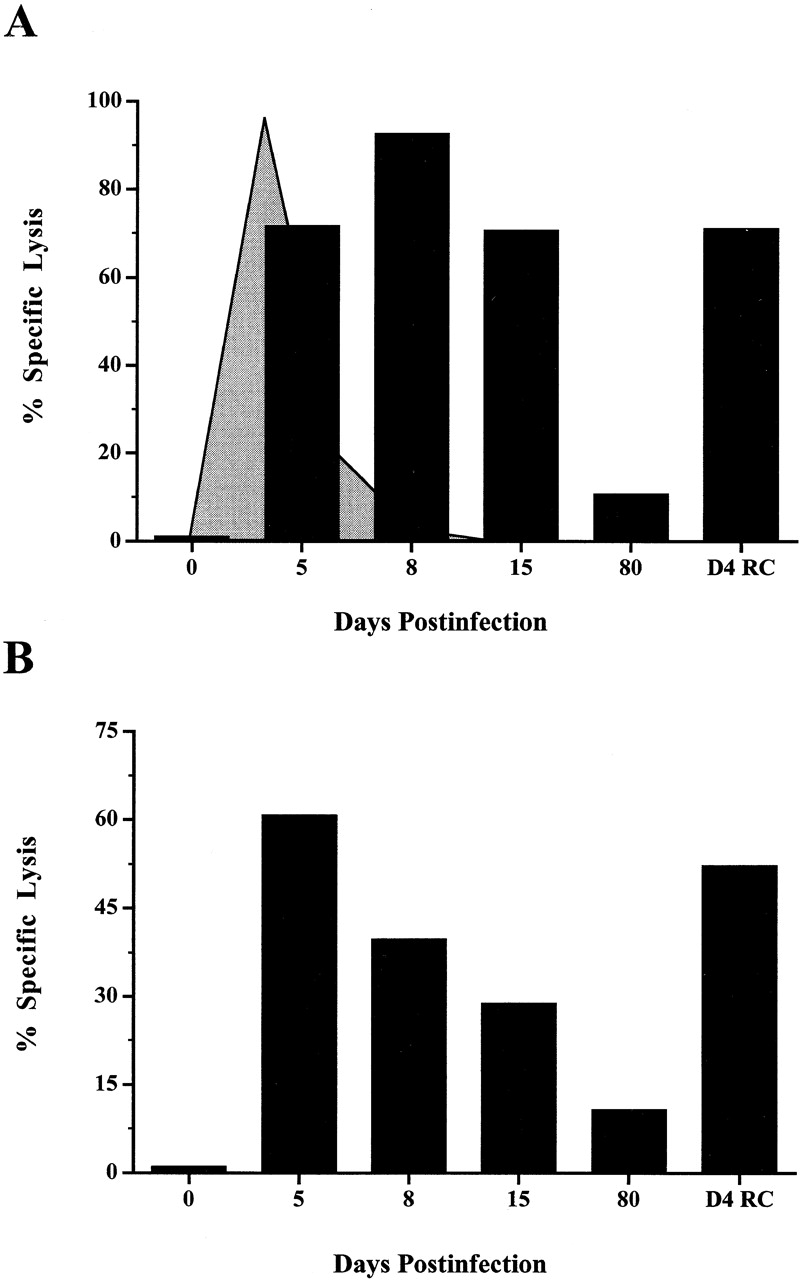
Kinetics of direct ex vivo virus-specific CTL activity during primary LCMV infection and after rechallenge. (A) Splenocytes from LCMV-infected mice were analyzed for direct ex vivo CTL activity (D4 RC = day 4 after LCMV rechallenge). Data shown are for an E/T ratio of 200:1. Lysis of noninfected target cells at the same E/T ratio was <5% (data not shown). The kinetics of viral clearance during acute LCMV infection are indicated by the shaded graph. The virus levels in the spleen peak between days 1 and 3 (at 107 pfu per spleen) and then drop precipitously between days 5 and 8 to <102 pfu per spleen. (B) The direct ex vivo CTL activity at different time points after infection after normalizing the number of antigen-specific CD8 T cells present in each sample. There were 104 NP118-specific CD8 T cells present in each sample, giving an E/T ratio of 1:1 based on virus-specific T cells. The data shown are representative of three experiments.
Expression of Cell Surface O-Glycans on Antigen-specific CD8 T Cells.
We have previously shown that activated T cells express increased neuraminidase activity and contain hyposialylated O-glycans 6. Based on these observations, we wanted to examine changes in O-glycosylation on the surfaces of antigen-specific effector and memory CD8 T cells as a potential marker to discriminate these two types of cells. To analyze changes in O-glycosylation on effector CD8 T cells, we costained day 5 and day 8 splenocytes with the 1B11 antibody and MHC class I tetramers. As shown in Fig. 2A and Fig. B, almost all of the antigen-specific CD8 T cells were 1B11hi (86 and 96%, respectively). When compared with the CD8 T cells from naive BALB/c mice, which do not exhibit any LCMV-specific CTL activity (Fig. 1 A) and have low levels of 1B11 binding (Fig. 2A and Fig. B), the effector phase CD8 T cells have a significant increase in the O-glycan epitope recognized by 1B11.
Figure 2.
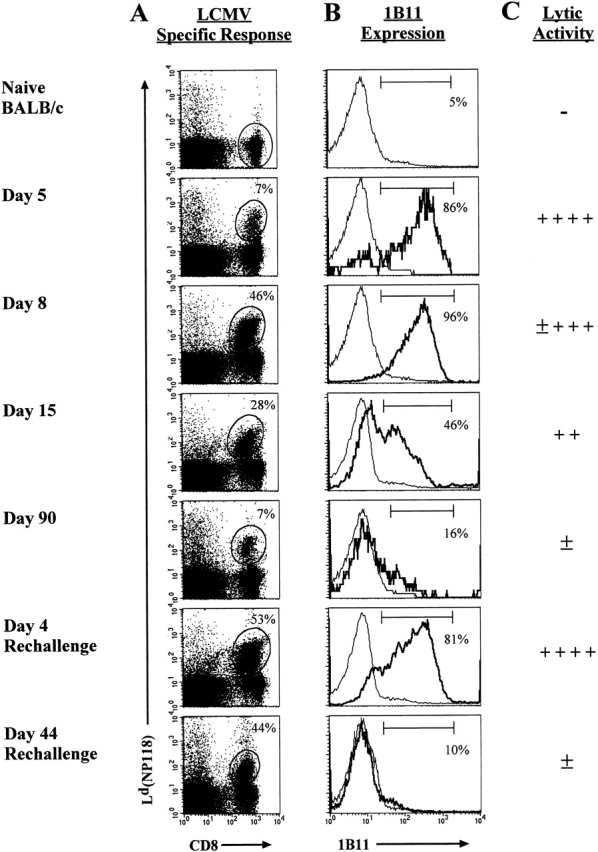
Kinetic analysis of 1B11 binding to antigen-specific CD8 T cells. (A) Splenocytes from LCMV-infected mice were stained with the Ld(NP118) tetramer and anti-CD8 and the percentage of CD8 T cells that were tetramer positive is noted. (B) Expression of cell surface O-glycans was examined with the mAb 1B11. Within the histograms, the bold line represents the 1B11 staining gated on the Ld(NP118)+ CD8 T cells at each time point, and the thin line corresponds to the 1B11 staining on naive BALB/c CD8 T cells. The percentage of 1B11hi cells within the gated population is noted. (C) This panel shows the direct ex vivo CTL activity as assessed on LCMV-infected targets.
After the LCMV infection is resolved, ≥95% of the CD8 T cells undergo cell death between days 8 and 30; the remaining cells comprise a stable LCMV-specific memory compartment 5 9. Splenocytes taken from mice during both of these phases (day 15 for the death phase and day 90 for the memory phase) were analyzed for LCMV-specific cytolytic activity and 1B11 binding. By day 15, there was a decrease in direct ex vivo CTL activity (Fig. 1A and Fig. B) and also a decrease in the percentage of antigen-specific cells that were 1B11hi (Fig. 2A and Fig. B). As previously described, the day 90 antigen-specific memory T cells displayed a low level of LCMV-specific cytotoxicity, and when the NP118-specific memory T cells were analyzed for 1B11 binding, very few of these cells were 1B11hi; instead, these cells showed a pattern of staining similar to that observed for naive CD8 T cells (Fig. 2A and Fig. B). These data demonstrate that cell surface O-glycan epitopes recognized by 1B11 are more highly expressed on effector CD8 T cells than on memory T cells.
Since it appeared that we had identified a marker that can be used to differentiate between effector and memory T cells, it was important to look at 1B11 binding on secondary effector and memory cells. If we were indeed marking effector T cells, 1B11 binding should again increase on the secondary effectors and then be downregulated on the secondary memory cells. To test this, LCMV immune mice were rechallenged with virus and analyzed at days 4 (secondary effector phase) and 44 (secondary memory phase) for 1B11 binding. At day 4 after rechallenge, splenocytes again displayed a high level of LCMV-specific CTL activity, and there was also a striking increase in 1B11 binding on antigen-specific secondary effector CD8 T cells (Fig. 2A and Fig. B). Antigen-specific memory CD8 T cells present on day 44 after rechallenge again displayed a low level of 1B11 binding, similar to that observed for memory CD8 T cells generated after primary infection. Therefore, binding of the 1B11 antibody, which primarily recognizes core 2 O-glycans on mucin-type glycoproteins, clearly distinguishes between effector and memory T cells.
We have shown that 1B11 binding is upregulated on antigen-specific CD8 T cells after LCMV infection, but due to the small number of antigen-specific T cells present in naive mice, it is not possible to analyze 1B11 binding on the naive NP118-specific precursors. To insure that there was not something intrinsically different about the LCMV-specific precursor cells, we examined P14 TCR-transgenic mice, which express a TCR specific for the H-2Db–restricted GP33-41 epitope of LCMV 2. By adoptively transferring 2 × 106 P14 splenocytes into nonirradiated, naive B6 mice and then giving the recipient mice an acute LCMV infection, we were able to analyze naive, effector, and memory LCMV-specific CD8-transgenic T cells for the binding of 1B11. As shown in Fig. 3A and Fig. B, naive GP33-specific CD8 T cells demonstrated very low levels of 1B11 binding. Similar to the BALB/c system, the majority of antigen-specific CD8 T cells from recipient mice at days 5 and 8 after infection were 1B11hi. Finally, the LCMV-specific memory CD8 T cells again displayed low levels of 1B11 binding. These experiments confirm that the O-glycan epitope recognized by 1B11 was upregulated on effector T cells and downregulated on memory T cells.
Figure 3.
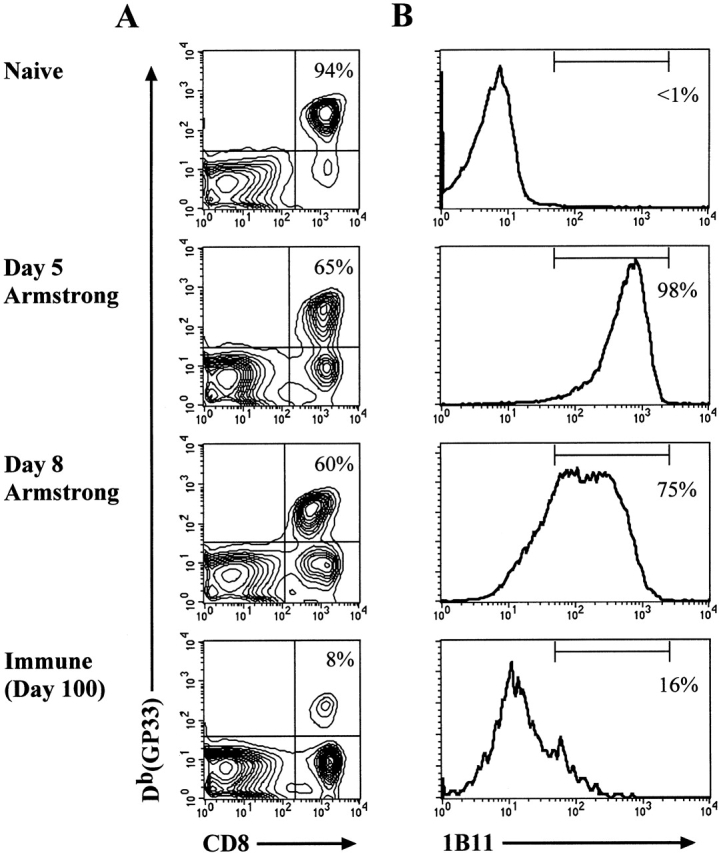
The 1B11 O-glycan epitope is upregulated on antigen-specific effector T cells. (A) LCMV-specific CD8 T cells were obtained from naive P14-transgenic mice or at days 5, 8, and 100 after adoptive transfer and infection. Splenocytes from these various groups of mice were stained with anti-CD8 and the Db(GP33) tetramer. The percent of CD8 T cells that are GP33 specific is noted in the upper right quadrant. (B) 1B11 staining of the GP33-specific CD8 T cells. The percent of cells which are 1B11hi within the gated Db(GP33)+CD8+ population is noted.
1B11 Epitope Expression Directly Correlates with CD8 T Cell Effector Function.
We have shown that the 1B11 epitope is upregulated on antigen-specific CD8 T cells during the effector phase of the immune response; therefore, this marker would be ideal for analyzing the recruitment of memory T cells into the secondary response. As shown in Fig. 4 A, when LCMV immune mice were rechallenged with increasing doses of virus, the percentage of antigen-specific CD8 T cells that became 1B11hi also increased, indicating that recruitment of memory CD8 T cells into the recall response is dependent on the antigen load. The higher the virus rechallenge dose, the greater the proportion of memory CD8 T cells that became effector cells: 32% after reinfection with 105 pfu, 60% with 3 × 105 pfu, and 80% with 2 × 106 pfu. In data not shown, along with the increase in the frequency of 1B11hi tetramer-positive cells, there was an increase in the LCMV-specific CTL activity, and this correlated with higher levels of intracellular perforin within the 1B11hi population.
Figure 4.

Using the 1B11 antibody to monitor recruitment of memory CD8 T cells into the secondary response (A) and to isolate antigen-specific effector CTLs (B). (A) LCMV immune mice were rechallenged with varying doses of LCMV-Armstrong intravenously, and recruitment of the antigen-specific CD8 T cells into the secondary response was monitored by 1B11hi staining. The contour plots show the Ld(NP118) tetramer-positive CD8 T cells at day 2 after reinfection with different doses of LCMV. The histograms demonstrate the 1B11 staining on the gated populations from the panels on the left, with the percentage of 1B11hi and 1B11lo tetramer-positive CD8 T cells noted. (B) LCMV immune mice were reinfected with 3 × 105 pfu LCMV-Armstrong intravenously, and 2 d later, Ld(NP118) tetramer-positive CD8 T cells were sorted into 1B11hi and 1B11lo populations. (C) The sorted 1B11hi and 1B11lo Ld(NP118) CD8 T cells were then analyzed for their LCMV-specific direct ex vivo CTL activity. The level of killing on uninfected target cells was <6%.
To show a direct relationship between 1B11 expression and effector function, we sorted 1B11hi and 1B11lo LCMV-specific CD8 T cells and used them in a direct ex vivo CTL assay. To obtain these two cell types from the same pool of cells, we rechallenged LCMV immune mice with a dose of LCMV that would recruit approximately half of the memory CD8 T cells to become activated. As shown in Fig. 4 B, ∼50% of the Ld(NP118)-specific CD8 T cells were 1B11hi, and 50% remained 1B11lo. These two populations were sorted to give pure populations of 1B11hi and 1B11lo Ld(NP118)+ CD8 T cells (Fig. 4 B) and then tested in a direct ex vivo CTL assay. Fig. 4 C shows that the 1B11hi tetramer-positive CD8 T cells exhibited high levels of killing on virus-infected targets, whereas the 1B11lo LCMV-specific CD8 T cells showed minimal killing. Thus, this data clearly establishes that the O-glycan epitope defined by the 1B11 antibody can be used to differentiate effector from memory CD8 T cells and that this antibody, in combination with MHC class I tetramers, can be used to obtain pure populations of antigen-specific effector CD8 CTLs.
Discussion
There are very few reliable cell surface markers that distinguish between effector and memory T cells. The lack of such markers has made it difficult to determine whether the “activated” T cells being examined are effector cells that were recently stimulated with antigen or whether they are true memory T cells. In this study, we have identified a new cell surface marker to distinguish between effector and memory T cells. We have shown that changes in cell surface O-glycosylation can be used to discriminate between these two cell populations and that these changes can be easily monitored by the mAb 1B11. We found a striking correlation between increased expression of cell surface O-glycans on antigen-specific CD8 T cells and the acquisition of CTL activity. Most importantly, we show that the 1B11 antibody in combination with MHC class I tetramers can be used to sort effector CD8 T cells. This should provide a convenient method of obtaining pure populations of antigen-specific memory and effector T cells for subsequent biological and molecular analysis.
It is worth pointing out that the 1B11 epitope is likely to be a better marker than CD69 or CD25 for distinguishing between effector and memory CD8 T cells because the expression of CD69 and CD25 is very transient and is rapidly downregulated once TCR stimulation wanes. In fact, the vast majority (>90%) of LCMV-specific (tetramer-positive) CD8 T cells that are present at day 8 after infection are both CD25 and CD69 negative (Murali-Krishna, K., and R. Ahmed, unpublished data). This is a somewhat surprising finding, since it is well established from numerous studies 5 that the day 8 LCMV-specific CD8 T cells exhibit high levels of killing ex vivo (i.e., are effector CTLs). However, as shown in this study, these day 8 virus-specific effector CTLs are still 1B11hi.
The new marker we have identified can be used to determine the proportion of memory CD8 T cells that are recruited into the secondary response upon reencounter with antigen. This information will be particularly useful in developing vaccination strategies for boosting CD8 T cell responses. Also, by using the 1B11 antibody and MHC class I tetramers in combination with antibodies against the different TCR Vβ families, it should be possible to determine if there is selective recruitment of antigen-specific memory CD8 T cells depending on the type of vaccination regimen used 10. This type of analysis may provide insight into the mechanism by which “high”-affinity T cells are selected during secondary immunization 11 12 13.
In this study, changes in O-glycosylation were detected with the mAb 1B11, which was initially characterized as recognizing an activation-associated isoform of CD43 7. The activation-associated CD43 isoform is a high-molecular-mass isoform that bears core 2 O-glycans, an oligosaccharide structure that can be created by the action of the core 2 GlcNAc transferase 14. Several studies have documented increased activity of core 2 GlcNAc transferase during murine and human T cell activation in vitro and in vivo 15. Increased expression of core 2 O-glycans has also been described in viral infection, autoimmune disease, and graft versus host disease. Mukasa et al. have recently described the binding of a core 2 O-glycan–specific mAb, 1D4, to a subset of activated human CD4+CD45RO+ peripheral T cells 16. This result appears to be different from our findings. However, it remains to be determined whether similar or different molecules are being recognized by the 1D4 and 1B11 antibodies. Also, it is possible that the glycosylation patterns of CD4 and CD8 T cells are different. In this context, it is worth noting that the S7 mAb that recognizes the low-molecular-mass isoform of CD43 reacts very differently with CD4 versus CD8 T cells 17. Both naive and memory CD8 T cells show high levels of binding to S7, whereas only memory CD4 T cells bind high levels of S7 (reference 17 and data not shown).
What are the functional consequences of these glycosylation changes on activated T cells? It is possible that these changes allow for better extravasation and migration of effector T cells to sites of infection 1 18. It is also possible that the glycosylation changes may be involved in downregulating the T cell response 19 20 21. It is well established that the majority of effector CD8 T cells undergo cell death, and it is tempting to speculate that some of the observed changes may play a role in the “death” phase of the T cell response. Future studies will be directed toward addressing these questions. It will also be of interest to determine more precisely the nature of the glycosylation changes. Core 2 O-glycans have been described on both CD43 and CD45. Although 1B11 was initially characterized as specific for CD43, it is now known that this antibody can bind both CD43 and CD45, and a recent report by Carlow et al. has shown that 1B11 recognition of CD45 occurs only in the absence of core 2 O-glycans 8. This is the opposite of CD43, as 1B11 only binds to CD43 when core 2 structures are expressed on the molecule. Work is in progress in our laboratories to identify the glycoproteins recognized by 1B11 on effector CD8 T cells and to characterize biochemically the changes in the glycosylation of these molecules during the naive to effector to memory transition.
Acknowledgments
We wish to thank M. Large and K. Madhavi-Krishna for their technical assistance, A. Zajac for his technical assistance and helpful discussion, and Robert Karaffa for help with cell sorting.
This work is supported by National Institutes of Health grants AI30048 (to R. Ahmed and L.G. Baum), NS21496 (to R. Ahmed), and CA09120-21 (to M. Galvan).
Footnotes
L. Harrington and M. Galvan contributed equally to this work.
References
- Ahmed R., Gray D. Immunological memory and protective immunityunderstanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- Zimmerman C., Brduscha-Riem K., Blaser C., Zinkernagel R.M., Pircher H. Visualization, characterization, and turnover of CD8+ memory T cells in virus-infected hosts. J. Exp. Med. 1996;183:1367–1375. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann D., Baars P.A., Rep M.H., Hooibrink B., Kerkhof-Garde S.R., Klein M.R., van Lier R.A. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali-Krishna K., Altman J.D., Suresh M., Sourdive D.J., Zajac A.J., Miller J.D., Slansky J., Ahmed R. Counting antigen-specific CD8 T cellsa reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- Buchmeier M.J., Zajac A.J. Lymphocytic choriomeningitis virus. In: Ahmed R., Chen I.S.Y., editors. Persistent Viral Infections. John Wiley & Sons; Chichester, UK: 1999. pp. 575–605. [Google Scholar]
- Galvan M., Murali-Krishna K., Ming L.L., Baum L., Ahmed R. Alterations in cell surface carbohydrates on T cells from virally infected mice can distinguish effector/memory CD8+ T cells from naive cells. J. Immunol. 1998;161:641–648. [PubMed] [Google Scholar]
- Jones A.T., Federsppiel B., Ellies L.G., Williams M.J., Burgener R., Duronio V., Smith C.A., Takei F., Ziltener H.J. Characterization of the activation-associated isoform of CD43 on murine T lymphocytes. J. Immunol. 1994;153:3426–3439. [PubMed] [Google Scholar]
- Carlow D.A., Ardman B., Ziltener H.J. A novel CD8 T cell-restricted CD45RB epitope shared by CD43 is differentially affected by glycosylation. J. Immunol. 1999;163:1441–1448. [PubMed] [Google Scholar]
- Razvi E.S., Welsh R.M. Programmed cell death of T lymphocytes during acute viral infectiona mechanism for virus-induced immune deficiency. J. Virol. 1993;67:5754–5765. doi: 10.1128/jvi.67.10.5754-5765.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourdive D.J., Murali-Krishna K., Altman J.D., Zajac A.J., Whitmire J.K., Pannetier C., Kourilsky P., Evavold B., Sette A., Ahmed R. Conserved T cell receptor repertoire in primary and memory CD8 T cell responses to an acute viral infection. J. Exp. Med. 1998;188:71–82. doi: 10.1084/jem.188.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.Y., Welsh R.M. Stability and diversity of T cell receptor repertoire usage during lymphocytic choriomeningitis virus infection of mice. J.Exp. Med. 1998;188:1993–2005. doi: 10.1084/jem.188.11.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch D.H., Pamer E.G. T cell affinity maturation by selective expansion during infection. J. Exp. Med. 1999;189:701–710. doi: 10.1084/jem.189.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage P.A., Boniface J.J., Davis M.M. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10:485–492. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- Barran P., Fellinger W., Warren C.E., Dennis J.W., Ziltener H.J. Modification of CD43 and other lymphocyte O-glycoproteins by core 2 N-acetylglucosaminyltransferase. Glycobiology. 1997;7:129–136. doi: 10.1093/glycob/7.1.129. [DOI] [PubMed] [Google Scholar]
- Piller F., Piller V., Fox R.I., Fukuda M. Human T-lymphocyte activation is associated with changes in O-glycan biosynthesis. J. Biol. Chem. 1988;263:15146–15150. [PubMed] [Google Scholar]
- Mukasa R., Homma T., Ohtsuki T., Hosono O., Souta A., Kitamura T., Fukuda M., Watanabe S., Morimoto C. Core 2-containing O-glycans on CD43 are preferentially expressed in the memory subset of human CD4 T cells. Int. Immunol. 1999;11:259–268. doi: 10.1093/intimm/11.2.259. [DOI] [PubMed] [Google Scholar]
- He Y.W., Bevan M.J. High level expression of CD43 inhibits T cell receptor/CD3-mediated apoptosis. J. Exp. Med. 1999;190:1903–1908. doi: 10.1084/jem.190.12.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellies L.G., Tsuboi S., Petryniak B., Lowe J.B., Fukuda M., Marth J.D. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9:881–890. doi: 10.1016/s1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- Tsuboi S., Fukuda M. Branched O-linked oligosaccharides ectopically expressed in transgenic mice reduce primary T-cell immune responses. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:6364–6373. doi: 10.1093/emboj/16.21.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum L.G., Pang M., Perillo N.L., Wu T., Delegeane A., Uittenbogaart C.H., Fukuda M., Seilhamer J.J. Human thymic epithelial cells express an endogenous lectin, galectin-1, which binds to core 2 O-glycans on thymocytes and T lymphoblastoid cells. J. Exp. Med. 1995;181:877–887. doi: 10.1084/jem.181.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser C., Kaufmann M., Muller C., Zimmermann C., Wells V., Mallucci L., Pircher H. Beta-galactoside-binding protein secreted by activated T cells inhibits antigen-induced proliferation of T cells. Eur. J. Immunol. 1998;28:2311–2319. doi: 10.1002/(SICI)1521-4141(199808)28:08<2311::AID-IMMU2311>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]