Residual Type 1 Immunity in Patients Genetically Deficient for Interleukin 12 Receptor β1 (IL-12Rβ1): Evidence for an IL-12Rβ1–Independent Pathway of IL-12 Responsiveness in Human T Cells (original) (raw)
Abstract
Genetic lack of interleukin 12 receptor β1 (IL-12Rβ1) surface expression predisposes to severe infections by poorly pathogenic mycobacteria or Salmonella and causes strongly decreased, but not completely abrogated, interferon (IFN)-γ production. To study IL-12Rβ1–independent residual IFN-γ production, we have generated mycobacterium–specific T cell clones (TCCs) from IL-12Rβ1–deficient individuals. All TCCs displayed a T helper type 1 phenotype and the majority responded to IL-12 by increased IFN-γ production and proliferative responses upon activation. This response to IL-12 could be further augmented by exogenous IL-18. IL-12Rβ2 was found to be normally expressed in the absence of IL-12Rβ1, and could be upregulated by IFN-α. Expression of IL-12Rβ2 alone, however, was insufficient to induce signal transducer and activator of transcription (Stat)4 activation in response to IL-12, whereas IFN-α/IFN-αR ligation resulted in Stat4 activation in both control and IL-12Rβ1–deficient cells. IL-12 failed to upregulate cell surface expression of IL-18R, integrin α6, and IL-12Rβ2 on IL-12Rβ1–deficient cells, whereas this was normal on control cells. IL-12–induced IFN-γ production in IL-12Rβ1–deficient T cells could be inhibited by the p38 mitogen-activated protein kinase (MAP) kinase inhibitor SB203580 and the MAP kinase kinase (MEK) 1/2 inhibitor U0126, suggesting involvement of MAP kinases in this alternative, Stat4-independent, IL-12 signaling pathway.
Collectively, these results indicate that IL-12 acts as a partial agonist in the absence of IL-12Rβ1. Moreover, the results reveal the presence of a novel IL-12Rβ1/Stat4–independent pathway of IL-12 responsiveness in activated human T cells involving MAP kinases. This pathway is likely to play a role in the residual type 1 immunity in IL-12Rβ1 deficiency.
Keywords: mycobacteria, Th1, interleukin 12 (receptor), interleukin 18 (receptor), interferon-γ
Introduction
The subdivision of CD4+ T lymphocytes into functionally distinct subsets, based on their cytokine production profile, has provided a basis for understanding the regulatory role of T cells in intracellular infections, allergic manifestations, and autoimmune diseases in both animals and humans 1 2 3 4 5. Th1 cells producing IFN-γ and IL-2 are known to sustain cell-mediated immunity against intracellular pathogens by activation of microbicidal mechanisms in infected macrophages. Th2 cells secrete IL-4 and IL-5, favoring allergic responses and immunity to extra-cellular pathogens. A key factor in human T cell differentiation is the presence of IL-12 and IL-4; whereas IL-12 promotes the generation of Th1 cells and additionally stimulates the growth and IFN-γ production of T cells and NK cells 6 7, IL-4 is the most potent stimulus for Th2 differentiation 8.
Recently, several studies in humans have shown that genetic deficiencies in type 1 cytokines (IL-12) or cytokine receptors (IL-12Rβ1, IFN-γRI, IFN-γRII) strongly affect cell-mediated immunity–dependent resistance to mycobacteria and Salmonellae 9 10 11. Other groups and ours have reported that IL-12Rβ1 deficiency, caused by deleterious genetic mutations in the IL-12Rβ1 gene, causes a predisposition to severe infections due to otherwise poorly pathogenic mycobacteria or Salmonella, and is associated with strongly reduced, yet not fully abrogated IFN-γ production by such patients' PBMCs 9 10. Interestingly, these patients did not develop unusual infections with other viral, bacterial, or fungal pathogens, suggesting that IL-12Rβ1 deficiency may selectively cause a predisposition to severe infections with certain intracellular bacteria. IL-12Rβ1–deficient patients develop severe, yet mostly curable infections; in contrast, patients with complete IFN-γR deficiencies often develop fatal and treatment-resistant infections 12 13. This suggests that IFN-γ/IFN-γR ligation is an obligatory step towards immunity to intracellular pathogens in humans, and that in IL-12Rβ1 deficiency low grade (type 1) immunity to intracellular pathogens persists in the absence of IL-12Rβ1. We have hypothesized that the observed low production of IFN-γ in IL-12Rβ1 deficiency is a key factor in controlling the residual type 1 immunity in these patients 11, but the IL-12Rβ1–independent pathway involved in this low IFN-γ remains unknown.
IL-12 binds with high affinity to heterodimeric IL-12Rβ1/β2 complexes on T cells and NK cells 14 15 16. In contrast to the IL-12Rβ1 chain, the human IL-12Rβ2 chain contains functional tyrosine-based signaling motifs in its cytoplasmic domain 15 16. The expression of the IL-12Rβ2 chain is therefore thought to be a critical factor in controlling T cell development and functioning. At present, several lines of evidence suggest that IL-12/IL-12R ligation signals through the Janus kinase (JAK)–signal tranducer and activator of transcription (Stat) signal transduction pathway in both mice and humans 17 18 19. In particular, the essential role of Stat4 in IL-12R signaling has been demonstrated in Stat4 knockout mice, in which IL-12 induced IFN-γ production and proliferation of T cells and NK cells were almost completely abrogated 20 21. Th1 cells can express both IL-12R subunits, but IL-12Rβ2 expression is lost in Th2 cells 22 23, and consequently, IL-12 induces tyrosine phosphorylation of JAK2 and Stat4 in human Th1, but not Th2 cells 22 24. One factor that is thought to contribute to the activity of IL-12 is IL-18, which strongly amplifies IFN-γ production of T cells in response to IL-12 25.
Previous transfection studies have shown that IL-12 can bind with low affinity to the independently expressed human IL-12Rβ2 chain when ectopically expressed in Ba/F3 or COS-7 cells, but a role for endogenous IL-12Rβ1 could not be excluded 15 16. Genetically IL-12Rβ1–deficient patients provide a unique opportunity to investigate IL-12Rβ2 expression and IL-12 functioning in the absence of IL-12Rβ1. We have isolated mycobacterium (myc)_-_responsive T cell clones (TCCs) from IL-12Rβ1–deficient patients and found that these cells display a Th1 phenotype and respond to IL-12 by increased proliferative responses and IFN-γ production upon activation.
Materials and Methods
Patients
Patients (P1, P3, P4, and P17) presented with (recurrent) severe mycobacterial and Salmonella infections in the absence of a recognized immunodeficiency. All patients failed to express cell surface IL-12Rβ1 due to individually different homozygous recessive mutations or deletions in the IL-12Rβ1 gene, leading to premature stop codons in the extracellular domain of the IL-12Rβ1 molecule. Genetic mutations or deletions have been described elsewhere for P1, P3 9, and P4 10. The genetic deficiency in the IL-12Rβ1 gene of P17 was identified more recently and will be described elsewhere. In brief, P1, a 27-yr-old female, developed a severe Salmonella paratyphi sepsis at the age of 3 yr and at the age of 22 yr presented with disseminated Mycobacterium avium infection. P3 is a 4-yr-old female who developed progressive Mycobacterium bovis BCG infection after vaccination at the age of 1 yr, followed by severe Salmonella typhimurium sepsis at the age of 2 yr. P4, a 31-yr-old man, suffered from a Salmonella enteritidis infection at the ages of 11 and 20 yr and developed M. avium infection at the age of 24 yr. P17, a 5-yr-old boy, who has now died, presented with disseminated BCG infection at the Hacettepe University Children's Hospital.
Generation and Cytokine Secretion Profile of Myc-responsive TCCs
To generate _myc_-responsive T cells, standard procedures were used 26. PBMCs from P1 and P3 were stimulated with an M. avium sonicate (2 μg/ml; Royal Tropical Institute) for 6 d, and further expanded in the presence of 10 U/ml IL-2 (Proleukin; Chiron Corp.) for another 7 d. Culture medium IMDM (Life Technologies) supplemented with 10% pooled heat-inactivated normal human serum (pooled screened serum from donors from The Netherlands), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). Expanding T cells were cloned in 96-well flat-bottomed plates by limiting dilution in the presence of 105 irradiated allogenic feeder cells from five random blood donors and M. avium sonicate (2 μg/ml). After 10–17 d, individual TCCs were further expanded in 24-well plates by standard mitogenic stimulation in the presence of irradiated allogenic feeder cells and 0.5% PHA (Murex Biotech Ltd.). After 3 d, 10 U/ml IL-2 was added to the cultures. 10 d later, TCCs were screened for their responsiveness to mycobacteria by testing 104 cells in 200 μl culture medium in a flat-bottomed 96-well plate for 3 d in the presence of whole sonicated M. avium, Mycobacterium tuberculosis, or purified protein derivate of M. tuberculosis (PPD, 2 μg/ml; Statens Serum Institute) in the presence of irradiated PBMCs (5 × 104 cell/well) from an HLA-matched donor. The cultures were incubated for an additional 16 h in the presence of [3H]thymidine (0.5 μCi/well). Cultures were harvested and incorporated radioactivity was measured by liquid scintillation counting. Control _myc_-responsive TCCs (R1E4 and R2F10) generated from leprosy patients were used 1.
Cytokine production of TCC was tested at least 10 d after the last restimulation. 105 cells were incubated in immobilized anti-CD3 mAb (OKT3) and soluble PMA in a total volume of 200 μl/well in a flat-bottomed 96-well plate. From a panel of TCCs, 5 × 104 cells were incubated with M. avium sonicate (2 μg/ml) in the presence of irradiated PBMCs (5 × 104 cell/well) from an HLA-matched donor. Control TCCs were incubated with a Mycobacterium leprae sonicate (5 μg/ml). After 24 h, 100 μl cell-free supernatants were collected, pooled, and stored at −20°C until testing. The remaining cultures were incubated for an additional 16 h in the presence of [3H]thymidine (0.5 μCi/well) to confirm stimulation. The results are expressed as mean cpm of triplicate cultures. Measurement of IFN-γ and IL-4 concentrations in the supernatants of stimulated or unstimulated TCCs was performed with a specific solid-phase sandwich ELISA for IFN-γ (Ucy-Tech; University of Utrecht), and for IL-4 (The Central Laboratory of The Netherlands Red Cross Blood Transfusion Service).
Stimulation of TCCs with IL-12 and IL-18
10 d after the last restimulation, TCCs were tested for IL-12 and IL-18 responsiveness upon activation. 5 × 104 cells were cultured in 200 μl culture medium in a flat-bottomed 96-well plate for 72 h in the presence or absence of 5 U/ml IL-12 (a gift from G. Trinchieri, Wistar Institute, Philadelphia, PA) or 2.5 ng/ml IL-12 (R&D Systems), and/or different concentrations of IL-18 (0–100 ng/ml; Medical and Biological Laboratories Co.). T cells were activated with M. avium sonicate (2 μg/ml) in the presence of irradiated PBMCs (5 × 104 cell/well) from an HLA-matched donor. Control TCCs were incubated with M. leprae sonicate (5 μg/ml). Mitogenic stimuli were soluble murine mAb to human CD28 (CLB-CD28/1), three murine mAbs to human CD2 (CLB-T11/1, CLB-T11.2/1, Hic 27), and immobilized antiCD3 mAb. mAbs to CD28 and CD2 were a gift from R.A.W. van Lier (The Central Laboratory of The Netherlands Red Cross Blood Transfusion Service, Amsterdam, The Netherlands). After 72 h, supernatants were collected, pooled, and stored at −20°C before determining IFN-γ levels. During the last 16 h, the cultures were incubated in the presence of 0.5 μCi/well [3H]thymidine to determine proliferation. The results are expressed as mean cpm of triplicate cultures.
Generation and Stimulation of PHA Blasts
PBMCs were isolated from heparinized blood by Ficoll-Amidotrizoate density gradient centrifugation. Cells (106/ml) were incubated in a 24-well plate in standard culture medium and 0.5% PHA. After 3 d, the cells were expanded in the presence of 10 U/ml IL-2. PHA blasts (106 cells/ml) on day 10 after activation were incubated in 24-well plates (Costar) with 1,000 U/ml IFN-α (Pharma Biotechnologie Hannover) for 16 h or with 25 U/ml IL-12 (Roche) for 20–24 h in the presence of 20 U/ml IL-2.
Cell Surface Staining
Cells (5 –20 × 104) were labeled with either FITC-conjugated mouse mAb to CD3 (Becton Dickinson), unlabeled purified rat anti–huIL-12Rβ1 mAb (2B10, 1 μg/ml) 29, rat anti–huIL-12β2 mAb (2B6, 1 μg/ml), mouse anti–huIL-18R (117-10C, 5 μg/ml) 30, FITC-conjugated mAb to α6-integrin (CD49f; BD PharMingen), or isotype-matched control antibodies (BD PharMingen) at 4°C for 1 h. mAbs 2B10 and 2B6 were provided by D. Presky (Department of Inflammation/Autoimmune Diseases, Hoffmann-La Roche, Nutley, NJ). The cells labeled with mAb 2B10, 2B6, or isotype control mAb were subsequently incubated with biotinylated sheep anti–rat Ig (Amersham Pharmacia Biotech) for 30 min at 4°C, followed by incubation with Avidin-FITC (1:500; Vector Laboratories) for 30 min. The cells labeled with mAb 117-10C were subsequently incubated with FITC-labeled anti–mouse IgM (Southern Biotechnology Associates Inc.) for 30 min. Cells were analyzed with a FACScan™ flow cytometer (Becton Dickinson).
Responsiveness of PHA Blasts to IL-12
PHA blasts were restimulated with irradiated allogenic adherent cells. To ∼0.25–0.5 × 106 adherent cells, 1 × 106 PHA blasts were added in 1 ml culture medium per well in a 24-well plate, and incubated in the presence of 0.5% PHA and IL-2 (10 U/ml). On day 3 after stimulation PHA blasts were collected, washed, and tested for IL-12Rβ1 and IL-12Rβ2 expression (as described above) and for responsiveness to IL-12 based on Stat4 activation.
Stat4 Activation
Cell Extracts.
Total proteins were extracted from PHA blasts (107 cells/ml) that were or were not exposed to 2.5 ng/ml IL-12 (R&D Systems) 1,000 U/ml IFN-α (Pharma Biotechnologie Hannover), or control medium for 20 min. Cells were washed with ice-cold PBS and packed cells were resuspended in 50 μl of lysis buffer (20 mM Hepes/KOH [pH 7.9], 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 5 mM dithiothreitol [DTT], 10 μg/ml aprotinin, 0.5% [vol/vol] of a saturated PMSF solution in ethanol, 20% [vol/vol] glycerol, and 1% [vol/vol] NP-40; Sigma-Aldrich). After 20 min incubation on ice, the lysate was cleared by 15 min centrifugation at 14,000 g. The supernatant was diluted with 1 vol of dilution buffer (20 mM Hepes/KOH [pH 7.9], 0.2 mM EDTA, 2 mM DTT, 0.5% [vol/vol] of a saturated PMSF solution in ethanol, 20% [vol/vol] glycerol, and 0.25% NP-40) to reduce salt concentration. The final protein concentration was determined by Bradford microassay (Bio-Rad Laboratories), using a calibrated BSA (Sigma-Aldrich) solution as a reference.
Electrophoretic Mobility Shift Assay.
All DNA binding reactions were conducted in a final volume of 20 μl. The reactions were started by adding 10 μg of total cell protein extract to a reaction mixture containing 2 μg of poly-deoxyinosinic-deoxycytidylic acid (Sigma-Aldrich), 2 μg of BSA, 2 μl of 10× binding buffer (100 mM Tris-HCl [pH 7.5], 500 mM NaCl, 75 mM EDTA, 50% glycerol, and 2 mM DTT), 0.5% (vol/vol) of a saturated PMSF solution in ethanol, 2 μl of dilution buffer (supplemented with 100 mM KCl), and ∼10,000 cpm (± 0.1 ng) of the [γ-32P] ATP–labeled double-stranded DNA oligonucleotide. For supershift experiments, binding reactions were incubated with affinity-purified rabbit anti–murine Stat4 (C-20; Santa Cruz Biotechnology, Inc.) during the last 20 min of incubation. All reactions were adjusted to the final volume of 20 μl with distilled water and incubated for 30 min at room temperature. The whole sample was then loaded on a 4% native polyacrylamide gel in 0.5× Tris-borate–EDTA buffer. After electropheresis for 2 h at room temperature at 10 V/cm, gels were dried, and separated protein–DNA complexes were visualized by autoradiography using XAR5 films (Eastman Kodak Co.).
Oligonucleotides.
The following double-stranded DNA oligonucleotides were used in electrophoretic mobility shift assay (EMSA) analyses: FcγRI (5′-AGCATGTTTCAAGGATTTGAGATGTATTTCCCAGAAAAG-3′), corresponding to the IFN-γ response region in the human FcγRI gene promoter 27, which is bound by various STAT family members, including Stat4, and a sequence (5′-CCGGCCCCGCCCATCCCCGGCCCCGCCCATCC-3′) containing the herpes simplex virus SP-1 site. All oligonucleotides were purchased from Biosource International and labeled by T4 polynucleotide kinase (Roche).
Inhibition of Mitogen-activated Protein Kinase Pathway
To assess the involvement of mitogen-activated protein kinases (MAPKs) and MAPK kinases (MEKs) in IL-12–induced IFN-γ production in IL-12Rβ1–deficient cells, _myc-_specific, mitogen-treated TCCs were incubated with IL-12 in the absence or presence of the p38 MAPK inhibitor SB203580 (Sigma-Aldrich), the mitogen-activated and extracellular signal–regulated kinase MEK1/MEK2 inhibitor PD098059 (Sigma-Aldrich) or the MEK1/MEK2 inhibitor U0126 (Promega). In brief, TCCs were stimulated with anti-CD2 and anti-CD28 mAbs (see above) for 18 h before the addition of SB203580, PD098059, or U0126 to a final concentration of 2.5, 25, and 2.5 μM, respectively. 30 min after addition of the inhibitors, TCCs were incubated for an additional period of 48 h, with or without exogenous IL-12. Supernatants were harvested and IFN-γ production was monitored by ELISA.
Results
Type 1 Cytokine Secretion Profiles (IFN-γ/IL-4) of TCCs Generated from IL-12Rβ1–deficient Patients.
Since IL-12 is known to be a key factor in Th1 cell development 6 7, T cell differentiation in IL-12Rβ1 deficiency might be anticipated to be Th2 skewed. To study this, _myc_-specific TCCs were generated from two IL-12Rβ1–deficient individuals with distinct recessive null mutations (P1 and P3), and cytokine profiles of 25 _myc_-responsive TCCs were determined after standard stimulation with anti-CD3 mAb plus PMA. All _myc_-responsive IL-12Rβ1–deficient TCCs were CD4+CD8− and showed a clear Th1 phenotype, producing significant levels of IFN-γ but low levels of IL-4 (shown in Table for 10 representative TCCs, compared with two control _myc_-responsive TCCs). Antigen-specific stimulation of these TCCs induced similar cytokine profiles, although lower levels of IFN-γ tended to be produced.
Table 1.
Cytokine Secretion Profile of Myc-responsive TCCs from IL-12Rβ1–deficient Patients
| Antigen-specific stimulation | Stimulation with anti-CD3/PMA | |||
|---|---|---|---|---|
| TCCs | IFN-γ | IL-4 | IFN-γ | IL-4 |
| Patients | ||||
| S8A5 | 226 | <7 | 6,300 | <7 |
| S3E8 | 554 | 25 | 109,915 | 54 |
| S7C4 | 649 | <25 | 13,700 | 16 |
| S8D3 | 1,119 | <25 | 6,827 | 33 |
| K2B10 | 1,063 | <25 | 26,036 | 252 |
| K8D1 | 426 | 8 | 31,400 | 4 |
| K3F12 | 365 | <7 | 49,500 | <7 |
| K8D10 | 2,129 | <7 | 26,400 | <7 |
| K4D4 | 246 | <25 | 5,563 | 36 |
| K2E9 | 4,559 | 23 | 2,900 | 154 |
| Controls | ||||
| R1E4 | 12,125 | 113 | 97,569 | 112 |
| R2F10 | 1,449 | 30 | 88,868 | 1,244 |
To exclude the influence of skewing by microenvironmental cytokines possibly present during the generation of TCCs, separate cloning experiments were performed by direct limiting dilution of PHA-stimulated PBMCs from P1 and P3. Cytokine secretion profiles of these TCCs (with unknown specificity) were determined after mitogenic stimulation. Also in these experiments TCCs with a clear Th1 phenotype were observed (data not shown).
Myc-reactive Th1 TCCs from IL-12Rβ1–deficient Patients Can Still Respond to IL-12.
Using the _myc_-responsive Th1 TCCs from genetically IL-12Rβ1–deficient patients, we next wanted to elucidate the pathway(s) involved in the remaining low IFN-γ production that was previously observed in PBMCs from these patients 9, and now also in TCCs (this study). Since IL-12 is a key factor in Th1 activation, we first determined whether IL-12Rβ1–deficient Th1 TCCs were still capable of responding to IL-12. To this end, IL-12Rβ1–deficient Th1 TCCs (n = 10) or IL-12Rβ1 wild-type (control) Th1 TCCs (n = 2) were stimulated independently with four different stimuli, in the presence or absence of exogenous IL-12, as indicated in Fig. 1. TCCs were stimulated with antigen presented by HLA-matched APCs (Fig. 1 A), or by mitogenic antibodies in the absence of any added APCs, using either a mix of two or three mAbs to CD2 (CD2d [Fig. 1B and Fig. C] or CD2t [Fig. 1 D], respectively) or a mAb to CD3 (Fig. 1 E) to which in each case costimulatory anti-CD28 mAb was added (Fig. 1B–E) 28. All experiments were repeated between two and seven times, and in each experiment TCCs were stimulated by one to four of these different stimuli (further details are given in the legend of Fig. 1).
Figure 1.
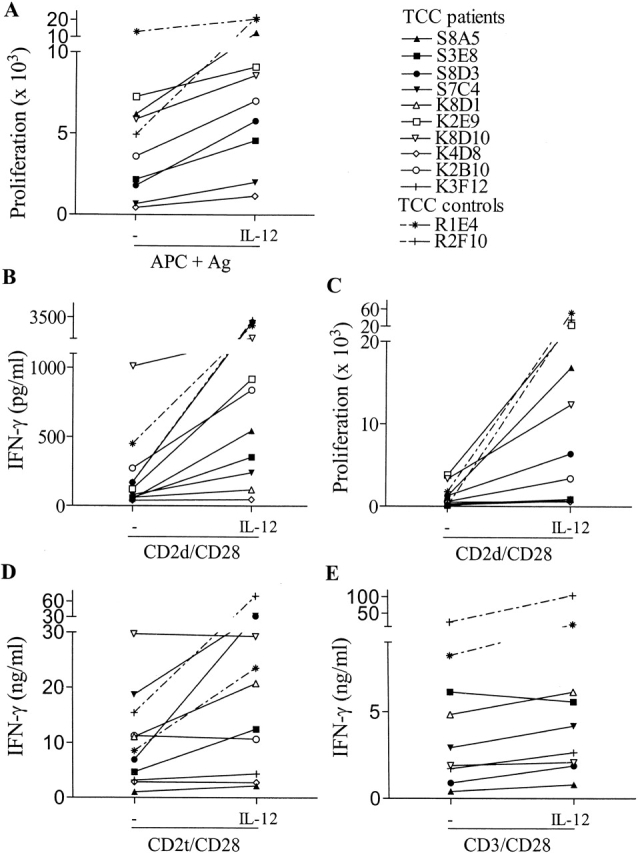
IL-12–enhanced proliferative responses and IFN-γ production by _myc_-responsive IL-12Rβ1–deficient Th1 TCCs and control TCCs upon activation. TCCs from P1 (filled symbols), P3 (open symbols), and control TCCs (symbols with dashed lines) were stimulated in the absence or presence of IL-12 with mycobacterial antigens and APCs from HLA-matched donors (A, showing proliferative responses), with two mAbs to CD2 and one mAb to CD28 (B and C, showing IFN-γ production and proliferative responses, respectively), with three mAbs to CD2 and one mAb to CD28 (D, showing IFN-γ production) and with one mAb to CD3 and one mAb to CD28 (E, showing IFN-γ production). Proliferation experiments shown in A and C were always performed in triplicate. The SD in these experiments did not exceed 20%. All TCCs were tested in two to seven independent experiments, and in each experiment, they were stimulated by at least one to four of the above described stimuli. TCC S8A5 was tested in six independent experiments, TCC S3E8 in five, TCC S8D3 in three, TCC S7C4 in three, TCC K8D1 in four, TCC K2E9 in two, TCC K8D10 in three, TCC K4D8 in two, TCC K2B10 in three, and TCC K3F12 in two independent experiments. The control TCCs R1E4 and R2F10 were tested in five and three independent experiments, respectively. The total number of stimulation assays in which each of the different TCCs was tested ranged from 7 to 19 (mean: 12.3).
As shown in Fig. 1, IL-12 was found to induce a significant response (defined as >50% increase in proliferation or IFN-γ production) in 8 out of the 10 tested IL-12Rβ1–deficient TCCs. The observed IL-12–induced responses were consistent and reproducible, and representative experiments are shown. The IL-12–mediated enhancement of proliferation and IFN-γ production in the case of stimulation with CD2d/CD28 (Fig. 1B and Fig. C) or CD2t/CD28 (Fig. 1 D) indicated a direct effect of IL-12 on IL-12Rβ1–deficient T cells in the absence of accessory cells. Although in some cases these stimuli already induced relatively high levels of IFN-γ secretion, this could be further augmented by IL-12. IL-12 also clearly enhanced the response of several IL-12Rβ1–deficient clones upon antigen-specific stimulation (Fig. 1 A) or activation by CD3/CD28 mAbs (Fig. 1 E). 2 of the 10 IL-12Rβ1–deficient TCCs (K4D8 and K3F12) repeatedly did not respond to IL-12 upon any of the stimulation modes used (Fig. 1A–E), whereas the two IL-12Rβ1 wild-type control TCCs (R1E4 and R2F10) responded strongly to IL-12 as expected (Fig. 1A–E).
Taken together, the results show that IL-12Rβ1–deficient patients are capable of developing Th1 T cells, and that the majority of the Th1 TCCs are capable of responding to IL-12 in a specific and consistent fashion by enhanced IFN-γ production and/or proliferation. Thus, these results reveal an IL-12Rβ1–independent pathway of IL-12 responsiveness in human T cells.
The IL-12–dependent Response of Myc-reactive, IL-12Rβ1–deficient Th1 TCCs Is Further Augmented by IL-18.
Since IL-18 is known to enhance IL-12–induced IFN-γ production in healthy individuals 25, we investigated the effect of IL-18 on IL-12–induced IFN-γ production also in IL-12Rβ1–deficient Th1 TCCs (n = 7). Representative results of one out of three independent experiments are shown in Fig. 2. In two IL-12Rβ1–deficient TCCs (S8A5 and K2E9), IL-18 synergized with IL-12 in a dose-dependent fashion by further increasing proliferation as well as IFN-γ production. A similar synergy was seen in control TCCs (R1E4) although the levels of increased proliferation and IFN-γ production were higher. In a third IL-12Rβ1–deficient TCC (K8D10), the response to IL-12 and IL-18 was additive, and this TCC already responded to IL-18 in the absence of IL-12. Thus, the IL-12–induced proliferation and IFN-γ production of IL-12Rβ1–deficient T cells can be further augmented by IL-18.
Figure 2.
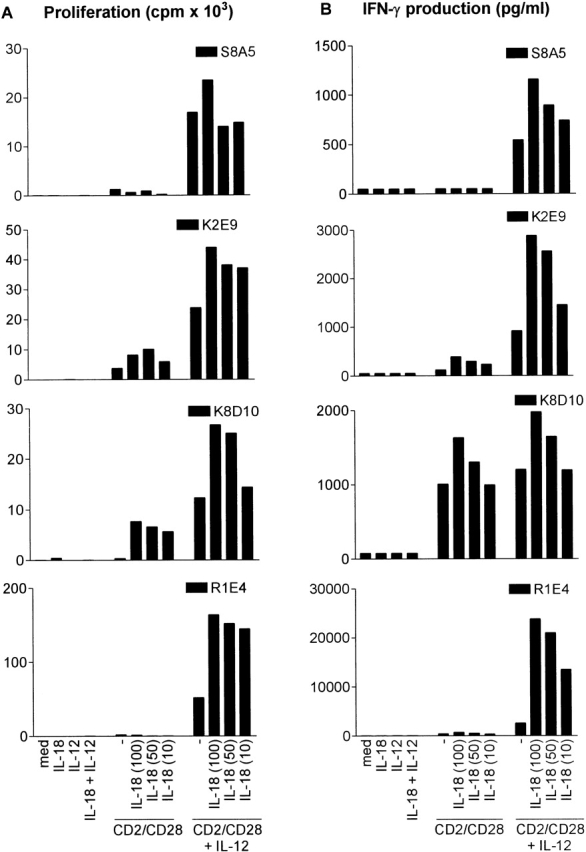
IL-18 further enhances IL-12–induced proliferative responses and IFN-γ production in _myc_-responsive IL-12Rβ1–deficient TCCs and control TCCs that were mitogenically stimulated. Proliferative responses (A) and IFN-γ production (B) of TCCs from P1 (S8A5), P3 (K2E9, K8D10), and control TCCs (R1E4, R2F10) were determined upon stimulation with two mAbs to CD2 and one mAb to CD28 in the absence or presence of IL-12 and/or IL-18 for 72 h and subsequent 16-h incubation with [3H]thymidine. R1E4 is representative for the results with R2F10. Proliferation experiments were always performed in triplicate, the SD of which did not exceed 20%.
Regulation of T Cell IL-12Rβ2 Expression in the Absence of IL-12Rβ1.
To further delineate this unexpected IL-12 responsiveness to IL-12Rβ1 deficiency, we determined surface expression of the IL-12Rβ2 signaling chain on IL-12Rβ1–deficient and control TCCs. At the same time, we studied the expression of the IL-18R that, in mice, has been suggested to be selectively expressed on Th1 cells 29. As can be seen in Fig. 3, PHA-stimulated IL-12Rβ1–deficient TCCs express IL-12Rβ2 and IL-18R. The level of expression differed among different TCCs, which may correlate with the activation state 23 and the capacity of the TCCs to respond to IL-12 and/or IL-18 as described above. Control TCCs expressed both receptors.
Figure 3.
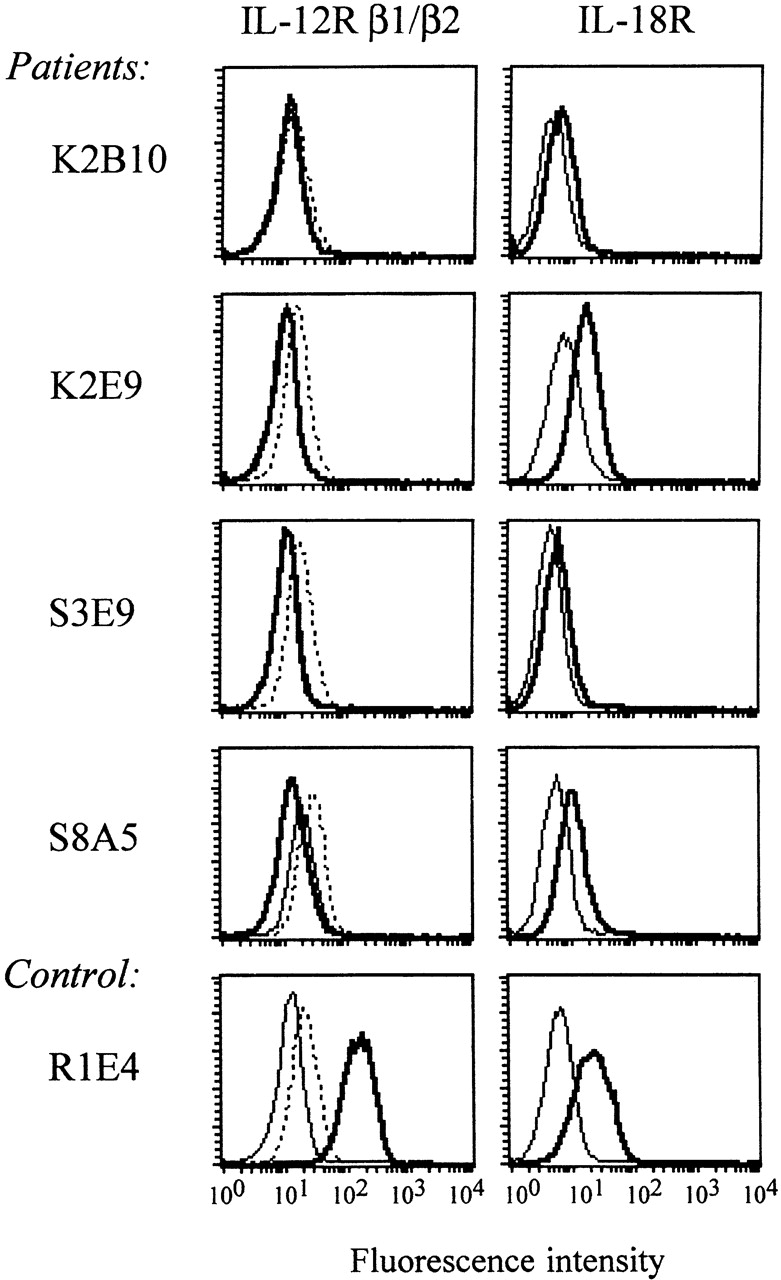
IL-12Rβ2 and IL-18R surface expression on mitogenically stimulated _myc_-responsive IL-12Rb1–deficient TCCs and control TCCs. TCCs from P1 (S3E9, S8A5), P3 (K2E9, K2B10), and control TCCs (R1E4) were stimulated with PHA and adherent-irradiated feeder cells for 72 h. Cell surface expression of IL-12Rβ1 (left, thick lines), IL-12Rβ2 (left, dotted lines) and IL-18R (right, thick lines) were examined by FACScan™ analysis. Isotype controls are presented by thin lines.
To investigate the dynamics of IL-12Rβ2 expression, PHA-activated PBMCs (PHA blasts) from four IL-12Rβ1–deficient patients and four healthy controls were assayed at different time points after stimulation (Fig. 4). No IL-12Rβ1 expression could be detected on patient-derived PHA blasts whereas control PHA blasts showed maximal levels of IL-12Rβ1 expression on day 3 after activation, declining thereafter to baseline expression. In line with the results in TCCs, PHA blasts from both patients and controls showed equally strong IL-12Rβ2 expression, which was highest on day 3 after activation and declined to low or undetectable levels on day 11 after activation. On day 3 after activation equal levels of IL-18R expression among patients and controls were seen (data not shown).
Figure 4.

Representative examples of IL-12Rβ2 surface expression on PHA-stimulated PBMCs from IL-12Rβ1–deficient patients and healthy controls. PBMCs from IL-12Rβ1–deficient patients (n = 4) and controls (n = 4) were stimulated with PHA for 72 h, followed by subsequent incubation in the presence of IL-2. Cell surface expression of IL-12Rβ1 (thick lines) and IL-12Rβ2 (dotted lines) were examined by FACScan™ analysis at different time points after activation. Isotype controls are presented by thin lines.
It has previously been reported that culturing T cells with IL-12 or IFN-α upregulates surface expression of IL-12Rβ2 receptor in the absence of TCR stimulation 22. Other molecules that have recently been described to be upregulated by IL-12 are IL-18R and integrin α6 30 31. To study the effect of IL-12 or IFN-α on IL-12Rβ2 expression in IL-12Rβ1 deficiency, PHA blasts of patients and controls (10 d after activation) were incubated with IL-12 or IFN-α for 20 and 16 h, respectively 23. Whereas IFN-α upregulated the surface expression of IL-12Rβ2 both in patients and in controls, exposure to IL-12 only induced the expression of IL-12Rβ2 on cells from healthy controls, but not from patients (Fig. 5). Similarly, IL-12 induced upregulation of IL-18R and integrin α6 expression in controls, but not in patients (Fig. 6).
Figure 5.

Representative examples of IL-12 or IFN-α upregulated expression of IL-12Rβ2 surface expression on PHA-stimulated PBMCs from IL-12Rβ1–deficient patients and healthy controls. PBMCs from IL-12Rβ1–deficient patients (n = 4) and controls (n = 3) were stimulated with PHA for 72 h, followed by subsequent incubation in the presence of IL-2. At day 10 after activation, cells were incubated with IL-2 in the absence or presence of IL-12 or IFN-α for another 20 or 16 h, respectively. Surface expression of IL-12Rβ1 (thick lines) and IL-12Rβ2 (dotted lines) was examined by FACScan™. Isotype controls are presented by thin lines.
Figure 6.
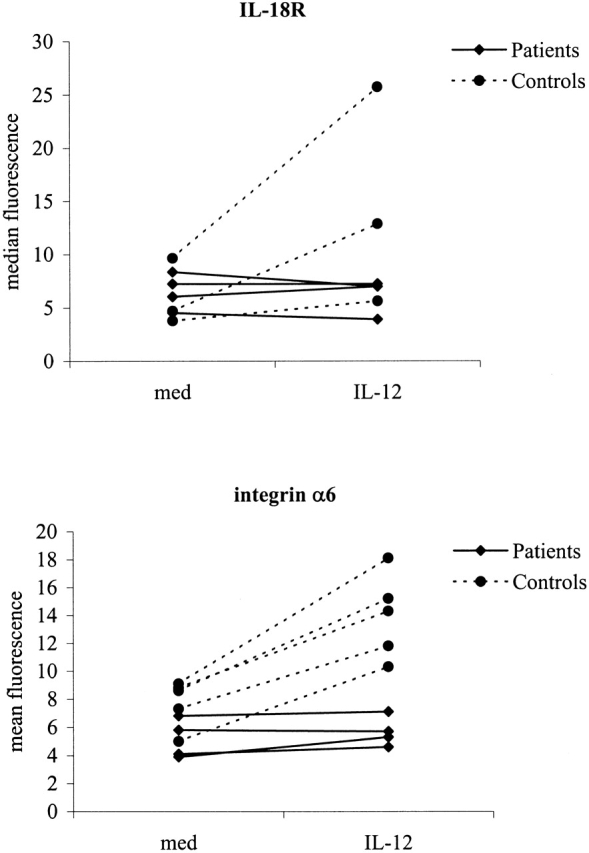
IL-12 does not upregulate the surface expression of IL-18R and integrin α6 on PHA-stimulated PBMCs from IL-12Rβ1–deficient patients compared with healthy controls. PBMCs from IL-12Rβ1–deficient patients (n = 4) and controls were stimulated with PHA for 72 h, followed by subsequent incubation in the presence of IL-2. At day 10 after activation, cells were incubated with IL-2 in the absence or presence of IL-12 for another 20 h. Surface expression of IL-18R and integrin α6 were examined by FACScan™ and represented as mean fluorescence.
Taken together these results demonstrate that (a) human IL-12Rβ2 expression can occur normally in the absence of IL-12Rβ1 and can be upregulated by IFN-α independent from IL-12Rβ1; and (b) IL-12Rβ2, IL-18R, and integrin α6 expression can only be upregulated by IL-12 in the presence of IL-12Rβ1, suggesting that IL-12Rβ2 expression alone is not sufficient to confer full IL-12 responsiveness to human T cells.
IL-12 Fails to Induce Stat4-containing Complexes in Human IL-12Rβ1 Deficiency.
Since the IL-12Rβ2 chain is essential in phosphorylating Stat4, a key signal transduction and activator of transcription in IL-12–induced T cell responses 17 19 20 21 22, we investigated whether IL-12 signaling in IL-12Rβ2–expressing, IL-12Rβ1–deficient T cells was involved in Stat4 activation. Total cell extracts of 3-d PHA-stimulated blasts of IL-12Rβ1–deficient patients (n = 4) and healthy controls (n = 2) that expressed optimum levels of IL-12Rβ2 (see above) were examined by EMSA for formation of Stat4-containing complexes and binding to the oligonucleotide probe FcγR1, which contains high affinity binding sites for Stat4. As shown in Fig. 7 A, IL-12 induced binding of the FcγR1 probe in the IL-12–responsive PHA blasts from controls (lanes 11 and 14), but no activity binding was found in cells from patients (lanes 2, 5, and 8). As a positive control, IFN-α induced similar levels of Stat4 activation in samples from patients and controls (lanes 3, 6, 9, 12, and 15). Unstimulated samples were always negative (lanes 1, 4, 7, 10, and 13). Competition analysis with unlabeled FcγR1 oligonucleotide (Fig. 7 B, lanes 1–3) indicated the specificity of this DNA–protein interaction, and this was further confirmed in supershift experiments showing that Stat4-specific antiserum interfered with the formation of IL-12–induced (Fig. 7 C, lane 8) as well as IFN-α–induced (Fig. 7 C, lanes 6 and 10) protein–DNA complexes. As an additional control for the quality of total cell extracts, no differences were found between patient versus controls and cytokine-stimulated versus unstimulated samples when DNA binding activity of the constitutively expressed transcription factor SP-1 to a specific SP-1 binding oligonucleotide was examined (data not shown).
Figure 7.
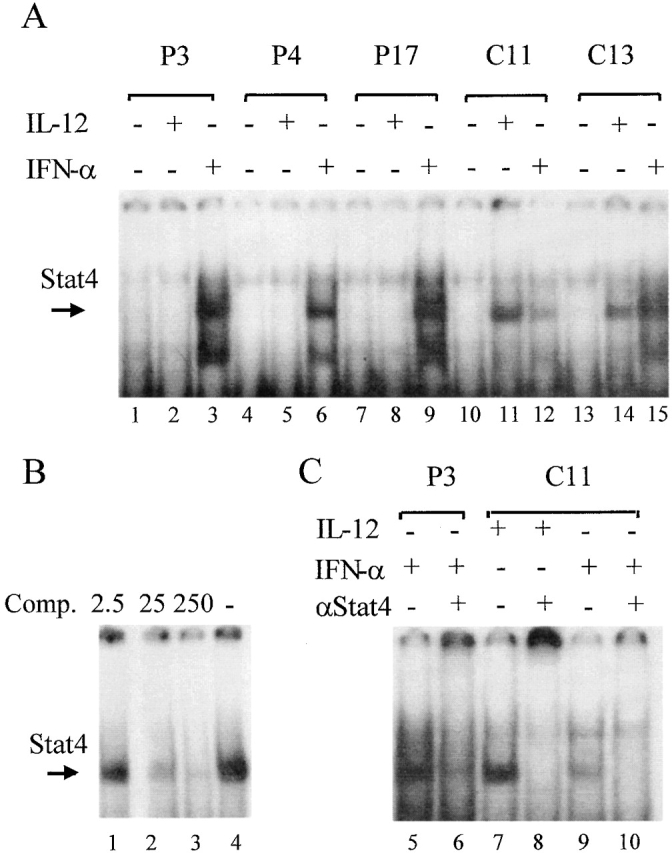
IL-12 fails to induce DNA binding of Stat4-containing complexes in IL-12Rβ1–deficient patients. PHA-activated PBMCs were (+) or were not (−) stimulated with IL-12 or IFN-α for 20 min. (A) Total cell extracts were examined by EMSA for formation of Stat4-containing complexes and binding to a labeled FcγR1 oligonucleotide. The arrow indicates the DNA–Stat4 complexes. (B) Competition analysis (Comp.) using unlabeled FcγR1 oligonucleotide (2.5, 5, or 250 times the amount of labeled FcγR1 oligonucleotide) indicated the specificity of this DNA–Stat4 interaction. (C) Supershift experiments were performed using Stat4-specific antiserum that interfered with the formation of IL-12– and IFN-α–induced DNA–Stat4 complex.
Involvement of MAPKs in IL-12 Responsiveness in IL-12Rβ1–deficient T Cells.
To assess the possible involvement of MAPK signaling in the residual, Stat4-independent IL-12 responsiveness in IL-12Rβ1-deficient T cells, we measured IL-12–induced IFN-γ production in three TCCs that were stimulated with anti-CD2/anti-CD28, either in the presence or absence of the following inhibitors: the p38 stress MAPK inhibitor SB203580, the MEK1/2 inhibitor PD098059, or the MEK1/2 inhibitor U0126. Whereas SB203580 blocks p38 MAPK–mediated stress responses, PD098059 and U0126 prohibit signaling via the extracellular signal-regulated kinase (ERK)/MAPK-mediated pathway by interfering with the upstream MAPK kinases MEK1/MEK2. Whereas U0126 inhibits the activated form of MEK directly, PD098059 merely prohibits activation of MEK by binding to its inactive form 32 33. As shown in Fig. 8, the IL-12–mediated enhancement of IFN-γ production was significantly reduced (∼50%) in the presence of SB203580 and U0126, but not PD098059, in the case of TCCs S8D3 and S8A5. In clone S3E8 there was only modest inhibition by SB203580 (20%), whereas U0126 again diminished IL-12–dependent IFN-γ production over 50%. No effects of these inhibitors were observed on cell viability, ruling out possibly toxic effects of these components. Thus, these results imply a significant role for the p38 MAPK, and the MEK1/2–p42 MAPK/ERK pathways in residual IL-12 responsiveness and IFN-γ production in IL-12Rβ1–deficient human T cells.
Figure 8.
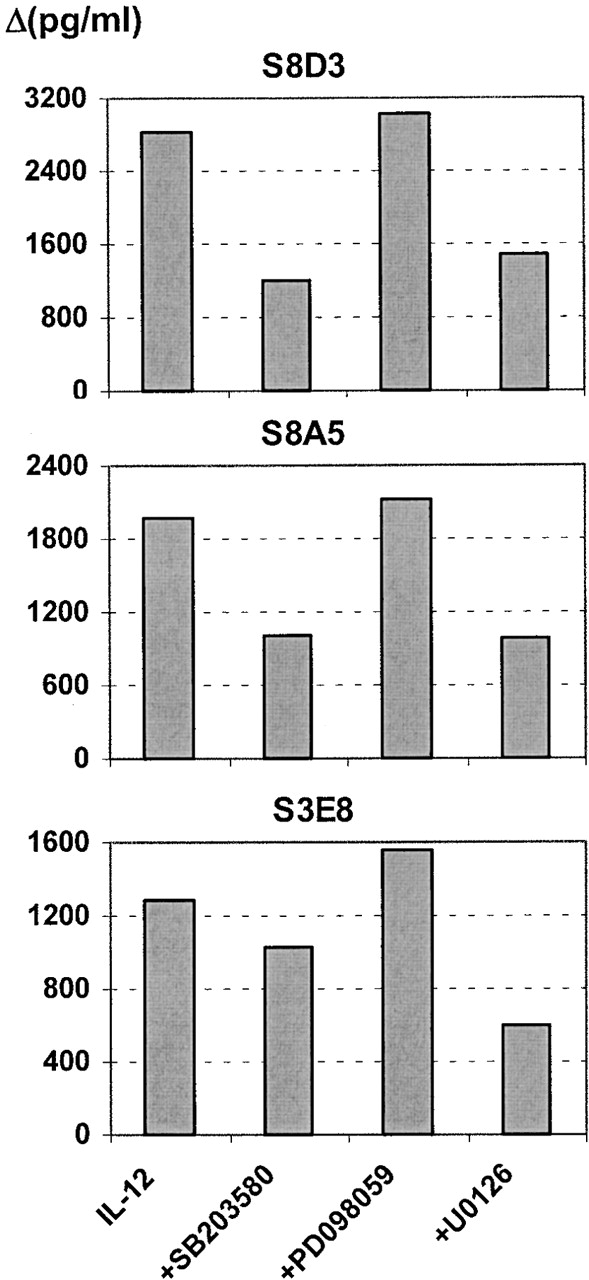
Inhibitors of MAPK signaling SB203580 and U0126 inhibit IL-12–enhanced IFN-γ production by IL-12Rβ1–deficient cells. _Myc_-specific TCCs were stimulated with anti-CD2/anti-CD28 for 18 h. Subsequently, cells were pretreated for 30 min with either the p38 MAPK–specific inhibitor SB203580, the MEK1/MEK2 (inactive form) inhibitor PD098059, or the MEK1/2 (active form) inhibitor U0126, before addition of IL-12. Control cultures were treated identically with mitogenic stimuli and inhibitors, but without addition of IL-12. After 48 h, IFN-γ secretion was measured by ELISA. Shown are control-corrected values of IFN-γ (in Δpg/ml), representing the effect of inhibiting MAPK signaling on IL-12–enhanced IFN-γ secretion in IL-12Rβ1 deficiency.
Discussion
The essential role of type 1 cytokines in protection against intracellular pathogens is illustrated by the extreme susceptibility of humans that are genetically deficient for type 1 cytokines (IL-12p40) or type 1 cytokine receptors (IL-12Rβ1, IFN-γRI, IFN-γRII) to severe infections with otherwise poorly pathogenic mycobacteria and Salmonella species 9 10 11. The different groups of genetic deficiencies, however, are associated with important differences in clinical appearances and severity of the disease outcome; while patients with complete IFN-γR deficiencies mostly develop fatal and treatment-resistant infections, patients with IL-12Rβ1 deficiency often develop less severe and mostly curable infections 11. This suggests that in IL-12Rβ1 deficiency, low grade (type 1) immunity to intracellular pathogens remains, independent of IL-12Rβ1. We have hypothesized that this may be due to the residual low level of IFN-γ production in these patients 9 11, but the pathway that controls this in the absence of functionally competent IL-12R complexes remains obscure.
Since IL-12 is known to be a key factor in Th1 cell development 6 7, IL-12Rβ1–deficient individuals might be anticipated to be skewed towards a Th2 phenotype. Surprisingly, all _myc_-responsive TCCs generated from IL-12Rβ1–deficient patients displayed a clear Th1 phenotype based on cytokines secretion profiles (high IFN-γ and low IL-4). Thus, they do not differ qualitatively from the IL-12R wild-type Th1 cytokine secretion profiles that were described previously 1 3 4, although their quantitative production of IFN-γ is clearly diminished. Perhaps most surprisingly, IL-12Rβ1–deficient Th1 TCCs still responded to IL-12 when activated, leading to both cellular proliferation and IFN-γ production. This IL-12 responsiveness of IL-12Rβ1−/− TCCs could be further augmented by IL-18. Although the biological role of IL-18 in humans is still largely unknown, a recent study showed a role for IL-18 in type 1 immunity against M. leprae, suggesting the involvement of IL-18 in mycobacterial immunity 34. Our data thus implicate that IL-12, either alone or together with IL-18, can regulate IL-12Rβ1–independent, low grade IFN-γ production by Th1 T cells. This low level of IFN-γ production presumably is necessary and sufficient to prohibit a fatal outcome of mycobacterial infectious disease in IL-12Rβ1–deficient patients.
This residual IL-12 response in IL-12Rβ1 deficiency compelled us to examine the expression and function of the IL-12Rβ2 signaling chain in the absence of the IL-12Rβ1. Previously, IL-12Rβ2 was suggested to bind IL-12 in the absence of IL-12Rβ1 14 15 16. In support of this suggestion, Ba/F3 cells expressing the huIL-12Rβ2 subunit alone bound IL-12 with low affinity and proliferated in response to IL-12 14 35. In these studies, however, a role for endogenous IL-12Rβ1 could not be excluded. To our knowledge, our results are the first to show that in humans IL-12Rβ2 can be expressed at the cell surface in the absence of IL-12Rβ1 and that this expression can be upregulated by IFN-α. The expression of IL-12Rβ2 as well as IL-18R on IL-12Rβ1−/− TCCs is in line with their observed Th1 phenotype, since both receptors have been described to be selectively expressed by Th1, but not Th2 T cells 23 29.
Although IL-12Rβ2–expressing IL-12Rβ1−/− T cells responded to IL-12 in the presence of antigens or mitogenic stimuli, IL-12 alone failed to induce upregulation of cell surface expression of IL-12Rβ2, IL-18R, and integrin α6, and failed to induce Stat4 activation, whereas in control cells all these markers of IL-12R signaling were present. Since several Stat4 binding sites have been identified on the IL-12Rβ2 promotor 36, the absence of IL-12–induced Stat4 activation in our patients may directly account for the lack of IL-12–induced upregulation of IL-12Rβ2. A similar explanation may account for the lack of IL-12–induced upregulation of IL-18R or integrin α6 expression. Although IL-12 activates multiple Stats (Stat1, Stat3, Stat4, and Stat5 [17, 19, 37]), the essential role of Stat4 in IL-12/IL-12R–induced signal transduction was demonstrated in Stat4 knockout mice that showed strongly reduced IL-12–dependent IFN-γ production and cellular proliferative responses in T cells and NK cells 20 21. Furthermore, the selective expression of the IL-12Rβ2 chain on human Th1, but not Th2 cells has been shown to correlate with the capacity of IL-12 to induce activation of Stat4 in Th1, but not Th2 cells 22 24. Here, however, we clearly show that in humans IL-12Rβ1 expression is also necessary for IL-12–dependent Stat4 activation. Taken together, our results show that IL-12 acts as a partial agonist in IL-12Rβ1 deficiency, inducing IFN-γ production and proliferation by activated T cells, whereas IL-12 alone failed to activate Stat4 and to upregulate expression of IL-12Rβ2, IL-18R, or integrin α6 to detectable levels.
Several lines of evidence have pointed to IL-12–triggered intracellular signaling pathways that are independent of Stat4. First, it has been shown that the cytoplasmic region of huIL-12Rβ2 could associate with JAK2, and that IL-12–induced JAK2 activation correlated with Stat5 phosphorylation and cellular proliferation, whereas huIL-12Rβ1 interacted directly with TYK2, and IL-12–induced activation of TYK2 correlated with Stat4 phosphorylation and IFN-γ production 35 38. IL-12 responsiveness may, however, also involve the activation of MAPKs that have also been implicated in T cell receptor CD3/CD28–mediated signaling. A 42–44-kD MAPK was found to be activated by IL-12 in mitogen-stimulated human T cells 39. Interestingly, a more recent study showed that a p38 MAPK was activated by the synergistic action of IL-12 and IL-2, and subsequently induced Stat1 and Stat3 phosphorylation on serine residues, which contributed to the IL-12–induced functional effects 40. Moreover, inhibition of p38 MAPK led to reduced IFN-γ production in mouse Th1 cells 41. In agreement with these recent reports, we here show that inhibiting p38 MAPK by SB203580, as well as inhibiting MEK1/2 by U0126, both significantly reduced IL-12–dependent IFN-γ production in IL-12Rβ1–deficient T cells. In contrast, a second MEK1/2 inhibitor, PD098059, had no inhibitory effect up to a concentration of 25 μM. Since PD098059 blocks the activation of MEK, but does not prohibit signaling by already activated MEK, the failure of PD098059 to inhibit IL-12–induced IFN-γ production in IL-12R–deficient cells may suggest that preactivation of the TCC with anti-CD2/anti-CD28 leads to activation of the MEK/ERK signaling pathway, which subsequently integrates with signaling via p28 MAPK in response to IL-12 to trigger IFN-γ production. However, future studies are required to unravel these signal transduction pathways and the putative cross-talk between various pathways involved in IL-12 responsiveness in IL-12Rβ1–deficient cells.
Finally, it cannot be ruled out that other cell surface molecules may associate with IL-12Rβ2 and engage in binding IL-12 and regulating IL-12 responsiveness. In one study an 85-kD protein was found to be associated with the IL-12Rβ1 subunit and suggested to represent a hitherto unidentified third component of the IL-12R complex 42.
The finding that all _myc_-responsive IL-12Rβ1–deficient TCCs appeared to be Th1 suggests significant Th1 cell development in IL-12Rβ1 deficiency. It is tempting to speculate that the residual IL-12 responsiveness shown in this paper may play a role in Th1 differentiation in these patients, although we cannot exclude that this is regulated by IL-12–independent mechanisms. Studies in IL-12p40−/− mice revealed polarized Th1 cell development in alloantigen responses or during viral infections 43 44 45, suggesting IL-12–independent pathways of Th1 induction. In humans, IL-12p40 protein deficiency was also found to be selectively associated with curable, but not fatal, disseminating M. bovis BCG and S. enteritidis infection 46. One alternative IL-12–independent Th1 sensitizing pathway has been shown to involve IFN-α. IFN-α, in humans, can directly induce Th1 development and induces IFN-γ production independently of IL-12 47. IFN-α was also found to synergize with IL-18 to induce IFN-γ production in human T cells 48. Here, we report normal responsiveness to IFN-α in IL-12Rβ1–deficient patients based on upregulated IL-12Rβ2 expression and Stat4 activation.
In conclusion, in this study we show that IL-12Rβ1−/− T cells can still develop into Th1 cells and can partially respond to IL-12 by cellular proliferation and IFN-γ production. Although IL-12Rβ2 can be expressed normally in the absence of IL-12Rβ1, this was most likely insufficient to confer IL-12 responsiveness to these cells, as judged by the lack of upregulation of Th1 cell surface markers and Stat4 activation. These results reveal a novel IL-12Rβ1–independent pathway of (residual) type 1 immunity in IL-12Rβ1 deficiency that may involve MAPK pathways. Thus, human IL-12Rβ1 deficiency provides a unique model to uncover novel intracellular signaling pathways and mechanisms involved in residual IFN-γ production and type 1 immunity.
Acknowledgments
These studies were supported by the European Commission, the Netherlands Leprosy Foundation (NLR), the Netherlands Organisation of Scientific Research (NWO), and the Royal Netherlands Academy of Arts and Sciences (KNAW).
Footnotes
Abbreviations used in this paper: DTT, dithiothreitol; EMSA, electrophoretic mobility shift assay; ERK, extracellular signal–regulated kinase; JAK, Janus kinase; MAP, mitogen-activated protein; MAPK, MAP kinase; MEK, MAPK kinase; myc, mycobacterium; Stat, signal transducer and activator of transcription; TCC, T cell clone.
References
- Haanen J., de Waal Malefijt R., de Vries R.R.P., Spits H. Selection of a human T helper type 1–like T cell subset by Mycobacterium leprae antigens. J. Exp. Med. 1991;174:583–592. doi: 10.1084/jem.174.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapsenberg M.L., Wierenga E.A., Bos J.D., Jansen H.M. Functional subsets of allergen-reactive human CD4+ T lymphocytes. Immunol. Today. 1991;12:392–395. doi: 10.1016/0167-5699(91)90137-I. [DOI] [PubMed] [Google Scholar]
- Parronchi P., Macchia D., Piccinni M.-P., Bisswas P., Simonelli C., Maggi E., Ricci M., Ansari A.A., Romagnani S. Allergen- and bacterial antigen-specific T cell clones established from atopic donors show a different profile of cytokine production. Proc. Natl. Acad. Sci. USA. 1991;88:4538–4542. doi: 10.1073/pnas.88.10.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen C.E., Wierenga E.A., Buffing A.A.M., Chand M.A., Faber W.R., Das P.K. Reversal reaction in borderline leprosy is associated with a polarized shift to type 1-like Mycobacterium leprae T cell reactivity in lesional skina follow-up study. J. Immunol. 1997;159:4474–4483. [PubMed] [Google Scholar]
- Abbas A.K., Murphy K.M., Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- Hsieh C.-S., Macatonia S.E., Tripp C.S., Wolf S.F., O'Garra A., Murphy K.M. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;269:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin 12a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- Swain S.L., Weinberg A.D., English M., Huston G. IL-4 directs the development of Th2-like helper effectors. J. Immunol. 1990;145:3796–3806. [PubMed] [Google Scholar]
- De Jong R., Altare F., Haagen I.-A., Elferink D.G., de Boer T., van Breda Vriesman P.J.C., Kabel P.J., Draaisma J.M.T., van Dissel J.T., Kroon F.P. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 1998;280:1435–1438. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- Altare F., Durandy A., Lammas D., Emile J.-F., Lamhamedi S., Le Deist F., Drysdale P., Jouanquy E., Döffinger R., Bernaudin F. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science. 1998;280:1432–1435. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- Ottenhoff T.H.M., Kumararatne D., Casanova J.-L. Novel human immunodeficiencies reveal the essential role of type-1 cytokines in immunity to intracellular bacteria. Immunol. Today. 1998;19:491–494. doi: 10.1016/s0167-5699(98)01321-8. [DOI] [PubMed] [Google Scholar]
- Newport M.J., Huxley C.M., Huston S., Hawrylowicz C.M., Oostra B.A., Williamson R., Levin M. A mutation in the interferon-γ-receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med. 1996;335:1941–1949. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- Dorman S.E., Holland S.M. Mutation in the signal-transducing chain of the interferon-γ receptor and susceptibility to mycobacterial infection. J. Clin. Invest. 1998;101:2364–2369. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua A.O., Chizzonite R., Desai B.B., Truitt T.P., Nunes P., Minetti L.J., Warrier R.R., Presky D.H., Levine J.F., Gately M.K., Gubler U. Expression cloning of a human IL-12 receptor component. A new member of the cytokine receptor superfamily with strong homology to pg130. J. Immunol. 1994;153:128–136. [PubMed] [Google Scholar]
- Presky D.H., Yang H., Minetti L.J., Chua A.O., Nabavi N., Wu C.Y., Gately M.K., Gubler U. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor units. Proc. Natl. Acad. Sci. USA. 1996;93:14002–14007. doi: 10.1073/pnas.93.24.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A.S., Gubler U., Presky D.H., Magram J. Structural and functional aspects of IL-12 receptor complex. Chem. Immunol. 1997;68:23–37. doi: 10.1159/000058692. [DOI] [PubMed] [Google Scholar]
- Bacon C.M., Petricoin E.F., III, Ortaldo J.R., Rees R.C., Larner A.C., Johnston J.A., O'Shea J.J. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc. Natl. Acad. Sci. USA. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon C.M., McVicar D.W., Ortaldo J.R., Rees R.C., O'Shea J.J., Johnston J.A. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2differential use of Janus family tyrosine kinases by IL-2 and IL-12. J. Exp. Med. 1995;181:399–404. doi: 10.1084/jem.181.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo S.J., Jacobson N.G., Dighe A.S., Gubler U., Murphy K.M. Developmental commitment to the Th2 lineage by extinction of IL-12 signalling. Immunity. 1995;2:665–675. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- Thierfelder W.E., van Deursen J.M., Yamamoto K., Tripp R.A., Sarawar S.R., Carson R.T., Sangster M.Y., Vignali D.A.A., Doherty P.C., Grosvels G.C., Ihle J.N. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- Kaplan M.H., Sun Y.-L., Hoey T., Grusby M.J. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Rogge L., Barberis-Maino L., Biffi M., Passini N., Presky D.H., Gubler U., Sinigaglia F. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J. Exp. Med. 1997;185:825–832. doi: 10.1084/jem.185.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge L., Papi A., Presky D.H., Biffi M., Minetti L.J., Miotto D., Agostini C., Semenzato G., Fabbri L.M., Sinigaglia F. Antibodies to the interleukin 12 receptor β2 chain mark human Th1 but not Th2 cells in vitro and in vivo . J. Immunol. 1999;162:3926–3932. [PubMed] [Google Scholar]
- Hilkens C.M.U., Messer G., Tesselaar K., van Rietschoten A.G.I., Kapsenberg M.L., Wierenga E.A. Lack of IL-12 signalling in human allergen-specific Th2 cells. J. Immunol. 1996;157:4316–4321. [PubMed] [Google Scholar]
- Micallef M.J., Ohtsuki T., Kohno K., Tanabe F., Ushio S., Namba M., Tanimoto T., Torigoe K., Fujii M., Ikeda M. Interferon-γ-inducing factor enhances T helper 1 cytokine production by stimulated human T cellssynergism with interleukin-12 for interferon-γ production. Eur. J. Immunol. 1996;26:1647–1651. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- Ottenhoff T.H.M., Klatser P.R., Ivanyi J., Elferink D.G., de Wit M.Y., de Vries R.R. Mycobacterium leprae-specific protein antigens defined by clones human helper T cells. Nature. 1986;319:66–68. doi: 10.1038/319066a0. [DOI] [PubMed] [Google Scholar]
- Pearse R.N., Feinman R., Ravetch J.V. Characterization of the promotor of the human gene encoding the high affinity IgG receptortranscription induced by γ-interferon is mediated through common DNA response elements. Proc. Natl. Acad. Sci. USA. 1991;88:11305–11309. doi: 10.1073/pnas.88.24.11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lier R.A.W., Brouwer M., Areden L.A. Signals involved in T cell activation. T cell proliferation induced through the synergistic action of ant-CD28 and anti-CD2 monoclonal antibodies. Eur. J. Immunol. 1988;1988. 18:167–172. doi: 10.1002/eji.1830180125. [DOI] [PubMed] [Google Scholar]
- Xu D., Chan W.L., Leung B.P., Hunter D., Schulz K., Carter R.W., McInnes I.B., Robinson J.H., Liew F.Y. Selective expression and function of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J. Exp. Med. 1998;188:1485–1492. doi: 10.1084/jem.188.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunikata T., Torigoe K., Ushio S., Okura T., Ushio C., Yamauchi H., Ikeda M., Ikegami H., Kurimoto M. Constitutive and induced IL-18 receptor expression by various peripheral blood cell subsets as determined by anti-huIl-18R monoclonal antibody. Cell. Immunol. 1998;189:135–143. doi: 10.1006/cimm.1998.1376. [DOI] [PubMed] [Google Scholar]
- Colantonio L., Iellem A., Clissi B., Pardi R., Rogge L., Sinigaglia F., D'Ambrosio D. Up-regulation of integrin α6/β1 and chemokine receptor CCR1 by IL-12 promotes the migration of human type 1 helper T cells. Blood. 1999;94:2981–2989. [PubMed] [Google Scholar]
- Dudley D.T., Pang L., Decker S.J., Bridges A.J., Saltiel A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favata M.F., Horiuchi K.Y., Manos E.J., Daulerio A.J., Stradley D.A., Feeser W.S., van Dyk D.E., Pitts W.J., Earl R.A., Hobbs F. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Garcia V.E., Uyemura K., Siegling P.A., Ochoa M.T., Morita C.T., Okamura H., Kurimoto M., Rea T.H., Modlin R.L. IL-18 promotes type 1 cytokine production from NK cells and T cells in human intracellular infection. J. Immunol. 1999;162:6114–6121. [PubMed] [Google Scholar]
- Zou J., Presky D.H., Wu C.-Y., Gubler U. Differential associations between the cytoplasmic regions of the interleukin-12 receptor subunits β1 and β2 and JAK kinases. J. Biol. Chem. 1997;272:6073–6077. doi: 10.1074/jbc.272.9.6073. [DOI] [PubMed] [Google Scholar]
- Sinigaglia F., D'Ambrosio D., Panina-Bordignon P., Rogge L. Regulation of the IL-12/IL-12R axisa critical step in T helper cell differentiation and effector function. Immunol. Rev. 1999;170:65–72. doi: 10.1111/j.1600-065x.1999.tb01329.x. [DOI] [PubMed] [Google Scholar]
- Gollob J.A., Murphy E.A., Mahajan S., Schnipper C.P., Ritz J., Frank D.A. Altered interleukin-12 responsiveness in Th1 and Th2 cells is associated with differential activation of Stat5 and Stat1. Blood. 1998;91:1341–1354. [PubMed] [Google Scholar]
- Ahn H.-J., Tomura M., Yu W.-G., Iwasaki M., Park W.-R., Hamaoka T., Fujiwara H. Requirement for distinct janus kinases and STAT proteins in T cell proliferation versus IFN-γ production following IL-12 stimulation. J. Immunol. 1998;161:5893–5900. [PubMed] [Google Scholar]
- Pignata C., Sanghera J.S., Cossette L., Pelech S.L., Ritz J. Interleukin-12 induces tyrosine phosphorylation and activation of 44-kD mitogen-activated protein kinase in human T cells. Blood. 1994;83:184–190. [PubMed] [Google Scholar]
- Gollob J.A., Schnipper C.P., Murphy E.A., Ritz J., Frank D.A. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J. Immunol. 1999;162:4472–4481. [PubMed] [Google Scholar]
- Rincon M., Enslen H., Raingeaud J., Recht M., Zapton T., Su M.S.-S., Penix L.A., Davis R.J., Flavell R.A. Interferon-γ expression by Th1 effector T cells mediated by the p38 MAP kinase signalling pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2817–2829. doi: 10.1093/emboj/17.10.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T., Kawasaki H., Kitamura T., Nojima Y., Morimoto C. Interleukin-12 induces tyrosine phosphorylation of an 85-kDa protein associated with the interleukin-12 receptor β1 subunit. Cell. Immunol. 1998;186:39–44. doi: 10.1006/cimm.1998.1294. [DOI] [PubMed] [Google Scholar]
- Magram J., Sfarra J., Connaughton S., Faherty D., Warrier R., Carvajal D., Wu C.T., Stewart C., Sarmiento U., Gately M.K. IL-12-deficient mice are defective but not devoid of Type 1 cytokine responses. Ann. NY Acad. Sci. 1996;795:60–70. doi: 10.1111/j.1749-6632.1996.tb52655.x. [DOI] [PubMed] [Google Scholar]
- Piccotti J.R., Li K., Chan S.Y., Ferrante J., Magram J., Eichwald E.J., Bishop D.K. Alloantigen-reactive Th1 development in IL-12-deficient mice. J. Immunol. 1998;160:1132–1138. [PubMed] [Google Scholar]
- Schijns V.E., Haagmans B.L., Wierda C.M.H., Kruithof B., Heijnen I.A., Alber G., Horzinek M.C. Mice lacking IL-12 develop polarized Th1 cells during viral infection. J. Immunol. 1998;160:3958–3964. [PubMed] [Google Scholar]
- Altare F., Lammas D., Revy P., Jouanguy E., Doffinger R., Lamhamedi S., Drysdale P., Scheel-Toellner D., Girdlestone J., Darbyshire P. Inherited interleukin 12 deficiency in a child with Bacille Calmette-Guerin and Salmonella enteritidis disseminated infection. J. Clin. Invest. 1998;102:2035–2040. doi: 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge L., D'Ambrosio D., Biffi M., Penna G., Minetti L.J., Presky D.H., Adorini L., Sinigaglia F. The role of STAT4 in species-specific regulation of Th cell development by type 1 IFNs. J. Immunol. 1998;161:6567–6574. [PubMed] [Google Scholar]
- Sareneva T., Matikainen S., Kurimoto M., Julkunen I. Influenza A virus-induced IFN-α/β and IL-18 synergistically enhance IFN-γ gene expression in human T cells. J. Immunol. 1998;160:6032–6038. [PubMed] [Google Scholar]