B Cells of HIV-1–Infected Patients Bind Virions through Cd21–Complement Interactions and Transmit Infectious Virus to Activated T Cells (original) (raw)
Abstract
The impact of HIV-associated immunopathogenesis on B cells has been largely associated with indirect consequences of viral replication. This study demonstrates that HIV interacts directly with B cells in both lymphoid tissues and peripheral blood. B cells isolated from lymph node and peripheral blood mononuclear cells (PBMCs) of 4 and 23 chronically infected patients, respectively, demonstrated similar capacities to pass virus to activated HIV-negative PBMCs when compared with CD4+ cells from the same patients. However, in contrast to T cells, virus associated with B cells was surface bound, as shown by its sensitivity to pronase and the staining pattern revealed by in situ amplification of HIV-1 RNA. Cell sorting and ligand displacing approaches established that CD21 was the HIV-binding receptor on B cells, and that this association was mediated through complement-opsonized virus. These B cells were also found to express significantly lower levels of CD21 compared with HIV-negative individuals, suggesting a direct perturbing effect of HIV on B cells. These findings suggest that B cells, although they themselves are not readily infected by HIV, are similar to follicular dendritic cells in their capacity to serve as extracellular reservoirs for HIV-1. Furthermore, B cells possess the added capability of circulating in peripheral blood and migrating through tissues where they can potentially interact with and pass virus to T cells.
Keywords: HIV-1, B cell, viral reservoir, complement, CD21
Introduction
HIV-1 replicates predominantly in activated CD4+ T cells. However, numerous other cell types contribute to HIV infection by acting as reservoirs or carriers for the virus. Intracellular reservoirs of HIV that have been well described include macrophages and latently infected resting CD4+ T cells, and extracellular sources of virus include dendritic cells (DCs) and follicular dendritic cells (FDCs; for review see references 1 2 3). Recently, the HIV capturing and transporting properties of DCs have been clarified with the identification of DC-SIGN, a lectin-based membrane protein that is uniquely expressed on DCs and demonstrates a specific capacity to interact with HIV envelope 4. Direct binding and stabilization of virions on cells that are not susceptible to infection have also been proposed to involve interactions between integrins expressed on the cell surface and adhesion molecules acquired by HIV-1 5. In contrast, the demonstration of HIV-trapping on FDCs has relied on ultrastructural analyses of germinal center sections stained for gag proteins or HIV-RNA 6 7 8, and in vitro reconstitution of HIV trapping on FDCs via immune complexes (ICs; 9, 10).
HIV-mediated perturbations of B cell functions have been well described 11 12 and are largely thought to reflect indirect effects of HIV replication. Several direct interactions between HIV and B cells have been suggested from in vitro studies, (for review see references 13, 14), yet in vivo correlates of these observations are lacking. Furthermore, perturbations of B cell function resulting from direct HIV infection of B cells have been suggested from numerous in vitro studies 15 16; however, few in vivo correlates have been identified.
It has been suggested that one of the major modes of interaction between HIV and FDCs in vivo involves binding of HIV IC to complement receptors expressed on FDCs 9. Interestingly, B cells also express complement receptors, traffic through follicles where HIV RNA signal is most intense 17 18, migrate to the periphery of follicles for interaction with T cells 19, and bind HIV in vitro through IC–CD21 interactions 20. In this study, we demonstrate that replication-competent virus binds directly to B cells isolated from lymphoid tissues and/or peripheral blood of patients chronically infected with HIV-1. This phenomenon was consistently demonstrated in chronically infected patients who have plasma viral loads >10,000 copies of HIV RNA per ml. By cell sorting, virus displacing, and complement-based capture assays we demonstrate that the association between HIV and B cells occurs through interactions between CD21 expressed on the surface of B cells and C3 fragments bound to HIV particles.
Materials and Methods
Study Subjects.
23 HIV-1–infected patients were studied; 19 were chronically viremic and 4 were recently viremic after interruption of antiretroviral therapy that previously had suppressed viremia successfully 21. Of the 19 chronically viremic individuals studied, patients 4, 7, 16, and 17 were antiretroviral drug naive, and the remaining patients were either not fully compliant with their antiretroviral regimens or were failing therapy, thus explaining the presence of detectable viremia. All protocols were carried out in accordance to the Institutional Review Board of the National Institute of Allergy and Infectious Diseases.
Antibodies.
FITC- or PE-conjugated mAbs specific for CD19, CD20, CD21, CD3, and CD14 were purchased from Becton Dickinson, and, in the case of CD4, CD1a, IgM, IgG, and CD32, from BD PharMingen. The C3-displacing anti-CD21 mAb FE8 was provided by Dr. W.M. Prodinger (University of Innsbruck, Innsbruck, Austria). The negative control, isotype-matched anti-CD21 mAb BL13, was purchased from Beckman Coulter. Rabbit anti–human C3d antibody, which recognizes native C3, C3b, iC3b, C3d, and C3dg, was purchased from Dako. Normal rabbit serum was obtained from Caltag Laboratories.
Cell Preparations.
PBMCs were obtained from lymphopheresis or standard blood draws by Ficoll-Hypaque density gradient centrifugation. Excisional inguinal lymph node biopsies were performed on a subset of patients to obtain lymph node mononuclear cells (LNMCs). LNMCs were obtained by gentle mechanical manipulation of the tissue. B cells were isolated by negative selection using a column-based purification technique (StemCell Technologies). Purity of B cell preparations was verified by flow cytometry (EPICS XL Flow cytometer; Beckman Coulter), with a panel of mAbs against CD19, CD20, and CD21. Typically these markers accounted for >95% percent of the cell suspensions, with the remainder of the cells expressing neither CD14 nor CD1a, nor coexpressing CD3 and CD4. CD4+ T cell populations were prepared by depleting PBMCs or LNMCs of CD8+ cells by magnetic bead depletion (Dynal); CD19+ cells were also removed for the data collected from patient 2 in Fig. 1 and all patients in Table (see below).
Figure 1.
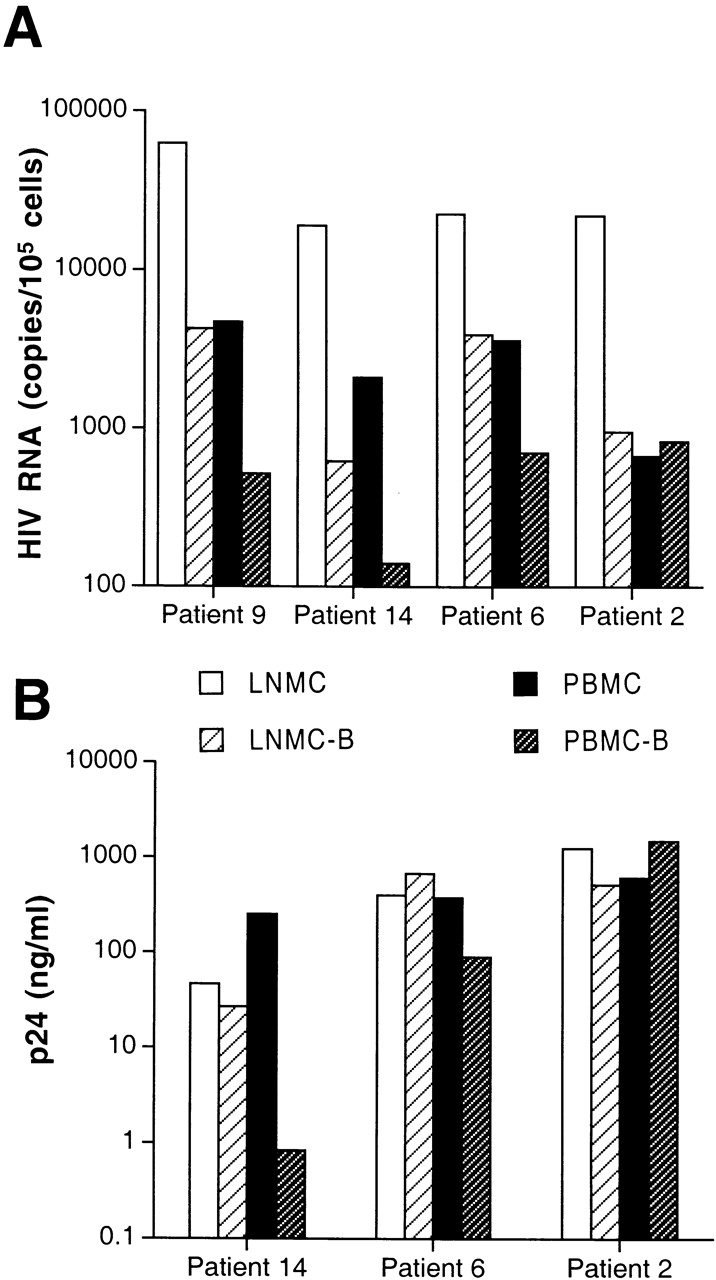
Levels of cell-associated HIV-1 RNA and replication-competent virus in various cell types isolated from patients chronically infected with HIV-1. Highly enriched B cells identified as LNMC-B and PBMC-B were isolated from LNMCs and PBMCs, respectively, of (A) four patients and compared with corresponding unfractionated cells for levels of cellular HIV RNA, and (B) in three patients compared with corresponding CD8-depleted cells for levels of replication-competent virus. Coculture conditions included 106 HIV-infected cells and 106 indicator T cells, with the exception of LNMCs of patient 6, where cocultures were performed on 0.2 × 106 input cells. Levels of HIV-1 p24 were measured in coculture supernatants on day 7.
Table 1.
Profiles of HIV-1–infected Patients with Capacity of B Cells to Bind Replication-competent Virus
| T cell–associated virus | B cell–associated virus | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | CD4 | CD19 | CD21− B Cells | Plasma viral load | HIV-1 RNA | Passage | Phenotype | HIV-1 RNA | Passage | Phenotype |
| per μl | % | % | copies per ml | |||||||
| 1 | 5 | 22 | 12 | 1,025,830 | 8,100 | Rapid/high | X4 > R5 | 630 | Mod/med | X4 > R5 |
| 2 | 6 | 47 | 20 | 194,638 | 670 | Rapid/high | R5, X4 | 830 | Rapid/high | R5, X4 |
| 3 | 7 | 76 | ND | 115,511 | 4,900 | Rapid/high | R5 > X4 | 450 | Mod/med | R5 > X4 |
| 4 | 8 | 14 | ND | 76,838 | 2,000 | Rapid/high | X4 | 370 | Rapid/high | X4 |
| 5 | 48 | 15 | ND | 169,014 | 1,900 | Rapid/high | R5 | 140 | Mod/med | R5 |
| 6 | 85 | 15 | 50 | 14,513 | 5,500 | Rapid/high | X4 > R5 | 1,200 | Rapid/med | R5 |
| 7 | 89 | 9 | ND | 1,105,380 | 7,800 | Rapid/high | X4 > R5 | 1,300 | Rapid/high | X4 > R5 |
| 8 | 92 | 12 | 23 | 16,989 | 540 | Rapid/high | X4 > R5 | 140 | Rapid/high | X4 > R5 |
| 9 | 108 | 10 | 8 | 133,822 | 4,400 | Mod/med | R5 | 1,700 | Mod/med | R5 |
| 10 | 114 | 20 | 42 | 58,211 | 1,200 | Rapid/high | X4 ≫ R5 | <LDL | Slow/high | X4 ≫ R5 |
| 11 | 135 | 10 | 30 | 37,606 | 420 | Mod/high | X4, R5 | 100 | Slow/high | X4 > R5 |
| 12 | 188 | 7 | 41 | 249,966 | ND | Rapid/high | R5 | ND | Mod/high | R5 |
| 13 | 202 | 21 | 38 | 11,232 | 2,600 | Rapid/med | R5 | 460 | Rapid/med | R5 |
| 14 | 227 | 16 | 37 | 176,481 | 5,500 | Rapid/high | X4 | 1,300 | Rapid/med | X4 |
| 15 | 350 | 13 | 28 | 41,183 | 2,400 | Rapid/high | R5 | 460 | Mod/high | R5 |
| 16 | 390 | 11 | 75 | 31,335 | 1,200 | Mod/med | R5 | 210 | Mod/low | R5 |
| 17 | 483 | 9 | 33 | 100,730 | 1,500 | Rapid/high | R5 | 470 | Mod/high | R5 |
| 18 | 542 | 15 | 39 | 217,208 | 1,800 | Rapid/high | R5 | 470 | Mod/high | R5 |
| 19 | 987 | 22 | 7 | 62,743 | 3,500 | Slow/med | R5 | 1,700 | Mod/med | R5 |
| 20 | 316 | 5 | 44 | 166,143 | 4,200 | Rapid/high | R5 | 1,400 | Mod/high | R5 |
| 21 | 562 | 3 | 32 | 89,442 | 300 | Slow/high | R5 | 140 | Slow/high | R5 |
| 22 | 1,177 | 9 | 18 | 254,150 | 640 | Slow/high | R5 | 310 | Mod/high | R5 |
| 23 | 1,573 | 3 | ND | 109,850 | <LDL | Mod/high | R5 | 100 | Mod/high | R5 |
HIV-1 Quantitation, Visualization, and Phenotyping Assays.
The number of copies of HIV-1 RNA was determined in each cell preparation by a nucleic acid sequence–based amplification (NASBA) technique, using an established protocol 22. The limit of detection of the assay varied between 40 and 80 copies of HIV RNA. For the in situ NASBA, cells were fixed (Permeafix™; Ortho Diagnostic Systems), cytospun onto silianated slides at a concentration of 5 × 104 cells per slide, and air-dried. The HIV NASBA reaction was carried out at 41°C for 2 h in the presence of an FITC-labeled probe that recognizes two separate areas of the amplified product and induces form concatamers. After the NASBA reaction, slides were washed and stained for DNA with DAPI (Sigma-Aldrich), according to the recommendations of the manufacturer. Images of the stained cells were acquired by confocal microscopy.
Replication-competent virus associated with each cell preparation was evaluated by coculturing 106 cells with 106 anti-CD3 stimulated HIV-negative PBMCs depleted of CD8+ T cells (indicator T cells). Surface-bound virus was removed by pronase (Roche Molecular Biochemicals) pretreatments as previously described 23, with the modification that 20 mg/ml of pronase was used in the assay. Productive infection was evaluated by ELISA determination of HIV-1 p24 (Beckman Coulter). Virus collected at the peak of replication in cocultures was analyzed for coreceptor usage by exposure to the human glioma cell lines U87/CD4 expressing the chemokine receptors CCR5 or CXCR4 (provided by D.R. Littman, New York University Medical Center, New York, NY).
B Cell Subset and Ligand-displacing Analyses.
Fresh or thawed PBMCs from HIV-infected individuals were depleted of CD2- and CD14-expressing cells using magnetic bead separation protocols, then sorted on a EPICS ELITE cell sorter (Coulter) into one of the following subset pairs: CD20+/CD21+ and CD20+/CD21− or CD20+/CD32+ and CD20+/CD32−. In other experiments, B cells prepared by negative selection (StemCell Technologies) were depleted of surface IgG– or surface IgM–expressing cells by incubation with biotinylated anti-IgG or anti-IgM antibodies (BD PharMingen), followed by incubation with streptavidin-conjugated magnetic beads (Dynal) and separation on a magnet. Subsets of B cells were tested for the presence of replication-competent HIV by coculturing equal numbers of B cells from each fraction with indicator T cells. For displacement of HIV-bound C3 fragments, B cells were isolated from PBMCs of HIV-infected patients by negative selection. The cells were incubated with 2 μg/ml of anti-CD21 C3 fragment–displacing mAb FE8 or control anti-CD21 mAb BL13 for 1 h at 37°C, washed once, incubated a second time with 2 μg/ml mAb for 30 min at 37°C, washed twice, and cocultured with indicator T cells. HIV virions displaced by FE8 were captured by sequential incubations with rabbit anti-C3d antibodies and sheep anti–rabbit–conjugated magnetic beads (Dynal), followed by separation of unbound virions by magnetic bead separation. Levels of HIV RNA in each fraction, including samples of FE8-displaced virus incubated with normal heat-inactivated rabbit serum (negative control), were determined by branched DNA assay (limit of detection, 50 copies of HIV-1 RNA; Chiron Corp.).
Results and Discussion
Association of HIV-1 with Tissue- and Blood-derived B Cells Isolated from Chronically Infected Patients.
We have previously reported that B cells are susceptible to CD4/CXCR4-dependent HIV-1 infection in vitro 24. To determine whether B cells are productively infected in vivo, we initially analyzed cultures of B cells derived from PBMCs of several HIV-1–infected patients. While there was little evidence of productive replication under these conditions, we found substantial amounts of HIV-1 RNA associated with freshly isolated LNMC- and PBMC-derived B cells of chronically infected patients harboring either R5 or X4 virus (see Fig. 1 A and Table for virus phenotypes). In agreement with previous studies, the levels of HIV RNA detected in each LNMC and LNMC-derived B cell sample were 5- to 15-fold higher than their corresponding PBMC samples 17 18, with the exception of levels from patient 2, who had similar levels of HIV RNA in both LMNC- and PBMC-derived B cells. The replication capacity of virus associated with each cell preparation for three of the four patients was also investigated by micro coculture analysis with anti-CD3 activated, CD8-depleted PBMCs from HIV-negative donors (referred to as indicator T cells). Under these conditions, all sources of B cells were found to harbor replication-competent virus (Fig. 1 B), the kinetics of which reflected the differences in cellular HIV RNA. Conversely, the high copy number of HIV RNA detected in unfractionated LNMC samples did not lead to correspondingly high levels of replication-competent virus in the cocultures. This probably was due to mechanical disruption and loss of FDC-trapped HIV particles, the form of virus thought to account for the majority of HIV RNA signal in lymphoid tissues 17 18. Interestingly, patient 2, who had a peripheral blood lymphocyte composition of 1% CD4+ T cells and 47% CD19 B cells (Table ), had higher levels of replication-competent virus per input cell associated with his PBMC-derived B cells than with his corresponding CD4+ T cells (1477 vs. 647 ng/ml p24; Fig. 1 B). Taken together, these data underscore the importance of the B cells of this patient as a reservoir of extracellular HIV.
Detailed Characterization of B Cell–associated HIV-1 in the Peripheral Blood of Chronically Infected Patients.
To establish the scope of patients whose B cells harbor HIV, we performed in depth analyses of peripheral blood B cells isolated from 19 chronically viremic patients (Table , patients 1–19) and 4 recently viremic patients (Table , patients 20–23). A parallel CD4+ T cell fraction was prepared from each patient studied by depleting PBMCs of CD8 and CD19 cells. For all patients studied, the level of HIV RNA associated with each cell fraction lysate was determined by NASBA analysis (Table ). The B cell–associated HIV-1 RNA levels ranged from undetectable to 1,700 copies/105 cells (median: 460 copies/105 cells), whereas T cell–associated HIV-1 RNA ranged from undetectable to 8,100 copies/105 cells (median: 2,000 copies/105 cells). The mean ratio of HIV RNA between B and CD4+ T cells was 0.23. To establish the cellular localization of the HIV RNA signal, we performed a qualitative in situ NASBA analysis for HIV-1 gag sequences. The B cell–associated HIV-1 RNA localized to the surface of the cells, as suggested by the staining pattern that was largely restricted to the periphery of the cells (Fig. 2A and Fig. B) and reduction of staining to background level (Fig. 2 E) when cells were treated with pronase before NASBA (data not shown). In contrast, the T cell–associated HIV-1 RNA was found to be dispersed throughout the cytoplasm (Fig. 2C and Fig. D) and largely insensitive to pronase treatment (data not shown). Varying degrees of brightness were observed throughout the B cell population (Fig. 2 A), ranging from rare bright green ring-like patterns to dimmer specks of green on the contour of the cells, indicated by arrows. This spectrum of HIV RNA staining could also be observed at higher magnification for a single B cell (Fig. 2 B), contrasting sharply with the dense intracellular staining associated with T cells (Fig. 2 D). These data suggest a nonuniform mode of interaction between HIV particles and B cells through molecules that are either unevenly expressed or vary in binding affinities.
Figure 2.
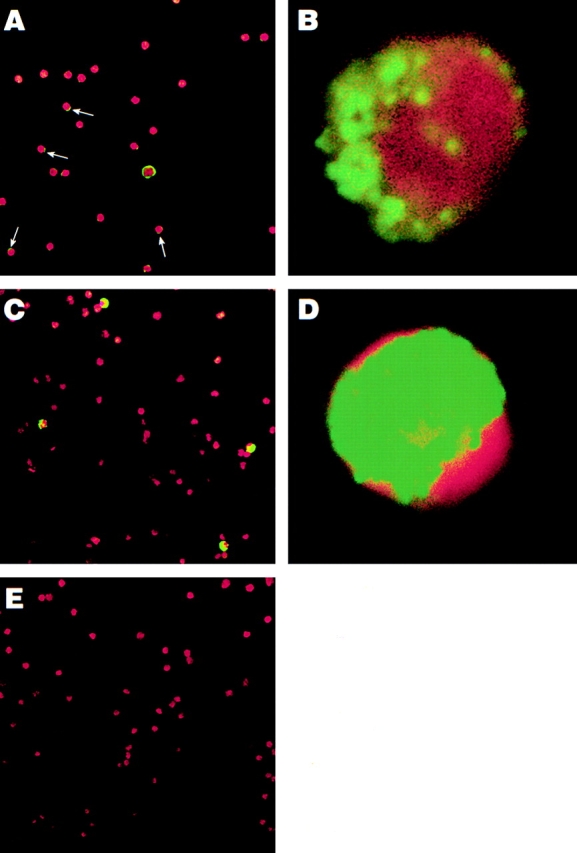
Location of HIV RNA in and on cells isolated from an HIV-infected individual. In situ amplification for HIV-1 RNA (gag) was carried out on highly purified B cells from patient 9 (A and B), CD8/CD19-depleted PBMCs from patient 9 (C and D), and HIV-negative PBMCs (E). The FITC stains (HIV RNA, in green) were overlaid onto the corresponding DAPI stains (cellular DNA, in red) to create the final image. Arrows indicate where weak positive signals were observed on the surface of B cells. Original magnification: (A, C, and E) ×400; (B and D) ×1,000.
To evaluate the replication capacity of the HIV RNA measured in each PMBC subset, B and CD4+ T cell preparations were cocultured with indicator T cells, as described for Fig. 1. As shown in Table , all 23 chronically and recently viremic patients had replication-competent virus associated with their B cells. The kinetics of virus passage generally correlated with the levels of HIV RNA detected in the B cells; however, other factors, including virus phenotype, were found to affect replication kinetics. Repeat analysis of several patients whose viral loads fluctuated over the course of the study revealed that levels of B cell–associated but not T cell–associated virus closely paralleled changes in plasma viremia (data not shown), supporting the notion of extracellular interactions between B cells and HIV virions. Furthermore, the replication-competent virus found on the surface of B cells of recently viremic patients (Table , patients 20–23) also suggests that the association between HIV and B cells is readily reestablished when the virus is rebounding after discontinuation of antiretroviral therapy after prolonged suppression of viremia.
Sensitivity of B and T Cell–associated HIV to Proteolytic Treatment of Cell Surfaces.
Inter-patient comparison of the capacities of B and T cells to pass virus to indicator T cells (B–T and T–T respectively) revealed that, overall, passage of virus was more efficient in T–T than B–T cocultures. However, the kinetics of passage of virus from input B cells to indicator T cells were surprisingly rapid considering the fivefold difference in RNA levels between CD4+ T cells and B cells. Based on these observations and the location of the pronase-sensitive RNA signal revealed by in situ NASBA (Fig. 2), we hypothesized that the HIV RNA detected on B cells represented readily available and infectious viral particles, whereas the signal in the T cells represented genomic or messenger RNA. To directly test this hypothesis, B and T cell suspensions from eight of the patients described in Table were treated with pronase before cocultivation. In all eight patients, as shown by three representative graphs in Fig. 3 A, proteolysis completely abrogated the passage of virus from the B cells to the indicator T cells, whereas the CD4+ T cell preparations were unaffected by pronase treatment. In seven out of eight patients a dramatic decrease in the levels of B cell–associated HIV-1 RNA, but not T cell–associated HIV-1 RNA as measured by NASBA, was also observed after pronase treatment (data not shown). Taken together, these data strongly suggest that the HIV-1 RNA associated with B cells of chronically HIV-1–infected patients is in the form of surface-bound virions that can readily initiate a spreading infection when placed in proximity to activated CD4+ T cell targets. Conversely, the HIV-1 RNA associated with T cells largely reflects pronase-insensitive intracellular HIV-1 RNA.
Figure 3.
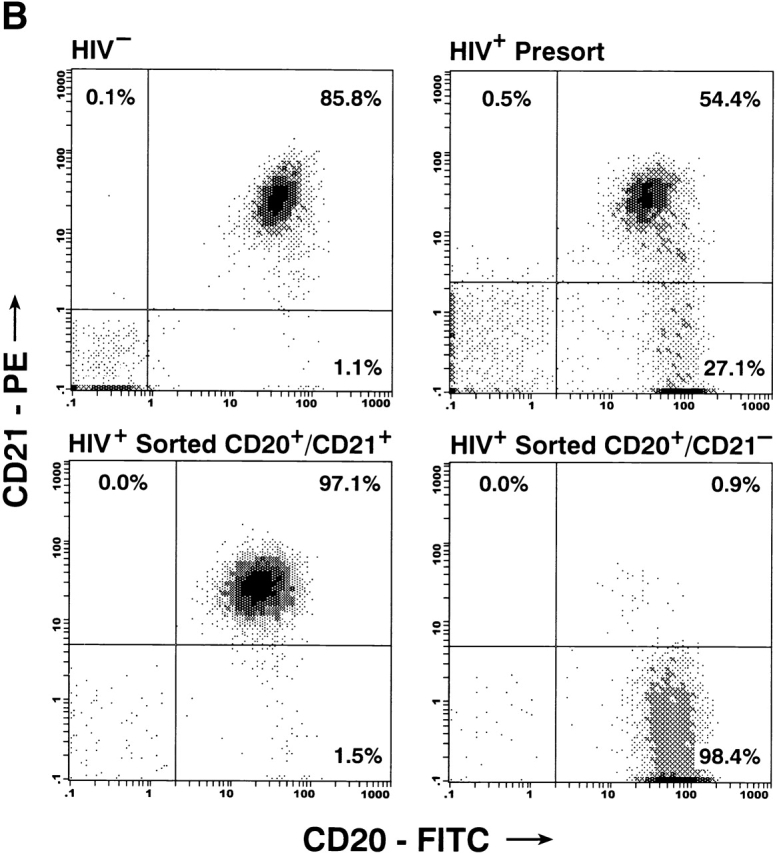
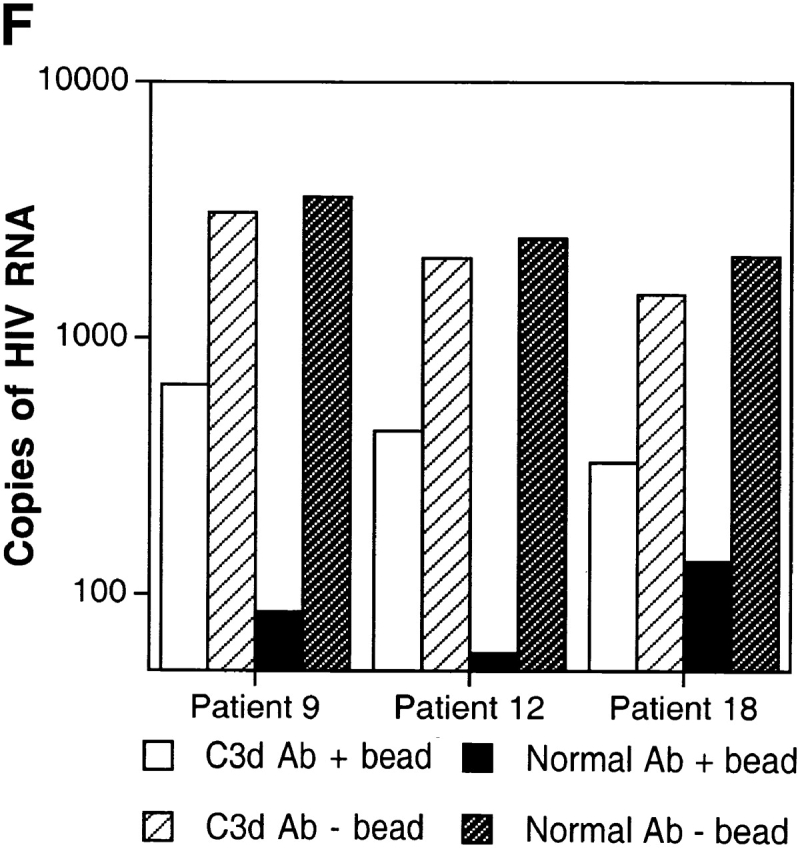
Characterization of the replication-competent virus associated with PBMC-derived B cells of HIV-infected patients. (A) B cells and T cells isolated from three HIV-infected individuals were pretreated with pronase followed by coculture with indicator T cells. (B, top) Flow cytometric comparison of CD21 expression on representative HIV-negative B cells and presorted B cells of patient 17, followed by (B, bottom) post-sorted CD20+/CD21+ and CD20+/CD21− fractions of patient 17. (C) B cells of three HIV-infected patients sorted into CD21+ and CD21− fractions (and CD32+ versus CD32− fractions in the case of patient 19) and cocultured with indicator T cells. Colors identify the sorted fraction and symbols identify the number of coculture wells plated for a given sorted fraction. (D) Flow cytometric comparison of CD21 expression on pronase-treated and -untreated B cells of patient 9. (E) B cells of three HIV-infected patients preincubated with mAbs FE8 and BL13 and cocultured with indicator T cells. (F) FE8-displaced HIV from 2 × 106 B cells of three HIV-infected patients, treated with rabbit anti–human C3d (C3d Ab) or normal rabbit serum (Normal Ab), and captured with magnetized anti-rabbit antibodies. Fractions captured on magnet (+ bead) and fractions not captured (− bead) were tested for HIV-1 RNA by bDNA.




Virus Phenotypes Associated with B Cells In Vivo.
Recently, we reported that X4 strains of HIV-1 can infect CD40-activated B cells in vitro through CD4–CXCR4 interactions, whereas a lack of CCR5 expression on these cells explains their resistance to R5 strains 24. In this study, we found that the majority of the virus associated with B cells of HIV-infected patients did not replicate when, in the absence of indicator T cells, the B cells were cultured under stimulatory conditions involving CD40 ligand, LPS, or the phorbol ester PDBu (data not shown). The reasons for the apparent discrepancy with our previous in vitro findings may be explained by difficulties in reproducing an in vivo environment in an in vitro setting, especially considering the low and variable expression of CD4 on B cells 25. Alternatively, and more likely, B cells may not be targets for productive HIV infection in vivo. Furthermore, we isolated both X4 and R5 viruses from the B cells of HIV-infected patients (Table ), and with the exception of patient 6 no detectable differences in coreceptor usage were observed between viruses recovered from the B cell and CD4+ T cell fractions (Table ). Since CCR5 expression on B cells is generally undetectable 26, and no R5-using HIV-1 has been found to infect primary B lymphocytes 24, these data suggest that B cells bind HIV in vivo by a mechanism that is independent of CD4 and chemokine receptors.
Mechanism of Interaction between HIV and B Cells.
To investigate the mechanism of interaction between HIV and B cells in vivo, we performed extensive B cell subset analyses using surface markers that have been associated with HIV binding to B cells in vitro 20 27 28, and/or with trapping of immune-complexed virus by FDCs in vivo 8 9 10. Accordingly, binding to HIV envelope–specific surface Ig was addressed by depleting the surface IgG– and IgM–expressing B cells of patients 8, 11, and 19; virus was passed to indicator T cells with kinetics similar to those described in Table (data not shown). The possibility that HIV binds B cells through Fc receptor interactions was addressed by sorting the B cells of patients 11, 13, and 19 into CD32+ and CD32− fractions; virus was passed to indicator T cells with kinetics similar to those described in Table ; data for patient 19 are shown in Fig. 3 C. The possibility that HIV binds B cells through IC–complement receptor interactions was addressed by sorting the B cells of patients 6, 11, 13, 16, 17, and 19 into CD21+ and CD21− fractions (typical FACS™ staining shown in Fig. 3 B). In all six patients, as shown by three representative graphs in Fig. 3 C, replication-competent virus in the CD21-depleted fractions was either absent or showed dramatically reduced replication kinetics compared with the CD21-enriched fractions. Finally, verification of surface markers after pronase treatment revealed a dramatic suppression of CD21 expression on B cells of HIV-infected patients (representative data shown in Fig. 3 D), providing a mechanistic explanation for the initial observation made in Fig. 3 A on the sensitivity of B cell–bound virus to pronase. We found no cosegregation of the CD21+ fraction with markers such as surface IgD/G/M that may be involved in superantigenic binding of HIV envelope to VH3 27 or CD4 (data not shown), suggesting that HIV-1 preferentially interacts with B cells through IC binding to CD21. However, there remained the possibility that HIV binds B cells through another CD21-cosegregating receptor, such as CD35, or a novel B cell marker.
Displacement of HIV-1 Particles from B Cells with a C3-displacing Anti-CD21 mAb.
To directly address the role of CD21 in HIV binding to B cells, we preincubated the B cells of six HIV-infected patients with the anti-CD21 mAb FE8, which has been shown to block and displace the binding of C3 fragment (C3d/C3dg, iC3b)–coated particles 29. In parallel, B cells were also preincubated with an isotype-matched, anti-CD21 mAb BL13, known to bind outside the ligand-binding domain for C3 fragments 30. In agreement with the sorting data, pre-incubation with FE8 but not BL13 either completely abrogated or dramatically reduced the levels of replication-competent HIV associated with the B cells of all six patients studied (Fig. 3 E shows three representative graphs). Furthermore, in order to demonstrate that the binding of HIV to CD21 was mediated by C3 fragments, we performed C3 capture assays on FE8-displaced virus using rabbit anti–human C3d and magnetized anti–rabbit IgG antibodies, followed by detection of HIV-1 RNA in the magnetic bead and soluble fractions. Although a high proportion of FE8-displaced HIV from the B cells of patients 9, 12, and 18 remained in the soluble fraction, anti–human C3d antibodies captured significant amounts of virus when compared with normal serum (Fig. 3 F). Considering that B cells incubated with the nondisplacing anti-CD21 mAb BL13 released undetectable levels of HIV RNA (data not shown), the virus released by FE8 but not captured by anti-C3d antibodies may represent a C3-independent mode of binding between HIV and CD21. Alternatively, virions may have become dissociated from C3 fragments during the incubation steps with FE8, or the HIV-C3 fragment association may have partially restricted recognition of the latter by anti-C3d. Nonetheless, these data confirm that C3 fragments contribute to the binding of HIV to CD21 on B cells.
This study has several implications for in vivo HIV pathogenesis, regarding direct effects of HIV on B cell function, enhancement of virus replication by complement products, and a potential role for B cells as a reservoir for HIV-1. CD21 has been directly associated with B cell activation 31, suggesting that the constant triggering of CD21 by HIV IC may contribute to the high levels of polyclonal B cell activation described in HIV disease and exacerbated when viremia is not controlled by antiretroviral therapy 32. Of note, CD21 expression on B cells of HIV-infected patients can be dramatically decreased compared with HIV-negative individuals (Table and Fig. 3 B; references 33, 34), suggesting a direct perturbation of B cells by HIV IC binding. Furthermore, in agreement with previous studies on HIV-mediated destruction of lymphoid tissues that are the major sites of HIV replication 7 8 17 18, we demonstrate that HIV–B cell interactions are amplified in lymphoid tissues. Our findings suggest that, similar to the situation in which HIV trapped on FDCs serves as a source of infection of CD4+ T cells migrating into lymphoid tissue germinal centers 10, HIV can be presented to activated CD4+ T cell targets as B cell immune-complexed virus. Furthermore, since B cells circulate through peripheral blood and migrate within lymphoid tissues to the border of follicles for cognate B–T interactions 19, they may have even greater opportunities than FDCs to transmit virus to T cells.
In summary, 23 out of 23 chronically HIV-1–infected patients with viral loads >10,000 copies of HIV-1 RNA per ml were found to have replication-competent, pronase-sensitive virus associated with their B cells that was readily transmitted to activated HIV-negative CD4+ T cells. The CD21-based sorting experiments and the C3-displacing assays performed on several patients indicate that HIV-1 interacts directly with B cells through CD21–IC interactions. These findings are underscored by the facts that a significant portion of virions circulate in the blood as ICs 35, and complement breakdown products increase with HIV infection 36. Furthermore, a recent in vitro study indicates that virions bound to B cells through IC interactions are far more infectious for T cells than are free virions 20. The role of HIV-specific antibodies in these ICs has not been addressed here; however, numerous studies suggest their presence contributes to the HIV-enhancing effects of complement 37 38 39 40. Thus, our findings provide new insights regarding the potential role of B cells as a unique extracellular reservoir for HIV.
Acknowledgments
We thank Bradley Foltz and Catherine Watkins for performing the flow cytometry assays, and Patricia Walsh for editorial assistance. We are thankful to James Arthos, Jan Orenstein, Claire Hallahan, and Mario Ostrowski for helpful discussions. We also thank NIAID study coordinators and case managers for recruiting patients, as well as Julie Metcalf and Robin Dewar for patient sample analyses. Finally, we thank the patients for their participation.
Footnotes
Abbreviations used in this paper: DCs, dendritic cells; FDCs, follicular dendritic cells; ICs, immune complexes; NASBA, nucleic acid sequence–based amplification.
S. Moir and A. Malaspina contributed equally to this paper.
Yuexia Li's present address is VIRxSYS, Inc., Gaithersburg, MD 20877.
References
- Burton G.F., Masuda A., Heath S.L., Smith B.A., Tew J.G., Szakal A.K. Follicular dendritic cells (FDC) in retroviral infectionhost/pathogen perspectives. Immunol. Rev. 1997;156:185–197. doi: 10.1111/j.1600-065x.1997.tb00968.x. [DOI] [PubMed] [Google Scholar]
- Chun T.W., Fauci A.S. Latent reservoirs of HIVobstacles to the eradication of virus. Proc. Natl. Acad. Sci. USA. 1999;96:10958–10961. doi: 10.1073/pnas.96.20.10958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase A.T. Population biology of HIV-1 infectionviral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 1999;17:625–656. doi: 10.1146/annurev.immunol.17.1.625. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek T.B., Kwon D.S., Torensma R., van Vliet S.J., van Duijnhoven G.C., Middel J., Cornelissen I.L., Nottet H.S., KewalRamani V.N., Littman D.R. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- Liao Z., Roos J.W., Hildreth J.E. Increased infectivity of HIV type 1 particles bound to cell surface and solid-phase ICAM-1 and VCAM-1 through acquired adhesion molecules LFA-1 and VLA-4. AIDS Res. Hum. Retroviruses. 2000;16:355–366. doi: 10.1089/088922200309232. [DOI] [PubMed] [Google Scholar]
- Tenner-Racz K., Racz P., Bofill M., Schulz-Meyer A., Dietrich M., Kern P., Weber J., Pinching A.J., Veronese-Dimarzo F., Popovic M. HTLV-III/LAV viral antigens in lymph nodes of homosexual men with persistent generalized lymphadenopathy and AIDS. Am. J. Pathol. 1986;123:9–15. [PMC free article] [PubMed] [Google Scholar]
- Fox C.H., Tenner-Racz K., Racz P., Firpo A., Pizzo P.A., Fauci A.S. Lymphoid germinal centers are reservoirs of human immunodeficiency virus type 1 RNA. J. Infect. Dis. 1991;164:1051–1057. doi: 10.1093/infdis/164.6.1051. [DOI] [PubMed] [Google Scholar]
- Spiegel H., Herbst H., Niedobitek G., Foss H.D., Stein H. Follicular dendritic cells are a major reservoir for human immunodeficiency virus type 1 in lymphoid tissues facilitating infection of CD4+ T-helper cells. Am. J. Pathol. 1992;140:15–22. [PMC free article] [PubMed] [Google Scholar]
- Joling P., Bakker L.J., Van Strijp J.A., Meerloo T., de Graaf L., Dekker M.E., Goudsmit J., Verhoef J., Schuurman H.J. Binding of human immunodeficiency virus type-1 to follicular dendritic cells in vitro is complement dependent. J. Immunol. 1993;150:1065–1073. [PubMed] [Google Scholar]
- Heath S.L., Tew J.G., Szakal A.K., Burton G.F. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature. 1995;377:740–744. doi: 10.1038/377740a0. [DOI] [PubMed] [Google Scholar]
- Lane H.C., Masur H., Edgar L.C., Whalen G., Rook A.H., Fauci A.S. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N. Engl. J. Med. 1983;309:453–458. doi: 10.1056/NEJM198308253090803. [DOI] [PubMed] [Google Scholar]
- Schnittman S.M., Lane H.C., Higgins S.E., Folks T., Fauci A.S. Direct polyclonal activation of human B lymphocytes by the acquired immune deficiency syndrome virus. Science. 1986;233:1084–1086. doi: 10.1126/science.3016902. [DOI] [PubMed] [Google Scholar]
- Chirmule N., Pahwa S. Envelope glycoproteins of human immunodeficiency virus type 1profound influences on immune functions. Microbiol. Rev. 1996;60:386–406. doi: 10.1128/mr.60.2.386-406.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S., Kohler H. B cell superantigens in HIV-1 infection. Int. Rev. Immunol. 1997;14:339–349. doi: 10.3109/08830189709116524. [DOI] [PubMed] [Google Scholar]
- Gras G., Richard Y., Roques P., Olivier R., Dormont D. Complement and virus-specific antibody-dependent infection of normal B lymphocytes by human immunodeficiency virus type 1. Blood. 1993;81:1808–1818. [PubMed] [Google Scholar]
- Poulin L., Paquette N., Moir S., Lapointe R., Darveau A. Productive infection of normal CD40-activated human B lymphocytes by HIV-1. AIDS. 1994;8:1539–1544. doi: 10.1097/00002030-199411000-00004. [DOI] [PubMed] [Google Scholar]
- Embretson J., Zupancic M., Ribas J.L., Burke A., Racz P., Tenner-Racz K., Haase A.T. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- Pantaleo G., Graziosi C., Demarest J.F., Butini L., Montroni M., Fox C.H., Orenstein J.M., Kotler D.P., Fauci A.S. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- Garside P., Ingulli E., Merica R.R., Johnson J.G., Noelle R.J., Jenkins M.K. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- Jakubik J.J., Saifuddin M., Takefman D.M., Spear G.T. Immune complexes containing human immunodeficiency virus type 1 primary isolates bind to lymphoid tissue B lymphocytes and are infectious for T lymphocytes. J. Virol. 2000;74:552–555. doi: 10.1128/jvi.74.1.552-555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey R.T., Jr., Bhat N., Yoder C., Chun T.W., Metcalf J.A., Dewar R., Natarajan V., Lempicki R.A., Adelsberger J.W., Miller K.D. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA. 1999;96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gemen B., van Beuningen R., Nabbe A., van Strijp D., Jurriaans S., Lens P., Kievits T. A one-tube quantitative HIV-1 RNA NASBA nucleic acid amplification assay using electrochemiluminescent (ECL) labelled probes. J. Virol. Methods. 1994;49:157–167. doi: 10.1016/0166-0934(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Marechal V., Arenzana-Seisdedos F., Heard J.M., Schwartz O. Opposite effects of SDF-1 on human immunodeficiency virus type 1 replication. J. Virol. 1999;73:3608–3615. doi: 10.1128/jvi.73.5.3608-3615.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S., Lapointe R., Malaspina A., Ostrowski M., Cole C.E., Chun T.W., Adelsberger J., Baseler M., Hwu P., Fauci A.S. CD40-mediated induction of CD4 and CXCR4 on B lymphocytes correlates with restricted susceptibility to human immunodeficiency virus type 1 infectionpotential role of B lymphocytes as a viral reservoir. J. Virol. 1999;73:7972–7980. doi: 10.1128/jvi.73.10.7972-7980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrona G., Leanza N., Ulivi M., Majolini M.B., Taborelli G., Zupo S., Baldari C.T., Roncella S., Ferrarini M. Apoptosis induced by crosslinking of CD4 on activated human B cells. Cell. Immunol. 1999;193:80–89. doi: 10.1006/cimm.1999.1455. [DOI] [PubMed] [Google Scholar]
- Wu L., Paxton W.A., Kassam N., Ruffing N., Rottman J.B., Sullivan N., Choe H., Sodroski J., Newman W., Koup R.A., Mackay C.R. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 1997;185:1681–1691. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberian L., Goodglick L., Kipps T.J., Braun J. Immunoglobulin VH3 gene productsnatural ligands for HIV gp120. Science. 1993;261:1588–1591. doi: 10.1126/science.7690497. [DOI] [PubMed] [Google Scholar]
- Golding H., Dimitrov D.S., Blackburn R., Manischewitz J., Blumenthal R., Golding B. Fusion of human B cell lines with HIV-1 envelope-expressing T cells is enhanced by antigen-specific Ig receptors. Possible mechanism for elimination of gp120-specific B cells in vivo. J. Immunol. 1993;150:2506–2516. [PubMed] [Google Scholar]
- Prodinger W.M., Schwendinger M.G., Schoch J., Kochle M., Larcher C., Dierich M.P. Characterization of C3dg binding to a recess formed between short consensus repeats 1 and 2 of complement receptor type 2 (CR2; CD21) J. Immunol. 1998;161:4604–4610. [PubMed] [Google Scholar]
- Cohen J.H., Fischer E., Kazatchkine M.D., Brochier J., Revillard J.P. Characterization of monoclonal antihuman-B-cell antibody BL13 as an anti-C3d-receptor (CR2) antibody. Scand. J. Immunol. 1986;23:279–285. doi: 10.1111/j.1365-3083.1986.tb01969.x. [DOI] [PubMed] [Google Scholar]
- Melchers F., Erdei A., Schulz T., Dierich M.P. Growth control of activated, synchronized murine B cells by the C3d fragment of human complement. Nature. 1985;317:264–267. doi: 10.1038/317264a0. [DOI] [PubMed] [Google Scholar]
- Morris L., Binley J.M., Clas B.A., Bonhoeffer S., Astill T.P., Kost R., Hurley A., Cao Y., Markowitz M., Ho D.D., Moore J.P. HIV-1 antigen-specific and -nonspecific B cell responses are sensitive to combination antiretroviral therapy. J. Exp. Med. 1998;188:233–245. doi: 10.1084/jem.188.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetto A., Di Caro A., Camporiondo M.P., Gallone D., Zaniratti S., Tozzi V., Elia G. Identification of a CD21 receptor-deficient, non-Ig-secreting peripheral B lymphocyte subset in HIV-seropositive drug abusers. Clin. Immunol. Immunopathol. 1992;62:139–147. doi: 10.1016/0090-1229(92)90066-w. [DOI] [PubMed] [Google Scholar]
- Scott M.E., Landay A.L., Lint T.F., Spear G.T. In vivo decrease in the expression of complement receptor 2 on B-cells in HIV infection. AIDS. 1993;7:37–41. doi: 10.1097/00002030-199301000-00006. [DOI] [PubMed] [Google Scholar]
- Nishanian P., Huskins K.R., Stehn S., Detels R., Fahey J.L. A simple method for improved assay demonstrates that HIV p24 antigen is present as immune complexes in most sera from HIV-infected individuals. J. Infect. Dis. 1990;162:21–28. doi: 10.1093/infdis/162.1.21. [DOI] [PubMed] [Google Scholar]
- Senaldi G., Peakman M., McManus T., Davies E.T., Tee D.E., Vergani D. Activation of the complement system in human immunodeficiency virus infectionrelevance of the classical pathway to pathogenesis and disease severity. J. Infect. Dis. 1990;162:1227–1232. doi: 10.1093/infdis/162.6.1227. [DOI] [PubMed] [Google Scholar]
- Szabo J., Prohaszka Z., Toth F.D., Gyuris A., Segesdi J., Banhegyi D., Ujhelyi E., Minarovits J., Fust G. Strong correlation between the complement-mediated antibody-dependent enhancement of HIV-1 infection and plasma viral load. AIDS. 1999;13:1841–1849. doi: 10.1097/00002030-199910010-00005. [DOI] [PubMed] [Google Scholar]
- Montefiori D.C., Lefkowitz L.B., Jr., Keller R.E., Holmberg V., Sandstrom E., Phair J.P. Absence of a clinical correlation for complement-mediated, infection-enhancing antibodies in plasma or sera from HIV-1-infected individuals. Multicenter AIDS Cohort Study Group. AIDS. 1991;5:513–517. doi: 10.1097/00002030-199105000-00006. [DOI] [PubMed] [Google Scholar]
- Robinson W.E., Jr., Montefiori D.C., Mitchell W.M. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection. Lancet. 1988;1:790–794. doi: 10.1016/s0140-6736(88)91657-1. [DOI] [PubMed] [Google Scholar]
- Sullivan B.L., Knopoff E.J., Saifuddin M., Takefman D.M., Saarloos M.N., Sha B.E., Spear G.T. Susceptibility of HIV-1 plasma virus to complement-mediated lysis. Evidence for a role in clearance of virus in vivo. J. Immunol. 1996;157:1791–1798. [PubMed] [Google Scholar]