Reversal of Spontaneous Autoimmune Insulitis in Nonobese Diabetic Mice by Soluble Lymphotoxin Receptor (original) (raw)
Abstract
One striking feature of spontaneous autoimmune diabetes is the prototypic formation of lymphoid follicular structures within the pancreas. Lymphotoxin (LT) has been shown to play an important role in the formation of lymphoid follicles in the spleen. To explore the potential role of LT-mediated microenvironment in the pathogenesis of insulin-dependent diabetes mellitus (IDDM), an LTβ receptor–immunoglobulin fusion protein (LTβR–Ig) was administered to nonobese diabetic mice. Early treatment with LTβR–Ig prevented insulitis and IDDM, suggesting that LT plays a critical role in the insulitis development. LTβR–Ig treatment at a late stage of the disease also dramatically reversed insulitis and prevented diabetes. Moreover, LTβR–Ig treatment prevented the development of IDDM by diabetogenic T cells in an adoptive transfer model. Thus, LTβR–Ig can disassemble the well established lymphoid microenvironment in the islets, which is required for the development and progression of IDDM.
Keywords: adhesion molecule, autoimmune diabetes, insulitis, lymphotoxin, lymphotoxin β receptor
Introduction
Insulin-dependent diabetes mellitus (IDDM) is a T cell–mediated autoimmune disease in which the insulin-secreting β cells are selectively destroyed after weeks or months of insulitis. Much of our current knowledge about the complex pathogenesis of IDDM derives from the studies of nonobese diabetic (NOD) mice 1 2 3 4 5. In NOD mice, infiltration of autoreactive T cells into the islets is essential for the development of IDDM. Studies performed in these animals have revealed that the influx of T cells into the pancreas is associated with an increased expression of adhesion molecules. However, factors that upregulate adhesion molecules in the pancreas have not been identified. Lymph node–like structures with de novo formation of lymphoid follicles are gradually established within the islets over 2–3 mo, with initial cellular infiltration starting at 3–4 wk of age 1 3 4 6 7. While the formation of lymphoid follicular structures is a prototypic feature of chronic progressive inflammation 6 7 8, the molecular mechanisms by which the lymphoid follicles are formed and the role of these follicles in the pathogenesis of IDDM have not been well defined.
The interaction between membrane lymphotoxin (LT) and its receptor is essential for the development and maintenance of the lymphoid microenvironment 9 10 11 12 13 14 15. LTα−/− mice and wild-type mice treated with LTβ receptor–immunoglobulin fusion protein (LTβR–Ig) exhibit altered lymphoid microenvironment. This effect is likely caused by a failure to induce lymphoid tissue chemokines and adhesion molecules 12 13 14 15. These results imply that LT may be a master cytokine responsible for the formation of lymphoid structures in chronic inflammation and autoimmune diseases, such as IDDM. Here, we report that administration of LTβR–Ig reverses the formation of lymphoid follicles, prevents β cell destruction by autoreactive T cells, and forestalls the development of IDDM.
Materials and Methods
Reagents and Mice.
The generation and production of recombinant LTβR–Ig has been described previously 15 16. The murine LTβR–human Ig in culture supernatants of BHK/VP16 15 or Chinese hamster ovary cells 16 was purified on a protein A column. No difference could be found between the two preparations. Human Ig was obtained from Biogen, Inc. or Sigma-Aldrich. Female NOD mice were purchased from The Jackson Laboratory and maintained under specific pathogen–free conditions at the University of Chicago. Antibodies to vascular cell adhesion molecule (VCAM), peripheral lymph node addressin (PNAd; MECA 79), mucosal addressin cell adhesion molecule 1 (MAdCAM-1; MECA367), B220, CD4 (GK1.5), CD8 (3.155), and CD11c (N418) were purchased from PharMingen.
LTβR–Ig Treatment and Measurement of Blood Glucose.
NOD mice were given 100 μg of LTβR–Ig or human Ig intraperitoneally once per week for 3 wk or as indicated. The glucose concentration in blood obtained from a tail vein was measured using SureStep® strips (Johnson and Johnson). Diabetes was monitored by levels of blood glucose. Animals were considered diabetic after two consecutive measurements of ≥250 mg/dl of glucose.
Adoptive Transfer.
Recipient NOD mice (7–9 wk) were irradiated with 6 Gy (199 rads/min) using a 137Cs irradiator. 2 h later, the mice were injected intravenously with 2 × 107 splenocytes from diabetic NOD mice in 0.2 ml of PBS, followed by intraperitoneal injection with 100 μg of LTβR–Ig, anti–MAdCAM-1, or control human Ig.
Histology and Immunohistochemistry.
Pancreatic tissue was collected 10 d after the last LTβR–Ig treatment. Hematoxylin and eosin staining was performed on 6-μm sections of 10% formalin-fixed tissue. Infiltration into islets was examined microscopically and counted by a third party pathologist. Sections were prepared on multiple levels, and 20–40 randomly chosen islets per mouse were semiquantitatively classified according to the severity of insulitis: moderate and severe insulitis are defined to be less or more than half of the structure was infiltrated, respectively. Additionally, some of the pancreata and spleen were frozen at –70°C in OCT compound for immunohistology. 6-μm cryostat sections were incubated with rat antibodies to CD3 (L363–29B), VCAM, PNAd (MECA 79), MAdCAM-1 (MECA367), B220, CD4 (GK1.5), CD8 (3.155), hamster antibody to CD11c (N418), or species- and isotype-specific nonreactive control mAbs overnight at 4°C. The next day, room temperature incubation with biotinylated rabbit anti–rat or goat anti–hamster secondary antibody (Vector Laboratories) for 30 min was followed by a 1-h incubation with streptavidin–horseradish peroxidase complex (Vector Laboratories).
Results
Prevention of Diabetes by the Administration of LTβR–Ig.
Typically, 65–75% of untreated NOD mice in our colony develop IDDM by 25 wk. To study the role of membrane LT at an early stage of the disease, 3–4-wk-old NOD female mice were treated weekly with LTβR–Ig (100 μg/wk) for 3 wk. Only one of the NOD mice treated with LTβR–Ig developed IDDM by 32 wk, while 67% of the NOD mice treated with control Ig developed IDDM by 26 wk (Fig. 1 A). To study whether LTβR–Ig treatment reduced IDDM after the initial phase, female NOD mice were treated with the soluble receptor at 6–7 wk of age, a time when many islets are infiltrated with autoreactive T cells. Treatment with LTβR–Ig at this time similarly prevented the development of IDDM, while the majority of control mice developed the disease by 25 wk (Fig. 1 B). These results suggest that LT is essential for the early development of IDDM.
Figure 1.



LTβR–Ig treatment prevents diabetes in young NOD mice. Young NOD mice (n = 10) 3–4 (A) and 6–7 wk of age (B), were treated intraperitoneally with LTβR–Ig or control human Ig for 3 wk. (C) Young CD28−/− NOD mice (n = 5 per group) of 3 wk of age were treated intraperitoneally with LTβR–Ig or PBS for 4 wk. Incidence of diabetes was evaluated at weekly intervals.
Previous studies have shown that CD4+CD25+ regulatory cells control spontaneous autoimmune diabetes, even at late stages, by limiting the destructive infiltrate in the islets 17. We previously reported that both B7−/− mice and CD28−/− NOD mice exhibited severe insulitis and accelerated disease as a consequence of a reduced number of these suppressor cells 17 18. To study whether LT was functioning by altering this regulatory T cell subset, CD28−/− NOD mice, deficient in CD4+CD25+ T cells, were treated weekly with LTβR–Ig or control Ig for 4 wk starting at 3 wk of age and examined for disease incidence. All of the control mice developed IDDM by 11 wk of age, similar to the time course observed in previous experiments. In contrast, there was a delay in disease onset in the LTβR–Ig-treated mice. In fact, some of the mice were free of IDDM for an additional 10–13 wk (Fig. 1 C). Similar retarded diabetes was found in CD28−/− NOD mice that were only treated twice with the fusion protein beginning at the ages of 4 and 5 wk. However, the number of CD4+CD25+ T cells in the spleens of LTβR–Ig-treated mice remained comparable with that in the control group (data not shown). Therefore, these results suggest a critical role for LT in the development of IDDM in this accelerated model (Fig. 1 C), which is independent of CD28 and cannot be attributed to the CD4+CD25+ T cell pathway.
Reversal of Islet Destruction by Preexisting Diabetic T Cells Using LTβR–Ig.
By 10 wk, most islets in NOD mice show severe infiltration by autoreactive T cells, accompanied by early signs of islet cell destruction. Very few reagents have been shown to block the development of IDDM at this late stage 5 19. To determine whether the effect of LTβR–Ig could prevent the development of IDDM at this late phase, 10-wk-old NOD mice were similarly treated weekly with LTβR–Ig over 3 wk. None of the LTβR–Ig-treated NOD mice became diabetic, whereas most of control NOD mice developed IDDM by 25 wk (Fig. 2 A). To study whether LTβR–Ig treatment just retarded the development of IDDM for a few more days or had a prolonged protection, we extended our observation up to 38 wk and found that none of the LTβR–Ig-treated NOD mice became diabetic. More astonishingly, two doses of LTβR–Ig blocked the development of diabetes in 85% of prediabetic NOD mice (11/13) treated as late as 14 wk of age, a time point when some NOD mice (two mice from each group) were already diabetic, with nearly all the islets attacked by autoreactive T cells (Fig. 2 B). Only 15% (2/13) of prediabetic mice treated with LTβR–Ig developed IDDM at 20 wk of age, whereas most of the control Ig–treated group (63%) developed IDDM by 18 wk of age. These findings suggest that LT also plays an important role in the late phases of the disease. However, treatment failed to reverse IDDM in the mice that were already diabetic at the time of treatment.
Figure 2.



LTβR–Ig treatment blocks autoreactive T cell–mediated islet destruction. (A) NOD (10 wk of age) female mice (n = 10) were given weekly intraperitoneal injection of 100 μg of LTβR–Ig or human Ig for 3 wk. (B) NOD (14 wk of age) female mice were given weekly intraperitoneal injection of 100 μg of LTβR–Ig (n = 13) or human Ig (n = 8) for only 2 wk. Blood glucose levels were checked weekly after the end of treatment (A and B). (C) Sublethally irradiated NOD female mice (10 mice per group at 7–9 wk of age) were injected intravenously with 2 × 107 splenocytes from mice with diabetes and treated with a single intraperitoneal injection of 100 μg of LTβR–Ig or human Ig. Blood glucose levels were checked weekly starting 1 wk after adoptive transfer.
The transfer of splenocytes from diabetic NOD mice into irradiated NOD recipient results in diabetes within a few weeks, due to acute infiltration of donor autoreactive T cells into islets. To further address whether the administration of LTβR–Ig can prevent diabetogenic T cells from destroying islet cells, splenocytes from diabetic NOD mice were transferred into irradiated NOD recipients treated with a single dose of LTβR–Ig. The development of IDDM in these irradiated mice was prevented (Fig. 2 C). These results support a model wherein the use of LTβR–Ig can prevent the development of IDDM mediated by preexisting autoreactive T cells in the circulation.
Prevention and Resolution of Insulitis after LTβR–Ig Therapy.
One striking feature of IDDM in NOD mice is the prototypic formation of lymphoid follicular structures within the pancreas. The profound blockade of different stages of IDDM by LTβR–Ig treatment may be attributed to the reduction or even reversal of LT-mediated lymphoid structures in the pancreas. To test this, 3–4- or 10-wk-old NOD mice were treated weekly with LTβR–Ig and control Ig for 3 wk. 10 d after the end of treatment, the pancreata were collected for hematoxylin and eosin staining. As seen in Fig. 3, the number with destructive insulitis and its severity was greatly reduced in both treated groups of mice during the inductive phase (Fig. 3 A). The percentage of islets in an early treated group of mice displaying overall infiltration with formation of lymphoid structures (28.4%) was considerably lower than that observed in the control mice (70.1%; Fig. 3 B). In NOD mice treated at the late stage (10 wk), only 15% of islets developed insulitis, and 3% of those showed the formation of lymphoid structure inside β cells by 14 wk. In contrast, 82% of the corresponding control group demonstrated insulitis and 69% exhibited severe insulitis (Fig. 3 B). The data also demonstrated that the number and scope of infiltrating cells in LTβR–Ig-treated mice at the age of 14 wk was reduced far below the level of untreated mice at the age of 8–9 wk (before the treatment). These results indicate that LTβR–Ig treatment not only prevents but also resolves the formation of lymphoid structures in the pancreas. The data also reveal that the infiltration of inflammatory cells and the development of lymphoid follicles may be a dynamic process and can be reversed once the influx of cells is blocked.
Figure 3.
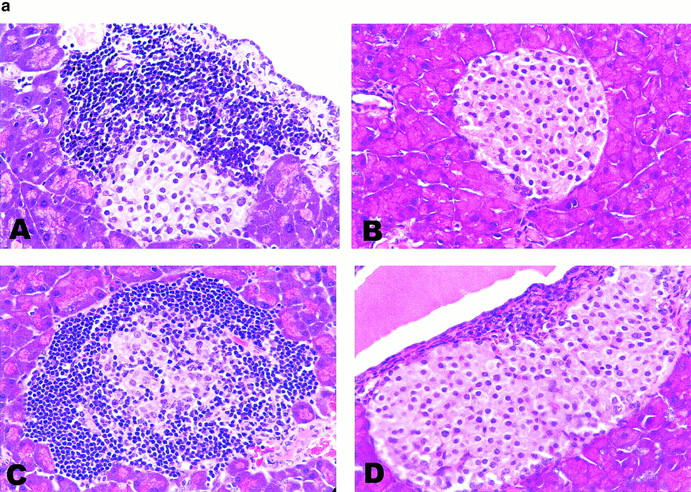

Reversal of insulitis by LTβR–Ig treatment. (a) 4- (A and B) or 10-wk-old (C and D) NOD mice were treated with control Ig (A and C) or LTβR–Ig (B and D) for 3 wk. The pancreata were collected 10 d after the end of treatment, and the sections were stained by hematoxylin and eosin. Representative fields (most common fields) are shown. (b) Individual islets from four mice per group were scored for insulitis at 8 or 14 wk, respectively.
Discussion
Our study demonstrates that the treatment of NOD mice with LTβR–Ig can be effective at blocking both the initiation and effector phases of diabetes. More importantly, such treatment reverses insulitis and prevents the formation of lymphoid follicles, even when insulitis is well established and diabetogenic T cells are clearly present. Such treatment at either early or late stage has prolonged the protection, suggesting that short-term treatment with LTβR–Ig could significantly alter the course of IDDM. Several possible mechanisms may be associated with this treatment. First, as both LT and LIGHT bind LTβR in vitro, these effects may arise from blockage of LIGHT signaling. LIGHT is a costimulatory molecule through its receptor, herpes virus entry mediator, that has been shown to promote T cell proliferation and IFN-γ production 20. Its biological activity in vivo, however, has not been well defined. Further dissection of the role of LIGHT is difficult due to the lack of reagents that effectively block LIGHT activity in vivo. Anti-LTβ antibody has an extremely short half-life of 1 d, while the half-life of LTβR–Ig is 7 d 16. Second, LT may be a critical cytokine for the development of the CD4+CD25+ T cells that regulate autoimmunity. Although LTβR–Ig inhibited the development of the disease in the CD4+CD25+ cell–reduced CD28 knockout NOD mice, there was no increase in the number of regulatory T cells in the spleen during or after treatment. There was also no increase of CD4+CD25+ cells in wild-type NOD mice after treatment. Thus, it appears unlikely that the effects of LTβR–Ig treatment are mediated through CD4+CD25+ T cells.
The most likely mechanism by which LTβR–Ig prevents the development of IDDM is through LT-mediated expression of adhesion molecules and chemokines and the formation of lymphoid tissues 9 10 11 12 13 14 15. Compared with other adhesion molecules, such as VCAM-1 and PNAd, the expression of MAdCAM-1 is more closely associated with the development of insulitis. In fact, treatment of NOD mice with anti–MAdCAM-1 antibody (MECA367) alone can reduce insulitis and prevent the development of IDDM 21. Interestingly, both LTβR–Ig and anti-LTβ antibody can very efficiently block the expression of MAdCAM-1 in the spleen 12 13. We found that the expression of MAdCAM-1 in the pancreas and spleen was readily reduced after the administration of LTβR–Ig. To directly test whether blocking MAdCAM-1 can prevent the migration of autoreactive T cells, NOD mice that received T cells from diabetic mice were treated with anti–MAdCAM-1 (MECA367) antibody. Such treatment could prevent or retard the development of IDDM (data not shown). Together, these data suggest that LT-mediated adhesion molecules, such as MAdCAM-1, are at least partially responsible for the development of IDDM. It is possible that the expression of MAdCAM-1 and other adhesion molecules is required for the formation of insulitis or lymphoid follicles in the pancreas. It has been proposed recently that the expression of membrane LT is required for the formation of large lymphoid follicles in the islets of B lymphocyte chemokine–mediated transgenic mice 8. We have previously showed that the migration of dendritic cells into the spleen is impaired in LTβR–Ig-treated mice 15. It is possible that the chemokines and adhesion molecules reduced by LTβR–Ig treatment slow the influx of inflammatory cells into the pancreas. In this study, we demonstrated that LTβR–Ig reduced the formation of lymphoid follicles and even reversed severe insulitis in NOD mice. Taken together, these data suggest that LT-mediated microenvironment is essential for the development of lymphoid structure that is required for the development of IDDM.
The generation of a genetic defect of the LT gene in NOD mice is another potential model that could be used to address the role of LT in the development of IDDM. Several disadvantages to this model exist, however. First, intercrossing of the NOD mice with LTα−/− mice to retain a defect in both LT and the MHC loci is an impractical task, as the locus of LT is very close to that of MHC class II, which is a key genetic locus for the development of IDDM. Second, LTα−/− mice lack organized lymph nodes, which may be critical for the development of IDDM by itself. Such compound defects may complicate the interpretation of the results. On the other hand, the use of soluble receptor has several advantages. We can correlate different phases of the disease with the timing of administration of fusion protein. Long-term impact after the termination of treatment can be readily assessed.
Resolution of severe insulitis by LTβR–Ig treatment suggests that the migration of inflammatory cells into the islets is a dynamic process in which these cells may constantly move in and out of the target tissue. Interference with this dynamic process may reduce inflammation, which prevents further activation of autoreactive T cells and tissue damage. Interestingly, alteration of the LT-mediated microenvironment by LTβR–Ig is a transient effect that ends once the treatment is terminated. For example, the expression of splenic MAdCAM-1 and the development of B cell follicles was gradually restored 4–5 wk after the termination of LTβR–Ig treatment. Thus, the observation that short-term LTβR–Ig treatment has a profound and prolonged impact on islet destruction may provide a new avenue for the study of pathogenesis and for the treatment of this disease or other autoimmune diseases. In the future, it will be interesting to explore whether LTβR–Ig treatment may prevent islet cell rejection by autoreactive or alloreactive T cells.
Acknowledgments
We thank Jennifer Arcella, Lesley Rhee, and Shihong Li for their expert technical support. We also thank the National Cell Culture Center for the generation of LTβR–Ig using its bioreactor.
This research was supported in part by Biogen, Inc., National Institutes of Health grants, and Juvenile Diabetes Foundation International (1-2000-875). Y.-X. Fu is a recipient of a clinical investigator award (AI01431). J.A. Bluestone, B. Salomon, and some of the research were supported by the Juvenile Diabetes Foundation as part of the JDFI Islet Transplant Center at University of Chicago and University of Minnesota.
Footnotes
Q. Wu and B. Salomon contributed equally to this study. Drs. Bluestone and Fu should both be considered senior authors of this article.
References
- Tisch R., McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- Salomon B., Bluestone J.A. Importance of co-stimulatory molecules in autoimmunity and transplantation. Annu. Rev. Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- Delovitch T.L., Singh B. The nonobese diabetic mouse as a model of autoimmune diabetesimmune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- Bach J.F., Mathis D. The NOD mouse. Res. Immunol. 1997;148:285–286. doi: 10.1016/s0923-2494(97)87235-5. [DOI] [PubMed] [Google Scholar]
- Atkinson M.A., Leiter E.H. The NOD mouse model of type 1 diabetesas good as it gets? Nat. Med. 1999;5:601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- Green E.A., Flavell R.A. Tumor necrosis factor-alpha and the progression of diabetes in non-obese diabetic mice. Immunol. Rev. 1999;169:11–22. doi: 10.1111/j.1600-065x.1999.tb01302.x. [DOI] [PubMed] [Google Scholar]
- Ludewig B., Odermatt B., Landmann S., Hengartner H., Zinkernagel R.M. Dendritic cells induce autoimmune diabetes and maintain disease via de novo formation of local lymphoid tissue. J. Exp. Med. 1998;188:1493–1501. doi: 10.1084/jem.188.8.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther S.A., Lopez T., Bai W., Hanahan D., Cyster J.G. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–481. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- Fu Y.X., Molina H., Matsumoto M., Huang G.M., Min J.J., Chaplin D.D. Lymphotoxin-α (LTα) supports development of splenic follicular structure that is required for IgG responses. J. Exp. Med. 1997;185:2111–2120. doi: 10.1084/jem.185.12.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.-X., Chaplin D.D. Development and maturation of secondary lymphoid tissues. Annu. Rev. Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- Iizuka K., Chaplin D.D., Wang Y., Wu Q., Pegg L.E., Yokoyama W.M., Fu Y.X. Requirement for membrane lymphotoxin in natural killer cell development. Proc. Natl. Acad. Sci. USA. 1999;96:6336–6340. doi: 10.1073/pnas.96.11.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F., Majeau G.R., Lawton P., Hochman P.S., Browning J.L. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur. J. Immunol. 1997;27:2033–2042. doi: 10.1002/eji.1830270830. [DOI] [PubMed] [Google Scholar]
- Mackay F., Browning J.L. Turning off follicular dendritic cells. Nature. 1998;395:26–27. doi: 10.1038/25630. [DOI] [PubMed] [Google Scholar]
- Ngo V., Korner H., Gunn M., Schmidt K., Riminton D.S., Cooper M., Browning J., Sedgwick J., Cyster J.G. Lymphotoxin α/β and tumor necrosis factor are required for stromal cells expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 1999;189:403–412. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., Wang Y., Wang J., Hedgeman E.O., Browning J.L., Fu Y.X. The requirement of membrane lymphotoxin for the presence of dendritic cells in lymphoid tissues. J. Exp. Med. 1999;190:629–638. doi: 10.1084/jem.190.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning J.L., Sizing I.D., Lawton P., Bourdon P.R., Rennert P.D., Majeau G.R., Ambrose C.M., Hession C., Miatkowski K., Griffiths D.A. Characterization of lymphotoxin-alpha-beta complexes on the surface of mouse lymphocytes. J. Immunol. 1997;159:3288–3298. [PubMed] [Google Scholar]
- Salomon B., Lenschow D.J., Rhee L., Ashourian N., Singh B., Sharpe A., Bluestone J.A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Lenschow D.J., Herold K.C., Rhee L., Patel B., Koons A., Qin H.Y., Fuchs E., Singh B., Thompson C.B., Bluestone J.A. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- Chatenoud L., Primo J., Bach J.F. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J. Immunol. 1997;158:2947–2954. [PubMed] [Google Scholar]
- Tamada K., Shimozaki K., Chapoval A.I., Zhu G., Sica G., Flies D., Boone T., Hsu H., Fu Y.X., Nagata S. Modulation of T-cell-mediated immunity in tumor and graft-versus-host disease models through the LIGHT co-stimulatory pathway. Nat. Med. 2000;6:283–289. doi: 10.1038/73136. [DOI] [PubMed] [Google Scholar]
- Michie S.A., Sytwu H.K., McDevitt J.O., Yang X.D. The roles of alpha 4-integrins in the development of insulin-dependent diabetes mellitus. Curr. Top. Microbiol. Immunol. 1998;231:65–83. doi: 10.1007/978-3-642-71987-5_5. [DOI] [PubMed] [Google Scholar]