A Coordinated Change in Chemokine Responsiveness Guides Plasma Cell Movements (original) (raw)
Abstract
Antibody-secreting plasma cells are nonrecirculatory and lodge in splenic red pulp, lymph node medullary cords, and bone marrow. The factors that regulate plasma cell localization are poorly defined. Here we demonstrate that, compared with their B cell precursors, plasma cells exhibit increased chemotactic sensitivity to the CXCR4 ligand CXCL12. At the same time, they downregulate CXCR5 and CCR7 and have reduced responsiveness to the B and T zone chemokines CXCL13, CCL19, and CCL21. We demonstrate that CXCL12 is expressed within splenic red pulp and lymph node medullary cords as well as in bone marrow. In chimeric mice reconstituted with CXCR4-deficient fetal liver cells, plasma cells are mislocalized in the spleen, found in elevated numbers in blood, and fail to accumulate normally in the bone marrow. Our findings indicate that as B cells differentiate into plasma cells they undergo a coordinated change in chemokine responsiveness that regulates their movements in secondary lymphoid organs and promotes lodgment within the bone marrow.
Keywords: CXCR4, CXCR5, CCR7, bone marrow, spleen
Introduction
Plasma cells play a critical role in humoral immunity by functioning as factories for antibody production. In the case of T cell–dependent antibody responses, antigen-engaged B cells receive T cell help in the region of the T zone–bordering follicles. A focus of differentiating B cell blasts soon appears and the cells move over the next day or two out of the lymphoid T zones. In the spleen, the cells move through marginal zone “bridging channels” and many of the cells lodge in foci near vessels or collagenous fibers in the red pulp 1 2 3. In lymph nodes, plasma cells are found distributed between the lymphatic sinuses of the medullary cords 4 5. Later in the response, plasma cells begin appearing in the bone marrow 6 7 8 9. While the plasma cell response is ongoing, small numbers of B cells colonize B cell follicles and differentiate into germinal center cells. After several days of development, germinal centers begin giving rise to memory B cells and more plasma cells 10 11. Plasma cells emanating from germinal centers are thought to preferentially home to the bone marrow 4 12 13. A similar series of events takes place in secondary immune responses except with faster kinetics and substantially greater accumulation of plasma cells in the bone marrow 6 8. Most of the plasma cells that lodge in splenic red pulp and lymph node medullary cords live for only a few days 8 14, whereas plasma cells that home to the bone marrow include cells that live for weeks or months 15 16.
Despite the well-characterized series of migration events, relatively little has been known about the cues directing plasma cell movements. Recent studies have highlighted the importance of members of the chemokine family in guiding cell movements within primary and secondary lymphoid organs 17. Chemokines form a family of >40 members and most are secreted proteins. All can act as chemoattractants and many have additional functions 17. Chemokine receptors are all members of the seven-transmembrane family of heterotrimeric G-protein–coupled receptors. Progenitor, immature, and mature B cells express the chemokine receptor CXCR4, and its ligand, CXCL12 (previously called stromal cell–derived factor [SDF]-1), is highly expressed by bone marrow stromal cells 18 19. Genetic studies have established that CXCL12 and CXCR4 are needed for retention of developing B cells in the bone marrow 20 21. As they mature to an IgD+ stage, B cells upregulate CXCR5 and acquire responsiveness to CXCL13 (BLC), a chemokine made in follicles of secondary lymphoid organs 22. Mature B cells also express CCR7, a receptor for the T zone chemokines CCL19 (ELC) and CCL21 (SLC), and they are able to migrate to these chemokines 17. Pointing toward the possibility that changes in chemokine responsiveness are important in directing plasma cell movements, an analysis of mouse and human plasmacytomas showed that, compared with naive B cells, they had downregulated CXCR5 23 24.
Previous studies have demonstrated that CXCL12 is made at high levels in bone marrow, a site of plasma cell homing. We demonstrate here that CXCL12 is also made in splenic red pulp and lymph node medullary cords. Plasma cells are shown to downregulate CXCR5 and CCR7 and have reduced responsiveness to B and T zone chemokines CXCL13, CCL19, and CCL21. By contrast, we find that plasma cells express CXCR4 and have increased sensitivity to CXCL12. Using CXCR4-deficient fetal liver chimeras, we show that CXCR4 plays an important role in regulating plasma cell positioning in spleen and plasma cell lodgment in bone marrow.
Materials and Methods
Mice and Chimeras.
C57BL/6 (B6), B6-Igha Thy1a GPi1a, and 129 mice were purchased from the Jackson ImmunoResearch Laboratories and B6-CD45.1 mice from Charles River Laboratories. CXCR4+/− mice 25 were on a 129 × B6 mixed genetic background. CXCR4+/− mice were intercrossed, embryos harvested on days 15–17, and livers isolated as described previously 26. To screen for CXCR4 genotype, 5% of the fetal liver cells were used as a source of DNA in PCR with primers: CXCR4-1 (5′-gatcctggtcatgggttacc-3′) and CXCR4-2 (5′-tgatgcagtaacaggagagg-3′) to detect the wild-type allele; and CXCR4-1 together with a neomycin gene specific primer (5′-cttgacgagttcttctgagggga-3′) to detect the targeted allele. For 100% chimeras, CXCR4−/−, +/− or, +/+ fetal liver cells were used to reconstitute 6–10-wk-old B6 or B6-CD45.1 mice that had been lethally irradiated by exposure to two 550 rad doses, separated by 3 h of γ irradiation from a Cesium source. During reconstitution, mice were maintained on water containing antibiotics as described previously 27. Sex-matched chimeras were used for immunizations after 5–6 wk of reconstitution. Wild-type/mutant-mixed chimeras were made using lethally irradiated 129 recipients and injecting 1–2 × 106 cells that were a mix of CXCR4-deficient IgHb fetal liver (90–95%) and CXCR4+/+ B6 IgHa bone marrow (5–10%). To make matching wild-type/wild-type control chimeras it was necessary to use lower amounts of CXCR4+/+ IgHb fetal liver (10%) in the mixture to ensure that the frequency of IgHb cells in the periphery was similar to the frequency of CXCR4−/− cells in the wild-type/mutant chimeras. Flow cytometric analysis of spleen cells from reconstituted animals using allele-specific reagents showed 5–50% of total B cells were CXCR4−/− or CXCR4+/+ fetal liver–derived and 50–95% were derived from the wild-type bone marrow.
Immunizations.
B6 mice were immunized with 100 μg of alum-precipitated nitro-phenyl (NP) chicken γ-globulin (CGG; Solid State Sciences) intraperitoneally in a volume of 200 μl. In some experiments, mice received a secondary immunization of the same type 3–5 wk later. For T cell–independent responses, mice received 15–20 μg of NP-Ficoll (Solid State Sciences) in 100 μl of PBS. Spleen, lymph nodes, blood, and bone marrow were harvested from mice at the indicated time points as described previously 27.
Chemokines and Chemotaxis Assays.
HIS6-tagged murine CXCL13/BLC was prepared as described previously 28. The HIS6-tagged murine CCL21 construct was provided by M. Gunn (Duke University, Durham, NC). The construct was transformed into Escherichia coli TAP302 cells and HIS6-tagged CCL21 was purified by the same protocol used for HIS6-tagged CCL19 29. Commercial mouse CCL21 (R&D Systems) was used in some experiments with similar results. Synthetic CXCL12 was from Gryphon Laboratories. Chemotaxis assays were performed as described previously 29 with the exception that splenocytes were incubated in 10% FCS and 5% CO2 at 37°C for 30 min before RBC lysis.
Generation of CCL19–Fc Fusion Protein.
CCL19–Fc was constructed by first using a PCR-based insertion to add a Kozak sequence and a 3′ splice recognition site to the full-length CCL19 cDNA 29. The fragment was cut with HindIII and SacI (NEB) and inserted into the Cγ1 vector (provided by Peter Lane, University of Birmingham, Birmingham, UK; reference 30). This construct was electroporated into the J558L plasmacytoma cell line and stable clones were selected using GPT selective media 31. Culture supernatants were tested for activity by their ability to induce a calcium flux in fluo-3–loaded spleen cells as described previously 32. Culture supernatant from a positive clone was purified over a protein A column and eluted using 0.1 M sodium citrate (Amersham Pharmacia Biotech), pH 4.0, and 0.2 M NaCl. On SDS-PAGE the purified CCL19–Fc protein ran at the expected size of ∼40 kD and in Western blotting it was reactive with goat anti–human Fc (Jackson ImmunoResearch Laboratories) and goat anti-CCL19 (R&D Systems). Cell lines transfected with murine CCR7 29, but not vector-control transfected cells, were stained by the CCL19–Fc fusion protein in flow cytometric analysis.
Flow Cytometry.
First, cells were incubated with rat anti-CD16/CD32 (BD PharMingen) to block Fc receptors. CCL19–Fc, CXCL12–Fc 33, hLFA3–Fc (provided by Jeff Browning, Biogen, Inc., Cambridge, MA), or rabbit anti-CXCR5 34 was then added, followed by PE-conjugated anti–human Fcγ (Jackson ImmunoResearch Laboratories) or goat anti–rabbit Ig (BD PharMingen) preincubated for 30–60 min with 2% normal rat and normal mouse serum (Sigma-Aldrich). Rat anti-B220–FITC, rat anti–syndecan-1 (CD138) –PE, and streptavidin cychrome (BD PharMingen) were added to the cells stained with chemokine receptor reagents. For CCL19–Fc and CXCL12–Fc specificity controls, soluble chemokine (5 μg/ml) was added at the initial step together with the Fc-receptor blocking antibody. For staining after chemotaxis assays, B220–FITC and syndecan-PE were used. Stained cells were analyzed on a FACScan™ (Becton Dickinson).
Histology and In Situ Hybridization.
Spleens and lymph nodes of unimmunized and immunized B6 mice were frozen in Tissue-Tek OCT compound (Baxter Scientific) and cryostat sections (7–8 μm) were fixed in acetone and stained as described previously 28 with the following reagents: goat anti-CXCL12 (Santa Cruz Biotechnology, Inc.); rat anti–syndecan–PE (BD PharMingen), or rat anti–IgM-biotin (Caltag). For some experiments, the anti-CXCL12 signal was amplified using a biotinylated donkey anti–goat antibody (Jackson ImmunoResearch Laboratories). Rat antibodies were detected using alkaline phosphatase (AP)–conjugated donkey anti–rat IgG or horseradish peroxidase (HRP)–conjugated donkey anti–rat IgG (Jackson ImmunoResearch Laboratories). Biotinylated reagents were detected using the SA-ABC AP or SA-ABC HRP kit (Vector Laboratories) or SA-AP (Jackson ImmunoResearch Laboratories). Goat antibodies were detected using AP-conjugated swine anti–goat IgG (Caltag). AP and HRP were detected as described previously 28 with the additional use of Fast Blue (Sigma-Aldrich). In situ hybridization was performed as described previously 29 using nucleotides −11 to +1,064 of SDF-1α 35 as a template for making digoxigenin-labeled riboprobes.
ELISPOT Assays.
Freshly isolated spleen, blood, bone marrow, and lymph node cells were used in ELISPOT assays as described previously 36. Plates were precoated with NP25-BSA (Solid State Sciences). IgM was detected with anti–mouse IgM biotin (Caltag) and IgG with anti–mouse IgG biotin (Jackson ImmunoResearch Laboratories) followed by SA-AP. IgMa, IgMb, IgG1a, and IgG1b were detected using biotyinylated allele-specific antibodies (BD PharMingen).
Results
CXCL12 Expression in Spleen and Lymph Nodes.
Northern blot analysis established that there is constitutive CXCL12 expression within primary and secondary lymphoid tissues 37 38. Using in situ hybridization we found that CXCL12 mRNA was expressed in the splenic red pulp and in marginal zone bridging channels of white pulp cords (Fig. 1A and Fig. C). Within lymph nodes, CXCL12 was expressed throughout the medullary cords (Fig. 1 B). Little or no hybridization was detected within follicles or T cell areas in spleen and lymph nodes. However, in several cases where large germinal centers were present, CXCL12 expression was detected in areas that reached close proximity with the germinal center (Fig. 1 B). As splenic red pulp and lymph node medullary cords are sites of plasma cell localization, we examined whether plasma cells localized in areas of CXCL12 expression. Comparison of CXCL12-hybridized and B220-stained spleen sections with nearby sections that were stained for the plasma cell marker, syndecan-1, revealed that plasma cell clusters were in areas that overlapped with regions of CXCL12 expression (Fig. 1C and Fig. D). Similar observations were made in lymph nodes (Fig. 1E and Fig. F). Northern blot analysis of spleen RNA from B cell–deficient mice showed CXCL12 expression was maintained at wild-type levels (data not shown), indicating that B-lineage cells do not make a substantial contribution to constitutive CXCL12 production. As most studies have indicated that CXCL12 is a SDF 18 35 39 40 41, we suggest that stromal cells in splenic red pulp and lymph node medullary cords are the predominant source of CXCL12 in these tissues.
Figure 1.
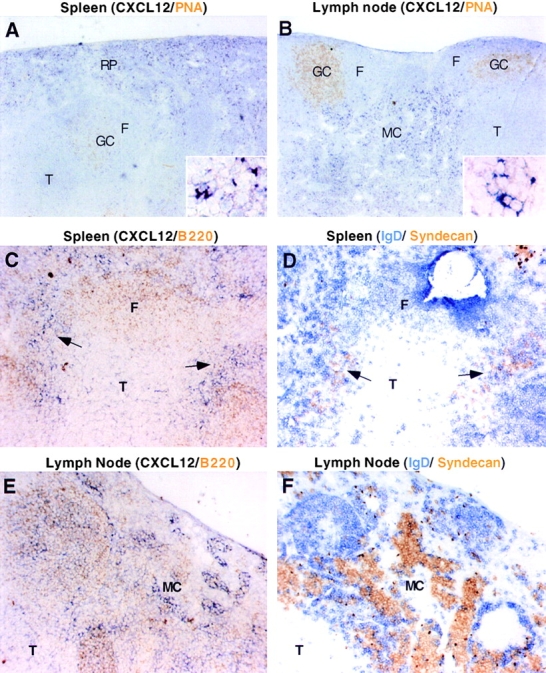
Expression pattern of CXCL12 (SDF-1) in spleen and lymph node and codistribution of CXCL12-expressing cells and plasma cells. In situ hybridization analysis of CXCL12 in spleen (A and C) and lymph node (B and E) in combination with peanut agglutinin to detect germinal centers (A and B) or B220 to localize B cell areas (C and E). D and F are from sections nearby in the tissue to those in C and E, respectively, and are stained to detect B cells (IgD, blue) and plasma cells (syndecan, brown). Insets in A and B show detail of in situ hybridization signal. Arrows in C and D point to locations where CXCL12 and syndecan staining colocalize. F, follicle; GC, germinal center; MC, medullary cords; RP, red pulp; T, T zone. (A–F) Original magnifications: 5×. (Insets) Original magnifications: 20×.
Plasma Cells Have Altered Chemokine Responsiveness.
The overlapping distribution of plasma cells and areas of CXCL12 expression led us to examine whether plasma cells could chemotax to CXCL12. Transwell chemotaxis assays were performed with spleen cells isolated from mice at day six after intraperitoneal immunization with NP-CGG in alum, near the peak of the plasma cell response. Consistent with previous findings 18 42, CXCL12 was an efficacious attractant of mature (B220+ syndecan−) B cells, with a maximum response occurring at ∼30 nM CXCL12 (Fig. 2a and Fig. b). Syndecan-1+ plasma cells also showed a chemotactic response towards CXCL12 and, notably, they exhibited greater dose sensitivity than mature B cells, responding maximally to ∼10 nM CXCL12 (Fig. 2a and Fig. c). Similar findings were made with plasma cells generated in vitro by incubating cells for 4 d with LPS (data not shown). Presently it is unclear whether the lower overall magnitude of the plasma cell response to CXCL12 compared with naive B cells is due to heterogeneity in the plasma cell population or a lower ability of the in vitro assay to support plasma cell migration. By contrast with their ability to respond to CXCL12, plasma cells had markedly reduced responsiveness to the B zone chemokine, CXCL13/BLC (Fig. 2 C), and the T zone chemokines, CCL21/SLC (Fig. 2 C) and CCL19/ELC (data not shown), across a broad range of concentrations.
Figure 2.


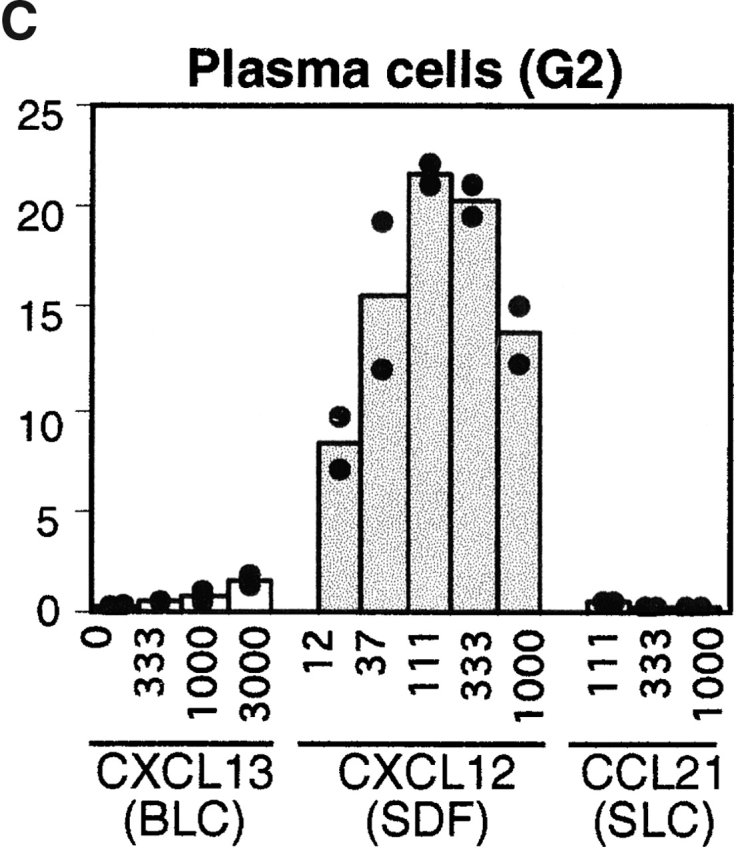
Chemotactic response profiles of B cells and plasma cells to CXCL12, CXCL13 (BLC), and CCL21 (SLC). (A) Flow cytometric analysis of input cells and cells that migrated to the lower well of a trans-well chamber in the absence of chemokine (no chemokine) or in response to 1 μg/ml CXCL13 or 0.1 μg/ml CXCL12. (B and C) Summary of chemotaxis data in response to the indicated concentrations of chemokine, expressed as the percentage of transmigrated input cells. B cells and plasma cells were identified using gates G1 and G2 shown in A. Bars represent means of duplicate transwells. Assays were performed with spleen cells from mice 6 d after primary NP-CGG/alum immunization. Data are from one experiment that is representative of more than four experiments for each chemokine.
Altered Chemokine Receptor Expression on Plasma Cells.
To determine whether the altered chemokine responsiveness of plasma cells reflected changes in chemokine receptor expression, cells were stained with CXCL12–Fc or CCL19–Fc, or with antibodies against CXCR5 (Fig. 3). Compared with naive B cells, plasma cells consistently exhibited elevated CXCL12–Fc staining (Fig. 3 A). Similar observations were made with plasma cells from lymph nodes and bone marrow (data not shown). Incubation of the cells with a molar excess of CXCL12, but not CCL19, reduced the CXCL12–Fc staining of plasma cells, confirming that the binding was chemokine specific (Fig. 3 B). To test the specificity for CXCR4, spleen cells were obtained from mice that had been irradiated and reconstituted with fetal liver cells from CXCR4+/− or CXCR4−/− embryos 25. CXCL12–Fc staining of CXCR4−/− B cells (Fig. 3 C) and plasma cells (Fig. 3 D) was reduced compared with the heterozygote controls, establishing that both cell types express CXCR4. However, CXCR4-deficient cells continued to show detectable staining with CXCL12–Fc, indicating that CXCL12 binds to surface molecules in addition to CXCR4. Previous work has shown that CXCL12 binds strongly to heparan sulfate 43 and, it is therefore possible that some of the CXCL12–Fc staining of plasma cells is due to binding to heparan sulfate-containing proteoglycans, such as syndecan-1 44. Nevertheless, CXCR4-deficient B cells and plasma cells were unable to respond to CXCL12 in chemotaxis assays (data not shown), indicating that CXCR4 is the only functional chemotactic receptor for CXCL12 on these cells.
Figure 3.

Increased CXCL12 binding and decreased CXCR5 expression and CCL19 (ELC) binding by plasma cells compared with B cells. Spleen cells from mice immunized with NP-CGG/alum 6 d before were stained with B220, syndecan, and CXCL12-Fc (A–D), anti-CXCR5 (E), or CCL19–Fc (F and G), and analyzed by flow cytometry. B cells and plasma cells were identified as in the legend to Fig. 2 A. Controls in A, B, D, and F are plasma cells and in C and G are B cells, stained with LFA-3–Fc (A–D, F and G), or no primary antibody (E). In B and F, cells were preincubated with the indicated chemokine before addition of the Fc fusion protein. In C and D, cells were from irradiated mice that had been reconstituted for 6 wk with CXCR4+/− or CXCR4−/− fetal liver cells before immunization. In all other panels, cells were from immunized wild-type mice.
By contrast with CXCR4, plasma cell expression of CXCR5 was substantially reduced (Fig. 3 E). To measure expression levels of CCL19 and CCL21 receptors, we generated a CCL19–Fc fusion protein (see Materials and Methods). CCL19–Fc staining was markedly reduced on plasma cells as compared with B cells (Fig. 3 F). The staining of B cells could be blocked by the addition of CCL19 and partially blocked with the addition of CCL21, but was not blocked at all by a variety of other chemokines (Fig. 3 G, and data not shown), establishing that the staining was specific and most likely a result of binding to CCR7. In summary, plasma cells appear to have decreased expression of CXCR5 and CCR7 and increased expression of CXCL12-binding molecules. These changes are consistent with the altered chemokine response profile of plasma cells compared with naive B cells shown in Fig. 2.
Altered Plasma Cell Localization in Spleen of CXCR4_−_ /− Fetal Liver Chimeras.
To test whether CXCR4 and CXCL12 play a role in plasma cell migration or lodgment, lethally irradiated mice that had been reconstituted with CXCR4-deficient fetal liver cells were immunized with NP-CGG, a T cell–dependent antigen. Intraperitoneal injections were performed to favor plasma cell responses within the spleen. Staining for a congenic marker, CD45.1, to distinguish donor and recipient cells established that >90% of B220+ B cells (97 ± 2%, n = 7) and syndecan-1+ plasma cells (95 ± 3%, n = 7) in CXCR4−/− fetal liver chimeras were of donor origin. Splenic B cell numbers in CXCR4-deficient chimeras were within threefold of controls, despite marked reductions in bone marrow pre-B and immature B cell numbers, as observed previously 20 21. Enumeration of anti-NP antibody-secreting cell frequencies in the spleen at day 4 after immunization revealed efficient induction of NP-specific IgM-secreting plasma cells in CXCR4−/− fetal liver chimeric mice (Fig. 4 A). Numbers of IgM-secreting cells remained within twofold of wild-type at day 6, while numbers of CXCR4-deficient IgG-secreting cells were significantly lower than in controls (Fig. 4 B).
Figure 4.

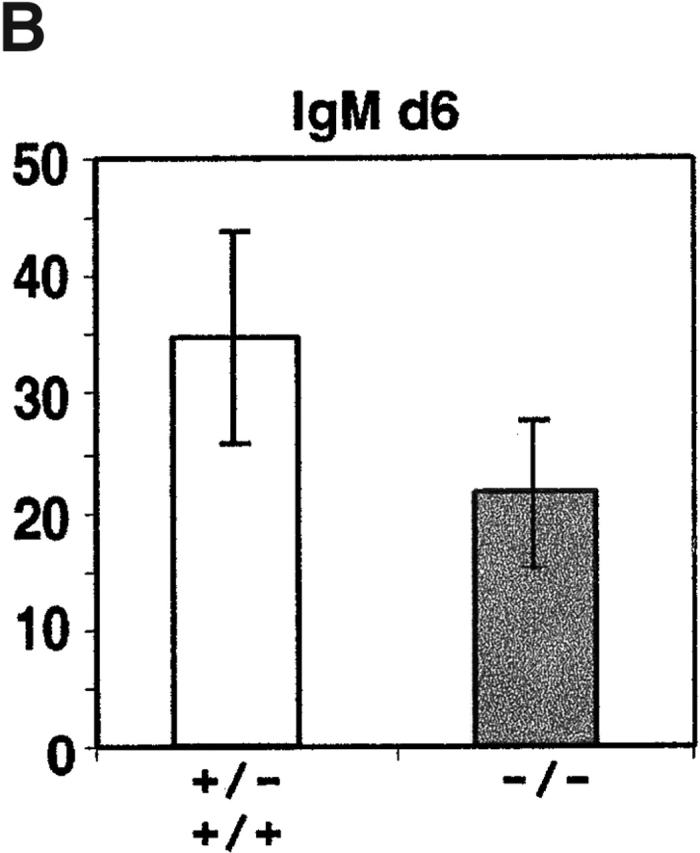
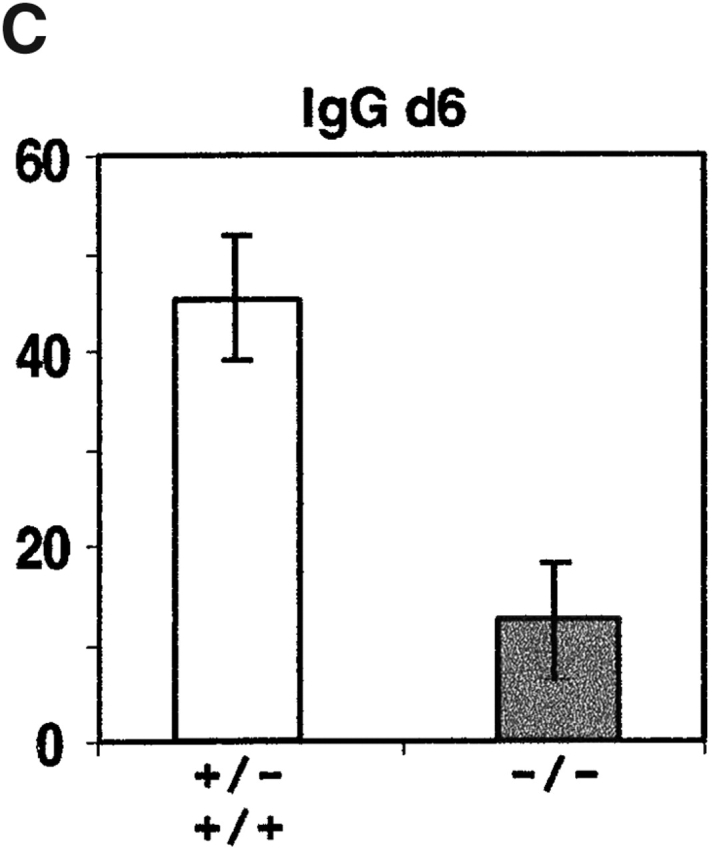
Antigen-specific IgM and IgG antibody-secreting cell numbers in spleen after primary immunization of CXCR4−/− and control (CXCR4+/− or CXCR4+/+) fetal liver chimeras. Chimeric mice were immunized intraperitoneally with NP-CGG in alum and 4 or 6 d later, spleen cells were isolated and antibody-secreting cell frequencies measured by anti–NP-ELISPOT assay. Bars represent mean number of ELISPOTs per spleen and error bars indicate 95% confidence intervals for data from groups of four mice at days 4 and 8 mice at day 6. No differences were observed between responses in +/− and +/+ animals. Average number of splenocytes per mouse: day 4, 9.4 × 107 +/+, 4.3 × 107 −/−; day 6, 1.1 × 108 +/+ and +/−, 7.0 × 107 −/−.
Examination of the plasma cell distribution within the spleen revealed a marked disturbance in CXCR4−/− fe-tal liver chimeras. Although CXCR4−/− and wild-type plasma cells appeared to exit white pulp areas with similar efficiency, CXCR4−/− cells did not lodge with normal efficiency in clusters near vessels or vessel-associated fibers in the red pulp (Fig. 5). Instead, the CXCR4-deficient plasma cells were more dispersed within the red pulp or were found associated with the marginal zone (Fig. 5A–D). Interestingly, in addition to the altered plasma cell distribution, the frequency of B220+ B cells within the red pulp of CXCR4−/− fetal liver chimeras appeared greatly reduced compared with the controls (Fig. 5A–D). Antibody responses to NP are dominated by cells expressing the λ light chain 45. As with the larger population of plasma cells detected with anti-IgG and syndecan-specific reagents, λ-positive plasma cells in CXCR4−/− chimeras were frequently found in the marginal zone area instead of localizing in the red pulp (Fig. 5E and Fig. F). Plasma cells induced in response to the T cell–independent antigen, NP-Ficoll, also showed an aberrant distribution within spleen (Fig. 5G and Fig. H). Therefore, CXCR4 is required for normal plasma cell localization in vessel- and fiber-associated clusters within the splenic red pulp.
Figure 5.
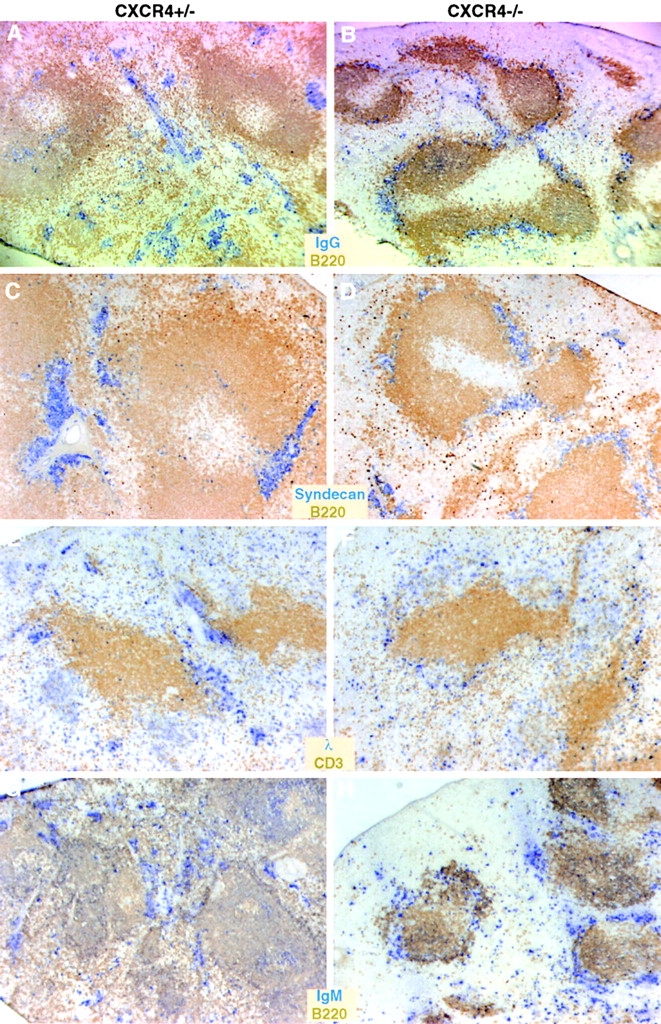
Altered plasma cell distribution in spleens of CXCR4−/− fetal liver chimeras. Spleen sections from CXCR4+/− or CXCR4−/− fetal liver chimeric mice immunized 6 d earlier with NP-CGG in alum (A–F) or with NP-Ficoll (G and H) were stained with the indicated antibodies (labels are the same color as the reaction product for that marker). Note that plasma cells contain large amounts of cytoplasmic antibody and stain strongly with the antibody-specific reagents (IgG, λ, or IgM, all in blue).
CXCR4 Is Required for Plasma Cell Accumulation in Bone Marrow.
While peripheral lymphoid tissues are important sites for lodgment of short-lived plasma cells, the bone marrow is the principal site of accumulation of long-lived plasma cells. Generation of long-lived plasma cells and redistribution of plasma cells from peripheral lymphoid tissue to bone marrow occurs to a substantially greater extent in secondary compared with primary responses 6 8. Therefore, to test whether CXCR4 functions in plasma cell trafficking to bone marrow, NP-CGG primed CXCR4−/− fetal liver chimeras and control chimeras were given secondary immunizations of NP-CGG, and plasma cell frequencies were measured 7 d later, at the peak of the bone marrow response 8. ELISPOT analysis showed that the number of NP-specific IgG-secreting plasma cells in the bone marrow was reduced more than threefold in CXCR4−/− chimeras compared with wild-type controls (Fig. 6 A).
Figure 6.

Reduced numbers of plasma cells in bone marrow and increased numbers in the blood of CXCR4−/− chimeras. (A–D) NP-specific IgG antibody-secreting cell frequencies in CXCR4−/− or control (CXCR4+/+) chimeras after secondary immunization with NP-CGG in alum. (E–H) NP-specific IgM antibody-secreting cell frequencies in CXCR4−/− or control (CXCR4+/+ or CXCR4+/−) chimeras after primary immunization with NP-CGG in alum. Tissues were isolated at the indicated time points after immunization and antibody-secreting cell frequencies determined by ELISPOT. IgM or IgG antibody-secreting cell frequencies per organ or per milliliter of blood were determined by NP-ELISPOT for four mice per group at days 3 and 7 of the secondary response, and four to seven mice per group at days 6 and 14 of the primary response. Average number of splenocytes per mouse: secondary day 3, 1.6 × 108 +/+ and 1.3 × 108 −/−; primary day 14, 8.9 × 107 +/+ and 4.3 × 107 −/−. Bone marrow cell numbers for each group differed by <30%.
Previous studies of plasma cell generation during secondary immune responses in wild-type mice have indicated that many of the cells that give rise to bone marrow plasma cells leave the spleen by days 3–4 after immunization 4 12 13. Enumeration of antibody-secreting cell frequencies in spleens of mutant and wild-type chimeras at day 3 revealed similar numbers, indicating that early events involved in plasma cell production in the spleen during the secondary immune response were intact (Fig. 6 B). Therefore, we asked whether the decreased number of plasma cells appearing in the bone marrow at day 7 could reflect a defect in cell emigration from the blood. At day 3 of the secondary response, high numbers (>2,000 per milliliter) of NP-specific IgG antibody-secreting cells were detected in the blood of wild-type animals (Fig. 6 C), consistent with this being an important period of plasma cell redistribution from spleen to bone marrow 4 12 13. Comparison of CXCR4−/− and wild-type fetal liver chimeras at this time point showed significantly higher numbers of antibody-secreting cells in the blood of CXCR4−/− chimeras (Fig. 6 C). By day 7, the number of cells in blood had decreased in both wild-type and mutant animals, but numbers in the blood of CXCR4−/− chimeras remained significantly higher than in controls (Fig. 6 D). Flow cytometric analysis with antibodies to syndecan confirmed that plasma cell frequencies were elevated in the blood of CXCR4−/− fetal liver chimeras (data not shown). These findings are consistent with a requirement for CXCR4 for IgG antibody-secreting cells to migrate from blood to bone marrow or to be retained within the marrow.
An analysis of IgM-secreting cells at day 6 of the primary NP-CGG response revealed a similar defect in CXCR4−/− chimeras compared with controls (Fig. 6E and Fig. F). Although the total number of antibody-secreting cells was lower than in the secondary response, CXCR4−/− chimeras again had elevated numbers in the blood (Fig. 6 E) and fewer in the bone marrow (Fig. 6 F), despite having similar numbers in spleen (Fig. 4 B). Similar findings were made in animals that had been given subcutaneous immunizations with NP-CGG in complete Freund's adjuvant to provoke T cell–dependent immune responses in lymph nodes, and in the response to the T cell–independent antigen, NP-Ficoll (data not shown). Analysis at day 14 of the NP-CGG/alum response showed an even more exaggerated difference in the number of IgM antibody-secreting cells in the bone marrow (Fig. 6 G). Again, numbers of anti-NP antibody-secreting cells in the spleen of mutants and controls were similar (Fig. 6 H). The frequency of plasma cells in the blood at this time was too low to measure.
Intrinsic Requirement of CXCR4 for Plasma Cell Accumulation in Bone Marrow.
To examine whether the increased number of antibody-secreting cells in the blood and decreased number in the bone marrow was due to an intrinsic defect in plasma cells or their precursors, or due to a defect in some other cell type, mixed bone marrow and fetal liver chimeras were generated. Lethally irradiated 129 (IgHa) strain mice were reconstituted with a mixture of IgHa congenic C57BL/6 bone marrow and CXCR4+/+ (IgHb) or CXCR4−/− (IgHb) fetal liver (Fig. 7 A). The mixing ratios were chosen to ensure that despite the low numbers of immature cells generated in the bone marrow, CXCR4−/− B cells made a significant contribution to the B cell compartment (see Materials and Methods). In several preliminary experiments we found that bone marrow from 129 (IgHa) mice efficiently outcompeted C57BL/6 cells during reconstitution (data not shown). Therefore, we used bone marrow from an IgHa congenic C57BL/6 strain as the wild-type partner. An analysis of mixed CXCR4+/+ and CXCR4−/− chimeras at day 6 after NP-CGG immunization revealed similar numbers of CXCR4+/+ (IgMa) and CXCR4−/− (IgMb) antibody-secreting cells in the spleen but strikingly elevated numbers of CXCR4−/− (IgMb) antibody-secreting cells in blood (Fig. 7 B). By contrast, in the control chimeras there were very few wild-type IgMb antibody-secreting cells in blood, their frequency being similar to the frequency of wild-type IgMa-secreting cells in the blood of both types of mixed chimera (Fig. 7 B). We also examined a further set of mixed chimeric animals for their IgG response 14 d after immunization, focussing on IgG1, the predominant IgG isotype produced in response to immunization with NP 45. This analysis revealed a striking deficit of CXCR4−/− IgG1b antibody-secreting cells in the bone marrow, whereas CXCR4+/+ IgG1a cells were readily detectable, despite there being similar numbers of both types of antibody-secreting cells within the spleen (Fig. 7 C). The control chimeras confirmed that IgG1b antibody-secreting cells were competent to localize within the bone marrow (Fig. 7 C) and that the deficiency, therefore, reflected an intrinsic requirement for CXCR4 in the plasma cells or their precursors.
Figure 7.
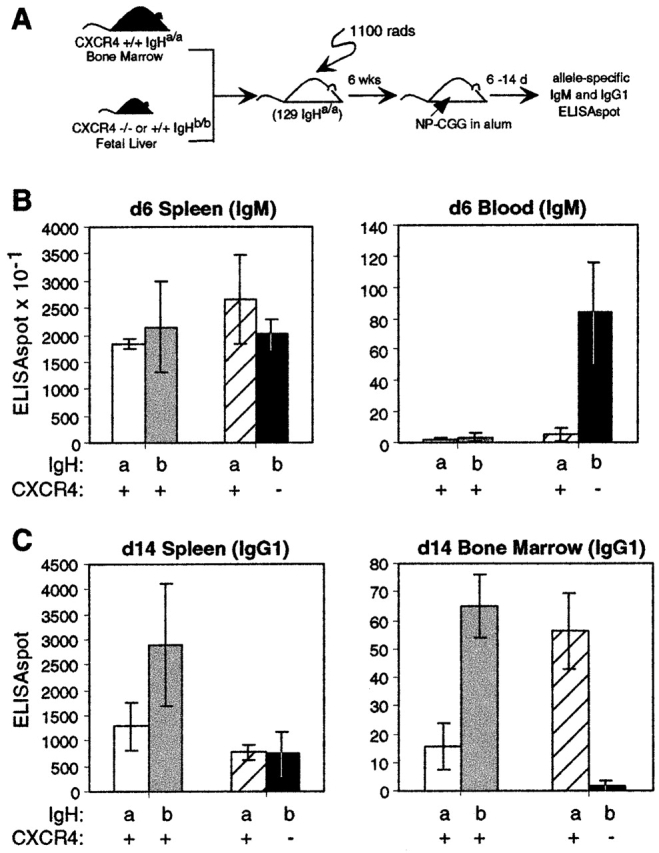
Intrinsic CXCR4 requirement for plasma cell accumulation in bone marrow. (A) Mixed bone marrow and fetal liver chimeras were generated by mixing bone marrow for IgHa B6 mice with CXCR4−/− or CXCR4+/+ IgHb fetal liver cells and reconstituting irradiated 129 (IgHa) recipients. (B and C) After 6 wk, chimeric mice were immunized with NP-CGG in alum and IgMa and IgMb (B) or IgG1a and IgG1b (C) antibody-secreting cells were measured 6 or 14 d later in spleen, blood, or bone marrow as indicated. Data from animals reconstituted with wild-type IgHa bone marrow and CXCR4+/+ IgHb fetal liver are shown in white (IgHa) and gray (IgHb) bars; data from animals reconstituted with wild-type IgHa bone marrow and CXCR4−/− IgHb fetal liver are shown in striped (IgHa) and black (IgHb) bars. Numbers of antibody-secreting cells per organ or per milliliter of blood in each animal were measured as indicated. Total splenocyte numbers for each group of animals differed by <30%. Bars represent mean and error bars indicate 95% confidence intervals for groups of three mice.
Discussion
The above findings establish that as B cells differentiate into plasma cells, they undergo a coordinated change in chemokine receptor expression and chemokine responsiveness. This includes decreased cell surface expression of the receptors for the B and T zone chemokines CXCL13, CCL19, and CCL21, and decreased chemotactic responsiveness to these chemokines. At the same time, the cells upregulate their sensitivity to the CXCR4 ligand CXCL12. CXCL12 is highly expressed in bone marrow, splenic red pulp, and lymph node medullary cords and our genetic studies have demonstrated that CXCR4 is required for normal plasma cell positioning within the spleen and for normal accumulation of plasma cells within the bone marrow. We suggest a model where decreased responsiveness to CXCL13, CCL19, and CCL21 helps direct plasma cells out of B and T zones, while the CXCR4/CXCL12 receptor–ligand pair regulates splenic red pulp localization and bone marrow lodgment.
The possibility that plasma cells would have altered chemokine receptor expression compared with B cells was suggested by the finding that plasmacytomas lacked CXCR5 expression 23 24. In a recent study published while our work was being completed, Wehrli and coworkers demonstrated that plasma cells downregulate CXCR5 mRNA and have reduced responsiveness to CXCL13, CCL19, and CXCL12 46. Consistent with these reports, we found that plasma cells downregulate surface CXCR5 and CCL19 receptors (presumably CCR7) and lose responsiveness to CXCL13, CCL19, and CCL21. Also in agreement with Wehrli et al., we observed that plasma cells maintain CXCR4 expression. However, in contrast to their report, we found that plasma cells exhibited a significant chemotactic response to CXCL12. The basis for this difference in our findings is unclear, although it may reflect differences in the sources of plasma cells or technical differences in the chemotaxis assays.
Genetic experiments have established that B cell entry to lymphoid follicles depends on CXCR5 and its ligand, CXCL13, 47 48 and entry of T cells and dendritic cells to the T zone depends on expression of CCR7 and ligands, CCL19 and CCL21 49 50. Therefore, downregulation of CXCR5 and CCR7, and decreased responsiveness to B and T zone chemokines, seems likely to be an important part of the mechanism promoting plasma cell movement out of the B and T cell areas in spleen and lymph node. Although we found CXCL12 is expressed in the splenic red pulp and lymph node medullary cords, our observations in mice reconstituted with CXCR4-deficient fetal liver cells suggest that CXCR4 does not play a limiting role in plasma cell movement out of follicles or T cell areas. Perhaps plasma cell exclusion from T and B zones occurs as a result of displacement by recirculating lymphocytes. Consistent with this, one study reported that in T cell–deficient mice, plasma cells tended to accumulate in the area corresponding to the T zone 51. However, the involvement of other cues in directing plasma cells out of T or B cell areas has not been excluded.
In the spleen, plasma cells leave the white pulp cords by marginal zone bridging channels, areas where the T zone interfaces with the red pulp 1. Plasma cells appear in these regions as large foci before later moving deeper into the red pulp where they become associated with vessels or connective tissue fibers 1. CXCL12 expression was detected in most areas of the splenic red pulp, including the bridging channels and in regions proximal to vessels and fibers (Fig. 1, and data not shown). We did not observe CXCL12 transcripts in follicular mantle zones, although CXCL12-expressing cells were sometimes observed in close proximity with germinal centers. In a study of human tonsils, it was reported that CXCL12 message could be detected in connective tissue cells surrounding germinal centers, including expression in follicular mantle zone areas 52. The basis for the discrepancy between our findings and those of Bleul et al. 52 is unclear but might reflect differences in chemokine expression between specific pathogen-free mouse tissue and inflammed human tonsil. In agreement with the mouse CXCL12 expression data, CXCR4-deficient B cells are able to form primary follicles that appear normal in structure (Fig. 5, and reference 20). It remains to be determined whether CXCR4 and CXCL12 have a function in the germinal center response. A role at some step preceding the plasma cell is suggested by the lower number of IgG-secreting cells generated in CXCR4−/− chimeras during the primary response. Interestingly, the numbers of IgD+ mature B cells in the red pulp of spleens from CXCR4−/− fetal liver chimeras was substantially reduced (Fig. 5), suggesting that lodgment of mature B cells in this area occurs at least in part in response to CXCL12. Whether CXCL12 is involved in promoting lodgment of other cell types in the splenic red pulp, such as activated CD8 T cells 53 and NK cells 54, requires further investigation.
In the absence of CXCR4, plasma cells tended to remain in bridging channels and were often observed in or adjacent to the splenic marginal zone (Fig. 5). The factors guiding cells to the marginal zone are unknown. In contrast to effects in the red pulp, CXCR4-deficient B cells were still efficient in populating the splenic marginal zone (Fig. 5 B, and data not shown). As previous findings have shown that B cell localization in the marginal zone is independent of CXCR5/CXCL13 28, but is sensitive to Pertussis toxin 55, a chemoattractant other than CXCL12 or CXCL13 is likely to be involved. Although CXCR4 is necessary for the normal distribution of plasma cells near vessels and fibers in the splenic red pulp, expression of CXCL12 is not limited to these locations (Fig. 1). Therefore, we speculate that additional factors, such as extracellular matrix proteins, adhesion molecules, or other chemokines, act together with CXCR4 and CXCL12 to regulate splenic plasma cell positioning.
In contrast to the disturbed plasma cell distribution within spleen, we have so far not observed effects of CXCR4 deficiency on plasma cell distribution or numbers in lymph nodes (data not shown). However, a possible role in microlocalization of cells within the medullary cords is not excluded. In this context, it is interesting that Wehrli and coworkers 46 focused on lymph node plasma cells and found that the cells did not migrate to CXCL12. Further work is needed to define whether CXCR4 and CXCL12 function in regulating the distribution or properties of plasma cells within lymph nodes.
The bone marrow is an important site of plasma cell accumulation in humans and rodents 6. Recent studies have indicated that bone marrow plasma cells may survive for extended periods, possibly many months, and it has been suggested that such long-lived cells make an important contribution to the maintenance of serum antibody levels long after antigen exposure 7 15 16. CXCR4 and CXCL12 have well-established roles within the bone marrow. Bone marrow stromal cells were identified as one of the most plentiful sources of CXCL12 18 35 39 40, and more recently bone marrow sinusoidal endothelial cells were found to express CXCL12 56 57 and to support CXCL12-dependent rolling and tethering of CXCR4+ cells 57. CXCR4 and CXCL12 play important roles in retaining pre-B cells and granulocytes in the bone marrow 20 21 25. The ability of this chemokine receptor–ligand pair to direct peripheral cell types to the bone marrow was suggested by the finding that transgenic overexpression of CXCR4 on CD4 T cells caused the cells to accumulate in the bone marrow 58. Our findings indicate that CXCR4 is needed for efficient plasma cell accumulation in the bone marrow. Presently, our studies do not distinguish between a role in promoting entry to this compartment or in promoting retention of cells after they have entered. They also do not exclude the possibility that CXCL12 functions to augment plasma cell survival within the marrow, although the elevated numbers of CXCR4-deficient antibody-secreting cells in circulation argues against CXCR4 functioning solely to transmit survival signals. We presently favor a model where the function of CXCR4 and CXCL12 in plasma cells is similar to its role in pre-B cells, promoting retention within the bone marrow. In support of a similar mechanism of action on these two cell populations, in in vitro chemotaxis assays, plasma cells (Fig. 2), and pre-B cells 19 59 60, both have elevated dose sensitivity to CXCL12 compared with mature B cells.
In addition to CXCL12, several adhesion molecules have been implicated in plasma cell localization within the bone marrow. Mice lacking CD22 have reduced numbers of bone marrow plasma cells 61 and recent conditional gene ablation experiments 62 63 have added to earlier evidence 64 implicating vascular cell adhesion molecule 1 and α4 integrins in B cell homing to bone marrow. Plasma cells have high levels of α4β1 integrin expression 46 65. As CXCL12 has been shown to activate α4 integrin binding 66 67, it seems likely that these molecules function together in promoting plasma cell accumulation within the bone marrow. Further investigation is required to determine the basis for the propensity of some plasma cells to remain in secondary lymphoid organs versus the propensity of others, particularly those generated early during the secondary response, to home to the bone marrow. Our studies suggest CXCR4 functions in both populations and therefore, we propose that other differences, perhaps responsiveness to other chemokines or differences in adhesion molecule expression, account for the differing tropisms.
In summary, we have demonstrated that CXCR4 is necessary for normal plasma cell accumulation in subcompartments of splenic red pulp and within bone marrow. As well as their implications for understanding normal antibody responses, our findings add to other studies implicating CXCR4 and CXCL12 in directing leukemia cells to bone marrow 40 68, and they suggest that in cases where CXCL12 is expressed at sites of chronic inflammation, such as within the joint synovium of rheumatoid arthritis patients 69, it will contribute to the local retention of plasma cells. This work also highlights the need to consider the impact of CXCR4-blocking treatments, such as those being investigated for anti-HIV therapy, on plasma cell homing, and antibody responses.
Acknowledgments
We thank Peter Lane for the Cγ1 vector and Sanjiv Luther for comments on the manuscript.
T.T. Lu is a National Institutes of Health Fellow of the Pediatric Scientist Development Program (NICHD grant award K12-HD00850). This work was supported by the Howard Hughes Medical Institute and by National Institutes of Health grant AI40098 to J.G. Cyster.
Footnotes
Abbreviations used in this paper: AP, alkaline phosphatase; CGG, chicken γ-globulin; HRP, horseradish peroxidase; NP, nitro-phenyl; SDF, stromal cell–derived factor.
D.C. Hargreaves and P.L. Hyman contributed equally to this work.
References
- van Rooijen N., Claassen E., Eikelenboom P. Is there a single differentiation pathway for all antibody-forming cells in the spleen? Immunol. Today. 1986;7:193–196. doi: 10.1016/0167-5699(86)90100-3. [DOI] [PubMed] [Google Scholar]
- Jacob J., Kassir R., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-J., Zhang J., Lane P.J.L., Chan E.Y.-T., MacLennan I.C.M. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur. J. Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- Kosco M.H., Burton G.F., Kapasi Z.F., Szakal A.K., Tew J.G. Antibody-forming cell induction during an early phase of germinal centre development and its delay with aging. Immunology. 1989;68:312–318. [PMC free article] [PubMed] [Google Scholar]
- Luther S.A., Gulbranson-Judge A., Acha-Orbea H., MacLennan I.C. Viral superantigen drives extrafollicular and follicular B cell differentiation leading to virus-specific antibody production. J. Exp. Med. 1997;185:551–562. doi: 10.1084/jem.185.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner R., Hijmans W., Haaijman J.J. The bone marrowthe major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin. Exp. Immunol. 1981;46:1–8. [PMC free article] [PubMed] [Google Scholar]
- Bachmann M.F., Kundig T.M., Odermatt B., Hengartner H., Zinkernagel R.M. Free recirculation of memory B cells versus antigen-dependent differentiation to antibody-forming cells. J. Immunol. 1994;153:3386–3397. [PubMed] [Google Scholar]
- Smith K.G.C., Hewitson T.D., Nossal G.J.V., Tarlinton D.M. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur. J. Immunol. 1996;26:444–448. doi: 10.1002/eji.1830260226. [DOI] [PubMed] [Google Scholar]
- Slifka M.K., Matloubian M., Ahmed R. Bone marrow is a major site of long-term antibody production after acute viral infection. J. Virol. 1995;69:1895–1902. doi: 10.1128/jvi.69.3.1895-1902.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan I.C.M. Germinal centers. Annu. Rev. Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- Kelsoe G. The germinal centera crucible for lymphocyte selection. Semin. Immunol. 1996;8:179–184. doi: 10.1006/smim.1996.0022. [DOI] [PubMed] [Google Scholar]
- Benner R., van Oudenaren A., de Ruiter H. Antibody formation in mouse bone marrow. IX. Peripheral lymphoid organs are involved in the initiation of bone marrow antibody formation. Cell. Immunol. 1977;34:125–137. doi: 10.1016/0008-8749(77)90235-0. [DOI] [PubMed] [Google Scholar]
- Dilosa R.M., Maeda K., Masuda A., Szakal A.K., Tew J.G. Germinal center B cells and antibody production in the bone marrow. J. Immunol. 1991;146:4071–4077. [PubMed] [Google Scholar]
- Ho F., Lortan J.E., MacLennan I.C., Khan M. Distinct short-lived and long-lived antibody-producing cell populations. Eur. J. Immunol. 1986;16:1297–1301. doi: 10.1002/eji.1830161018. [DOI] [PubMed] [Google Scholar]
- Manz R.A., Thiel A., Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- Slifka M.K., Ahmed R. Long-lived plasma cellsa mechanism for maintaining persistent antibody production. Curr. Opin. Immunol. 1998;10:252–258. doi: 10.1016/s0952-7915(98)80162-3. [DOI] [PubMed] [Google Scholar]
- Cyster J.G. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–2102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- Bleul C.C., Fuhlbrigge R.C., Casasnovas J.M., Aiuti A., Springer T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J. Exp. Med. 1996;184:1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Apuzzo M., Rolink A., Loetscher M., Hoxie J.A., Clark-Lewis I., Melchers F., Baggiolini M., Moser B. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur. J. Immunol. 1997;27:1788–1793. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- Ma Q., Jones D., Springer T.A. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- Kawabata K., Ujikawa M., Egawa T., Kawamoto H., Tachibana K., Iizasa H., Katsura Y., Kishimoto T., Nagasawa T. A cell-autonomous requirement for CXCR4 in long-term lymphoid and myeloid reconstitution. Proc. Natl. Acad. Sci. USA. 1999;11:5663–5667. doi: 10.1073/pnas.96.10.5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster J.G., Ngo V.N., Ekland E.H., Gunn M.D., Sedgwick J.D., Ansel K.M. Chemokines and B-cell homing to follicles. Curr. Top. Microbiol. Immunol. 1999;246:87–92. doi: 10.1007/978-3-642-60162-0_11. [DOI] [PubMed] [Google Scholar]
- Kaiser E., Forster R., Wolf I., Ebensperger C., Kuehl W.M., Lipp M. The G-protein coupled receptor BLR1 is involved in murine B cell differentiation and is also expressed in neuronal tissues. Eur. J. Immunol. 1993;23:2532–2539. doi: 10.1002/eji.1830231023. [DOI] [PubMed] [Google Scholar]
- Forster R., Emrich T., Kremmer E., Lipp M. Expression of the G-protein-coupled receptor BLR1 defines mature, recirculating B cells and a subset of T-helper memory cells. Blood. 1994;84:830–840. [PubMed] [Google Scholar]
- Zou Y.R., Kottmann A.H., Kuroda M., Taniuchi I., Littman D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- Birnbaum R.A., O'Marcaigh A., Wardak Z., Zhang Y.Y., Dranoff G., Jacks T., Clapp D.W., Shannon K.M. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol. Cell. 2000;5:189–195. doi: 10.1016/s1097-2765(00)80415-3. [DOI] [PubMed] [Google Scholar]
- Cyster J.G., Goodnow C.C. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 1995;3:691–701. doi: 10.1016/1074-7613(95)90059-4. [DOI] [PubMed] [Google Scholar]
- Ansel K.M., McHeyzer-Williams L.J., Ngo V.N., McHeyzer-Williams M.G., Cyster J.G. In vivo–activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J. Exp. Med. 1999;190:1123–1134. doi: 10.1084/jem.190.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo V.N., Tang H.L., Cyster J.G. Epstein-Barr virus-induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J. Exp. Med. 1998;188:181–191. doi: 10.1084/jem.188.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane P., Burdet C., Hubele S., Scheidegger D., Muller U., McConnell F., Kosco-Vilbois M. B cell function in mice transgenic for mCTLA4-H γ1lack of germinal centers correlated with poor affinity maturation and class switching despite normal priming of CD4+ T cells. J. Exp. Med. 1994;179:819–830. doi: 10.1084/jem.179.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traunecker A., Oliveri F., Karjalainen K. Myeloma based expression system for production of large mammalian proteins. Trends Biotechnol. 1991;9:109–113. doi: 10.1016/0167-7799(91)90038-j. [DOI] [PubMed] [Google Scholar]
- Matloubian M., David A., Engel S., Ryan J.E., Cyster J.G. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat. Immunol. 2000;1:298–304. doi: 10.1038/79738. [DOI] [PubMed] [Google Scholar]
- Suzuki G., Nakata Y., Dan Y., Uzawa A., Nakagawa K., Saito T., Mita K., Shirasawa T. Loss of SDF-1 receptor expression during positive selection in the thymus. Int. Immunol. 1998;10:1049–1056. doi: 10.1093/intimm/10.8.1049. [DOI] [PubMed] [Google Scholar]
- Schmidt K.N., Hsu C.W., Griffin C.T., Goodnow C.C., Cyster J.G. Spontaneous follicular exclusion of SHP1-deficient B cells is conditional on the presence of competitor wild-type B cells. J. Exp. Med. 1998;187:929–937. doi: 10.1084/jem.187.6.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro K., Tada H., Heilker R., Shirozu M., Nakano T., Honjo T. Signal sequence trapa cloning strategy for secreted proteins and type I membrane proteins. Science. 1993;261:600–603. doi: 10.1126/science.8342023. [DOI] [PubMed] [Google Scholar]
- Goodnow C.C., Crosbie J., Jorgensen H., Brink R.A., Basten A. Induction of self-tolerance in mature peripheral B lymphocytes. Nature. 1989;342:385–391. doi: 10.1038/342385a0. [DOI] [PubMed] [Google Scholar]
- Jiang W., Zhou P., Kahn S.M., Tomita N., Johnson M.D., Weinstein I.B. Molecular cloning of TPAR1, a gene whose expression is repressed by the tumor promoter 12-O-tetradecanoylphorbol 13-acetate (TPA) Exp. Cell Res. 1994;215:284–293. doi: 10.1006/excr.1994.1344. [DOI] [PubMed] [Google Scholar]
- Ngo V.N., Korner H., Gunn M.D., Schmidt K.N., Riminton D.S., Cooper M.D., Browning J.L., Sedgwick J.D., Cyster J.G. Lymphotoxin-α/β and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 1999;189:403–412. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa T., Kikutani H., Kishimoto T. Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc. Natl. Acad. Sci. USA. 1994;91:2305–2309. doi: 10.1073/pnas.91.6.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger J.A., Burger M., Kipps T.J. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94:3658–3667. [PubMed] [Google Scholar]
- Arai J., Yasukawa M., Yakushijin Y., Miyazaki T., Fujita S. Stromal cells in lymph nodes attract B-lymphoma cells via production of stromal cell-derived factor-1. Eur. J. Haematol. 2000;64:323–332. doi: 10.1034/j.1600-0609.2000.90147.x. [DOI] [PubMed] [Google Scholar]
- Gunn M.D., Ngo V.N., Ansel K.M., Ekland E.H., Cyster J.G., Williams L.T. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- Amara A., Lorthioir O., Valenzuela A., Magerus A., Thelen M., Montes M., Virelizier J.L., Delepierre M., Baleux F., Lortat-Jacob H., Arenzana-Seisdedos F. Stromal cell-derived factor-1α associates with heparan sulfates through the first β-strand of the chemokine. J. Biol. Chem. 1999;274:23916–23925. doi: 10.1074/jbc.274.34.23916. [DOI] [PubMed] [Google Scholar]
- Sanderson R.D., Sneed T.B., Young L.A., Sullivan G.L., Lander A.D. Adhesion of B lymphoid (MPC-11) cells to type I collagen is mediated by integral membrane proteoglycan, syndecan. J. Immunol. 1992;148:3902–3911. [PubMed] [Google Scholar]
- Jack R.S., Imanishi-Kari T., Rajewsky K. Idiotypic analysis of the response of C57BL/6 mice to the (4-hydroxy-3-nitrophenyl)acetyl group. Eur. J. Immunol. 1977;7:559–565. doi: 10.1002/eji.1830070813. [DOI] [PubMed] [Google Scholar]
- Wehrli N., Legler D.F., Finke D., Toellner K.-M., Loetscher P., Baggiolini M., MacLennan I.C.M., Acha-Orbea H. Changing responsiveness to chemokines allows medullary plasmablasts to leave lymph nodes. Eur. J. Immunol. 2001;31:609–616. doi: 10.1002/1521-4141(200102)31:2<609::aid-immu609>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Forster R., Mattis A.E., Kremmer E., Wolf E., Brem G., Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- Ansel K.M., Ngo V.N., Hyman P.L., Luther S.A., Forster R., Sedgwick J.D., Browning J.L., Lipp M., Cyster J.G. A chemokine driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- Gunn M.D., Kyuwa S., Tam C., Kakiuchi T., Matsuzawa A., Williams L.T., Nakano H. Mice lacking expression of secondary lymphoid-organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster R., Schubel A., Breitfeld D., Kremmer E., Renner-Muller I., Wolf E., Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Garcia de Vinuesa C., O'Leary P., Sze D.M., Toellner K.M., MacLennan I.C. T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. Eur. J. Immunol. 1999;29:1314–1323. doi: 10.1002/(SICI)1521-4141(199904)29:04<1314::AID-IMMU1314>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Bleul C.C., Schultze J.L., Springer T.A. B lymphocyte chemotaxis regulated in association with microanatomic localization, differentiation state, and B cell receptor engagement. J. Exp. Med. 1998;187:753–762. doi: 10.1084/jem.187.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potsch C., Vohringer D., Pircher H. Distinct migration patterns of naive and effector CD8 T cells in the spleencorrelation with CCR7 receptor expression and chemokine reactivity. Eur. J. Immunol. 1999;29:3562–3570. doi: 10.1002/(SICI)1521-4141(199911)29:11<3562::AID-IMMU3562>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Biology of natural killer cells. Adv. Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard R., Okigaki M., Schlessinger J., Ravetch J.V. Absence of marginal zone B cells in Pyk-2 deficient mice define their role in the humoral response. Nat. Immunol. 2000;1:31–36. doi: 10.1038/76882. [DOI] [PubMed] [Google Scholar]
- Imai K., Kobayashi M., Wang J., Shinobu N., Yoshida H., Hamada J., Shindo M., Higashino F., Tanaka J., Asaka M., Hosokawa M. Selective secretion of chemoattractants for haemopoietic progenitor cells by bone marrow endothelial cellsa possible role in homing of haemopoietic progenitor cells to bone marrow. Br. J. Haematol. 1999;106:905–911. doi: 10.1046/j.1365-2141.1999.01644.x. [DOI] [PubMed] [Google Scholar]
- Peled A., Grabovsky V., Habler L., Sandbank J., Arenzana-Seisdedos F., Petit I., Ben-Hur H., Lapidot T., Alon R. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34+ cells on vascular endothelium under shear flow. J. Clin. Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada S., Gowrishankar K., Kitamura R., Suzuki M., Suzuki G., Tahara S., Koito A. Disturbed CD4+ T cell homeostasis and in vitro HIV-1 susceptibility in transgenic mice expressing T cell line-tropic HIV-1 receptors. J. Exp. Med. 1998;187:1439–1449. doi: 10.1084/jem.187.9.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedyk E.R., Ryyan D.H., Ritterman I., Springer T.A. Maturation decreases responsiveness of human bone marrow B lineage cells to stromal-derived factor 1 (SDF-1) J. Leukoc. Biol. 1999;66:667–673. doi: 10.1002/jlb.66.4.667. [DOI] [PubMed] [Google Scholar]
- Honczarenko M., Douglas R.S., Mathias C., Lee B., Ratajczak M.Z., Silberstein L.E. SDF-1 responsiveness does not correlate with CXCR4 expression levels of developing human bone marrow B cells. Blood. 1999;94:2990–2998. [PubMed] [Google Scholar]
- Nitschke L., Floyd H., Ferguson D.J., Crocker P.R. Identification of CD22 ligands on bone marrow sinusoidal endothelium implicated in CD22-dependent homing of recirculating B cells. J. Exp. Med. 1999;189:1513–1518. doi: 10.1084/jem.189.9.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni P.A., Joshi S.K., Temann U.A., Olson D., Burkly L., Flavell R.A. Conditional vascular cell adhesion molecule 1 deletion in mice. Impaired lymphocyte migration to bone marrow. J. Exp. Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuker C.E., Labow M., Muller W., Wagner N. Neonatally induced inactivation of the vascular cell adhesion molecule 1 gene impairs β cell localization and T cell–dependent humoral immune response. J. Exp. Med. 2001;193:755–768. doi: 10.1084/jem.193.6.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin-Rufenach C., Otto F., Mathies M., Westermann J., Owen M.J., Hamann A., Hogg N. Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1–deficient mice. J. Exp. Med. 1999;189:1467–1478. doi: 10.1084/jem.189.9.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H., Kawano M.M., Huang N., Harada Y., Iwato K., Tanabe O., Tanaka H., Sakai A., Asaoku H., Kuramoto A. Phenotypic difference of normal plasma cells from mature myeloma cells. Blood. 1993;81:2658–2663. [PubMed] [Google Scholar]
- Grabovsky V., Feigelson S., Chen C., Bleijs D.A., Peled A., Cinamon G., Baleux F., Arenzana-Seisdedos F., Lapidot T., van Kooyk Y. Subsecond induction of α4 integrin clustering by immobilized chemokines stimulates leukocyte tethering and rolling on endothelial vascular cell adhesion molecule 1 under flow conditions. J. Exp. Med. 2000;192:495–506. doi: 10.1084/jem.192.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled A., Kollet O., Ponomaryov T., Petit I., Franitza S., Grabovsky V., Slav M.M., Nagler A., Lider O., Alon R. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34+ cellsrole in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95:3289–3296. [PubMed] [Google Scholar]
- Mohle R., Schittenhelm M., Failenschmid C., Bautz F., Kratz-Albers K., Serve H., Brugger W., Kanz L. Functional response of leukaemic blasts to stromal cell-derived factor-1 correlates with preferential expression of the chemokine receptor CXCR4 in acute myelomonocytic and lymphoblastic leukaemia. Br. J. Haematol. 2000;110:563–572. doi: 10.1046/j.1365-2141.2000.02157.x. [DOI] [PubMed] [Google Scholar]
- Buckley C.D., Amft N., Bradfield P.F., Pilling D., Ross E., Arenzana-Seisdedos F., Amara A., Curnow S.J., Lord J.M., Scheel-Toellner D., Salmon M. Persistent induction of the chemokine receptor CXCR4 by TGF-β1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J. Immunol. 2000;165:3423–3429. doi: 10.4049/jimmunol.165.6.3423. [DOI] [PubMed] [Google Scholar]