Requirement for Transforming Growth Factor β1 in Controlling T Cell Apoptosis (original) (raw)
Abstract
Transforming growth factor (TGF)-β1, a potent immunoregulatory molecule, was found to control the life and death decisions of T lymphocytes. Both thymic and peripheral T cell apoptosis was increased in mice lacking TGF-β1 (TGF-β1−/−) compared with wild-type littermates. Engagement of the T cell receptor enhanced this aberrant T cell apoptosis, as did signaling through either the death receptor Fas or the tumor necrosis factor α receptor in peripheral T cells. Strikingly, TGF-β was localized within the mitochondria of normal T cells, and the absence of TGF-β1 resulted in disruption of mitochondrial membrane potential (Δψm), which marks the point of no return in a cell condemned to die. This TGF-β–dependent regulation of viability appears dissociable from the TGF-β1 membrane receptor–Smad3 signaling pathway, but associated with a mitochondrial antiapoptotic protein Bcl–XL. Thus, TGF-β1 may protect T cells at multiple sites in the death pathway, particularly by maintaining the essential integrity of mitochondria. These findings may have broad implications not only for T cell selection and death in immune responses and in the generation of tolerance, but also for defining the mechanisms of programmed cell death in general.
Keywords: TCR, mitochondrial membrane potential, Fas/TNF-α receptor, Smad3, Bcl–XL
Introduction
Null mutation of the murine TGF-β1 gene (TGF-β1−/−) results in multiorgan lymphocyte and macrophage infiltration, massive inflammation, and untimely death of the animals within 3–4 wk 1 2, consistent with the critical role of TGF-β1 in regulating immune and inflammatory responses 3 4 5 6. Paradoxically, however, attempts to induce peripheral null T cell proliferative responses by triggering the TCR with anti-CD3 monoclonal antibody or with mitogens in vitro were unsuccessful 7 8. Moreover, these mice have smaller thymuses and spleens than those of age-matched TGF-β1+/+ littermates 1 7, indicative of T cell depletion, perhaps through enhanced apoptosis.
The apoptotic process can be divided into at least three functionally overlapping phases: initiation, effector, and degradation 9 10 11 12. In T cells, the initiation phase includes ligation of death receptors, particularly Fas 13 14 15 16 17 and TNF-α receptors 18. However, normal T cells 19 20 are typically resistant to apoptosis induced with the counterreceptor Fas ligand (FasL), despite expression of Fas antigen. Only extensive antigenic stimulation in the presence of IL-2 (generation of T cell blasts) is reportedly able to sensitize peripheral T cells to FasL and TNF-α, rendering them susceptible to activation-induced cell death (AICD). During the subsequent effector phase, death receptor–mediated signals can be translated into a regular pattern of metabolic reactions and the “decision to die” is taken. In this chain of events, changes in mitochondrial membrane potential (Δψm) appear irreversibly lethal for the cell and can lead to release of death factors 9 10 11 12. How normal Δψm is maintained is not fully understood, although antiapoptotic Bcl-2 family members reportedly play a part in preventing both Δψm dissipation and the release of cytochrome c and/or apoptosis-inducing factor 21 22 23.
Although TGF-β1 has been implicated in influencing AICD in T cells, the mechanism remains unclear 24 25 26. Whereas one report demonstrated that TGF-β1 inhibited FasL expression without affecting Fas-mediated death signaling, another indicated that TGF-β1 did reduce Fas-mediated apoptosis. These controversial findings are based on the effects of exogenous TGF-β1 on normal T cells or hybridoma cell lines, which produce their own TGF-β 8 27 28 29. Another intriguing observation was the identification of TGF-β within mitochondria 30, but its function in this context is totally unknown. We investigated how TGF-β1 influences apoptotic processes by examining the death behavior of T cells deficient in TGF-β1 (TGF-β1−/−) or lacking an intact TGF-β signaling pathway (Smad3−/−).
Materials and Methods
Mice.
TGF-β1−/− (C57BL/6×Sv129; reference 2) and age-matched TGF-β1+/+ mice were housed in a specific pathogen-free rodent facility at the National Institute of Dental and Craniofacial Research. DO11.10 OVA peptide TCR transgenic mice were purchased from The Jackson Laboratory.
In Situ Detection of Apoptotic Cells in Tissues with Terminal Deoxynucleotidyl Transferase (TdT)-mediated dUTP-biotin Nick End-labeling Method.
Thymuses and spleens from TGF-β1−/− and age-matched TGF-β1+/+ mice were fixed with 10% buffered formalin and 5–6-μ paraffin sections placed on slides. DNA fragmentation was detected with a terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL) kit (ApopTag@Plus Peroxidase, S7101-kit; Intergen Co.) after 1 h treatment with proteinase K and counterstained with 0.5% (wt/vol) methyl green for photomicroscopy with a Leica microscope.
7-Amino-Actinomycin D Staining of Apoptotic Cells.
Staining of apoptotic cells with 7-amino-actinomycin D (7-AAD; Calbiochem) was performed as described previously 8 31. In brief, cells were first stained with FITC–anti-CD8 and PE–anti-CD4 mAbs (Caltag), washed, and incubated with 7-AAD (20 μg/ml) in PBS containing 2% FBS and 0.1% sodium azide at 4°C for 20 min protected from light. Multiparameter data analysis was performed with Lysis II software. Cell subsets were gated, and 7-AAD staining on fluorescence channel-3 (FL-3) versus forward scatter channels was displayed.
Anti-CD3 mAb Treatment In Vitro and In Vivo.
In vitro, freshly isolated thymocytes or spleen cells were stimulated with soluble anti-CD3 mAb (5 μg/ml 145-2C11; BD PharMingen) in complete DMEM for 12–16 h. In vivo, TGF-β1−/− and TGF-β1+/+ mice were injected intraperitoneal with anti-CD3 antibody (50 μg per mouse) in 50 μl PBS or PBS only. After 6–16 h, tissues were processed and stained with TUNEL or isolated cells were stained with 7-AAD.
Assessment of mRNA for Apoptosis-related Molecules.
Total RNA was isolated from freshly isolated thymocytes or spleen cells using a modification of guanidinium isothiocyanate method (RNeasy kit; QIAGEN) for RNase Protection assay 32. A multiple template mAPO-3 or mAPO-2 set (BD PharMingen) was used, and radiolabeled probes were generated using [33P]dUTP (New England Biolabs, Inc.). Individual mRNAs were normalized as the ratio of mRNA for death receptors (FasL, Fas, TNF–related apoptosis-inducing ligand, and TNF-α p55) to the constitutively expressed L32 or glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH).
Induction of T Cell Apoptosis In Vitro.
Spleen cells were cultured in complete DMEM with recombinant human TNF-α (50 ng/ml; R&D Systems), recombinant murine TNF-α (40 ng/ml; R&D Systems), or anti-Fas antibody (1 μg/ml Jo2; BD PharMingen) for 16 or 56 h. Some spleen cells were cultured with anti-CD3 and anti-Fas (Jo2) antibodies for 16–24 h. Cells were then stained with FITC–anti-CD8 and PE–anti-CD4 together with 7-AAD for evaluation of apoptosis. Number of viable cells (7-AAD−) in parallel untreated cultures was considered as 100%. In some experiments, peripheral T cells were isolated from spleens as described previously 8. Purified T cells were incubated with cross-linked anti-CD3 (2 μg/ml) or plus anti-CD28 (5 μg/ml; BD PharMingen) antibodies by goat anti–hamster IgG (20 μg/ml) (Pierce Chemical Co.) for 16 h. Cells were then stained with 7-AAD for detection of cell death. CD8+CD44−/low T cells (naive) were purified (≥99%) from spleen cells of TGF-β1 null (6–10-d-old, pooled from five mice) or age-matched wild-type (n = 5) mice by flow cytometry using FITC-conjugated anti-CD8 and PE-conjugated anti-CD44 (antibodies were depleted of sodium azide by dialysis in PBS overnight) on a FACStarplus™ Cell Sorter (Becton Dickinson).
In some experiments (see Fig. 9), lymph node cells (10–11-d-old mice, n = 5) were pooled and stimulated with plate-coated anti-CD3 (10 μg/ml) in the presence of soluble anti-CD28 (5 μg/ml) antibodies or medium overnight. Some cells were harvested and adjusted for equal number of live cells in each treatment (30 × 106) for RNA and protein extraction. For Western blot analysis, an anti-Bcl–XL antibody (BD PharMingen) was used as described previously 33. Bcl–XL mRNA analysis was performed by RPA by using the mAPO-2 set template. Some cells were washed three times and then irradiated (1,200 rad) with a γ-irradiator (Gammacell 1000; Atomic Energy of Canada) and cultured in complete DMEM for an additional 6–24 h. For viability, cells were assessed by Trypan blue exclusion or by 7-AAD staining.
Figure 9.
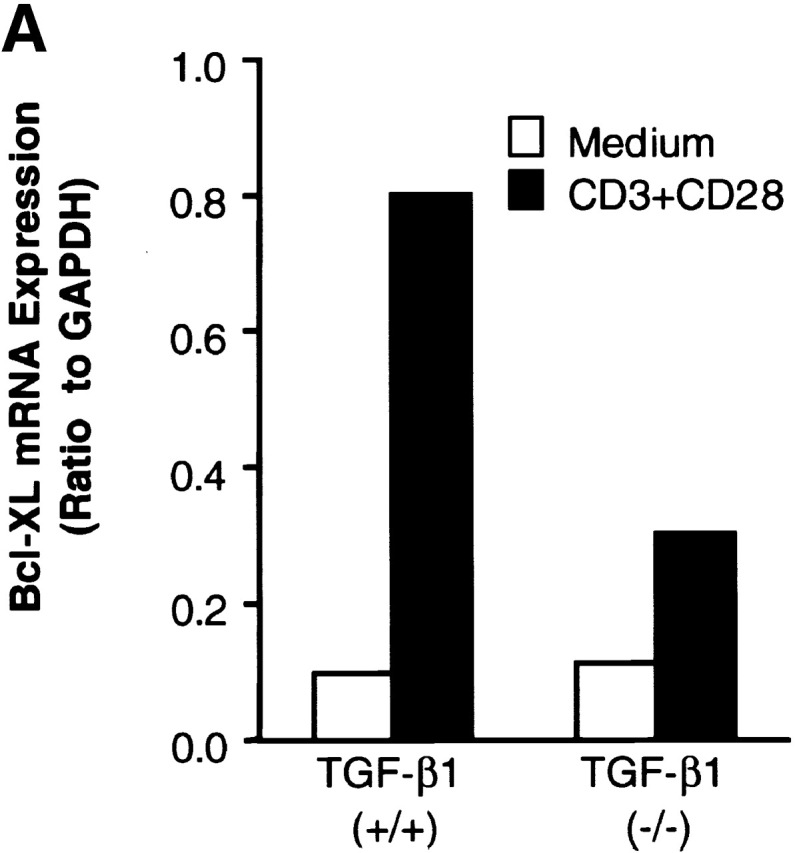


Prestimulation of TGF-β1−/− T cells with anti-CD3 plus CD28 failed to upregulate Bcl–XL or to prevent subsequent T cell apoptosis induced by γ-irradiation. (A) Bcl–XL mRNA expression in T cells. Lymph node cells were stimulated with anti-CD3 and CD28 antibodies or medium only overnight. RNA was extracted as described in the Methods and Materials. Bcl–XL mRNA was assayed with RPA by using mAPO-2 set template. 5 μg of RNA was loaded into each lane of gel. The corrected ratio of mRNA of BcL–XL to GAPDH is shown. (B) Western blot analysis of Bcl–XL in lymph node T cells. Expression of Bcl–XL was detected with anti-Bcl–XL monoclonal antibody (BD PharMingen). 100 μg of protein was loaded into each lane. (C) γ-irradiation induced T cell apoptosis. Lymph node cells were prestimulated with anti-CD3 and CD28 antibodies (black bar) or medium only (white bar) overnight, washed, and then irradiated. After 6 h, cells were stained with 7-AAD together with surface PE-CD4 and FITC-CD8 antibodies. Cells were analyzed by FACS®. CD4+ or CD8+ T cells were gated and the 7-AAD+ (apoptotic/dead) and 7-AAD− (live) on FL3 channel were calculated. Data shown are percentages of 7-AAD+ cells within the CD4+ or CD8+ T cells. The shown data are a representative of two experiments.
Flow Cytometry Analysis.
Cell staining for surface markers was performed as described previously 8. In brief, cells were incubated with FITC–anti-CD8, PE–anti-CD4, and biotin-conjugated anti-Fas (Jo2), anti-FasL, anti-CD44, anti-CD45RB, anti-CD69, or anti-CD62L antibodies (from BD PharMingen or Caltag), followed by streptavidin-tricolor (Caltag). Multiparameter data analysis was performed with Lysis II software.
Isolation of the Mitochondria and Cytosol from Cells.
Isolation of the mitochondria from cells was done according to the published protocol 22. In brief, T cell pellets were washed with ice-cold PBS and resuspended in buffer A (20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM dithiothreitol, and 0.1 mM phenylmethysulfonyl fluoride) containing 250 mM sucrose (thymocytes: 100 × 106 cells in 500 μl; peripheral T cells: 15–30 × 106 cells in 200–250 μl). The cells were homogenized with 10 strokes of a Teflon homogenizer, and the homogenates were centrifuged twice at 750 g for 10 min at 4°C. The pellets were discarded and the supernatants were centrifuged at 10,000 g for 15 min at 4°C. The resulting mitochondrial-enriched pellets were resuspended in the original volumes of buffer A and frozen at −80°C. The supernatants of the 10,000 g spin were frozen at –80°C. Some supernatants were further centrifuged at 100,000 g for 1 h at 4°C, and the resulting supernatants were frozen at −80°C. An aliquot of mitochondrial sample was fixed with 2.5% glutaraldehyde for examination by an electron microscope (Zeiss EMIO microscope). For analysis of TGF-β by ELISA, samples were treated with 1N HCl (1 μl per 50 μl supernatant) for 15 min at room temperature and neutralized with an equal amount of 1N NaOH. Samples were diluted (1:5) with the sample dilution buffer included in the TGF-β1 ELISA kit (Promega).
Western Blot Analysis.
Aliquots of mitochondria and cytosol preparations (20 μg protein) were subjected to 12.5% SDS-PAGE and transferred to a nitrocellulose filter (GIBCO BRL). The nitrocellulose membrane was blocked for 1 h with 5% instant skim milk powder in TBS. The filter was probed with an antibody to LAP-TGF-β1 (0.2 μg/ml anti-rhLAP; R&D Systems), washed, and incubated with horseradish peroxidase linked with Protein A as recommended by the manufacturer (Amersham Pharmacia Biotech). Finally, the washed blots were exposed to an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech) and recorded on an autoradiograph (Kodak X-Omat film).
Immunostaining of TGF-β in T Cells.
Splenic CD4+ and CD8+ T cells were incubated with mitochondrion-selective dye Mito-Tracker® Red CMXRos (50 nM; Molecular Probes) in complete DMEM at 37°C for 40 min. After washing with warm PBS, cells were fixed with Cytofix/Cytoperm® (Cytofix/Cytoperm Plus™ kit; BD PharMingen) at 4°C for 20 min. 106 cells were washed with Perm/WashTM buffer (BD PharMingen) and incubated with chicken anti–TGF-β (10–20 μg, AB-101-NA; R&D Systems) or with goat anti–human LAP (TGF-β1) (50 μg AB-246-NA; R&D Systems) at 4°C for 30 min. Cells were washed twice with Perm/Wash™ buffer and stained with cyanine (Cy2)-conjugated F(ab)2 donkey anti–chicken IgY or FITC-conjugated F(ab)2 donkey anti–goat IgG antibodies (Jackson ImmunoResearch Laboratories) at 4°C for 30 min. For negative staining controls, MitoTracker® Red-stained cells were incubated with secondary Cy2- or FITC-conjugated antibodies as above. After two washes, cells were resuspended in staining buffer (PBS-BSA[0.5%]–Az[0.1%]) and subjected to fluorescence microscopy.
Detection of Δψm and Reactive Oxygen Species in T Cells.
To detect Δψm, cells were first stained with PE–anti-CD4 or PE–anti-CD8 antibodies, washed, and resuspended in complete DMEM containing 25 nM dye 3,3′-dihexyloxacarbocyanine iodide (DiOC6; Molecular Probes) for 40 min at 37°C as described previously 34. For determination of intracellular reactive oxygen species, cells were incubated with dihydroethidium (2.5 μM) in complete DMEM at 37°C for 40 min 34. For dual labeling, FITC-conjugated anti-CD4 or -CD8 staining was performed at 4°C, before incubation with the dye. CD4+ T cells or CD8+ T cells were gated and the histograms of DiOC6 are displayed on FL-1 or of dihydroethidium on FL-2.
Electron Microscopy.
Thymus tissues were fixed in a solution containing 0.5% glutaraldehyde and 4% paraformaldehyde, stained with 1% osmium tetroxide, dehydrated with ethanol, and embedded in Epon/Araldite resin. Thin sections were cut, stained with 3% aqueous uranyl acetate and Reynolds lead citrate, and observed in a Zeiss EM10-A transmission electron microscope.
Immunoelectronmicroscopy.
Spleen cells were fixed with 4% paraformaldehyde in PBS containing 0.1% glutaraldehyde for 30 min and cell pellets were stabilized in 2% agar in PBS. Pellets were rinsed with PBS, followed by a graded ethanol series (50, 70, 95%). Pellets were then infiltrated with LR White (hard grade, Ted Pella, Inc.) at 1:1 (95% ethanol:LR White), then two changes of pure LR White and left in pure LR White overnight at room temperature. After one change of pure LR White, the cell pellets were placed into BEEM capsules, filled with LR White, and the capsules sealed with parafilm and the capsule top. Polymerization took place at 70°C for 16 h.
Thin sections were cut on an RMC MT-7 and placed on formvar/carbon-coated 200 nickel grids. All immunolabeling was done in a microwell staining mold (Ted Pella, Inc.) placed in a humid chamber. Grids were placed on a drop of blocking buffer containing BSA, Tween 20, cold water fish gelatin, and serum from either normal goat or rabbit. Grids were immunolabeled with 80 μg of goat anti-LAP (TGF-β1; R&D Systems). Negative controls were exposed to blocking buffer in place of the primary antibody. Secondary antibodies were either rabbit anti–goat conjugated to a 10-nm gold particle in a 1:20 dilution with blocking buffer. Grids were subsequently stained with 2% aqueous uranyl acetate and Reynold's lead citrate and examined using a Zeiss 10A transmission electron microscope operating at 60 kV.
Results
Increased Thymic Apoptosis in Mice Lacking TGF-_β_1.
To define a role for TGF-β in apoptosis of T cells, we first examined thymocyte apoptosis in situ with the TUNEL method 35 36. In the normal TGF-β1+/+ thymus, apoptotic cells were scattered throughout the cortex as reported 36 and also found in the thymic medulla, although with less frequency (Fig. 1 A). In the TGF-β1−/− mice, a significantly enhanced number of TUNEL-positive cells was observed in both thymic cortex and medulla (Fig. 1 B). To quantify the apoptotic cells, freshly isolated thymocytes were stained with DNA dye 7-AAD, which discriminates live cells from apoptotic and/or dead cells, in combination with antibodies which recognize CD4+ and CD8+ T lymphocytes for flow cytometry 31. In the total population, 2–4% of the TGF-β1−/− thymocytes were apoptotic, more than double the number in the TGF-β1+/+ thymocytes (0.5–1.5%) 31 36. Increased apoptosis occurred in both CD4+CD8+ double positive (DP) and CD4+CD8− or CD4−CD8+ single positive thymocytes (Fig. 1 F), but not in the CD4−CD8− double negative (DN) thymocytes (data not shown).
Figure 1.
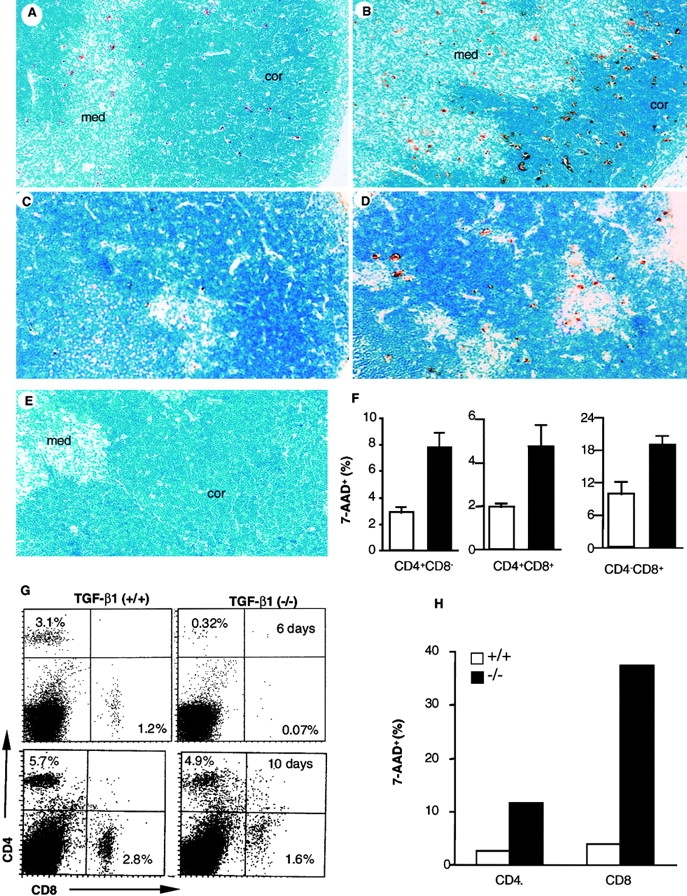
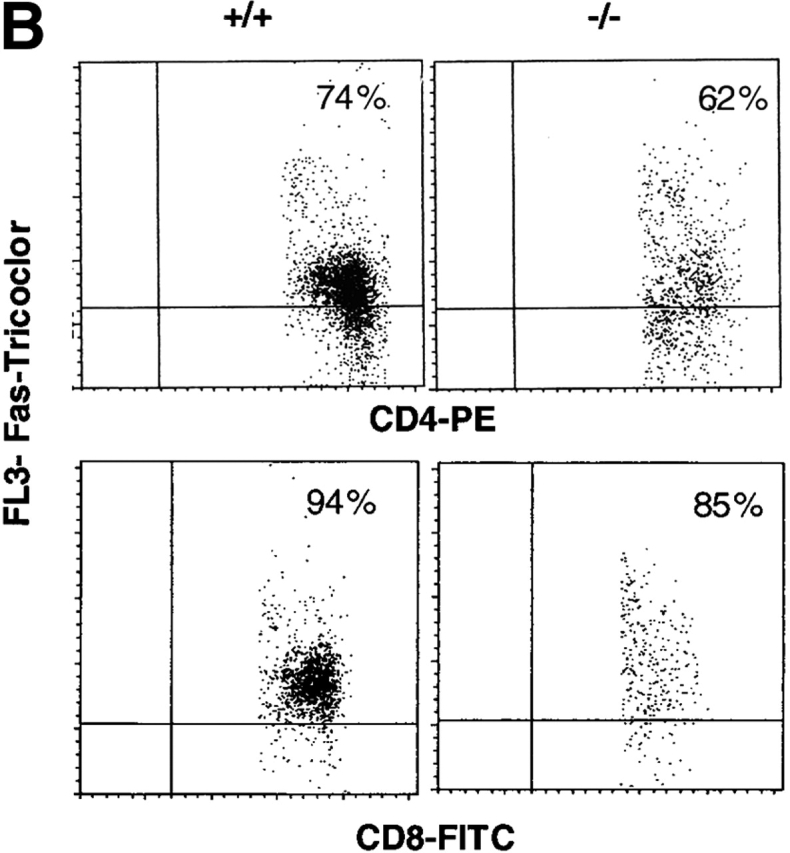
Apoptotic cells in thymus and spleen of TGF-β1+/+ and TGF-β1−/− mice. (A–E) In situ detection of apoptotic cells in thymus sections with a modified TUNEL method. Original magnification: 40×. (A) TGF-β1+/+ thymus (11 d). (B) TGF-β1−/− thymus (11 d). (C) Fetal gestation day 18 TGF-β1+/+ thymus: stained cells are seldom seen. (D) Fetal gestation day 18 TGF-β1−/− thymus: stained cells are clearly evident. (E) TGF-β1+/+ thymus stained in the absence of TdT (negative control), no stained cells. (F) Detection of apoptotic cells by staining with 7-AAD in thymocyte subsets. The values are mean ± SD of three experiments (six mice per group, 11–16 d old). TGF-β1+/+ (open); TGF-β1−/−(solid). (G–H) Enhanced apoptosis in peripheral TGF-β1−/− T cells. (G) Profiles of surface CD4+ versus CD8+ staining of spleen T cells. (6, 10 d). (H) 7-AAD+ cells within CD4+ or CD8+ splenocyte T cell subsets (7 d).
Since TGF-β1−/− mice develop extensive tissue inflammation between 2–4 wk, one may argue that increased apoptosis occurs secondary to the inflammatory response. However, a significant decrease in the total number of thymocytes was observed before symptoms appeared (9–10-d-old TGF-β1−/− mean: 149 × 106 thymocytes; TGF-β1+/+ 278 × 106, n = 2). Subsequent peripheral inflammation may further increase thymocyte apoptosis (≥3-wk-old TGF-β1−/− mean: 70 × 106, n = 4; TGF-β1+/+: 345 × 106, n = 2). To determine whether inflammation and/or cytokines contributed to this early apoptotic phenotype, thymocytes and spleen cells from asymptomatic null (6–10 d old) and wild-type mice were evaluated for cytokine mRNA levels. No significant increase in cytokine mRNA (TNF-α or IFN-γ) was found in freshly isolated thymocytes or spleen cells. Furthermore, sera from neither asymptomatic null nor age-matched wild-type mice had detectable levels of TNF-α, IFN-γ, IL-2, or IL-4 (data not shown). Importantly, we examined the thymuses of fetal mice as early as gestation day 18, namely at a stage when thymocyte positive selection is just beginning and cell death is very limited 36 37. While TUNEL staining was seldom seen in TGF-β1+/+ thymuses (Fig. 1 C, TUNEL+ cells: 0–1 per high power field, original magnification: 40×), increased apoptosis was already evident in fetal TGF-β1−/− thymocytes (Fig. 1 D, TUNEL+ cells: 6–20 per high power field, original magnification: 40×). The results indicate a requirement in vivo for TGF-β1 in T cell development through its ability to influence thymocyte apoptosis, which appears to occur after thymocytes acquire expression of CD4 and CD8. Moreover, this is the first evidence that the absence of TGF-β1 exerts an effect on the immune system in utero and is associated with aberrant immunological development.
Peripheral T Cell Apoptosis in TGF-_β_1 Null Mice.
Because the absence of TGF-β1 has such a profound influence on thymocyte survival, we next examined the impact of TGF-β1 deficiency on peripheral T cells. Although by flow cytometry, reduction of both CD4+ and CD8+ T cell numbers occurred in the spleens of young, asymptomatic TGF-β1−/− mice (Fig. 1 G), the decrease was more significant in CD8+ T cells, skewing the CD4/CD8 ratio toward CD4 (4.5 in TGF-β1 null versus 2.5 in wild-type, 6 d). With increasing age, the percentage of both CD4+ and CD8+ T cells increased (Fig. 1 G, 10 d), but the disproportionate reduction in CD8+ T cells remained. A similar profile was also evident in lymph node cells (data not shown). Increased apoptotic cells (7-AAD+) were evident in CD4+ and particularly CD8+ TGF-β1−/− T cells (Fig. 1 H). Although this enhanced apoptosis became more pronounced as the TGF-β1−/− mice developed symptomatic inflammation (≥3 wk old, data not shown), increased peripheral T cell apoptosis was clearly evident within 6–7 d of birth (Fig. 1G and Fig. H), before evidence of inflammation or enhanced cytokine expression. This increased sensitivity to apoptosis appears to be a basic feature of TGF-β1 gene–deleted T cells.
TGF-_β_1 Controls Activation-induced T Cell Death.
The impact of TCR stimulation on T cell death in the absence of TGF-β1 was even more impressive. Activation of thymocytes with anti-CD3 antibody exacerbated thymocyte apoptosis, affecting CD4+CD8+ DP cells more than CD4+CD8− and CD4−CD8+ single positive cells (Fig. 2). Apoptotic DP thymocytes also downregulated CD4 and CD8 surface expression 31 38 concomitant with positive staining for 7-AAD (Fig. 2 A), yielding more CD4+/low CD8+/low DP thymocytes in TGF-β1−/− mice (41%) than in TGF-β1+/+ thymocytes (21%, Fig. 2 A, R2). Unprecedented, however, was the response to intraperitoneal injection of anti-CD3 mAb (50 μg per mouse). Although anti-CD3 injection into 11–12-d-old wild-type mice triggered apoptosis of thymocytes, primarily in the cortex (Fig. 2 D) as reported 36 39, and total thymocyte numbers decreased two to threefold compared with a mock PBS injection (PBS mean: 300 × 106, n = 2; mAb CD3 mean: 156 × 106, n = 2), TGF-β1 null thymuses manifested enormous numbers of TUNEL-stained cells within hours after anti-CD3 antibody injection (Fig. 2 E). Moreover, large aggregates of apoptotic cells, particularly in the cortex, were apparent and often localized within distended macrophages, and surprisingly, TUNEL-stained cells were also seen in thymic medulla (Fig. 2 E). Total thymocyte numbers in anti-CD3 mAb–treated TGF-β1−/− mice dropped precipitously by 10-30-fold (PBS mean: 120 × 106, n = 2; mAb CD3 mean: 5 × 106, n = 2). By flow cytometry, substantially more CD4+/lowCD8+/low (24 vs. 7%) and 7-AAD+ thymocytes were present in anti-CD3–treated null mice compared with wild-type (data not shown). Thus, the absence of TGF-β1 has a profound influence on the sensitivity of thymocytes to TCR-mediated apoptosis, particularly in the CD4+ CD8+ DP population, to influence their negative selection.
Figure 2.

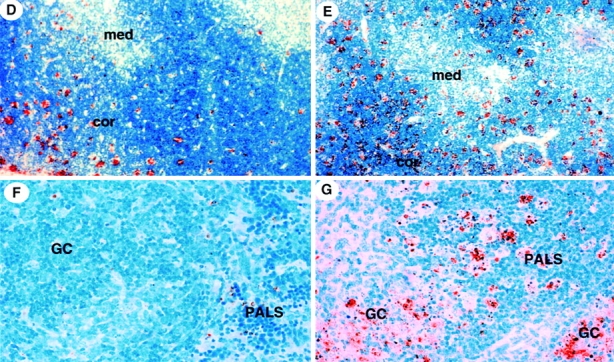
Detection of apoptotic cells induced by anti-CD3 mAb in vitro and in vivo. (A) Anti-CD3 mAb (16 h) induced more apoptosis of CD4+CD8+ DP thymocytes in TGF-β1−/− than TGF-β1+/+ mice in vitro. CD4+/hi CD8+/hi (R1) and CD4+/lowCD8+/low (R2) DP thymocytes were gated and the percentages are displayed in lower right corner. The histograms of 7-AAD staining of R1 (open) and R2 (filled) on FL-3 are overlayed in right panel. (B) Anti-CD3–induced cell death in TGF-β1−/− peripheral T cells in vitro. Viable CD8+ T cells (2 × 105) purified from spleens of mice (9–10 d old) were stimulated with the indicated antibodies cross-linked with goat anti–hamster IgG antibody for 16 h. Cells were then stained with 7-AAD for detection of cell death (7-AAD+: apoptotic; 7-AAD−: viable). Spleen cells (five mice per group) were pooled before CD8+ T cell isolation. (C) Increased apoptosis of CD8+ CD44−/low naive null T cells in vitro. Flow cytometry purified live CD8+CD44−/low peripheral T cells (105) were cultured with the indicated antibodies (2 μg/ml anti-CD3; 5 μg/ml anti-CD28; 1 μg/ml anti-Fas) and cross-linked with goat anti–hamster IgG antibody (20 μg/ml) for 16 h. Cells were then stained with 7-AAD for apoptosis detection. Numbers shown in the figure represent the percentage of viable cells (7-AAD−). (D–G) In situ detection of apoptotic cells with TUNEL staining in tissues 16 h after anti-CD3 mAb injection. (D) TGF-β1+/+ thymus; (E) TGF-β1−/− thymus; (F) TGF-β1+/+ spleen; (G) TGF-β1−/− spleen. Cor, cortex; GC, germinal center; Med, medulla.
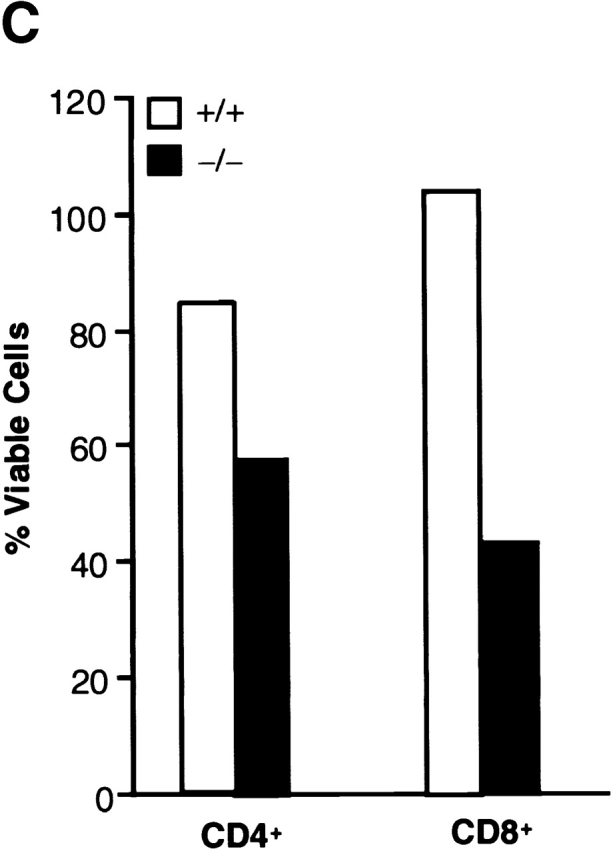
Extending these studies to the peripheral lymphoid tissues, purified peripheral viable CD8+ T cells (Fig. 2 B) and CD4+ T cells (data not shown) were stimulated with anti-CD3 alone or together with anti-CD28 antibodies for 16 h. TGF-β1 null T cells exhibited enhanced spontaneous cell death and the anti-CD3 stimulation further increased this aberrant T cell apoptosis. Even more dramatically, cross-linking of CD3 together with CD28 promoted extensive cell death in the purified CD8+ TGF-β1 null population (Fig. 2 B). Although many freshly isolated peripheral null T cells expressed CD44hi (Fig. 3), suggestive of activation, when CD8+CD44−/low (naive) null T cells were purified and cultured in media (Fig. 2 C), they still manifested profound spontaneous T cell death (57% survival, 7AAD−) compared with wild-type (85% survival). Treatment with anti-CD3 or anti-CD3 plus CD28 antibodies further exacerbated this abnormal naive null cell death (13 and 21% survival, respectively), whereas the same treatment only minimally induced or failed to drive cell death in the wild-type T cells (80 and 86%, respectively). Moreover, unstimulated TGF-β1 null T cells failed to release detectable TNF-α, IFN-γ, or IL-2. Anti-CD3 treatment in vitro induced these cytokines, but the levels in the null T cell cultures were typically lower than the wild-type controls. Inclusion of neutralizing anti–TNF-α and/or anti–IFN-γ antibodies did not reverse null T cell death (data not shown). Thus, by multiple criteria, prior activation and secondary cytokines do not appear to represent the mechanism underlying null T cell apoptosis.
Figure 3.

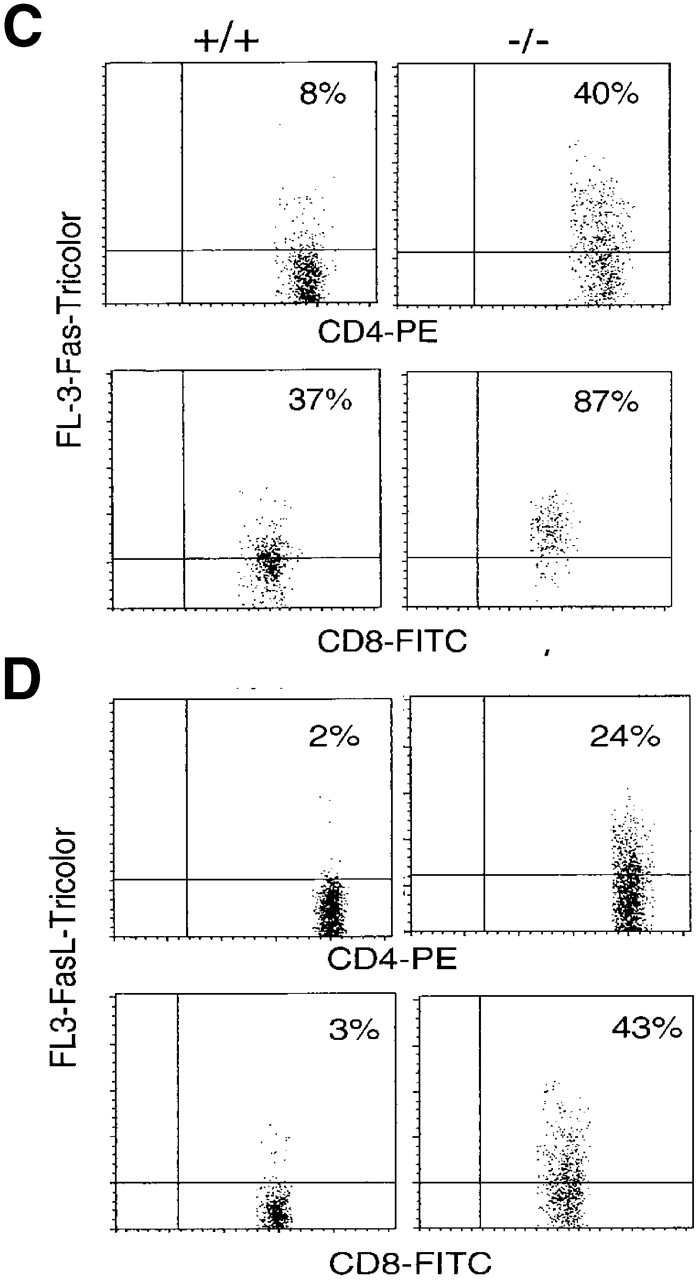
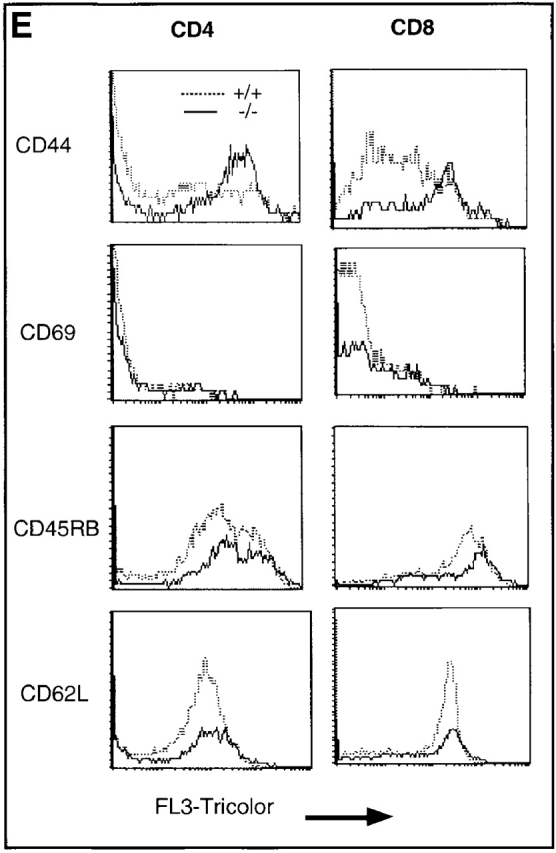
Upregulation of apoptosis-related molecules in TGF-β1−/− peripheral T cells (10–11-d-old mice). (A) mRNA detection in spleens (pooled cells from five mice per group) by RPA. The experiment represents one of five experiments. (B) The ratio of mRNA of death receptors was normalized as a ratio to the constitutively expressed GAPDH. Data presented are mean ± SEM (n = 5). (C and D) Increased surface expression of Fas and FasL on peripheral TGF-β1−/− CD4+ and CD8+ T cells. CD4+ or CD8+ T cells are gated and the profiles of CD4 or CD8 versus Fas (C) or FasL (D) are shown. (E) Histograms of CD44, CD45RB, CD69, and CD62L of CD4+ and CD8+ T cells in lymph nodes are shown on FL-3.
In vivo injection of anti-CD3 mAb induced only sparse TUNEL-positive cells in wild-type spleen, mainly in the T cell–dependent regions (periarteriolar lymphatic sheaths [PALSs]), but seldom in the B cell–rich germinal centers (Fig. 2 F). In marked contrast, in mice with the TGF-β1 gene deletion, massive apoptosis occurred throughout all regions of the spleen. The larger and more intense aggregates of TUNEL-positive cell clusters were found scattered not only throughout the PALSs, but also in the B cell–dependent germinal centers (Fig. 2 G). Within hours after anti-CD3 mAb injection, tricolor fluorescence revealed a significant increase in apoptotic (7-AAD+) cells in both CD4+ and CD8+ T cell populations (data not shown). This dramatic effect on activated T cell death, whether in vitro or in vivo, occurs independent of mouse age (7–20 d old) and underscores a requirement for TGF-β1 in the maintenance of activated lymphoid cell viability.
TGF-β1 Regulates Fas-mediated Death Signaling in Peripheral, but Not Thymic T Cells.
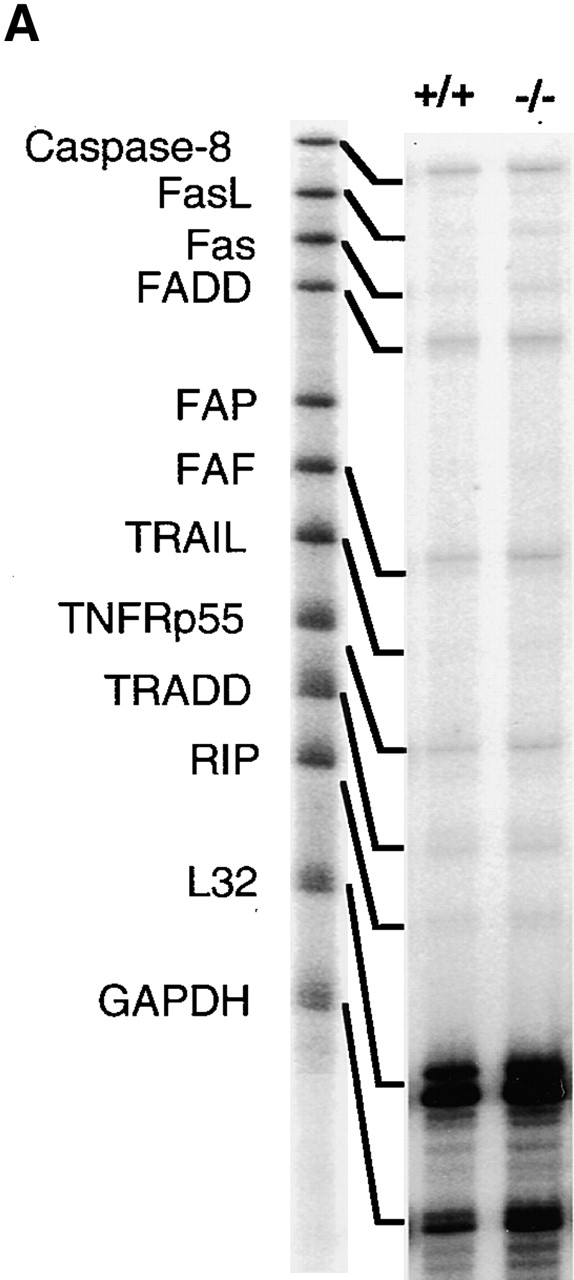
Whether the ligand-death receptor pair, FasL–Fas, was involved in the dysregulated apoptosis in T cells of TGF-β1−/− mice was assessed by multiple parameters. By RNase Protection assay 32, comparison of thymocytes from null and wild-type mice, with or without anti-CD3 mAb stimulation, revealed no significant differences in transcripts of the multiple death-related molecules including FasL and Fas (data not shown), despite the remarkable increase in numbers of TGF-β1−/− apoptotic cells. Additionally, by flow cytometry, both wild-type and null thymocytes expressed nondiscriminating levels of Fas antigen on their surface (data not shown).
In contrast to the thymocytes, the TGF-β1−/− spleen cells exhibited clearly increased levels of both Fas and FasL mRNA compared with their TGF-β1+/+ counterparts (Fig. 3a and Fig. b). Consistent with the mRNA, surface expression of Fas (Fig. 3 C) and FasL (Fig. 3 D) was significantly elevated in both CD4+ and CD8+ T cells, perhaps reflecting an activated state 40 in the absence of TGF-β1. Paradoxically, peripheral CD4+ and CD8+ T cells, despite their upregulation of FasL and Fas, expressed a unique hybrid phenotype, including CD44+, CD62Llow, CD69−, and CD45RBhi in symptom-free 10-d-old mice (Fig. 3 E). With the spontaneous development of inflammation and autoimmune-like lesions (≥3 wk), FasL–Fas expression on peripheral T cells further increased, along with upregulation of CD69 and CD25 (data not shown). Increased FasL–Fas expression appears to be a phenotypic characteristic of TGF-β1−/− peripheral T cells, which may be augmented by secondary lymphocyte activation, pointing to an involvement of TGF-β1 in the regulated expression of FasL–Fas.
When we examined whether the absence of TGF-β1 influenced the Fas-mediated death signaling pathway, we found that anti–mouse Fas monoclonal antibody (1 μg/ml Jo2), in the absence of the protein synthesis inhibitor cycloheximide 41, triggered minimal apoptosis of TGF-β1+/+ CD4+ CD8+ DP thymocytes (10–20% increase) and only modestly enhanced apoptosis in CD4+ CD8+ DP null thymocytes (20–30% increase). In peripheral T cells, however, differences in anti-Fas–induced death were more clearly evident. Fas ligation with mAb Jo2 markedly induced apoptosis of both CD4+ and CD8+ peripheral TGF-β1−/− T cells (40–60%), without affecting viability of wild-type spleen cells (Fig. 4 A). Since TGF-β1−/− T cells expressed more Fas than TGF-β1+/+ T cells (Fig. 3), one might reasonably consider that the killing induced by anti-Fas antibody was solely due to the upregulated expression of Fas. Surprisingly, however, when we stimulated TGF-β1+/+ T cells with anti-CD3 mAb overnight to upregulate equivalent Fas expression (Fig. 4 B), mAb Jo2 still induced only marginal apoptosis of CD4+ T cells (15%), and failed to engage specific cell death of wild-type CD8+ T cells (Fig. 4 C). This specific Fas-induced apoptosis in null T cells was apparently not mediated by secondary cytokines in the cultures, since no detectable levels of IL-2, IFN-γ, or TNF-α were observed. Addition of anti–TNF-α and/or anti–IFN-γ failed to rescue and/or prevent anti-Fas induced TGF-β1−/− T cell death (data not shown). Importantly, when flow cytometry sorted CD8+CD44−/low naive T cells were exposed to anti-Fas antibody for 16 h, only TGF-β1 null, but not wild-type, T cells were specifically killed by Fas-mediated signaling (Fig. 2 C). While these observations are consistent with the evidence that normal murine T cells are typically resistant to anti-Fas antibody–induced apoptosis 19, TGF-β1−/− T cells, on the other hand, are exquisitely sensitive to this route of death.
Figure 4.

Enhanced sensitivity to Fas and TNFR2-mediated cell death in TGF-β1−/− peripheral T cells. (A) Anti-Fas antibody (Jo2)-induced apoptosis of both CD4+ and CD8+ T cells. Mouse TNF-α specifically killed CD8+, but not CD4+, T cells in TGF-β1−/− spleens. Data are expressed as percentages of viable cells compared with control (cells in medium alone) after 16 h culture (mean ± SD; n = 4 experiments). Typically, the background viability of cells cultured with complete DMEM only (untreated cells) for 16 h is: TGF-β1+/+: CD4+ T cells 7-AAD−, 76.1%; 7-AAD+, 22%; CD8+ T cells 7-AAD−, 60%; 7-AAD+, 36.9%; TGF-β1−/− CD4+ T cells 7-AAD−, 42.8%; 7-AAD+, 56%; CD8+ T cells 7-AAD−, 42.6%; 7-AAD+, 55%. *P < 0.05 compared with medium-treated null cells. (B) Fas expression on splenic T cells after anti-CD3 mAb (5 μg/ml) stimulation overnight. (C) Failure of anti-Fas mAb to induce apoptosis of TGF-β1+/+ peripheral T cells expressing Fas antigen. Percentages of viable cells within CD4+ or CD8+ T cells from TGF-β1+/+ (open) or TGF-β1−/− (solid) are displayed. Data are representative of three experiments (five mice per group).
TGF-β1 Controls TNF-α Receptor 2(p75)-mediated Apoptosis in Peripheral CD8+, but Not CD4+ T Cells.
In addition to Fas-mediated apoptosis, T cell death can also be orchestrated through TNFR. Murine TNF-α binds to TNFR2(p75), which is constitutively expressed on murine peripheral T cells 42 and signals by a pathway distinct from TNFR1(p55)-driven apoptosis of nonlymphoid cells 43 44 to cause death of mature CD8+ T cell blasts after 48 or more h in culture 18. As a receptor discriminating tool, human TNF-α binds exclusively to mouse TNFR1 44 45. In normal wild-type peripheral T cells, neither murine nor human TNF-α–induced specific apoptosis after 16 h in culture (Fig. 4 A). A striking difference occurred when TGF-β1 null spleen cells were exposed to murine, but not human TNF-α, which drove rapid and significant TNFR2-dependent apoptosis of CD8+ (20–40%), but not CD4+ T cells (Fig. 4 A). After longer intervals (by 56 h), >60% of TGF-β1−/− CD8+ T cells were specifically killed by murine TNF-α, whereas CD4+ T cells were spared, implicating a CD8+-specific mechanism of TGF-β–mediated protection from TNFR2-mediated loss of life.
Exogenous TGF-β1 Failed to Rescue or Prevent Dysregulated TGF-β1 null T Cell Death.
To dissect the mechanism(s) whereby TGF-β1 controls T cell life and death decisions, we added exogenous TGF-β1 to the cells in an attempt to restore TGF-β1 signaling pathways regulating viability. TGF-β1 null mice express normal TGF-β receptors (unpublished data), yet exogenous TGF-β1 (2 ng/ml) added to the cell cultures failed to rescue or prevent either spontaneous or death receptor-induced apoptosis in null CD4+ and CD8+ T cells (Fig. 5 A). In fact, addition of TGF-β1 slightly enhanced null T cell apoptosis, whereas it did not affect cell death in parallel 16 h wild-type T cell cultures (Fig. 5 A). The bioactivity of exogenous TGF-β1 was verified by its profound suppression of antigen-specific proliferation in OVA peptide TCR transgenic (DO11.10) T cells: OVA (100 μg/ml) 18, 212 ± 2761 cpm TdR3H incorporation; OVA plus TGF-β1 (2 ng/ml) 1,538 ± 519.
Figure 5.


(A) Failure of exogenous TGF-β1 to prevent and/or rescue T cell death in TGF-β1−/− mice. Spleen cells were cultured with anti-Fas mAb (1 μg/ml) or medium in the absence of presence of recombinant human TGF-β1 (2 ng/ml; R&D Systems) for 16 h. Profiles of 7-AAD+ cells on FL3 versus cell sizes on forward scatter channels are displayed. The percentages are presented as 7-AAD+ cells within the CD4+ or CD8+ T cell subsets. (B) Smad3−/− T cells show normal apoptosis. (B) Profiles of CD4 versus CD8 surface staining in freshly isolated spleen cells (n = 2 per pooled group; 4 wk old).
The failure to reverse aberrant apoptosis behavior in TGF-β1 null T cells with exogenous TGF-β1, consistent with our failure to rescue null mice from lethality with TGF-β, prompted exploration of whether traditional TGF-β signaling pathways were involved in the control of cell death. In this regard, ligand binding to TGF-β receptors induces a signaling cascade which involves intracellular Smads 46 47. Due to the disruption of downstream signaling from the receptor, mutation of the Smad3 gene (Smad3−/−) manifests in uninhibited spontaneous T cell activation. These Smad3 null T cells, when activated by anti-CD3 antibody, were insensitive to the inhibitory effects of TGF-β 47, even though TGF-β production in the mice was intact. Analysis of death and depletion of the Smad3−/− T cells revealed that in marked contrast to TGF-β1−/− T cells, neither CD4+ nor CD8+ T cell populations were reduced in Smad3−/− spleens (Fig. 5 B). By 7-AAD staining, freshly isolated Smad3−/− CD4+ T cells had little or no increase in apoptotic cells (14%) compared with wild-type (11%), whereas no difference in apoptosis was observed in CD8+ T cells (Smad3−/−, 5.0%; wild-type, 5.5%). The absolute numbers of splenic CD4+ and CD8+ T cells were not reduced in Smad3−/− mice compared with the wild-types. In fact, the total numbers of spleen T cells, particularly CD8+ T cells, increased in Smad3−/− mice, which may reflect lack of TGF-β inhibitory signals (Smad3−/− CD4+, 18 ± 2.6 × 106; CD8+, 12 ± 2.1 × 106, n = 5; wild-type CD4+, 11 ± 0.2 × 106; CD8+, 5.8 ± 0.1 × 106, n = 6). The lack of difference in apoptosis was apparent even though the majority of the T cells also expressed CD62Llow 47, more like memory cells. Culture of the Smad3−/− T cells for 16 h revealed a similar rate of spontaneous death between the null and control cells. Anti-Fas failed to induce specific CD4+ or CD8+ T cell apoptosis (data not shown), which was quite distinct from the TGF-β1−/− T cells (Fig. 2 and Fig. 4). In addition, Smad3 null mice had normal development of thymocytes 47. Thus, the enhanced apoptosis evident in TGF-β1−/− T cells appeared not to be a function of the TGF-β1 receptor–Smad3 signal transduction pathway, suggesting an alternative, perhaps intracellular site for TGF-β1–mediated control of apoptosis.
TGF-β1–dependent Δ_ψ_ m.
Previous reports had demonstrated an intracellular site for TGF-β. Because TGF-β was localized within the mitochondria of rat liver and myocardial cells 30, we investigated this possibility in T cells. After isolating the mitochondria and cytosol from wild-type T cells, we compared the levels of TGF-β within the mitochondria-enriched and the mitochondria-depleted cytosol. By Western blot analysis, higher levels of TGF-β were detected in the mitochondrial preparations than in the cytosol (Fig. 6 A). This was TGF-β1 specific, because the same Western blot analysis showed no band with the mitochondrial fraction derived from TGF-β1 null cells (Fig. 6 A). By ELISA, a three to fivefold higher level of total TGF-β was found within the mitochondrial-enriched preparation than in the cytosol of wild-type peripheral T cells, particularly in the CD8+ population (Fig. 6 B). The purity of the mitochondrial preparations was assessed by electron microscopy, but potential contamination by endoplasmic reticulum (ER) that contains TGF-β could not be completely excluded. Although an ER-specific enzyme like glucose-6 phosphatase (G-6-P) can be used to detect ER contamination in some cells such as liver, G-6-P does not appear to be detectable in ER of mouse lymphocytes (data not shown) and no other ER-specific enzyme has been reported in lymphocytes. Consequently, we directly examined intact lymphocyte mitochondria for TGF-β. We first stained peripheral T cells with anti-LAP (TGF-β1) or anti–TGF-β (data not shown) antibodies together with a mitochondria-tracking dye (Fig. 6 C). As evident in Fig. 6 C, the anti-LAP stained the peripheral cytoplasm in a punctate pattern. MitoTracker® was observed in a similar orientation and the overlays revealed a colocalization (yellow) pattern of the two stains. To further document this association between TGF-β and the mitochondria, we performed immunogold labeling of spleen T cells with anti-LAP (TGF-β1) antibody (Fig. 7). Gold grains are localized over mitochondria (Fig. 7B–D, arrowheads), whereas ER and nucleus show little or no labeling in these freshly isolated spleen cells. This anti-LAP (TGF-β1) immunogold labeling was specific for TGF-β1, since omission of the primary antibody resulted in no labeling (Fig. 7 A). By multiple approaches, the data are consistent with TGF-β1 localization in T cell mitochondria and indicate that TGF-β in this intracellular compartment might bypass membrane TGF-β receptors to influence T cell apoptosis.
Figure 6.



Localization of TGF-β in the mitochondria of wild-type T cells. (A) Western blot analysis of TGF-β1 in mitochondria and cytosol. Mitochondria and cytosol preparations were isolated from pooled spleen cells (S) or thymocytes (T) in wild-type (+/+) and TGF-β1 null−/− mice (three to five mice per group). 20 μg of protein was loaded into each lane and probed with anti-LAP (TGF-β1) antibody. (B) Total TGF-β from the mitochondrial and cytosol preparations of peripheral CD4+ T cells (1–2 × 106 cells), CD8+ T cells (1–2 × 106 cells) is shown as mean ± SD of duplicate ELISA measurements. Cyto, cytosol; Mito, mitochondrial preparations. (C) Immunostaining for TGF-β1. Spleen T cells were stained with MitoTracker® Red for mitochondria (red) and goat anti-LAP (TGF-β1) followed by FITC-conjugated F(ab)2 donkey anti–goat IgG antibody (green). Colocalization of TGF-β and mitochondria is represented as yellow. (a) Green fluorescence of anti-LAP (TGF-β1) in T cells, showing a punctate staining pattern in the cytoplasm around the nucleus. (b) Red fluorescence of mitochondria stained by MitoTracker® Red. (c) Overlay of a and b to demonstrate colocalization of TGF-β1(LAP) in the mitochondria represented by yellow. A representative cell is shown. (d–f) A nonspecific control for anti–TGF-β1 staining in which the primary anti-LAP(TGF-β1) antibody was omitted. No green fluorescence was seen (d), but mitochondria stained with MitoTracker® Red (e). No yellow color was observed in the overlay of d and e (f). This experiment was repeated at least five times with similar results. The profiles of TGF-β1 and mitochondrion staining in both CD4+ and CD8+ T cells were similar.
Figure 7.
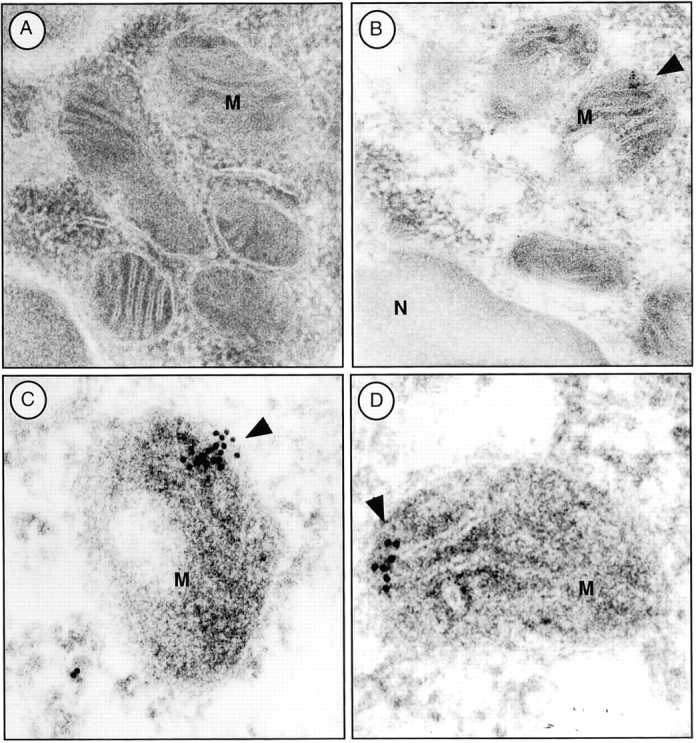
Immunogold labeling for TGF-β in mouse spleen cells. (A) Control gold labeling in the absence of the primary antibody. No immunoreaction was observed over the mitochondria (M). (B) Specific labeling of TGF-β1 in mitochondria (M) with anti-LAP (TGF-β1). Gold grains are localized predominantly over mitochondria (arrowheads). ER and nucleus show little or no labeling. (C and D) Representative immunogold labeling for LAP (TGF-β1) in the mitochondria from additional cells. Cell fixation and preparation as described in Materials and Methods. Original magnification: 106,000×.
Since mitochondria function as intracellular integrators of discrete proapoptotic stimuli and intact membrane potential (Δψm) is indispensable for normal mitochondrial function 9 10 11 48, the effect of TGF-β1 deletion on Δψm in T cells was monitored. In concert with their higher rate of apoptosis, both TGF-β1−/− thymocytes (data not shown) and peripheral T cells exhibited markedly decreased Δψm as determined by dye DiOC6 34 which accumulates in intact mitochondria (Fig. 8 A, Fresh). Significantly increased numbers of TGF-β1−/− T cells had reduced Δψm (DiOC6 low) compared with the wild-type T cells after 16 h in culture (Fig. 8 A, Med). Promotion of death by anti-Fas antibody Jo2 further dampened their Δψm (increase in DiOC6 low cells), but the wild-type T cells were resistant to changes in Δψm (Fig. 8 A) and to Fas execution (Fig. 4). Parallel with the apoptotic profile, murine TNF-α reduced the Δψm in peripheral TGF-β1−/− CD8+ (data not shown), but not CD4+ T cells (Fig. 8 A). Consistent with the failure to rescue the cells from death, exogenous TGF-β1 (2 ng/ml) also failed to prevent either spontaneous or death receptor-induced loss of Δψm. Moreover, the absence of Smad3 did not appear to influence Δψm (data not shown). These data again favor a nontraditional receptor-dependent and/or intracellular role for TGF-β1 in maintaining the integrity of Δψm.
Figure 8.


Loss of Δψm in TGF-β1−/− T cells. (A) Spleen cells from TGF-β1−/− (9 d old, n = 2) (lower) or age-matched TGF-β1+/+ (n = 2) (upper) mice were harvested and pooled. Cells were assayed immediately (fresh) or cultured with indicated reagents as in Fig. 4 A for 16 h. To detect Δψm, cells were first stained with PE–anti-CD4 or PE–anti-CD8 antibodies, washed, and resuspended in complete DMEM containing 25 nM DiOC6 (Molecular Probes) for 40 min at 37°C as described previously (reference 34). CD4+ T cells were gated and the profiles of DiOC6 on FL-1 versus CD4 on FL-2 are displayed. DiOC6 staining in CD8+ T cells (data not shown) was similar to CD4+ T cells except for a significant increase in DiOC6 low cells after mTNF-α treatment. The experiment was repeated three times with similar results. (B and C) Electron micrograph of a fetal thymocyte from TGF-β1−/− mouse as in Fig. 1 D (gestation day 18). Original magnification: 16,000×. Abnormal mitochondria (Boxed, higher magnification, original magnification: 25,000× in C) appeared in TGF-β1 null T cells. Electron density in mitochondrial matrix was increased and cristae lost with dilated intracristal space. M, mitochondria; N, nucleus.
Changes of Mitochondrial Morphology in TGF-β1_−_ /− T Cells.
Consistent with the reduced Δψm in TGF-β1–deficient T cells, we observed altered mitochondrial structural integrity in the cells lacking intracellular TGF-β1. Ultrastructural examination of mitochondrial structure revealed aberrancies as early as gestation day 18 in fetal thymus, when thymocyte positive selection was just beginning and cell death was undetectable in normal mice (Fig. 1 C; references 36 and 37), but discernible in the fetal null thymus (Fig. 1 D, and data not shown). Null thymocytes with abnormal mitochondria were frequently detected even before nuclear morphological changes indicative of apoptosis emerged (Fig. 8B and Fig. C), whereas mitochondrial architecture was protected in the TGF-β1 wild-type mice.
Failure of Bcl–XL Upregulation in TGF-β1_−_ /− T Cells.
Since Bcl-2 family members influence apoptotic events and are integral membrane proteins associated with the outer membrane of mitochondria 22 23, and since TGF-β1 was also demonstrated in mitochondria 30 (Fig. 6 and Fig. 7), we next explored potential functional consequences of deletion of TGF-β1 on Bcl-2 family molecules. We focused on inducible Bcl–XL, which inhibits translocation of death factors from the mitochondria to prevent apoptosis in T cells 33 49. Prestimulation of normal T cells with anti-CD3 plus anti-CD28 upregulates Bcl–XL (Fig. 9 A) to prevent subsequent apoptosis induced by nonspecific γ-irradiation 33. In marked contrast, in TGF-β1 null T cells, anti-CD3 and anti-CD28 failed to significantly increase either Bcl–XL mRNA or protein (Fig. 9 B) and thus, many fewer cells were rescued from the secondary apoptosis induced by γ-irradiation (Fig. 9 C). It appears that the presence of TGF-β1 is associated with intact Δψm and expression of Bcl–XL to control T cell viability.
Discussion
Our findings have uncovered an apparent essential role for TGF-β1 in central thymocyte development and selection, and in the maintenance of peripheral lymphocyte homeostasis, by virtue of its ability to control their survival. The unexpected increase in apoptosis in TGF-β1−/− thymocytes indicates a previously unrecognized link between TGF-β1 and T cell development within the thymus. Although the secondary inflammation may exacerbate the dysregulated thymocyte apoptosis, significant increases in apoptosis and decreased total thymocyte number occur before overt signs of inflammation and immunopathology. Furthermore, our in situ apoptosis study with TUNEL (Fig. 1 D) in day 18 fetal thymuses strongly supports TGF-β1 as an inherent factor in controlling thymocyte death and selection, since at this stage, normal thymocyte death is undetectable 36 37 (Fig. 1 C). Despite extensive analyses, there has to date, been no evidence that inflammation occurs before birth, or even perinatally, in the TGF-β1 null mice, in large part due to the maternal-fetal transfer of TGF-β1 50. Thus, even in the presence of maternal TGF-β1 (exogenous) and without evidence of inflammation, gestation day 18 TGF-β1 null T cells already exhibit increased apoptosis. Thymocyte death due to absence of inherent TGF-β1 is further amplified by anti-CD3 both in vitro and in vivo, documenting that TGF-β1−/− thymocytes are exquisitely sensitive to TCR-mediated cell death.
The increased sensitivity to apoptosis appears also to be an essential feature of TGF-β1 gene deleted–peripheral T cells. Several unexpected findings emerged. First, although both CD4+ and CD8+ T cells exhibit increased sensitivity to apoptosis, the decrease in CD8+ peripheral T cells is more dramatic, leading to an abnormal skewing of the CD4/CD8 ratio, irrespective of inflammation. While only Fas-mediated apoptosis increased in CD4+ T cells, TGF-β1–null CD8+ T cells are more sensitive to both Fas and TNFR-mediated death signals, which may offer a clue for the increased ratio of CD4 to CD8. Secondly, TGF-β1−/− lymphocytes undergo spontaneous as well as TCR-mediated apoptosis. The increased sensitivity to cell death of TGF-β1−/− T cells cannot be attributed primarily to prior activation and/or secondary cytokines, although some influence of these factors cannot be absolutely excluded.
Another surprising finding in the current study is that anti-Fas antibody (Jo2) directly kills peripheral CD4+ and CD8+ T cells of TGF-β1−/− mice within 16–24 h. It is unusual for anti-Fas antibody to induce freshly isolated peripheral T cells to become apoptotic after only overnight incubation, since normal T cells do not become susceptible to cell death in response to Fas cross-linking until they have been activated for extended periods of time 33 51 52. TGF-β1−/− peripheral T cells express higher levels of death receptors including Fas, which appears to be a phenotypic characteristic, but not necessarily linked to inflammation. Although the rapid cell death by anti-Fas antibody in these null T cells may be attributed to the upregulation of Fas and FasL, parallel cultures of wild-type T cells stimulated with anti-CD3 antibody overnight to express equivalent levels of Fas 33 do not succumb to anti-Fas–induced lethality (Fig. 4b and Fig. c). Thus, it is unlikely that the supersensitivity to anti-Fas–induced apoptosis in TGF-β1−/− T cells is merely due to their increased expression of death receptors. Since anti-Fas antibody specifically kills null peripheral naive (CD44−/low) T cells (Fig. 2 C) and neutralizing anti–TNF-α and IFN-γ antibodies fail to prevent their Fas-mediated death, the mechanism is likely independent of cellular activation and/or cytokine release. Consequently, the increased susceptibility to death receptor mediated–T cell death appears to be a characteristic of TGF-β1 gene–deleted T cells.
On the basis of this novel observation that enhanced T cell apoptosis is a fundamental characteristic of TGF-β1–deleted T cells, one immediate question is how TGF-β1 normally regulates T cell apoptosis. One plausible explanation would be that the lack of TGF-β1 production and release would mitigate TGF-β receptor–mediated signaling, and previous studies have found that inclusion of exogenous TGF-β in cell culture during the activation phase protects normal T cells from AICD 24 25 26 51. Although not disputing that TGF-β1 receptor–mediated signaling contributes to protection, this may represent only the tip of the iceberg in a very complex picture. Previous studies adding TGF-β to normal T cells or hybridoma cells have not excluded the possibility that endogenous T cell–derived TGF-β might also play an important role, since TGF-β upregulates its own production in both macrophages and T cells 53 54. In our study, exogenous TGF-β1 failed to rescue or prevent spontaneous and death receptor–mediated T cell apoptosis in TGF-β1−/− T cells, whereas these null T cells express normal levels of TGF-β1 receptors. Secondly, T cells deficient in Smad3, a key mediator in the TGF-β–receptor signaling cascade, have no significant spontaneous or Fas-mediated–T cell death compared with wild-type T cells, although these null T cells express an effector/memory phenotype 47. Thus, a pathway distinct from the TGF-β1–TGF-β receptor-mediated signaling cascade was considered in the dysregulated T cell death in the TGF-β1−/− mice.
Mitochondria are considered the common effector of cell death and TGF-β is located within the mitochondria of T cells (Fig. 6 and Fig. 7), suggesting a connection in the pathway to death. The data are consistent with the previous demonstration of TGF-β within the mitochondria in both liver and heart cells 30 and favor a potential role for TGF-β in maintaining Δψm. Indeed, deletion of the TGF-β1 gene was associated with a loss of Δψm. In this regard, the presence of TGF-β1 must normally promote the integrity of Δψm to dissociate the death signals from the initiation phase. The significant increase in disrupted Δψm (DiOC6 low) in T cells in asymptomatic TGF-β1−/− mice and particularly, the multiple forms of abnormal mitochondria in the day 18 fetal thymuses strongly indicate an inherent defect of mitochondrial structure and function. Whether TGF-β is involved in the process of ATP synthesis is conceivable, but aberrant mitochondrial Δψm must promote increased spontaneous apoptosis. The defect also sensitizes to death signals derived from the selective or “private” phase such as FasL–Fas receptors. Anti-Fas antibody causes further reduction of Δψm and apoptosis. Whether Fas-mediated null T cell Δψm disruption involves caspase-1 55 and/or other caspases remains to be determined. This finding may offer an intriguing clue as to why anti-Fas antibody fails to induce apoptosis in TGF-β1 intact normal T cells, despite the expression of Fas receptors on the surface. Therefore, it is not difficult to conceive why exogenous TGF-β1 fails to rescue both spontaneous and death receptor-induced apoptosis, because it is unlikely that exogenous TGF-β1–TGF-β receptor signaling could restore the disrupted Δψm, a cornerstone that marks the point of no return in a cell condemned to die 9 10 11 34 48. The possibility of protection by endogenous TGF-β1 induced by exogenous TGF-β1 is also absent in TGF-β1−/− mice. Moreover, the fact that cyclosporin A, an agent known to induce TGF-β in normal T cells 29 56, inhibits loss of Δψm 57 strengthens the connection between TGF-β and the integrity of Δψm..
The failure to upregulate the antiapoptotic protein Bcl–XL after anti-CD3 plus CD28 challenge in TGF-β1−/− T cells is fascinating, since Bcl–XL is located predominantly in the outer mitochondrial membrane and its induction prevents normal T cells from the subsequent programmed cell death induced by γ-irradiation 33 and by Fas-induced apoptosis 49. On the one hand, it offers an explanation for the failure of anti-CD3 plus CD28 prestimulation to rescue the secondary irradiation-induced cell death in TGF-β1−/− T cells. More importantly, this finding may indicate a previously unrecognized link between TGF-β1, mitochondrial proteins such as Bcl–XL and the control of Δψm. How TGF-β1 regulates Bcl–XL expression remains to be determined, but exacerbation of TGF-β1−/− T cell apoptosis by anti-CD3 plus anti-CD28 or anti-Fas may be the consequence of the inability to upregulate this Bcl–XL protein to protect Δψm.
This protective mechanism of TGF-β1 in normal T cells may involve prevention of the release of other death factors such as apoptosis-inducing factor, cytochrome c, and/or caspase-9 10 11 12 57, which in turn are responsible for the killing of the cells, and is under extensive investigation. Our preliminary data indicate that increased reactive oxygen species and cytochrome c are found in the cytoplasm of TGF-β1−/− T cells, which are downstream of the loss of Δψm (data not shown). Whether these findings are generalizable to nonlymphoid cells and involve key regulatory elements such as nuclear factor κB 44 and/or other Bcl-2 family protein-dependent sequelae 21 22 23 remains to be pursued.
Finally, a paradox emerges from the fact that increased apoptosis of TGF-β1 null T cells is accompanied with or followed by massive inflammation and untimely death of all TGF-β1 null mice 1 2. Resolution of this paradox may rely on a well-known fact that apoptosis and clearing of apoptotic cells are noninflammatory events, involving immunoregulatory cytokines such as TGF-β produced by macrophages during their phagocytosis of apoptotic cells 58 to dampen inflammation. With increasing cell death and lacking requisite TGF-β release during phagocytosis, the balance of the immune response shifts toward the uncontrolled production of proinflammatory cytokines such as TNF-α by phagocytically activated macrophages (unpublished data). The death of these null animals becomes unavoidable.
In summary, we have provided evidence for TGF-β1 as a pivotal regulator in the control of the death of T cells. While TGF-β may play a role at several levels, an underlying mechanism may be the ability of TGF-β to maintain the integrity of mitochondria by protecting Δψm. This protective mechanism may occur independently of the classic TGF-β1 receptor-mediated signaling pathway and irrespective of inflammation. Our data also indicate that TGF-β1 may serve as a moderator in the normal apoptosis process, interfering between the death signaling selective or private phase and the mitochondrial effector phase. Nonetheless, our data offer new clues for defining T cell resistance and susceptibility to death in immunologic diseases, as exemplified in the TGF-β null mouse. Manipulation of TGF-β1 and its stronghold on death may enable rescue and/or deletion of appropriate target cells not only in autoimmune-like diseases, but also in tumors, and potentially, HIV infection.
Acknowledgments
We thank George McGrady for maintaining the mice and E. Clark for technical assistance, Dr. C. Deng, at National Institute of Diabetes and Digestive Kidney Diseases, National Institutes of Health for the Smad3−/− mice, Drs. G. Ashcroft and K. Lei for valuable insight, and Drs. N. McCartney-Francis and X. Song for critical review of the manuscript.
Footnotes
Abbreviations used in this paper: 7-AAD, 7-amino-actinomycin D; Δψm, mitochondrial membrane potential; AICD, activation-induced cell death; DiOC6, 3,3′-dihexyloxacarbocyanine iodide; DP, double positive; ER, endoplasmic reticulum; FasL, Fas ligand; GAPDH, glyceraldehyde 3-phosphate dehydrogenase gene; PALS, periarteriolar lymphatic sheath; TUNEL, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling.
References
- Shull M.M., Ormsby I., Kier A.B., Pawlowski S., Diebold R.J., Yin M., Allen R., Sidman C., Proetzel B., Calvin D. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A.B., Huh C.-H., Becker D., Gerser A., Lyght M., Flanders K.C., Roberts A.B., Sporn M.B., Ward J.M., Karlsson S. Transforming growth factor-β null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl S.M. Transforming growth factorthe good, the bad, and the ugly. J. Exp. Med. 1994;180:1587–1590. doi: 10.1084/jem.180.5.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney-Francis N.L., Wahl S.M. Transforming growth factor βa matter of life and death. J. Leukoc. Biol. 1994;55:401–409. doi: 10.1002/jlb.55.3.401. [DOI] [PubMed] [Google Scholar]
- Weiner H.L. Oral toleranceimmune mechanisms and treatment of autoimmune diseases. Immunol. Today. 1997;18:335–343. doi: 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]
- Strober W., Kesall B., Fuss I., Marth T., Ludviksson B., Ehrhardt R., Neurath M. Reciprocal IFN-γ and TGF-β response regulate the occurrence of mucosal inflammation. Immunol. Today. 1997;18:61–64. doi: 10.1016/s0167-5699(97)01000-1. [DOI] [PubMed] [Google Scholar]
- Christ M., McCartney-Francis N.L., Kulkarni A.B., Ward J.M., Mizel D.E., Mackall C.L., Gress R.E., Hines K.L., Tian H., Karlsson S., Wahl S.M. Immune dysregulation in TGF-β1 deficient mice. J. Immunol. 1994;153:1936–1946. [PubMed] [Google Scholar]
- Chen W.J., Jin W.W., Wahl S.M. Engagement of CTLA-4 induces transforming growth factor-β production by murine CD4+ T cells. J. Exp. Med. 1998;188:1849–1857. doi: 10.1084/jem.188.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G., Zamzami N., Susin S.A. Mitochondrial control of apoptosis. Immunol. Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Mignotte B., Vayssiere J.-L. Mitochondria and apoptosis. Eur. J. Biochem. 1998;252:1–15. doi: 10.1046/j.1432-1327.1998.2520001.x. [DOI] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Nagata S., Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Dhein J., Walczak H., Baumler C., Debatin K.-M., Krammer P.H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- Brunner T., Mogil R.J., Laface D., Yoo N.J., Mahboubl A., Echeverrl F., Martin S.J., Force W.R., Lynch D.H., Ware C.F., Green D.R. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediated activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- Ju S.-T., Panka D.J., Cul H., Ettinger R., El-Khatib M., Sherr D.H., Stanger B.Z., Marshak-Rothstein A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- Singer G.G., Abbas A.K. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–371. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Zheng L., Fisher G., Miller R.E., Peschon J., Lynch D.H., Lenardo M.J. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- Nishimura Y., Ishii A., Kobayashi Y., Yamasaki Y., Yonehara S. Expression and function of mouse Fas antigen on immature and mature T cells. J. Immunol. 1995;154:4395–4403. [PubMed] [Google Scholar]
- Suda T., Hashimoto H., Tanaka M., Ochi T., Nagata S. Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J. Exp. Med. 1997;186:2045–2050. doi: 10.1084/jem.186.12.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Marchetti P., Castedo M., Decaudin D., Macho A., Hirsch T., Susin S.A., Petit P.X., Mignotte B., Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Cerwnka A., Kovar H., Majdic O., Holter W. Fas-and activation-induced apoptosis are reduced in human T cells preactivated in the presence of TGF-β1. J. Immunol. 1996;156:459–464. [PubMed] [Google Scholar]
- Zhang X., Giangreco L., Broome H.E., Dargan C.M., Swain S.L. Control of CD4 effector fatetransforming growth factor β1 and interleukin 2 synergize to prevent apoptosis and promote effector expansion. J. Exp. Med. 1995;182:699–709. doi: 10.1084/jem.182.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genestier L., Kasibhatla S., Brunner T., Green D.R. Transforming growth factor β1 inhibits Fas ligand expression and subsequentactivation-induced cell death in T cells via downregulation of c-Myc. J. Exp. Med. 1999;189:231–239. doi: 10.1084/jem.189.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrl J.H., Wakefield L.M., Roberts A.B., Jakowelw S., Alvarez-Mon M., Derynck R., Sporn M.B., Fauci A.S. Production of transforming growth factor β by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 1986;163:1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Kuchroo V.K., Inobe J., Hafler D.A., Weiner H.L. Regulatory T cell clones induced by oral tolerancesuppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- Li B., Sehajpal P.K., Khanna A., Vlassara H., Cerami A., Stenzel K.H., Suthanthiran M. Differential regulation of transforming growth factor β and interleukin 2 genes in human T cellsdemonstration by usage of novel competitor DNA constructs in the quantitative polymerase chain reaction. J. Exp. Med. 1991;174:1259–1262. doi: 10.1084/jem.174.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine U.I., Burmester J.K., Flanders K.C., Danielpour D., Munoz E.F., Roberts A.B., Sporn M.B. Location of transforming growth factor-β1 in mitochondria of murine heart and liver. Cell. Regulation. 1991;2:467–477. doi: 10.1091/mbc.2.6.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.J., Sayegh M.H., Khoury S.J. Mechanisms of acquired thymic tolerance in vivointrathymic injection of antigen induces apoptosis of thymocytes and peripheral T cell anergy. J. Immunol. 1998;160:1504–1508. [PubMed] [Google Scholar]
- Chen W.J., Jin W., Cook M., Weiner H.L., Wahl S.M. Oral delivery of group A streptococcal cell walls augments circulating TGF-β and suppresses SCW arthritis. J. Immunol. 1998;161:6297–6304. [PubMed] [Google Scholar]
- Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Hildeman D.A., Mitchell T., Teague T.K., Hanson P., Day B.J., Kappler J., Marrack P.C. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y., Y.Sherman, Ben-Sasson S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh C.D., Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. 1994;372:100–103. doi: 10.1038/372100a0. [DOI] [PubMed] [Google Scholar]
- Robey E., Fowlkes B.J. Selective events in T cell development. Annu. Rev. Immunol. 1994;12:675–705. doi: 10.1146/annurev.iy.12.040194.003331. [DOI] [PubMed] [Google Scholar]
- Sprent J., Webb S.R. Intrathymic and extrathymic clonal deletion of T cells. Curr. Opin. Immunol. 1995;7:196–205. doi: 10.1016/0952-7915(95)80004-2. [DOI] [PubMed] [Google Scholar]
- Smith C.A., Williams G.T., Kingston R., Jenkinson E.J., Owen J.J. Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature. 1989;337:181–184. doi: 10.1038/337181a0. [DOI] [PubMed] [Google Scholar]
- Suda T., Takahashi T., Golstein P., Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- Ogasawara J., T.Suda, Nagata S. Selective apoptosis of CD4+ CD8+ thymocytes by the anti-Fas antibody. J. Exp. Med. 1995;181:485–491. doi: 10.1084/jem.181.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Trageser C.L., Willerford D.M., Lenardo M.J. T cell growth cytokines cause the superinduction of molecules mediating antigen-induced T lymphocyte death. J. Immunol. 1998;160:763–769. [PubMed] [Google Scholar]
- Smith C.A., Farrah T., Goodwin R.G. The TNF receptor superfamily of cellular and viral proteinsactivation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Beg A.A., Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Lewis M., Tartaglia L.A., Lee A., Bennett G.L., Rice G.C., Wong G.H., Chen E.Y., Goeddel D.V. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA. 1991;88:2830–2834. doi: 10.1073/pnas.88.7.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFβ signalingreceptors, transducers, and Mad proteins. Cell. 1996;85:947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- Yang X., Letterio J.J., Lechleider R.J., Chen L., Hayman R., Gu H., Roberts A.B., Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Brenner C., Larochette N., Prevost M.C., Alzari P.M., Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise L.H., Thompson C.B. Bcl-x(L) can inhibit apoptosis in cells that have undergone Fas-induced protease activation. Proc. Natl. Acad. Sci. USA. 1997;94:3759–3764. doi: 10.1073/pnas.94.8.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letterio J.J., Geiser A.G., Kulkani A.B., Roche N.S., Sporn M.B., Roberts A.B. Maternal rescue of transforming growth factor-β null mice. Science. 1994;264:1936–1938. doi: 10.1126/science.8009224. [DOI] [PubMed] [Google Scholar]
- Dutton R.W., Bradley L.M., Swain S.L. T cell memory. Annu. Rev. Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- Klas C., Debatin K.M., Jonker R.R., Krammer P.H. Activation interferes with the APO-1 pathway in mature human T cells. Int. Immunol. 1993;5:625–630. doi: 10.1093/intimm/5.6.625. [DOI] [PubMed] [Google Scholar]
- Wahl S.M., McCartney-Francis N., Allen J.B., Dougherty E.B., Dougherty S.F. Macrophage production of TGF-β and regulation by TGF-β. Ann. NY Acad. Sci. 1990;593:188–196. doi: 10.1111/j.1749-6632.1990.tb16111.x. [DOI] [PubMed] [Google Scholar]
- Seder R.A., Marth T., Sieve M.C., Strober W., Letterio J.J., Roberts A.B., Kelsall B. Factors involved in the differentiation of TGF-β-producing cells from naive CD4+ T cellsIL-4 and IFN-γ have opposing effects, while TGF-β positively regulates its own production. J. Immunol. 1998;160:5719–5728. [PubMed] [Google Scholar]
- Susin S.A., Zamzami N., Castedo M., Daugas E., Wang H.G., Geley S., Fassy F., Reed J.C., Kroemer G. The central executioner of apoptosismultiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J. Exp. Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin G.T., Khanna A., Ding R., Sharma V.K., Lagman M., Li B., Suthanthiran M. In vivo expression of transforming growth factor-β1 in humansstimulation by cyclosporine. Transplantation. 1998;65:313–318. doi: 10.1097/00007890-199802150-00003. [DOI] [PubMed] [Google Scholar]
- Kroemer G. The mitochondrion as an integrator/coordinator of cell death pathways. Cell Death Differ. 1998;5:547. doi: 10.1038/sj.cdd.4400387. [DOI] [PubMed] [Google Scholar]
- Fadok V.A., Bratton D.L., Konowal A., Freed P.W., Westcott J.Y., Henson P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]