Essential Role of the Prosurvival bcl-2 Homologue A1 in Mast Cell Survival After Allergic Activation (original) (raw)
Abstract
Mast cells reside in tissues, where upon activation through the high-affinity-IgE-receptor (FcεRI) they degranulate and orchestrate the allergic reaction. Mast cells survive this activation and can thus be reactivated. In this study we demonstrate that this process depends on the pro-survival gene A1. Activation of mast cells through FcεRI resulted in degranulation, strong induction of A1 mRNA and protein, and cell survival. In contrast, A1-deficient mast cells released granule mediators similar to the wild-type control, but the cells did not survive an allergic activation. Furthermore, A1−/− mice that had been sensitized and provocated with allergen exhibited a lower number of mast cell compared with littermate controls. The induction of A1 was dependent on calcium, as EDTA prevented A1 expression. The calcium ionophore, ionomycin, induced A1 expression and mast cell survival, whereas compound 48/80, a well-known mast cell secretagogue, did not. This study uncovers the importance of A1 for mast cell survival in allergic reactions, and it proposes A1 as a potential target for the treatment of allergic diseases.
Keywords: allergy, apoptosis, IgE, inflammation, FcεRI
Introduction
Mast cells are central effector and regulatory cells in allergic diseases. They are long-lived cells that are widely distributed throughout vascularized tissues and certain epithelia, where they play a fundamental role in the pathogenesis of immediate hypersensitivity reactions (1–3). Especially in diseases such as allergy and asthma, an increased number of tissue mast cells can be documented (2), and a correlation between the number of mast cells in the tissue and the severity of the symptoms has been described (4–7). In allergy, multivalent antigens bind and cross-link IgE molecules bound to the high-affinity IgE-receptor (FcεRI)* expressed on mast cells (8). Receptor aggregation induces multiple signaling pathways that control diverse cellular functions (9). One of the key events induced by cross-linking of FcεRI is the rapid release of a broad spectrum of vasoactive and proinflammatory mediators causing the symptoms associated with an allergic reaction (1, 3, 10). Thus, mast cell activation and hyperplasia are key components that contribute to allergic diseases.
A unique feature of mast cells is that they withstand the exhaustive degranulation process, survive, regranulate, and can thereafter be activated again (11, 12). Therefore, a fundamental question in mast cell biology is how the cells survive the FcεRI-mediated degranulation process, which is an important feature of these cells for the perpetuation of an allergic reaction.
One important mechanism for protecting cells from apoptosis is through the regulation of a number of proto-oncogenes whose products interact to determine the final outcome of apoptotic signals. A major class of such intracellular regulators is the bcl-2 family of proteins (13). The bcl-2 family includes a number of apoptosis-regulatory genes, which may either be death antagonists (bcl-2, bcl-XL, bcl-w, mcl-1, and A1/bfl1) or death agonists (bax, bak, bcl-XS, bad, bid, and bik) (14, 15). Although the precise mechanism by which bcl-2 family members influence apoptosis is unknown, several lines of evidence suggest that bcl-2 proteins function at a critical decision point immediate upstream of an irreversible commitment to cell death (16).
The role of survival/apoptosis-regulatory genes in mast cell activation has not been fully defined. There are some reports on the expression and regulation of bcl-2 in mast cells (17–20). Despite the wide-ranging ability of bcl-2 to promote cell survival, it is clear that in a number of circumstances bcl-2 is not responsible for protecting cells from apoptosis (21, 22). Therefore, it is possible that other members of the bcl-2 family may provide protective effects in some specific biological processes.
One of the pro-survival bcl-2 homologues is A1, which was originally identified from mouse bone marrow culture induced with GM-CSF (23). A1 was described as an early-response gene, expressed in multiple tissues such as thymus, spleen, and bone marrow, and also expressed in a number of hemotopoietic cell lineages including T- and B lymphocytes, macrophages, neutrophils, and endothelial cells (23–26). A1 is the only bcl-2 family member that is inducible by inflammatory cytokines such as TNF-α and IL-1β, which suggests a possible role for A1 in inflammatory responses (27). The present study addresses the question of a possible role for bcl-2 family genes in the regulation of mast cell survival after activation by FcεRI cross-linking. Our results demonstrate a critical role for A1 in mast cell survival upon allergic activation.
Materials and Methods
Mast Cell Culture.
Bone marrow–derived cultured mouse mast cells (BMCMCs) were obtained by culturing mouse bone marrow cells from C57BL/6 and BALB/c (Bommice). BMCMCs were also obtained from mice lacking A1-a (C57BL/6 genetic background) (28), one of the A1 subtypes (29). Bone marrow cells were cultured in 10% WEHI-3 enriched conditioned RPMI 1640, supplemented with 10% heat-inactivated fetal bovine serum, 4 mM L-glutamine, 50 μM 2-mercaptoethanol, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 10 mM Hepes, and 100 μg/ml penicillin/streptomycin (Sigma-Aldrich). Mast cell development was confirmed by toluidine blue staining and the expression of Kit on the cell surface. The growth factor–dependent mouse mast cell lines MCP5/L (30) (obtained from Dr. D.D. Metcalfe, National Institutes of Health, Bethesda, MD) and MC/9 (American Type Culture Collection) were maintained in the same medium as described above. The growth-factor independent cell line C57 (31, 32) (obtained from Dr. S.J. Galli, Stanford University, Stanford, CA) were maintained in RPMI medium supplemented with 10% FCS and 50 μM 2-mercaptoethanol.
Cell Survival Assay.
For cell viability assays, the cell suspension was mixed with the vital dye, trypan blue, and the number of live cells was enumerated. Cell apoptosis was measured by Cell Death Detection ELISA (Boehringer), quantitatively detecting the mono- and oligo-nucleosomes released into culture supernatant.
Mast Cell Activation.
For FcεRI-dependent activation, mast cells were sensitized using a monoclonal murine IgE anti-TNP antibody (IgEl-b4; American Type Culture Collection; used as 15% hybridoma supernatant) for 90 min. After two washings, the cells were subjected to challenge with 1 μg/ml TNP-BSA (coupling ratio: 4.8, provided by Dr. Birgitta Heyman, Uppsala University) for time periods as indicated. For cell viability and β-hexosaminidase release assays, both the antibody sensitization and the antigen challenge were performed in RPMI supplemented with 0.5% BSA at 37°C. For gene regulation studies, all the incubations were carried out in RPMI supplemented with 5% FCS or as indicated in the text. To determine the effects of ionomycin or compound 48/80 (Sigma-Aldrich) on A1 regulation, cells were resuspended in medium containing ionomycin or compound 48/80 and incubated for 6 h at 37°C. For detection of β-hexosaminidase, an enzymatic colorimetric assay was used as described (33). In brief, supernatant or cell lysate was mixed with an identical volume of substrate solution (7.5 mM p-nitrophenyl N-acetyl-β-D-glucosaminide (Sigma-Aldrich) in 80 mM citric acid, pH 4.5) and incubated at 37°C for 2 h. The reaction was stopped by the addition of glycine (0.2 M, pH 10.7) into each well and the absorbance was measured. For inhibition studies, cells were cross-linked in medium containing EDTA (1 mM), wortmannin (100 nM), dexamethasone (1 μM), or cycloheximide (10 μg/ml; Sigma-Aldrich). EDTA was added at the same time as the addition of IgE. Dexamethasone was added 14 to 16 h before IgE sensitization. The other inhibitors were added at the same time as FcεRI aggregation. In some experiments, mast cells were treated with cytokines which include rmTNF-α (10 ng/ml), rm GM-CSF (10 ng/ml), rm stem cell factor (SCF; 100 ng/ml), rmIL-4 (10 ng/ml; PeproTech), and mouse nerve growth factor (NGF; 100 ng/ml; Promega), for 1, 3, 6, and 18 h.
Ribonuclease Protection Assay.
Total cellular RNA was isolated using the TriPure isolation reagent (Boehringer). Ribonuclease protection assay (RPA) was performed using the mAPO-2 multi-probe set from the RiboQuant System (BD PharMingen) following the supplier's recommended protocol. Quantification of mRNA expression was determined using a PhosphorImaging device, and levels of each gene transcript were quantified by MacBas V2.2 (Fuji Photo Film Co. Ltd.).
Western Blot.
BMCMCs were activated in complete medium through FcεRI cross-linking and cells were harvested at 6, 12, 18, 30, 48, and 72 h postactivation, washed once in PBS, and lysed in hot 2× SDS gel sample buffer (20 mM dithiothreitol, 6% SDS, 0.25 M Tris, pH 6.8, 10% glycerol, and bromophenyl blue). The protein concentration in the samples was measured using a Bradford assay (Bio-Rad Laboratories) and 50 μg of protein was resolved on SDS-10% polyacrylamide gels. Protein was then electroblotted to nitrocellulose membrane. Equal protein loading was confirmed by Ponceau S (Sigma-Aldrich) staining of the gel after transfer. A1 protein was detected using a rat monoclonal antiserum against murine A1 (R&D Systems) and bcl-XL was detected using a rabbit anti-bcl-XL (Santa Cruz Biotechnology, Inc.), followed by a horseradish peroxidase–conjugated sheep anti–rat or goat anti–rabbit antibody (Amersham Pharmacia Biotech) at a 1:2,000 dilution. Western blots were then visualized by chemoluminescence using enhanced chemoluminescence (ECL).
Immunization and Histochemical Staining.
Female A1-a knockout and littermate control mice (three mice/group) between 8 and 12 wk of age, which were maintained on OVA-free diet, were used. Immunization was conducted weekly for 4 wk by injections of 20 μg intraperitoneally chicken OVA (grade V; Sigma-Aldrich) adsorbed to 1 mg alum in 0.2 ml PBS. 2 d after the last exposure the mice were provocated with an identical dose of OVA plus alum intraperitoneally. 2 d later skin samples were collected from the footpad.
Skin biopsies from the footpad were fixed in 4% paraformaldehyde overnight and embedded in paraffin blocks. Skin sections were cut (5 μm) and mounted on Superfrost® Plus slides. After deparaffinization steps the sections were stained with toluidine blue for 10 min. Mast cell number was assessed in three skin sections for each animal. Quantification was performed in coded slides in three randomly selected areas of each section.
Statistics.
Statistics were calculated using an analysis of variance (ANOVA), followed by multiple comparison using Fisher's method. * denotes P < 0.05, ** P < 0.01. Values presented are the means ± SEM.
Results
Cross-linking of FcεRI Prevents Apoptosis Induced by Growth Factor Deprivation.
IL-3 is one of the primary growth factors for murine mast cells. Both mouse mast cell line MCP5/L and BMCMCs are growth factor dependent, and they require the inclusion of IL-3 in their media for survival. As shown in Fig. 1, A and B, withdrawal of WEHI, which forms the source of IL-3, resulted in a progressive decrease in the number of live cells as determined by trypan blue exclusion staining. To examine whether mast cell activation would influence the survival of cells deprived of growth factor, we activated MCP5/L cells and BMCMCs through FcεRI aggregation. The survival percentage of activated MCP5/L increased compared with control cells after 2 d of incubation, and on days 4 and 5 FcεRI aggregation led to cell survival promotion by ∼70 and 120%, respectively, over control cells (Fig. 1 A). For BMCMCs, which are more sensitive to growth factor withdrawal, FcεRI aggregation resulted in cell survival promotion by 74 and 600% on the second and third day, respectively (Fig. 1 B).
Figure 1.
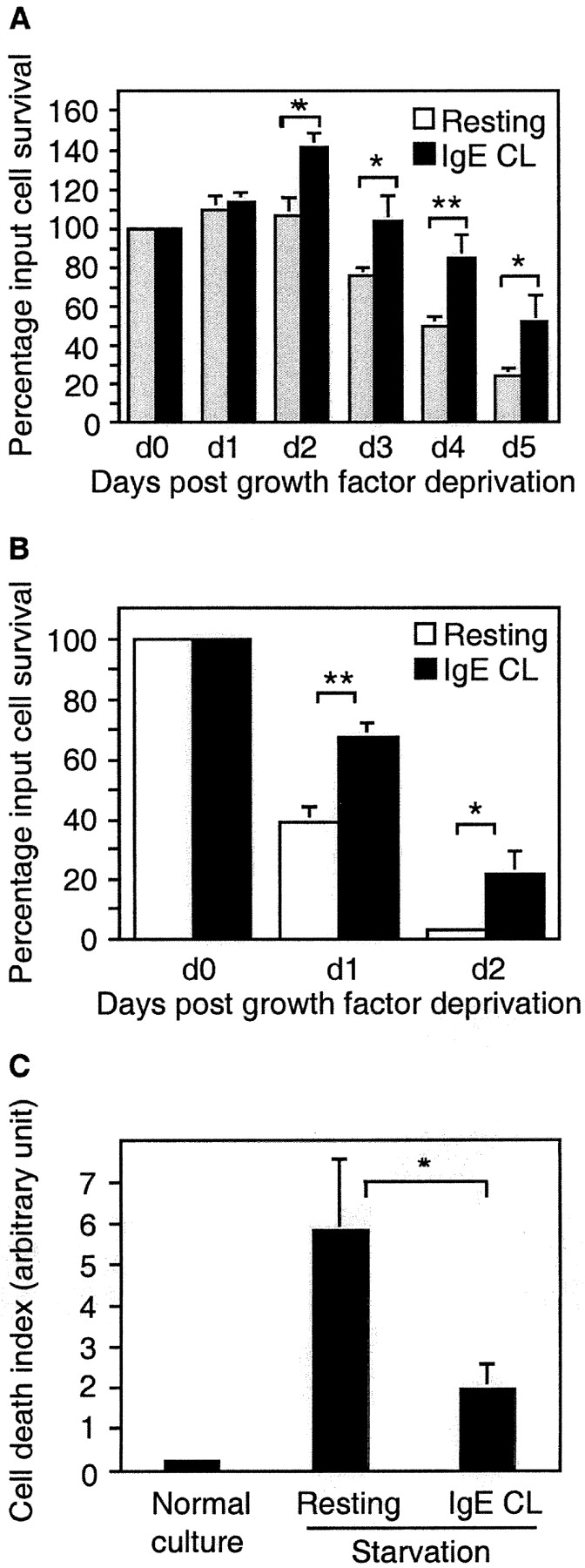
Survival promotion after mast cell activation by FcεRI cross-linking. MCP5/L cells (A) or BMCMCs from C57BL/6 (B and C) were either stimulated through cross-linking of FcεRI (IgE CL) or left untreated in RPMI deprived of serum and growth factors. Viability was determined by trypan blue exclusion and presented as the percentage of input cells that are still alive when examined every 24 h (A and B). In C, BMCMC apoptosis was assessed by ELISA measuring the release of nucleosomes into the culture supernatant after 24 h. Data are presented as the mean ± SEM from three to five separate experiments.
One distinct feature indicating apoptosis is the activation of an endogenous endonuclease which cleaves double-stranded DNA at the most accessible internucleosomal linker region, generating mono- and oligo-nucleosomes. By measuring the release of mono- and oligo-nucleosomes, we could confirm that cross-linking of FcεRI rescued the cells from undergoing apoptosis (Fig. 1 C). Thus, the activation of mast cells through FcεRI initiates a cellular response that directly prevents apoptosis, without the addition of exogenous growth factors.
A1 Is Upregulated upon FcεRI Activation.
Apoptosis is regulated by members of the bcl-2 family that can either be prosurvival or proapoptotic. Our finding that cross-linking of FcεRI promotes mast cell survival upon growth factor withdrawal, led us to investigate whether this activation induced transcriptional regulation of any of the bcl-2 family members.
RPA was performed on MCP5/L cells either at resting condition or after activation by FcεRI, using the mAPO-2 multi-probe set from BD PharMingen which allows the simultaneous analysis of multiple bcl-2 family genes. What attracted our initial attention was a striking up-regulation of the pro-survival bcl-2 homologue A1 (Fig. 2 A). A1 was absent in resting cells but substantially upregulated after FcεRI aggregation for 6 h. For the other genes analyzed, no obvious regulation was observed directly from the PhosphorImaging picture. Therefore we quantified the bands by densitometric analysis (Fig. 2 B). The A1 mRNA level in cells activated by FcεRI aggregation had increased >26-fold when the signal was compared with that of control cells (Fig. 2 B). Bcl-XL was upregulated by an increase of 1.8-fold over control cells. The identity of bcl-XL was confirmed by Western blot (data not shown). As for other genes, FcεRI aggregation did not modulate the regulation of gene expression levels (Fig. 2 B). The expression of A1 in activated mast cells was confirmed in BMCMCs from C57BL/6 and BALB/c mice, as well as in other mast cell lines (MC/9 and C57; Fig. 2 C, and data not shown). We also performed control experiments for the FcεRI aggregation where either IgE-anti-TNP or the antigen was omitted. β-hexosaminidase release, promotion of mast cell survival, or A1 induction could not be determined in any of these experiments (data not shown).
Figure 2.

bcl-2 family gene expression in mast cells after cross-linking of FcεRI. (A) RPA was performed to analyze the expression levels of the indicated genes on total RNA isolated from MCP5/L cells either resting or activated for 6 h through FcεRI cross-linking (IgE CL). Lane 1, resting cells cultured in the presence of WEHI-3–conditioned medium. Lane 2, resting cells that were incubated and washed through the same procedures needed for FcεRI cross-linking (deprived of growth factor). Lane 3, cells that were stimulated through FcεRI cross-linking. A representative experiment of at least six performed independently is shown. (B) Phosphor-Imaging signals presented in A (lanes 2 and 3) are shown as gene expression relative to the average expression of the house keeping genes GAPDH and L32. Data are normalized such that the densitometric level of each gene from the control cells is given a value of 1. (C) RPA was performed as in A to analyze A1 induction in MCP5/L and C57 cells activated for 6 h in the presence or absence of FCS and WEHI as a source of IL-3.
As the mast cell activation was performed in low-serum and growth factor–deprived medium, which is not a physiological condition we next investigated if A1 could be induced also in the presence of serum with or without IL-3. As shown in Fig. 2 C, A1 was indeed induced after FcεRI aggregation also in the presence of IL-3. Furthermore, C57 cells that are growth factor independent also exhibit A1 induction upon activation further supporting that IL-3 is not a prerequisite for the expression of A1 (Fig. 2 C).
We next investigated the kinetics of A1 expression in mast cells upon FcεRI cross-linking. Kinetic studies revealed that A1 transcript could be detected 2 h after activation, peaked around 6 h, and was no longer detectable by 24 h (Fig. 3 A). A similar expression pattern has been observed in macrophages and endothelial cells stimulated with cytokines or LPS (23, 26).
Figure 3.



Kinetics of A1 mRNA and protein expression in mast cells activated by FcεRI cross-linking. (A) RPA was performed on RNA isolated from MCP5/L cells either at resting state or activated through FcεRI cross-linking (IgE CL) for various time points as indicated to detect A1 transcript levels. (B) Accumulation of A1 protein in resting and activated BMCMCs was analyzed by Western blot analysis. (C) MCP5/L cells were activated through FcεRI cross-linking (1st CL) up to 24 h, and then were washed and incubated under normal culture condition. 48 h after the first activation, cellular IgE receptors were cross-linked again (2nd CL). A1 induction at time points both in the first and the second activation periods were analyzed by RPA. Data shown are representative of three separate experiments.
To confirm the translation of A1 mRNA into protein after mast cell activation, we performed Western blotting to measure the intracellular A1 protein expression. The results indicated that A1 protein expression in BMCMCs was detectable at 6 h and peaked at around 48 h after FcεRI-mediated activation (Fig. 3 B).
Under physiological conditions, mast cells do not die after degranulation triggered by FcεRI aggregation, and previously activated mast cells can be activated again. If A1 plays a role in mast cell survival after activation, it would be interesting to explore whether A1 expression is repeatedly induced in mast cells after repeated activation of FcεRI. MCP5/L cells were therefore first activated by IgE and antigen, washed, and then activated again 48 h after the initial activation. The kinetics demonstrated that A1 appeared 2 h after the second FcεRI aggregation, peaked at around 6 h, after which the expression declined (Fig. 3 C), which is similar to the initial activation (Fig. 3 A). The release of the granule-associated β-hexosaminidase was measured upon the second activation to indicate mast cell degranulation, which is also similar to the initial activation (data not shown).
Thus, through cross-linking of FcεRI mast cells can be repeatedly activated to degranulate, and this process is accompanied by the repeated induction of A1 expression.
Regulation of A1 Expression.
In mast cell activation, the sequence of events leading from receptor cross-linking to exocytosis has been well elucidated, and one of the hallmarks of cellular activation is an increase in concentrations of free intracellular calcium that is derived from both extracellular and intracellular stores (9). Therefore we were interested to reveal how A1 regulation would be affected when mast cell activation was performed in the presence of the Ca2+ and Mg2+ chelating agent EDTA. Our results showed that neither the release of β-hexosaminidase (data not shown) nor the induction of A1 (Fig. 4) could be detected in activated MCP5/L when EDTA was present.
Figure 4.

Effects of a number of inhibitors on A1 induction in mast cells after activation by FcεRI cross-linking. RPA was performed on RNA from MCP5/L cells which were activated through FcεRI cross-linking (IgE CL) in the absence or presence of various inhibitors as indicated. Data shown represent comparable results from at least three separate experiments.
To investigate the regulatory control of A1 expression in more detail, we treated MCP5/L cells with dexamethasone, wortmannin, and cycloheximide. As shown in Fig. 4, all inhibitors completely blocked A1 induction.
Ionomycin but Not Compound 48/80 Induces A1 Expression and Mast Cell Survival.
To determine whether the regulation of A1 transcript was only seen with FcεRI aggregation as a physiological stimulus, or if A1 induction could also be achieved in response to other stimuli, MCP5/L cells were exposed to ionomycin for 6 h. Ionomycin is a calcium ionophore, which induces increased levels of intracellular calcium, capable of mimicking the activation of FcεRI. Similar to FcεRI aggregation, activation with ionomycin led to induction of A1, release of β-hexosaminidase, and mast cell survival in a dose-dependent manner (Fig. 5, A–C). Release of β-hexosaminidase after ionomycin treatment as well as FcεRI aggregation of MCP5/L cells was 10–15%. The conclusion from these data, as well as the fact that EDTA could block A1 induction (Fig. 4), is that A1 is induced through a calcium-dependent mechanism.
Figure 5.

The effects of ionomycin and compound 48/80 on mast cell A1 expression, mediator release, and survival. RPA was performed on RNA derived from MCP5/L cells incubated for 6 h in the presence of ionomycin or compound 48/80 at concentrations indicated (A and D). Release of β-hexosaminidase (β-hex) after 30 min was measured using an enzymatic colorimetric method and was indicative of activation and degranulation of mast cells (B and E). Survival rate of mast cells after 4 days in medium deprived of serum and growth factors was measured (C and F) as explained in Fig. 1. Data shown in A and D are a representative of three separate experiments and data shown in the rest are expressed as the mean ± SEM from three experiments.
Another well-known mast cell secretagogue is compound 48/80, which induces exocytosis without elevation of cytosol Ca2+ and activates G-proteins directly (34). Furthermore, GTP-binding proteins are located downstream of signal transduction pathways leading from receptor activation to exocytosis, so that the release of mast cell granules can be triggered by direct activation of such proteins (35). Prompted by these descriptions, we then explored whether compound 48/80 would induce A1 expression in mast cells. As shown in Fig. 5 D, compound 48/80 at concentrations of up to 100 μg/ml failed to upregulate A1 induction, although it triggered a substantial release of β-hexosaminidase (Fig. 5 E). What is compatible with our hypothesis that A1 is responsible for mast cell survival during its degranulation process, is that the treatment of cells with compound 48/80 failed to rescue cells from dying after growth factor deprivation (Fig. 5 F). These data support the hypothesis that the difference in survival promotion potential between ionomycin and compound 48/80 is related to A1 regulation.
Cytokines Fail to Induce A1 in Mast Cells.
A1 was initially described as an early-response gene whose expression was associated with a variety of inflammatory stimuli such as GM-CSF and TNF-α (23, 26). We therefore analyzed whether cytokines previously shown to induce A1 in other cell types had a similar effect on mast cells. In addition we tested IL-4, SCF, and NGF, which have been implicated in regulating mast cell survival/apoptosis (18, 20, 36–38). As shown in Fig. 6, none of these cytokines induced expression of A1 in growth factor–deprived mast cells. We also analyzed the effect of IL-3 on A1 induction. Both BMCMCs and MCP5/L are growth factor dependent and were cultured in the presence of IL-3, although the activation studies were performed in growth factor–deprived medium. Culturing the cells in the presence of IL-3 did not induce expression of A1 (Fig. 2 A, 1st lane), and the presence of IL-3 during the activation did not affect the induction of A1 (Fig. 2 C). Furthermore, treatment of the growth factor–independent mast cell line C57 with IL-3 did not cause any induction of A1 (data not shown).
Figure 6.

Absence of A1 induction in cells treated with cytokines. MCP5/L cells were incubated in the presence of various cytokines as indicated for 6 h and A1 expression was measured by RPA. Cells that were activated by FcεRI cross-linking (IgE CL) were presented as positive control.
Mast Cells Deficient in A1 Do Not Survive an Allergic Activation.
To address the issue of whether A1 is a prerequisite for mast cell survival upon cross-linking of FcεRI, or if the up-regulation of A1 is just a parallel phenomenon, we used mast cells deficient in A1-a (28). A1-a−/− mice are healthy and fertile, but have an increased spontaneous apoptosis of neutrophils (28). They have normal numbers of mast cells in the skin, lung, and spleen (data not shown). The A1-a−/− BMCMCs had a morphology similar to wild-type mast cells, stained with toluidine blue, expressed Kit on the surface, and the cell growth rate was similar to the wild-type control. Thus, A1 seems not to have any obvious role in mast cell homeostasis or in vitro development. Upon activation with IgE and antigen, the A1-a−/− BMCMCs released granule-associated β-hexosaminidase similar to the wild-type control (Fig. 7 A). Cross-linking of FcεRI did not promote survival of these cells; instead they died at a similar rate as resting control cells (Fig. 7 B). Thus, this finding confirms the hypothesis that A1 expression is necessary for mast cell survival upon allergic activation.
Figure 7.



Absence of survival promotion of mast cells from A1−/− mice after FcεRI cross-linking (IgE CL). (A) β-hexosaminidase (β-hex) release from A1−/− mast cells was assessed as explained in Fig. 5 to demonstrate that the cells were properly activated. (B) BMCMCs from A1 knockouts and wild-type littermates were stimulated and viability was determined as explained in Fig. 1. (C) The mast cell number was quantified in the footpad skin sections of A1-a−/− and wild-type control mice before and after OVA treatment as detailed in Materials and Methods. Values presented are the means ± SEM from three to six separate experiments.
We also examined the number of mast cells in vivo after allergen provocation. Mice deficient in A1-a expressed the same number of mast cells in the skin of the footpad as wild-type control (∼5 mast cells/mm2). However, after allergen challenge and provocation, a significant difference in mast cell number could be observed in the skin of A1-a−/− compared with the littermate control mice (Fig. 7 C). The mast cell number in the skin of A1-a−/− was decreased by ∼50% compared with A1-a+/+.
Discussion
The current study demonstrates that A1 is central to FcεRI-induced mast cell survival, while not having any obvious role in mast cell development. Mice deficient in A1 expressed normal numbers of mast cells in the skin, lung, and spleen. Furthermore, there was no difficulty culturing mast cells from the bone marrow of A1-deficient mice. However, A1−/− mast cells did not survive an allergic activation by cross-linking of the high-affinity IgE-receptor in vitro, and the mast cell number was reduced in vivo in A1−/− mice after allergen sensitization and provocation (Fig. 7).
One of many interesting features of mast cells is that they, upon degranulation, can regranulate and be activated again. In ultrastructural studies it has been shown that mast cells, upon IgE-dependent activation, enter a stage manifested by very dramatic exocytosis, including the release of nearly all cell granules and the shedding of large amounts of membranes, surface processes, and cytoplasmic fragments (12, 39). One characteristic feature of the acute allergic reaction is that it can be initiated repeatedly, for example during a pollen season. This is most likely due to the de- and regranulation capacity of mast cells. The involvement of bcl-2 family members in regulating this process, although logical, has hitherto been unconfirmed.
Several previous studies have indicated the possible role of bcl-2 in mast cell biology. For instance, overexpression of bcl-2 cDNA prolongs the survival of mast cells upon withdrawal of IL-3 (40), and overexpression of bcl-2 by human bone marrow mast cells is found in mast cell leukemia (41). Bcl-2 is also induced upon treatment of mast cells with IL-3 or NGF (17, 18). However, in our mast cell FcεRI-dependent activation model, we have not found any significant regulation of bcl-2 transcript levels (Fig. 2). Although bcl-2 is the most extensively studied death antagonist and has been shown to enhance cell survival by inhibiting apoptosis under a wide variety of circumstances, bcl-2–independent prevention of apoptosis has also been reported (22). Therefore, it should not be surprising that A1 but not bcl-2 is upregulated and promotes mast cell survival after IgE-dependent activation.
A pronounced upregulation, like an all-or-none event, was seen in A1 (Fig. 2), which strongly suggests that A1 might play a key role in mast cell activation-dependent survival promotion and hence in the long life-span of mast cells. A similar on/off effect has been shown for A1 expression after treatment of macrophages and endothelial cells with inflammatory cytokines and LPS (23, 26, 42). A1 has been demonstrated to exert a prominent role in the prevention of apoptosis in a variety of cell systems. A1 prolongs cell survival during myeloid differentiation (42) and protects endothelial cells from apoptosis induced either by serum starvation (43) or by TNF-α treatment (26).
In addition to A1, we could detect an increase of bcl-XL after FcεRI aggregation (Fig. 2). It has been shown that activation of v-Abl protein tyrosine kinase in pre-mast cells results in the suppression of apoptosis after withdrawal of IL-3, and the activation is followed by an approximately twofold increase at the mRNA level of bcl-XL (44). Our observation of an approximately 1.8-fold increase of bcl-XL (Fig. 2 B) is compatible with the above finding, and suggests that bcl-XL might also be functional in mast cell activation.
It is not clear from our results if the survival role of A1 in mast cells is mediated through direct or indirect mechanisms. Several studies have shown that members of the bcl-2 family can interact with each other and form dimers (45). The heterodimerization between a prosurvival and a pro-death member may be one of the most important mechanisms in the regulation of apoptosis (15). A1 has been described to interact with bax (46), which is a pro-death gene expressed in mast cells (Fig. 2). Furthermore, bax has previously been suggested to be involved in the regulation of mast cell apoptosis (47). Whether A1 acts as an antagonist for bax in mast cells or if A1 promotes mast cell survival through other mechanisms remains to be elucidated.
Both calcium ionophores, such as ionomycin, and compound 48/80 are mast cell secretagogues which can efficiently stimulate exocytosis. Ionomycin mimics FcεRI aggregation signals by stimulating calcium influx. Our data show that ionomycin stimulated A1 induction in a dose-dependent manner (Fig. 5 A), which suggests that calcium influx is required in the signaling pathways for A1 induction. The requirement for calcium mobilization has been further substantiated by the fact that addition of EDTA completely blocked A1 induction after FcεRI cross-linking (Fig. 4). In contrast, compound 48/80 induces exocytosis in a receptor-independent manner by interacting directly with heterotrimeric G proteins that are located downstream of calcium mobilization (34). In our observations, although compound 48/80 could induce β-hexosaminidase release (Fig. 5 E), it failed to induce A1 transcript expression (Fig. 5 D) and the cells did not survive (Fig. 5 F). Not surprisingly, compound 48/80 is used to deprive mast cells in vivo (e.g., reference 48), and our findings may explain the mechanisms by which mast cell depletion is achieved.
We observed a strong inhibition of A1 expression after treatment with dexamethasone (Fig. 4). Repression of nuclear factor (NF)-κB–dependent gene expression is one of the key characteristics by which glucocorticoids exert their antiinflammatory effects. Recent studies show that the promoter region of A1 contains NF-κB binding sites, and survival mechanisms mediated by NF-κB–dependent expression of A1 have been reported (49–51). When we take into account these observations, it is tempting to speculate that FcεRI-induced expression of A1 and mast cell survival is mediated via NF-κB, although this has yet to be proven.
It has been reported that TNF-α and GM-CSF can potentiate A1 induction in macrophages and endothelial cells (23, 26). We were therefore interested in characterizing whether these or other cytokines were capable of upregulating A1 transcript in mast cells. The cytokines that we screened included the proinflammatory cytokines such as TNF-α and GM-CSF, as well as cytokines that have strong implications in the survival, differentiation, and functional maintenance of mast cells such as SCF, NGF, and IL-4 (18, 20, 36–38). The result indicated that none of the cytokines tested could induce A1 induction. In a recent publication by Yoshikawa et al. they described that FcεRI-induced mast cell survival is mediated by autocrine release of cytokines, i.e., IL-4 (52). In contrast to their findings, we could not confirm that activation-induced mast cell survival depends on factors released from the cells. Supernatants from activated mast cells did not induce A1 expression, but prevented apoptosis of cells that had been deprived of growth factors probably because of the presence of cytokines in the supernatant (unpublished data). Furthermore, compound 48/80 did not induce A1 expression or mast cell survival, although it caused substantial degranulation (Fig. 5). Taken together, these lines of evidence would indicate that A1 induction in mast cells may be a direct effect of FcεRI aggregation that requires calcium influx as a prerequisite condition. Further studies elucidating the regulation of A1 expression and function in mast cells are currently in progress.
This study describes that FcεRI aggregation induces the expression of A1 that is needed for mast cells to survive the degranulation process during an allergic reaction. Recently it was described that FcεRI aggregation also increases the expression of Fas-associated death domain–like IL-1–converting enzyme (FLICE), a protein involved in the protection against Fas-induced apoptosis (53). Although mast cells express Fas and undergo apoptosis upon activation through Fas (54), the physiological role for Fas–FasL interaction and its importance for regulating mast cell number in vivo has not been clarified. However, it appears that allergic activation by FcεRI aggregation can regulate mast cell survival through different mechanisms.
Our finding that A1 plays a pivotal role in allergy-dependent mast cell survival fits well with the characteristic of A1 as a pro-survival gene, acting rapidly upon activation. A direct inhibition of A1 may prove beneficial in reducing mast cell numbers in tissues affected by allergic reactions. The challenge is now to determine whether A1 can be used as a target for the development of new allergy therapies. It is also possible that dysregulation of A1 in humans may increase the susceptibility to mast cell–mediated disorders.
Acknowledgments
The authors would like to thank Dr. Birgitta Heyman for reagents, and Dr. Jaroslaw Dastych for helpful discussions.
This work was supported by the Swedish Medical Research Council, the Swedish Cancer Society, the Swedish Society of Medicine, the King Gustaf V:s 80-years Foundation, the Ollie and Elof Ericsson Foundation, the Agnes and Mac Rudbergs Foundation, the Lilly and Ragnar Åkerham Foundation, the network for inflammation research funded by the Swedish Foundation for Strategic Research, the Göran Gustavsson Foundation, and Innoventus Uppsala Life Science AB.
Footnotes
*
Abbreviations used in this paper: BMCMC, bone marrow–derived cultured mast cells; FcεRI, high-affinity IgE-receptor; NF, nuclear factor; NGF, nerve growth factor; RPA, ribonuclease protection assay; SCF, stem cell factor.
References
- 1.Metcalfe, D.D., D. Baram, and Y.A. Mekori. 1997. Mast cells. Physiol. Rev. 77:1033–1079. [DOI] [PubMed] [Google Scholar]
- 2.Nilsson, G., J.J. Costa, and D.D. Metcalfe. 1999. Mast cells and basophils. Inflammation: Basic Principles and Clinical Correlates. J.I. Gallin and R. Snyderman, editors. Lippincott-Raven publications, Philadelphia, PA. 97–117.
- 3.Galli, S.J. 2000. Allergy. Curr. Biol. 10:R93–R95. [DOI] [PubMed] [Google Scholar]
- 4.Otsuka, H., J. Denburg, J. Dolovich, D. Hitch, P. Lapp, R.S. Rajan, J. Bienenstock, and D. Befus. 1985. Heterogeneity of metachromatic cells in human nose: Significance of mucosal mast cells. J. Allergy Clin. Immunol. 76:695–702. [DOI] [PubMed] [Google Scholar]
- 5.Wardlaw, A.J., S. Dunnette, G.J. Gleich, J.V. Collins, and A.B. Kay. 1988. Eosinophils and mast cells in bronchoalveolar lavage in subjects with mild asthma. Relationship to bronchial hyperreactivity. Am. Rev. Respir. Dis. 137:62–69. [DOI] [PubMed] [Google Scholar]
- 6.Holgate, S.T., C. Hardy, C. Robinson, R. Agius, and P.H. Howarth. 1986. The mast cell as a primary effector cell in the pathogenesis of asthma. J. Allergy Clin. Immunol. 77:274–280. [DOI] [PubMed] [Google Scholar]
- 7.Koshino, T., Y. Arai, Y. Miyamoto, Y. Sano, M. Itami, S. Teshima, K. Hirai, T. Takaishi, K. Ito, and Y. Morita. 1996. Airway basophil and mast cell density in patients with bronchial asthma: relationship to bronchial hyperresponsiveness. J. Asthma. 33:89–95. [DOI] [PubMed] [Google Scholar]
- 8.Kinet, J.-P. 1999. The high-affinity IgE receptor (FcεRI): From physiology to pathology. Annu. Rev. Immunol. 17:931–972. [DOI] [PubMed] [Google Scholar]
- 9.Turner, H., and J.-P. Kinet. 1999. Signalling through the high-affinity IgE receptor FcεRI. Nature. 402[Suppl.]:B24–B30. [DOI] [PubMed] [Google Scholar]
- 10.Ishizaka, T., and K. Ishizaka. 1984. Activation of mast cells for mediator release through IgE receptors. In Mast Cell Activation and Mediator Release_._ K. Ishizaka, editor. Karger, Basel, Switzerland. 188–235. [PubMed]
- 11.Kobayasi, T., and G. Asboe-Hansen. 1969. Degranulation and regranulation of human mast cells. Acta Derm. Venerol. 49:369–381. [PubMed] [Google Scholar]
- 12.Dvorak, A.M., R.P. Schleimer, and L.M. Lichtenstein. 1987. Morphologic mast cell cycles. Cell. Immunol. 105:199–204. [DOI] [PubMed] [Google Scholar]
- 13.Gross, A., J.M. McDonnell, and S.J. Korsmeyer. 1999. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899–1911. [DOI] [PubMed] [Google Scholar]
- 14.Raff, M. 1998. Cell suicide for beginners. Nature. 396:119–122. [DOI] [PubMed] [Google Scholar]
- 15.Kroemer, G. 1997. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 3:614–620. [DOI] [PubMed] [Google Scholar]
- 16.Dragovich, T., C.M. Rudin, and C.B. Thompson. 1998. Signal transduction pathways that regulate cell survival and cell death. Oncogene. 17:3207–3213. [DOI] [PubMed] [Google Scholar]
- 17.Yee, N.S., I. Peak, and P. Besmer. 1994. Role of c-kit ligand in proliferation and suppression of apoptosis in mast cells. Basis for radiosensitivity of white spotting and steel mutants. J. Exp. Med. 179:1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bullock, E.D., and E.M. Johnson. 1996. Nerve growth factor induces the expression of certain cytokine genes and bcl-2 in mast cells - potential role in survival promotion. J. Biol. Chem. 271:27500–27508. [DOI] [PubMed] [Google Scholar]
- 19.Mekori, Y.A., C.K. Oh, and D.D. Metcalfe. 1995. The role of c-Kit and its ligand, stem cell factor, in mast cell apoptosis. Int. Arch. Allergy Immunol. 107:136–138. [DOI] [PubMed] [Google Scholar]
- 20.Yeatman, C.F., S.M. Jacobs-Helber, P. Mirmonsef, S.R. Gillespie, L.A. Bouton, H.A. Collins, S.T. Sawyer, C.P. Shelburne, and J.J. Ryan. 2000. Combined stimulation with the T helper cell type 2 cytokines interleukin (IL)-4 and IL-10 induces mouse mast cell apoptosis. J. Exp. Med. 192:1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hueber, A.O., G. Raposo, M. Pierres, and H.T. He. 1994. Thy-1 triggers mouse thymocyte apoptosis through a bcl-2-resistant mechanism. J. Exp. Med. 179:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gottschalk, A.R., L.H. Boise, C.B. Thompson, and J. Quintans. 1994. Identification of immunosuppressant-induced apoptosis in a murine B- cell line and its prevention by bcl-x but not bcl-2. Proc. Natl. Acad. Sci. USA. 91:7350–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin, E.Y., A. Orlofsky, M.S. Berger, and M.B. Prystowsky. 1993. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J. Immunol. 151:1979–1988. [PubMed] [Google Scholar]
- 24.Chuang, P.I., E. Yee, A. Karsan, R.K. Winn, and J.M. Harlan. 1998. A1 is a constitutively and inducible Bcl-2 homologue in mature human neutrophils. Bichem. Biophys. Res. Commun. 249:361–365. [DOI] [PubMed] [Google Scholar]
- 25.Gerber, H.P., V. Dixit, and N. Ferrara. 1998. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 273:13313–13316. [DOI] [PubMed] [Google Scholar]
- 26.Karsan, A., E. Yee, K. Kaushansky, and J.M. Harlan. 1996. Cloning of a human Bcl-2 homologue: Inflammatory cytokines induce human A1 in cultured endothelial cells. Blood. 87:3089–3096. [PubMed] [Google Scholar]
- 27.Orlofsky, A., R.D. Somogyi, L.M. Weiss, and M.B. Prystowsky. 1999. The murine antiapoptotic protein A1 is induced in inflammatory macrophages and constitutively expressed in neutrophils. J. Immunol. 163:412–419. [PubMed] [Google Scholar]
- 28.Hamasaki, A., F. Sendo, K. Nakayama, N. Ishida, I. Negishi, K.-i. Nakayama, and S. Hatakeyama. 1998. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the _bcl-2_–related A1 gene. J. Exp. Med. 188:1985–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatakeyama, S., A. Hamasaki, I. Negishi, D.Y. Loh, F. Sendo, K. Nakayama, and K.-i. Nakayama. 1998. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. Int. Immunol. 10:631–637. [DOI] [PubMed] [Google Scholar]
- 30.Arora, N., K.U. Min, J.J. Costa, J.S. Rhim, and D.D. Metcalfe. 1993. Immortalization of mouse bone marrow-derived mast cells with Ad12-SV40 virus. Int. Arch. Allergy Immunol. 100:319–327. [DOI] [PubMed] [Google Scholar]
- 31.Tsai, M., J. Hunt, J.P. Arm, C. London, M. Gurish, and S.J. Galli. 1996. The Cl.MC/C57.1 (C57) mouse mast cell line is of BALB/c origin and is tumorigenic in BALB/c mice. FASEB J. 10:A1268. [Google Scholar]
- 32.Young, J.-E., C.-C. Liu, G. Butler, Z.A. Cohn, and S.J. Galli. 1987. Identification, purification, and characterization of a mast cell-associated cytolytic factor related to tumor necrosis factor. Proc. Natl. Acad. Sci. USA. 84:9175–9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dastych, J., M.C. Hardison, and D.D. Metcalfe. 1997. Aggregation of low affinity IgG receptors induces mast cell adherence to fibronectin. Requirement for the common FcR γ-chain. J. Immunol. 158:1803–1809. [PubMed] [Google Scholar]
- 34.Aridor, M., L.M. Traub, and R. Sagi-Eisenberg. 1990. Exocytosis in mast cells by basic secretagogues: evidence for direct activation of GTP-binding proteins. J. Cell Biol. 111:909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomperts, B.D., Y. Churcher, A. Koffer, T.H. Lillie, P.E. Tatham, and T.D. Whalley. 1991. Intracellular mechanisms regulating exocytotic secretion in mast cells. Int. Arch. Allergy Appl. Immunol. 94:38–46. [DOI] [PubMed] [Google Scholar]
- 36.Mekori, Y.A., C.K. Oh, and D.D. Metcalfe. 1993. IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3. Prevention of apoptosis by c-kit ligand. J. Immunol. 151:3775–3784. [PubMed] [Google Scholar]
- 37.Inemura, A., M. Tsai, A. Ando, B.K. Wershil, and S.J. Galli. 1994. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J. Pathol. 144:321–328. [PMC free article] [PubMed] [Google Scholar]
- 38.Kawamoto, K., T. Okada, Y. Kannan, H. Ushio, M. Matsumoto, and H. Matsuda. 1995. Nerve growth factor prevents apoptosis of rat peritoneal mast cells through the trk proto-oncogene receptor. Blood. 86:4638–4644. [PubMed] [Google Scholar]
- 39.Xiang, Z., M. Block, C. Löfman, and G. Nilsson. 2001. IgE-mediated mast cell degranulation and recovery monitored by time-lapse photography. J. Allergy Clin. Immunol. 108:116–121. [DOI] [PubMed] [Google Scholar]
- 40.Mekori, Y.A., C.K. Oh, J. Dastych, J.P. Goff, S. Adachi, P.J. Bianchine, A. Worobec, T. Semere, J.M. Pierce, and D.D. Metcalfe. 1997. Characterization of a mast cell line that lacks the extracellular domain of membrane c-kit. Immunology. 90:518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cervero, C., L. Escribano, J.F. San Miguel, B. Diaz-Agustin, P. Bravo, J. Villarrubia, R. Garcia-Sanz, J.L. Velasco, P. Herrera, M. Vargas, et al. 1999. Expression of Bcl-2 by human bone marrow mast cells and its overexpression in mast cell leukemia. Am. J. Hematol. 60:191–195. [DOI] [PubMed] [Google Scholar]
- 42.Lin, E.Y., A. Orlofsky, H.G. Wang, J.C. Reed, and M.B. Prystowsky. 1996. A1, a Bcl-2 family member, prolongs cell survival and permits myeloid differentiation. Blood. 87:983–992. [PubMed] [Google Scholar]
- 43.Noble, K.E., R.G. Wickremasinghe, C. DeCornet, P. Panayiotidis, and K.L. Yong. 1999. Monocytes stimulate expression of the Bcl-2 family member, A1, in endothelial cells and confer protection against apoptosis. J. Immunol. 162:1376–1383. [PubMed] [Google Scholar]
- 44.Chen, Q., J. Turner, A.J. Watson, and C. Dive. 1997. v-Abl protein tyrosine kinase (PTK) mediated suppression of apoptosis is associated with the up-regulation of Bcl-XL. Oncogene. 15:2249–2254. [DOI] [PubMed] [Google Scholar]
- 45.Yin, X.M., Z.N. Oltvai, and S.J. Korsmeyer. 1994. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 369:321–323. [DOI] [PubMed] [Google Scholar]
- 46.Zhang, H., S.W. Cowan-Jacob, M. Simonen, W. Greenhalf, J. Heim, and B. Meyhack. 2000. Structural basis of BFL-1 for its interaction with BAX and its anti- apoptotic action in mammalian and yeast cells. J. Biol. Chem. 275:11092–11099. [DOI] [PubMed] [Google Scholar]
- 47.Maurer, M., M. Tsai, M. Metz, S. Fish, S.J. Korsmeyer, and S.J. Galli. 2000. A role for Bax in the regulation of apoptosis in mouse mast cells. J. Invest. Dermatol. 114:1205–1206. [DOI] [PubMed] [Google Scholar]
- 48.McLean, P.G., A. Ahluwalia, and M. Perretti. 2000. Association between kinin B(1) receptor expression and leukocyte trafficking across mouse mesenteric postcapillary venules. J. Exp. Med. 192:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grumont, R.J., I.J. Rourke, and S. Gerondakis. 1999. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 13:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zong, W.X., L.C. Edelstein, C. Chen, J. Bash, and C. Gelinas. 1999. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 13:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang, C.Y., D.C. Guttridge, M.W. Mayo, and A.S. Baldwin, Jr. 1999. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol. Cell. Biol. 19:5923–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshikawa, H., Y. Nakajima, and K. Tasaka. 1999. Glucocorticoid suppresses autocrine survival of mast cells by inhibiting IL-4 production and ICAM-1 expression. J. Immunol. 162:6162–6170. [PubMed] [Google Scholar]
- 53.Yoshikawa, H., Y. Nakajima, and K. Tasaka. 2000. Enhanced expression of Fas-associated death domain-like IL-1-converting enzyme (FLICE)-inhibitory protein induces resistance to Fas-mediated apoptosis in activated mast cells. J. Immunol. 165:6262–6269. [DOI] [PubMed] [Google Scholar]
- 54.Hartmann, K., A.L. WagelieSteffen, E. vonStebut, and D.D. Metcalfe. 1997. Fas (CD95,APO-1) antigen expression and function in murine mast cells. J. Immunol. 159:4006–4014. [PubMed] [Google Scholar]