Identification of a Novel Major Histocompatibility Complex Class II–restricted Tumor Antigen Resulting from a Chromosomal Rearrangement Recognized by CD4+ T Cells (original) (raw)
Abstract
CD4+ T cells play an important role in antitumor immune responses and autoimmune and infectious diseases. Although many major histocompatibility complex (MHC) class I–restricted tumor antigens have been identified in the last few years, little is known about MHC class II– restricted human tumor antigens recognized by CD4+ T cells. Here, we describe the identification of a novel melanoma antigen recognized by an human histocompatibility leukocyte antigen (HLA)-DR1–restricted CD4+ tumor-infiltrating lymphocyte (TIL)1363 using a genetic cloning approach. DNA sequencing analysis indicated that this was a fusion gene generated by a low density lipid receptor (LDLR) gene in the 5′ end fused to a GDP-l-fucose:β-d-galactoside 2-α-l-fucosyltransferase (FUT) in an antisense orientation in the 3′ end. The fusion gene encoded the first five ligand binding repeats of LDLR in the NH2 terminus followed by a new polypeptide translated in frame with LDLR from the FUT gene in an antisense direction. Southern blot analysis showed that chromosomal DNA rearrangements occurred in the 1363mel cell line. Northern blot analysis detected two fusion RNA transcripts present only in the autologous 1363mel, but not in other cell lines or normal tissues tested. Two minimal peptides were identified from the COOH terminus of the fusion protein. This represents the first demonstration that a fusion protein resulting from a chromosomal rearrangement in tumor cells serves as an immune target recognized by CD4+ T cells.
Keywords: melanoma antigens, fusion proteins, antitumor immunity, immunotherapy, chromosomal rearrangement
The adoptive transfer of CTLs, derived from tumor- infiltrating lymphocytes (TILs),1 along with IL-2 into autologous patients with cancer resulted in the objective regression of tumor, indicating that these CTLs could recognize cancer rejection antigens on tumor cells. In the last few years, many tumor antigens recognized by CD8+ T cells have been identified in melanomas as well as in other types of cancers using a cDNA library expression system and direct sequencing of peptides eluted from the tumor cell surface (1–3). These findings led to several clinical trials using peptides derived from the molecularly defined tumor antigens recognized by CD8+ T cells. Although the first clinical trial using a modified peptide derived from gp100 has shown evidence of therapeutic efficacy for the treatment of patients with metastatic melanoma and achieved a 42% clinical response rate (4), increasing evidence has indicated that optimal cancer vaccines require the participation of both CD4+ and CD8+ T cells (5, 6). CD4+ T cells play a central role in antitumor immune responses and autoimmune and infectious diseases (7–10). Identification of antigens recognized by CD4+ T cells is thus important for the development of cancer vaccines and new strategies for the treatment of autoimmune and infectious diseases.
CD4+ T cells recognize a peptide bound on MHC class II molecules on the surface of APCs (11, 12). Attempts to isolate antigenic peptides recognized by CD4+ T cells from MHC class II–peptide complexes using a combination of HPLC and mass spectrometric sequence analysis have been unsuccessful due to the heterogeneous lengths of the antigenic peptides. The biochemical purification of antigenic proteins from tumor cell lysates has been used successfully (13), but this strategy largely depends on the abundance of the Ag of interest and its efficient uptake by APCs. Tyrosinase is a human MHC class II–restricted melanoma Ag recognized by CD4+ T cells, which was identified by testing the reactivity of several candidate Ags pulsed onto MHC class II–matched EBV-B cells (14). Because of these difficulties, much less is known about MHC class II– restricted human tumor antigens recognized by CD4+ T cells. To facilitate the identification of these MHC class II– restricted antigens, we recently developed a novel genetic approach to identifying MHC class II–restricted tumor antigens. In this report, we describe the identification of a new tumor antigen generated from a chromosomal rearrangement and recognized by CD4+ T cells.
Materials and Methods
Chemicals and Reagents.
The following chemicals and reagents were used and were purchased from the sources indicated: RPMI 1640, AIM-V medium, Lipofectamine, and G418 (GIBCO BRL); the eukaryotic expression vector pcDNA3.1 (Invitrogen); anti– HLA-DR1 mAb (One Lambda); and anti-IgM antibody conjugated with FITC (Vector Labs., Inc.).
Cell Lines and Cultures.
CD4+ TILs were cultured from a subcutaneous metastasis resected from patient 1363. T cell clones or lines were grown in AIM-V medium containing 10% human AB serum and rIL-2 (1,000 IU/ml; Chiron). Melanoma cell lines and EBV-transformed B cell lines were maintained in RPMI 1640 with 10% FCS. COS-7, 293, 293IMDR1, and 293IMDR4 cell lines were grown in the DMEM containing 10% FCS. The T cell clones or cloids were generated by limiting dilution methods (at 1 cell/well) from the CD4+ TIL1363 cell line. After 12 d, the T cell clones were expanded in AIM-V medium containing 6,000 IU/ml IL-2. To obtain an optimal expansion, we used the OKT3 expansion method as previously described (15, 16). In brief, on day 0, 5 × 104–5 T cells were cocultured with HLA-DR1+ PBLs (PBL/T cell ratio, 500:1) and 586 EBV B cells (EBV/T cell ratio, 100:1) in 25 ml RPMI 1640 containing 11% human sera, 30 ng/ml OKT3 antibody, and antibiotics. On day 1, IL-2 was added at a final concentration of 180 IU/ml. The medium was changed with fresh medium containing 11% human sera and 180 IU/ml IL-2 on day 5. The medium was then changed every 3 d. On days 12–14, T cells were harvested, counted, and cryopreserved.
Melanoma cell lines 397mel, 1088mel, 1359mel, 1363mel, and 1558mel and EBV-transformed B cell lines 586EBV, 1363EBV, 1359EBV, and 1558EBV were established in our laboratory and cultured in RPMI 1640 containing 10% FCS.
cDNA Library Construction.
Total RNA was extracted from 1363mel using Trizol reagent from GIBCO BRL. Poly(A) RNA was purified from total RNA by polyATract system (Promega Corp.) and converted to cDNA using a cDNA construction kit (GIBCO BRL) with an oligo-dT primer. To create an invariant chain (Ii) fusion library, an Ii fragment (amino acids 1–80) was inserted into a mammalian expression vector, pEAK8 (Edge BioSystem), to generate a pTi80 vector. The cDNA inserts were then ligated to pTi80, and cDNA libraries were electroporated into DH10B cells (GIBCO BRL). Plasmid DNAs for cDNA library pools were prepared from bacteria, each consisting of ∼100 cDNA clones.
cDNA Library Screening and GM-CSF Secretion Assay.
DNA transfection and GM-CSF assays were performed as previously described (15, 16). In brief, 200 ng of cDNA pools was mixed with 2 μl of Lipofectamine in 100 μl of serum-free DMEM for 15–45 min. The DNA/Lipofectamine mixture was then added to the 293IMDR1 (5 × 104) cells and incubated overnight. The following day, cells were washed twice with AIM-V medium. CD4+ TIL1363 cells were added at a concentration of 5 × 104 cells/well in AIM-V medium containing 120 IU/ml of IL-2. After 18–24 h of incubation, 100 μl of supernatant was collected and GM-CSF concentration was measured in a standard ELISA assay (R&D Systems). For testing peptide recognition, 1363EBV cells were incubated with peptides at 37°C for 90 min, then washed three times with AIM-V medium containing 120 IU/ml of IL-2. T cells were added and incubated for an additional 18– 24 h. In some experiments, peptides were directly incubated with CD4+ TIL1363 for 90 min, then washed three times. 5 × 104 T cells were then mixed with the peptide-binding T cells and incubated for an additional 18–24 h. 100 μl of supernatant was collected for GM-CSF assay.
Northern Blot Analysis.
Total RNA was isolated using Trizol reagent from GIBCO BRL. Total RNA from human normal tissue was purchased from Clontech. 20 μg of total RNA was subjected to electrophoresis in a 1.2% formaldehyde agarose gel and transferred to a nylon membrane. DNA fragments of the LDLR-FUT (low density lipid receptor–GDP-l-fucose:β-d-galactosidase 2-α-l-fucosyltransferase) fusion gene for probe A, the LDLR-specific probe B, and the FUT-specific probe C were labeled with [α-32P]CTP by the random priming method. Prehybridization and hybridization were performed according to the QuickHyb protocol (Stratagene). Membranes were washed twice with 2× SSC/0.1% SDS at room temperature for 15 min and twice with 0.1× SSC/0.1% SDS at 60°C for 30 min. The autoradiography was performed at −70°C.
Southern Blot Analysis.
Genomic DNAs were isolated from tumor and EBV-B cell lines using a DNA Isolation Kit (Boehringer Mannheim), and digested with BamHI, HindIII, or EcoRI. The digested DNA fragments were separated from an 0.8% agarose gel, and transferred to nitrocellulose membrane. Membranes were prehybridized for 1 h, then hybridized with [α-32P]CTP– labeled probes using QuickHyb solution (Stratagene). After washing twice with 2× SSC/0.1% SDS at room temperature for 15 min and twice with 0.1× SSC/0.1% SDS at 65°C for 15 min, the membrane was exposed to an x-ray film at −70°C.
5′ RACE.
Total RNA was extracted from tumor cell lines as described above. 5′ RACE (rapid amplification of cDNA ends) was performed according to the manufacturer's procedure (GIBCO BRL). PCR products were cloned into the TA cloning vector (Invitrogen). Recombinant DNA was prepared and used for DNA sequencing analysis using an automatic sequencer (Applied Biosystems, Inc.).
Peptides Synthesis and T Cell Epitopes.
The peptides were synthesized by a solid-phase method using a peptide synthesizer (model AMS 422; Gilson Co., Inc.). Some peptides were purified by HPLC and had >98% purity. The mass of some peptides was confirmed by mass spectrometry analysis. Identification and characterization of peptides reactive with CD4+ TIL1363 were conducted as previously described (15, 16).
Results
Recognition of Autologous Tumor Cells by HLA-DR–restricted CD4+ T Cells.
CD4+ TIL1363 cells were generated from a melanoma metastasis of patient 1363 and were found to recognize the autologous tumor cell line 1363mel, but did not recognize autologous EBV-B cells (1363EBV), other EBV-B cells, MHC class II–matched or –mismatched tumor cell lines, nor 293-expressing DR molecules (Fig. 1 A). Furthermore, CD4+ TIL1363 cells were capable of recognizing cell lysates of 1363mel pulsed onto DR-matched EBV B cells, but did not recognize cell lysates derived from other tumors, EBV-B cells, or fibroblasts (data not shown). These results suggested that TIL1363 recognized a unique tumor antigen from the autologous tumor. T cell recognition was specifically blocked by an mAb against HLA-DR, but not by mAbs against HLA-DQ or MHC class I (Fig. 1 B). FACS® analysis of 1363mel indicated that only HLA-DR1 was expressed in 1363mel (Fig. 1 C), which was consistent with the result of HLA genotyping analysis. These results suggested that CD4+ TIL1363 recognized a tumor antigen presented by HLA-DR1.
Figure 1.

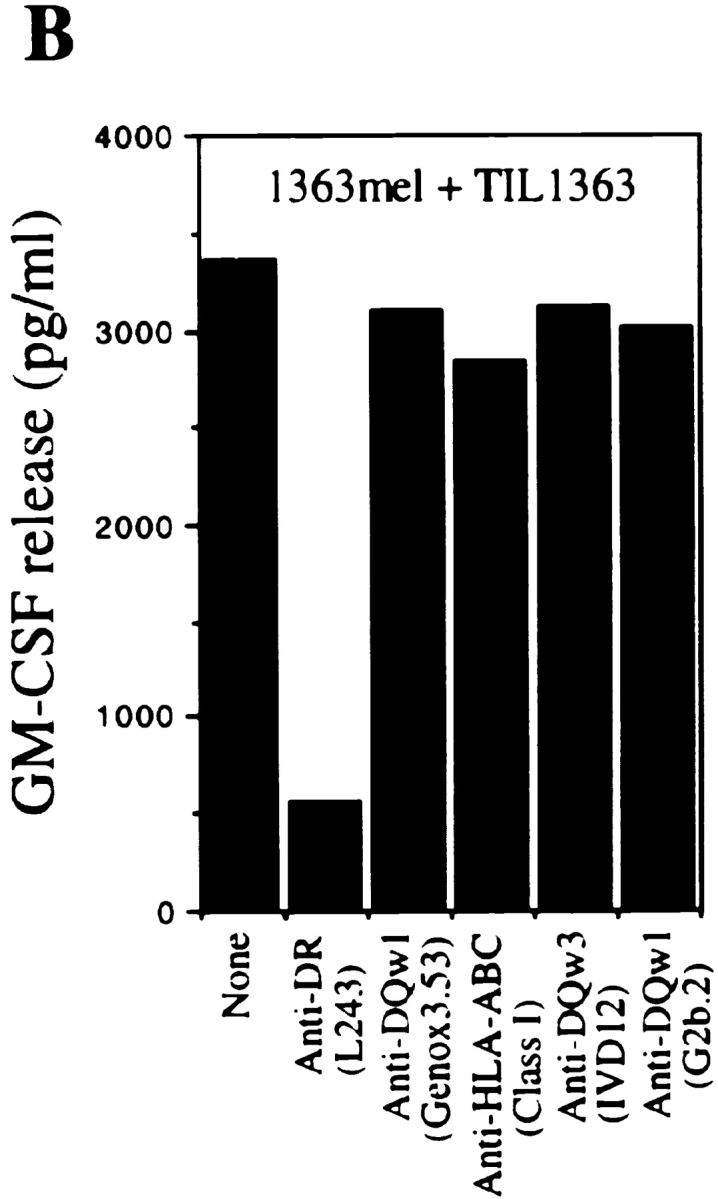

Recognition of the autologous melanoma cell line by CD4+ TIL1363. (A) Specific antitumor recognition of CD4+ TIL1363. TIL1363 recognized the autologous 1363mel, but did not recognize autologous 1363EBV, other EBV-B cell lines, or allogeneic melanoma cell lines, nor 293-derived cell lines. 1558mel shared the DR molecule β1*0101 with 1363mel. T cell recognition was evaluated based on GM-CSF release from CD4+ TIL1363. (B) HLA restriction of T cell recognition. CD4+ TIL1363 cells were cocultured with autologous 1363mel cells in the presence or absence of various anti-MHC antibodies. GM-CSF release was determined after an 18-h incubation. T cell recognition of 1363mel was specifically blocked by an anti-DR antibody, but not by anti–MHC class I or anti-DQ antibodies. HLA genotyping of 1363mel was HLA-DR β1*0101. (C) FACS® analysis of 1363mel and TIL1363 for HLA-DR1 expression.
Identification of a Fusion Protein Recognized by CD4+ TIL1363.
Although many MHC class I–restricted tumor antigens have been identified by the expression cloning approach, this conventional expression approach cannot be applied to isolating genes encoding MHC class II–restricted Ags, because of differences in the endogenous and exogenous Ag presentation pathways. It has been reported that Ii-fused antigens could be endogenously processed and presented to CD4+ T cells (17–20). Our strategy is to target Ii fusion proteins translated from the Ii fusion library to the endosomal/lysosomal compartment for efficient antigen processing and presentation. Although EBV-B and dendritic cells are professional APCs, they were poorly transfectable and cDNA libraries could not be efficiently introduced into these APCs. Thus, we generated 293IMDR1 cells by introducing cDNAs encoding DRα, DRβ, DMA, DMB, and Ii into 293 cells, a transformed human kidney embryonic cell line, and used them as “professional” APCs. This novel approach has been successfully used to isolate a mutated CDC27 as an MHC class II–restricted tumor antigen recognized by CD4+ T cells (20a).
To isolate the gene encoding a tumor antigen recognized by CD4+ TIL1363, we constructed a cDNA library with the fusion of a targeting sequence of Ii in the 5′ end of cDNAs derived from 1363mel. cDNA subpools with ∼100 cDNA clones per pool were prepared. The 1363 cDNA library was introduced into 293IMDR1 expressing DMA, DMB, Ii, and DR1 molecules. After screening a total of 3.5 × 105 cDNA clones, we identified >10 positive cDNA pools that conferred T cell recognition by CD4+ TIL1363 when transfected into 293IMDR1. The individual positive clones were then isolated from the positive cDNA pools and tested for recognition by CD4+ TIL1363. Representative data is shown in Fig. 2.
Figure 2.

Screening of a cDNA library from RNA of 1363mel using CD4+ TIL1363. After screening 2.1 × 105 Ii-cDNA library clones generated from 1363mel RNA, positive cDNA pools were identified based on GM-CSF release from CD4+ TIL1363. CD4+ TIL1363 recognized 293IMDR1 transfected with the cDNA clone 7, but not with a control cDNA or green fluorescence protein. Recognition of 1363mel by TIL1363 was used as a positive control.
DNA sequencing analysis showed that cDNA clone 7 encoded a fusion protein consisting of 363 amino acids (Fig. 3). A database search revealed that the first 880 nucleotides were identical to a published sequence of the LDL receptor (21, 22). However, the DNA sequence in the 3′ end of cDNA clone 7 was found to be identical to the previously published sequence of FUT with the exception of a one-nucleotide (G) deletion at position 1049 (23). Furthermore, the LDLR sequence in the 5′ end of the cDNA was fused to the intron/exon 3 of the FUT sequence in an antisense orientation such that the translational reading frame of the LDLR fusion protein continued for an additional 131 amino acids until a stop codon was reached. Therefore, the LDLR-FUT fusion protein contained the first five ligand binding repeats of LDLR followed by a novel polypeptide of 131 amino acids at the COOH terminus. A database search did not reveal any sequence homologue to the 131 amino acid polypeptide.
Figure 3.
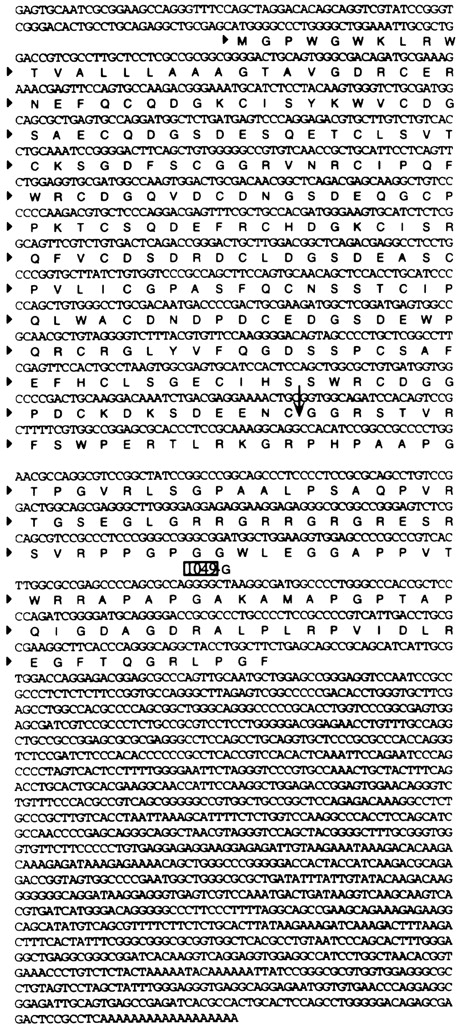
The nucleotide and amino acid sequences of the human LDLR-FUT fusion gene. The numbering of amino acid sequence of the LDLR-FUT protein starts with the first amino acid Met. The 5′ untranslated region was isolated by 5′ RACE and was found to be identical to the previously published sequence of LDLR after sequencing analysis. The fusion junction between LDLR and FUT was indicated by an arrow. The EMBL/GenBank/DDBJ accession number for the fusion gene is AF117899.
The Fusion cDNA Encoded a Secretory Protein.
To confirm if T cell recognition was restricted by HLA-DR1, the cDNA clone 7 was transfected into both 293IMDR1 and 293IMDR4 and then tested for recognition by CD4+ TIL1363. To our surprise, CD4+ TIL1363 cells were capable of recognizing both HLA-DR1 and DR4 positive cell lines transfected with cDNA clone 7, but not with other control cDNA clones (data not shown). FACS® analysis revealed that TIL1363 expressed a high level of HLA-DR1 molecules on the cell surface, suggesting that these T cells may function as APCs (Fig. 1 C). To further explore how TIL1363 recognized an antigen expressed by 293IMDR4 cells lacking DR1 molecules, we tested the possibility that T cells may capture an antigen from cell supernatants of cells transfected with cDNA clone 7, and process and present antigenic peptides to each other. Results presented in Fig. 4 showed that CD4+ TIL1363 cells were capable of recognizing an antigen derived from the cell culture supernatants of COS-7, 293, and 293IMDR4 cells transfected with cDNA clone 7 and supernatants from 1363mel. Recognition of the supernatants by CD4+ TIL1363 cells were blocked by an anti-DR antibody, but not by a control antibody. Treatment of the cDNA clone 7–transfected COS-7, 293, and 293IMDR4 cells with the fixative agent PFA alone or PFA combined with the anti-DR antibody completely abrogated the ability of conferring T cell recognition (Fig. 4), suggesting that blocking antigen secretion from cells transfected with cDNA clone 7 abolished antigen recognition by T cells. T cell recognition of 1363mel by CD4+ TIL1363 was partially inhibited by PFA. This may be due to the existence of different subsets of CD4+ T cells in the TIL1363 population that recognized additional antigens presented by 1363mel. These results indicated that COS-7, 293, and 293IMDR4 cells transfected with cDNA clone 7, as well as the untransfected 1363mel, secreted the LDLR-FUT fusion protein into the culture medium, which was in turn presented by MHC class II–positive T cells to themselves.
Figure 4.

Recognition of the secreted fusion protein by CD4+ TIL1363. COS-7, 293, and 293IMDR4 were transfected with cDNA clone 7. 48 h after transfection, cell supernatants were harvested from the transfected COS-7, 293, and 293IMDR4 cells as well as the untransfected 1363mel, and cocultured with CD4+ TIL1363 for an additional 18–24 h. Antibody blocking was done by the pretreating supernatants or transfected COS-7, 293, and 293IMDR4 cells with either anti-DR or control antibodies for 30 min before coculture with CD4+ TIL1363. To block the secretion of the LDLR-FUT from the cDNA clone 7–transfected cells or 1363mel, these transfectants and 1363mel were treated with 1% PFA for 20 min, followed by three time washes with PBS. CD4+ TIL1363 cells were added and incubated for 18–24 h. Supernatants were used to measure GM-CSF release from CD4+ TIL1363. CD4+ TIL1363 reacted with 1363mel and the cDNA clone 7–transfected cells and their supernatants. T cell recognition could be blocked with an anti-DR antibody.
Generation of a Unique Fusion Tumor Antigen by Chromosomal Rearrangement.
To determine whether the fusion protein was a consequence of a chromosomal rearrangement, we did Southern blot analysis using the LDLR-FUT fusion cDNA as probe A (Fig. 5 A). No difference was found in the DNA band pattern of 1363mel genomic DNA digested with either HindIII or EcoRI, compared with genomic DNA derived from other cell lines digested with the same enzymes. However, an additional band was observed in 1363mel genomic DNA digested with BamHI when compared with the DNA patterns of 293, SK23, 1359mel, 624mel, and 586mel (Fig. 5 B). These results suggested that a DNA rearrangement occurred in 1363mel. To further evaluate if the fusion cDNA was expressed in 1363mel, Northern blot analyses were performed. Two unique bands were detected only in 1363mel by the fusion cDNA probe A (Fig. 5 C). No corresponding bands were observed from any other tumor cell lines or normal tissues tested (Fig. 5 C). Using specific DNA probes derived from either LDLR cDNA or FUT (Fig. 5 A), we found that probe B, which was specific for LDLR, detected two identical bands observed in the 1363mel RNA sample using probe A, but not in the RNA samples of 1363EBV, 1359mel, or 293 (Fig. 5 D). However, the DNA probe C, which was specific for FUT, only detected a single band, suggesting that the 5′ portion of two RNAs from 1363mel was the same and contained the LDLR fragment, but the 3′ portion was different, which might result from an alternative splicing.
Figure 5.

Northern and Southern blot analysis of genomic DNAs derived from different tumor cell lines. (A) A schematic presentation of the LDLR-FUT cDNA and probes used for Southern and Northern blot analyses. (B) Southern blot analysis of genomic DNAs from 293 and melanoma cell lines. Genomic DNAs were digested with HindIII (H), BamHI (B), or EcoRI (R), and separated on a 0.8% agarose gel. After transfer to a membrane, DNA bands were detected with a 32P-labeled probe A. In addition to bands seen in all cell lines, 1363mel displayed an additional band indicated by an arrow when digested with BamHI. (C) Northern blot analysis of the LDLR-FUT RNA transcripts in normal human tissues and tumor cell lines. Two distinct RNA bands were detected in 1363mel RNA, but not in RNA from other tumor cell lines or normal tissues. (D) Northern blot analysis of RNA samples using three different probes. Probe A was used to detect the whole fusion cDNA; probe B was specific for the detection of LDLR transcript; and probe C was specific for the intron 1 sequence of FUT gene. Hybridization of blots with the probe A or B detected two RNA bands in 1363mel, but not in other cell lines. However, only a single band was seen in 1363mel when hybridized with the probe C.
Both LDLR and FUT had been mapped on human chromosome 19, but LDLR was located on 19p and FUT on 19q (24, 25). In Fig. 6 we propose a possible gene fusion resulting from a chromosomal rearrangement. The LDLR gene was fused to the FUT gene in an opposite direction to LDLR. The fusion junction was located between the exon 4 of LDLR and the intron/exon 3 of FUT. Two RNA species were generated, probably by an alternative splicing.
Figure 6.

A schematic presentation of a chromosomal rearrangement between LDLR and FUT. LDLR is located in 13p and FUT in 13q of human chromosome 19. Only the first 5 exons of LDLR (total 18 exons) and exons 2 and 3 of FUT were shown with their own promoters. Chromosomal rearrangement resulted in the fusion between LDLR and FUT. The DNA sequence of the fusion RNA 1 was identical to that shown in Fig. 3. DNA sequence of the fusion RNA 2 was a variant of RNA 1, which lacked the intron 1 sequence of FUT based on the Northern blot analysis shown in Fig. 5.
Characterization of Peptides Recognized by CD4+ TIL1363.
To determine the antigenic epitopes recognized by CD4+ TIL1363, we first tested whether CD4+ TIL1363 recognized 293IMDR1 transfected with the wild-type LDLR cDNA. No T cell recognition was observed with the LDLR-transfected 293IMDR1, suggesting that T cell epitopes were located either in the new polypeptide translated from the sequence of FUT in an antisense direction or in the junction region of the fusion protein.
34 15-mer peptides overlapping by eight amino acids were synthesized based on the predicted amino acid sequence of the new polypeptide derived from the FUT sequence, and were tested for T cell recognition. Two of these peptides were found to be recognized by CD4+ TIL1363 (Fig. 7). These two peptides shared eight amino acids (WRRAPAPG). Further truncations of the peptides from either the NH2 or COOH terminus defined Trp (W) at amino acid position 315 and Ala (A) at amino acid position 320 as critical amino acids required for T cell recognition (Fig. 8 A). All the peptides containing the core sequence WRRAPA were recognized by CD4+ TIL1363. One 9-mer peptide LRFP312–320 (PVTWRRAPA), the shortest peptide used in this experiment, was found to be active in the stimulation of T cells (Fig. 8 A). Further truncations of this 9-mer peptide LRFP312–320 from the NH2 terminus resulted in a loss of the ability to stimulate CD4+ TIL1363 for GM-CSF release (data not shown). We also tested T cell recognition of several truncated forms of the 15-mer peptide (WRRAPAPGAKA) from the COOH terminus, and found that the shortest active peptide was the 9-mer peptide LRFP315–323 (WRRAPAPGA) (Fig. 8 B). Therefore, these studies defined two 9-mer peptides, LRF312–320 and LRFP315–323, recognized by CD4+ TIL1363. These two peptides shared a core sequence WRRAPA, and Trp (W) at amino acid position 315 may serve as the P1 anchor residue for MHC binding, conforming to the HLA-DR1 binding motif (26). The minimal peptide length is 9-mer, and peptides shorter than 9-mer failed to activate CD4+ TIL1363 for GM-CSF release even though they contained the core sequence (Fig. 8 B). Peptide titration experiments showed that both LRFP312–320 and LRFP315–323 peptides exhibited a very similar peptide binding avidity to DR1 and could be detected by T cells at 100 ng/ml peptide concentration (Fig. 8 C).
Figure 7.

Identification of peptides recognized by CD4+ TIL1363 from the fusion protein. 34 15-mer peptides overlapping by 8 amino acids were synthesized, and verified by mass spectrometry analysis. These peptides were pulsed onto 1363EBV B cells for 90 min followed by three washes. T cells were added, and after incubation for 18–24 h, GM-CSF release was measured to identify the reactive peptides.
Figure 8.

Characterization of the antigenic peptides recognized by CD4+ TIL1363. (A) Truncated forms of the peptides were synthesized based on the peptide sequence identified as reactive in Fig. 7, and tested for T cell recognition. A core peptide sequence (WRRAPA) was defined based on T cell recognition. A 9-mer peptide was identified as a minimal antigenic determinant required for T cell reactivity. (B) Further truncations of the reactive peptides were synthesized to define the minimal peptide determinant. 1363mel and 1359mel were used as positive and negative controls, respectively. (C) T cell secretion of GM-CSF in response to peptide stimulation was measured. LRFP312–320 (▪) and LRFP315–323 (▴) peptides exhibited a similar avidity for T cell recognition. Background release of GM-CSF by TIL1363 cultured with 1363EBV B cells in the absence of peptide was undetectable.
Discussion
In this study we identified a novel tumor antigen recognized by CD4+ TIL1363 using a novel genetic strategy. Although a biochemical approach has been used to identify MHC class II–restricted tumor antigens, it is limited to the CD4+ T cells that are capable of recognizing tumor lysates pulsed onto APCs such as EBV B cells. Several CD4+ T cell lines established in our laboratory recognized whole tumor cells, but did not recognize cell lysates of autologous tumor due to either low amounts of a particular antigen or low efficient uptake by APCs (data not shown). We recently cloned a mutated CDC27 as a tumor antigen recognized by CD4+ TIL1359 using this novel cloning strategy (20a). Here, we demonstrate that a secreted protein encoded by a LDLR-FUT fusion cDNA is a tumor antigen recognized by CD4+ TIL1363. Despite the fact that a special cell line expressing MHC class II was not required in this case, this genetic approach can be useful for the isolation of genes encoding MHC class II–restricted tumor antigens recognized by CD4+ T cells.
The LDLR-FUT fusion gene was generated by a chromosomal rearrangement as evidenced by our Southern and Northern blot analyses (Fig. 3). Human LDL receptor is a cell surface glycoprotein that regulates plasma cholesterol levels. The ligand-binding domain of LDLR comprises seven imperfect repeats in the extracellular region of the protein. Each domain consists of ∼40 amino acids. LDL receptors specifically bind LDL, the receptor–lipoprotein complex is internalized by the cells via coated pits and vesicles, and the entire LDL particle is delivered to lysosomes, where it is disassembled by enzymatic hydrolysis, releasing cholesterol for further cellular metabolism (24). More than 250 LDLR mutations including deletions, insertions, and single point mutations have been reported to affect LDLR function associated with familial hypercholesterolemia (24). The FUT gene product regulates the expression of the H antigen mainly on erythrocyte membranes. Koda et al. reported that the FUT gene was expressed in gastric cancer and ovarian cancer cells (23). Although the fusion protein may still have the ability to bind LDL, we do not know the biological significance of the fusion protein resulting from a chromosomal rearrangement in 1363mel at this time.
There were several unique features associated with the fusion antigen. (a) To our knowledge, this is the first report that a secretory protein functions as a tumor antigen recognized by CD4+ T cells. Most antigens identified as MHC class I–restricted tumor antigens are membrane proteins and the previously identified MHC class II–restricted tumor antigen, tyrosinase, is a membrane protein as well (14). (b) It was previously reported that CTLs could be generated in vitro against the junction region of an oncogenic fusion protein resulting from a chromosomal translocation (27, 28). We show here that natural CD4+ T cells from the patient 1363 were capable of recognizing a melanoma antigen consisting of a fusion protein, which allowed us to detect a chromosomal rearrangement in chromosome 19 in 1363mel. The chromosomal rearrangement observed may be a consequence of intrachromosomal recombination between the Alu repeats in the LDLR and FUT genes (24). Lehrman et al. reported that deletions in the LDLR gene by recombination of Alu repeats produced a truncated, secreted LDLR protein (29). (c) DR1-positive TIL1363 cells were capable of presenting the LDLR-FUT fusion protein to each other though it is not clear whether the fusion protein directly bound to the DR1 molecules on TIL1363 for recognition. Alternatively, TIL1363 may capture the antigen from the culture medium and process and present it to each other by the exogenous antigen presentation pathway.
Our results indicate that CD4+ TIL1363 recognized a unique fusion antigen expressed in 1363mel (Fig. 1). Several CD4+ TILs have been established and characterized in our laboratory. Most CD4+ TILs recognized unique antigens, but not nonmutated shared antigens, as is the case for the majority of the MHC class I–restricted tumor antigens recognized by CD8+ CTLs that have been characterized (1–3). Tyrosinase is the only shared human melanoma antigen identified thus far and is recognized by an HLA-DR4–restricted CD4+ TIL1088 (14). Pieper et al. reported a mutated triosephosphate isomerase as a tumor antigen recognized by CD4+ TIL1558 (30). The point mutation of triosephosphate isomerase created a T cell epitope that required a lower peptide concentration necessary for T cell reactivity compared with the wild-type peptide. Recently, we identified a mutated CDC27 as a tumor antigen recognized by a DR4-restricted CD4+ TIL1359 (20a). CDC27 is an important component of the anaphase promoting complex and plays a role in cell cycle regulation (31, 32). Interestingly, the mutation itself in CDC27 did not constitute a T cell epitope, rather it affected the posttranslational modification (phosphorylation) and protein trafficking, which led to targeting of the mutated CDC27 protein to the MHC class II presentation pathway. Peptides identified from the LDLR-FUT fusion by CD4+ TIL1363 cells were located in the new polypeptide translated from the FUT sequence in an antisense orientation. These T cell epitopes were present in the 1363mel tumor cells, but not in the normal cells. The requirement for peptides to be recognized by CD4+ TIL1363 was the antigenic peptides containing a core sequence (WRRAPA) with a minimal length of 9 residues.
Although many tumor cells are MHC class II negative, the specific role of CD4+ T cells in antitumor immunity has been demonstrated in a MHC class II–negative tumor model (33). Moreover, in addition to providing help for CD8+ CTLs (34), CD4+ T cells may play a far broader role in orchestrating the host response to tumor (35) and autoimmune diseases (36, 37). Our studies indicate the importance of incorporating CD4+ reactive antigens in immunotherapy strategy. Definition of these MHC class II–presented antigens may provide new opportunities for developing more effective immunotherapies against cancer.
Acknowledgments
We are grateful to Gang Zeng, Samuel L. Johnston, and Alicia C. Atwood for assistance in DNA pool preparation; John Wunderlich and the TIL lab (University of Texas Southwestern Medical Center, Dallas, TX) for providing TIL1363; Maria Parkhurst and Ellen Fitzgerald for peptide synthesis; Arnold Mixon for FACS® analysis; and David W. Russell for providing the LDLR cDNA (University of Texas Southwestern Medical Center).
Abbreviations used in this paper
FUT
GDP-l-fucose:β-d-galactoside 2-α-l-fucosyltransferase
Ii
invariant chain
LDLR
low density lipid receptor
RACE
rapid amplification of cDNA ends
TIL
tumor-infiltrating lymphocyte
References
- 1.Boon T, Cerottini J-C, Van Den Eynde B, Van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18:175–182. doi: 10.1016/s0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- 3.Wang RF. Tumor antigens discovery: perspectives for cancer therapy. Mol Med. 1997;3:716–731. [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, et al. Immunologic and therapeutic evaluation of a synthetic tumor-associated peptide vaccine for the treatment of patients with metastatic melanoma. Nature Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrand-Rosenberg S. Tumor immunotherapy: the tumor cell as an antigen-presenting cell. Curr Opin Immunol. 1994;6:722–727. doi: 10.1016/0952-7915(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 6.Pardoll DM, Topalian SL. The role of CD4+T cell responses in antitumor immunity. Curr Opin Immunol. 1998;10:588–594. doi: 10.1016/s0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- 7.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 8.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 9.Bona CA, Casares S, Brumeanu TD. Towards development of T-cell vaccines. Immunol Today. 1998;19:126–133. doi: 10.1016/s0167-5699(97)01218-8. [DOI] [PubMed] [Google Scholar]
- 10.Riddell SR, Greenberg PD. Principles for adoptive T cell therapy of human viral diseases. Annu Rev Immunol. 1995;13:545–586. doi: 10.1146/annurev.iy.13.040195.002553. [DOI] [PubMed] [Google Scholar]
- 11.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 12.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 13.Monach PA, Meredith SC, Siegel CT, Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2:45–59. doi: 10.1016/1074-7613(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 14.Topalian SL, Gonzales MI, Parkhurst M, Li YF, Southwood S, Sette A, Rosenberg SA, Robbins PF. Melanoma-specific CD4+T cells recognize nonmutated HLA-DR–restricted tyrosinase epitopes. J Exp Med. 1996;183:1965–1971. doi: 10.1084/jem.183.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R-F, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R-F, Johnston SL, Zeng G, Schwartzentruber DJ, Rosenberg SA. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol. 1998;161:3596–3606. [PubMed] [Google Scholar]
- 17.Sanderson S, Frauwirth K, Shastri N. Expression of endogenous peptide–major histocompatibility complex class II complexes derived from invariant chain–antigen fusion proteins. Proc Natl Acad Sci USA. 1995;92:7217–7221. doi: 10.1073/pnas.92.16.7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Bergen J, Schoenberger SP, Verreck F, Amons R, Offringa R, Koning F. Efficient loading of HLA-DR with a T helper epitope by genetic exchange of CLIP. Proc Natl Acad Sci USA. 1997;94:7499–7502. doi: 10.1073/pnas.94.14.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujii S, Senju S, Chen YZ, Ando M, Matsushita S, Nishimura Y. The CLIP-substituted invariant chain efficiently targets an antigenic peptide to HLA class II pathway in L cells. Hum Immunol. 1998;59:607–614. doi: 10.1016/s0198-8859(98)00058-5. [DOI] [PubMed] [Google Scholar]
- 20.Malcherek G, Wirblich C, Willcox N, Rammensee HG, Trowsdale J, Melms A. MHC class II–associated invariant chain peptide replacement by T cell epitopes: engineered invariant chain as a vehicle for directed and enhanced MHC class II antigen processing and presentation. Eur J Immunol. 1998;28:1524–1533. doi: 10.1002/(SICI)1521-4141(199805)28:05<1524::AID-IMMU1524>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 20a.Wang, R.-F., X. Wang, A.C. Atwood, S.L. Topalian, and S.A. Rosenberg. 1999. Genetic approach to cloning genes encoding MHC class II-restricted antigens: mutated human CDC27 as a melanoma antigen. Science. In press. [DOI] [PubMed]
- 21.Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russell DW. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38. doi: 10.1016/0092-8674(84)90188-0. [DOI] [PubMed] [Google Scholar]
- 22.Russell DW, Schneider WJ, Yamamoto T, Luskey KL, Brown MS, Goldstein JL. Domain map of the LDL receptor: sequence homology with the epidermal growth factor precursor. Cell. 1984;37:577–585. doi: 10.1016/0092-8674(84)90388-x. [DOI] [PubMed] [Google Scholar]
- 23.Koda Y, Soejima M, Kimura H. Structure and expression of H-type GDP-L-fucose:beta-D-galactoside 2-alpha-L-fucosyltransferase gene (FUT1). Two transcription start sites and alternative splicing generate several forms of FUT1 mRNA. J Biol Chem. 1997;272:7501–7505. doi: 10.1074/jbc.272.11.7501. [DOI] [PubMed] [Google Scholar]
- 24.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 25.Rouquier S, Lowe JB, Kelly RJ, Fertitta AL, Lennon GG, Giorgi D. Molecular cloning of a human genomic region containing the H blood group alpha(1,2)fucosyltransferase gene and two H locus-related DNA restriction fragments. Isolation of a candidate for the human Secretor blood group locus. J Biol Chem. 1995;270:4632–4639. doi: 10.1074/jbc.270.9.4632. [DOI] [PubMed] [Google Scholar]
- 26.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 27.Cheever MA, Disis ML, Bernhard H, Gralow JR, Hand SL, Huseby ES, Qin HL, Takahashi M, Chen W. Immunity to oncogenic proteins. Immunol Rev. 1995;145:33–59. doi: 10.1111/j.1600-065x.1995.tb00076.x. [DOI] [PubMed] [Google Scholar]
- 28.Yotnda P, Garcia F, Peuchmaur M, Grandchamp B, Duval M, Lemonnier F, Vilmer E, Langlade-Demoyen P. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J Clin Invest. 1998;102:455–462. doi: 10.1172/JCI3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehrman MA, Russell DW, Goldstein JL, Brown MS. Alu-Alu recombination deletes splice acceptor sites and produces secreted low density lipoprotein receptor in a subject with familial hypercholesterolemia. J Biol Chem. 1987;262:3354–3361. [PubMed] [Google Scholar]
- 30.Pieper R, Christian RE, Gonzales MI, Nishimura MI, Gupta G, Settlage RE, Shabanowitz J, Rosenberg SA, Hunt DF, Topalian SL. Biochemical identification of a mutated human melanoma antigen recognized by CD4+T cells. J Exp Med. 1999;189:757–766. doi: 10.1084/jem.189.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tugendreich S, Tomkiel J, Earnshaw W, Hieter P. CDC27Hs colocalizes with CDC16Hs to the centrosome and mitotic spindle and is essential for the metaphase to anaphase transition. Cell. 1995;81:261–268. doi: 10.1016/0092-8674(95)90336-4. [DOI] [PubMed] [Google Scholar]
- 32.King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- 33.Ossendorp F, Mengede E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med. 1998;187:693–702. doi: 10.1084/jem.187.5.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalams SA, Walker BD. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med. 1998;188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4+T cells in the antitumor immune response. J Exp Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olivares-Villagomez D, Wang Y, Lafaille JJ. Regulatory CD4+T cells expressing endogenous T cell receptor chains protect myelin basic protein–specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med. 1998;188:1883–1894. doi: 10.1084/jem.188.10.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van de Keere F, Tonegawa S. CD4+T cells prevent spontaneous experimental autoimmune encephalomyelitis in anti–myelin basic protein T cell receptor transgenic mice. J Exp Med. 1998;188:1875–1882. doi: 10.1084/jem.188.10.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]