Identification of Lymphomyeloid Primitive Progenitor Cells in Fresh Human Cord Blood and in the Marrow of Nonobese Diabetic–Severe Combined Immunodeficient (NOD-SCID) Mice Transplanted with Human CD34+ Cord Blood Cells (original) (raw)
Abstract
Transplantation of genetically marked donor cells in mice have unambiguously identified individual clones with full differentiative potential in all lymphoid and myeloid pathways. Such evidence has been lacking in humans because of limitations inherent to clonal stem cell assays. In this work, we used single cell cultures to show that human cord blood (CB) contains totipotent CD34+ cells capable of T, B, natural killer, and granulocytic cell differentiation. Single CD34+ CD19−Thy1+ (or CD38−) cells from fresh CB were first induced to proliferate and their progeny separately studied in mouse fetal thymic organotypic cultures (FTOCs) and cocultures on murine stromal feeder layers. 10% of the clones individually analyzed produced CD19+, CD56+, and CD15+ cells in stromal cocultures and CD4+CD8+ T cells in FTOCs, identifying totipotent progenitor cells. Furthermore, we showed that totipotent clones with similar lymphomyeloid potential are detected in the bone marrow of nonobese diabetic severe combined immunodeficient (NOD-SCID) mice transplanted 4 mo earlier with human CB CD34+ cells. These results provide the first direct demonstration that human CB contains totipotent lymphomyeloid progenitors and transplantable CD34+ cells with the ability to reconstitute, in the marrow of recipient mice, the hierarchy of hematopoietic compartments, including a compartment of functional totipotent cells. These experimental approaches can now be exploited to analyze mechanisms controlling the decisions of such primitive human progenitors and to design conditions for their ampification that can be helpful for therapeutic purposes.
Keywords: stem cells, fetal thymic organotypic culture, nonobese diabetic-severe combined immunodeficient, lymphopoiesis, hematopoiesis
Syngeneic transplantation approaches using genetically marked stem cells (1, 2) or limited numbers of them (3, 4) have led to the identification of murine progenitor cells with multiple differentiative potentials. These strategies have been exploited to analyze the contribution of stem cell populations to short- and long-term hematopoietic reconstitution (5) and define conditions for their amplification (or self-renewal) (6). Both issues have an obvious clinical relevance, but experimental strategies used in mice cannot be easily applied to human cells (7). Although engraftment is the rule after injection of human cells into immunodeficient mice (8–12), there is no definite proof as yet that the entity defined as nonobese diabetic (NOD)1-SCID–repopulating cells (13, 14) is a reliable indicator of long-term reconstituting human stem cell function. One important limitation both in vitro and in vivo has been the difficulty in obtaining efficient lymphoid differentiation from primitive human progenitors. Thus, in NOD-SCID recipients, lymphoid differentiation is restricted to the development of the B cell lineage (8, 9, 12), and mature T or NK cells are produced very rarely or not at all (9). It is presently unknown whether this defect is accounted for by the fact that regulatory controls do not operate in these chimeras or because engrafted human CD34+ cells have an impaired T and NK potential. In the case of T cells, differentiation can be rescued by the coimplantation of human thymic tissue (15), and data in favor of a migration defect have recently been provided (16). In contrast to the chimeric in vivo situation, high numbers of functional, mature NK and dendritic cells can be obtained in vitro from various cell sources if appropriate cytokines are provided (17, 18). Human B cell differentiation requires stromal feeders, and, unexpectedly, decisive progress in the production of CD19+ B cells from human primitive progenitors has resulted from the use of murine (as opposed to human) stromal cells (19, 20). The T cell potential of primitive CD34+ human cells can also be successfully identified in fetal thymic organotypic cultures (FTOCs) using embryonic thymic lobes from both normal (21) or immunodeficient mice (including NOD-SCID mice) (22–24). However, even though FTOC conditions can be manipulated to obtain B, NK, and myeloid cells in addition to T cells, at least in mice (25, 26), cocultures of human CD34+ populations on stromal feeders, even if they are derived from the thymus, do not reproducibly allow T cell differentiation (27), and the identification of individual cells with all possible lymphoid and myeloid potentials still requires the use of separate assays (28).
In this study, we show for the first time that in vitro conditions exist that allow investigators to reproducibly assess all lymphoid (T, B, NK) and some myeloid potentials from single CD34+CD19−Thy1+ human cord blood (CB) cells. The strategy first requires induction of cell proliferation with cytokines to separately assess T cell potential in FTOCs and NK cell, B cell, and myeloid potentials in a unique coculture assay using MS5 murine stromal feeder layers. We also detected totipotent cells in the marrow of NOD-SCID recipient mice 4 mo after their transplantation with CD34+ CB cells. Taken together, our data demonstrate that human CB contains totipotent cells and in vivo– transplantable cells with similar lymphomyeloid differentiative potentials. This strongly suggests that cells exist in CB that have some characteristics of true stem cells.
Materials and Methods
Collection and Fractionation of Cells.
CB samples were collected with the informed consent of the mothers involved in our study. Low density CB mononuclear cells were first subjected to a standard CD34 immunomagnetic bead separation using the miniMACS® system (Miltenyi Biotec). Bead-separated CD34+ cells (purity >75%) were either injected into NOD-SCID mice (105 cells/mouse) or further fractionated in CD34+CD19− CD38− or CD34+CD19−Thy1+ fractions by cell sorting using a FACS Vantage™ equipped with an argon ion laser (Innova 70-4-coherent radiation) tuned to 488 nm and operating at 500 Mw. A morphological gate including all of the CD34+ cells was first defined on two-parameter histograms, side scatter versus forward scatter. Positivity or negativity for CD19, Thy1, and CD38 among the CD34+ cells was determined using cells labeled with CD34-PE-Cy5 (Immunotech) and an irrelevant IgG1 mAb. The Thy1+ and CD38− subsets of CD34+ cells were obtained in two steps. CD34+CD19− cells were sorted and relabeled with CD34-PE-Cy5 (Immunotech) and either CD38-PE (Becton Dickinson), or Thy1 (CD90)-PE (PharMingen). Limits of the sorting gates for Thy1+ and CD38− cells were set as illustrated (see Fig. 1). Single cell cultures were initiated by directly sorting cells into 96-well tissue culture plates precoated with MS5 murine stromal cells using an automatic cloning design unit (Becton Dickinson). The standardized distribution of one cell per well was controlled by examining over 4,000 wells individually in parallel experiments initiated with human cells.
Figure 1.


Selection of human cell populations. Human CD34+ cells purified from fresh CB (A and B) and NOD-SCID marrow (C, D, and E) were first depleted of CD19+ cells (A and C). CD34+CD19− cells were further enriched for Thy1+ cells (B and D) or CD38− cells (E). Gates selected for cell sorting are indicated in B (CD34+CD19−Thy1+), D (CD34+ CD19−Thy1+), and E (CD34+CD19−CD38−). Limits of the quadrants were determined by analyzing cells labeled with isotype-control Ig.
For in vivo experiments, 105 CB CD34+ cells were intravenously injected into irradiated (3–3.5 Gy; cobalt-60 Eldorado S irradiator; AECL Medical) NOD-LtSz-scid/scid (hereafter called NOD-SCID) mice anesthesized briefly with ether. 10–16 wk later, marrow cells were flushed from the femurs and tibias of recipient mice as previously described (12), and human CD34+ cells were isolated and fractionated into CD34+CD19−, CD34+ CD19−Thy1+, or CD34+CD19−CD38− as indicated above (see Fig. 1).
Assessment of Human T Cell Potential in FTOC.
Isolation of murine embryonic thymic lobes, incubation with human cells after the hanging drop procedure, and organotypic cultures were performed following standard procedures initially described to analyze mouse T lymphoid differentiation and adapted to the identification of human T cell potential (22, 29). Our slight modifications of the technique included the use of NOD-SCID embryonic (day 14) thymic lobes and the addition of recombinant human (rhu) IL-2 (5 ng/ml; Diaclone), 20 ng/ml rhu-IL-7 (Diaclone), and 50 ng/ml rhu-stem cell factor (SCF; provided by Amgen) during the hanging drop procedure. Thymic lobes were incubated onto floating filters (Isopore membrane, 25-mm diameter, 8-μm pore size; Millipore SA) at 37°C and 5% CO2 in medium without cytokines. Cells recovered from the thymic lobes after 28–35 d were labeled with anti–human CD4-PE and CD8-FITC (Becton Dickinson), TCR-α/β-PE, and CD3-FITC (Immunotech). Lack of reactivity of the mouse anti–human mAbs with NOD-SCID murine cells was verified in pilot experiments (12, 24).
Simultaneous Assessment of B, NK, and Granulomonocytic (Gr/M) Differentiation.
The different cell fractions were incubated in 24- or 96-well plates precoated with confluent murine marrow–derived MS5 cells (30) in RPMI supplemented with 10% human serum, 5% FCS, and the following six rhu cytokines: rhu-SCF (50 ng/ml; Amgen), rhu-Flt3-ligand (50 ng/ml; Diaclone), pegylated (PEG)- rhu-megakaryocyte growth and differentiation factor (MGDF) (50 ng/ml; Amgen), rhu-IL-2 (5 ng/ml; Diaclone), rhu-IL-15 (10 ng/ ml; Diaclone), and rhu-IL-7 (20 ng/ml; Diaclone). Wells with significant cell proliferation (usually >500 cells after 3–6 wk) were selected, and cells were collected and their phenotype assessed by flow cytometry after labeling with the following mAbs: CD19-PE (Becton Dickinson), CD15-FITC (Dako Corp.), and CD56-PE-Cy5, CD34-PE-Cy5, CD1a-PE, and HLA-DR-FITC (all from Immunotech). In some experiments, the intracellular expression of myeloperoxidase (MPO; Caltag Labs.) was determined using a cell permeabilization kit (Harlan, Sera-Lab Ltd.). Analysis was performed on a FACScan™ (Becton Dickinson) using CellQuest software.
Analysis of the Lymphoid and Gr/M Potentials of Single Progenitors.
Individual CD34+CD19−, CD34+CD19−Thy1+, or CD34+ CD19−CD38− cells from either fresh CB or the marrow of chimeric NOD-SCID mice 4 mo posttransplant were induced to proliferate in 96-well plates precoated with MS5 cells in the presence of the six growth factors listed above. Wells were carefully monitored for 7–21 d, and clones containing ≥1,000 cells were divided: for each clone, half of the cells were cultured on new MS5 feeders as described above and the other half were incubated in NOD-SCID FTOC together with 5,000 irradiated (15 Gy) CD34+CD38+ CB cells, as accessory cells have been shown to help engraftment in FTOC seeded with small numbers of cells (24). At the end of the FTOC, cells collected from each lobe were analyzed individually for the presence of CD4bright and/or CD4+ CD8+ human cells. In three control experiments, 25 NOD-SCID fetal thymic lobes were reconstituted with 10,000 irradiated, fresh CD34+CD38+ cells/lobe, but none of them yielded human T cells. In contrast, all lobes incubated with unirradiated CD34+ CD38+ cells produced CD4bright and CD4+CD8+ T cells (not shown).
Results
Selection of Human Cells from Fresh CB and from the Marrow of NOD-SCID Mice Transplanted with CB CD34+ Cells.
Because primitive lymphomyeloid cells most likely represent only a minor fraction of human CD34+ cells collected from both fresh CB and NOD-SCID marrow, we performed an enrichment step. We first removed CD34+ CD19+ committed pre-B cells, which represented 13 ± 8% (n = 6) of fresh CB cells (Fig. 1 A) but 83 ± 6% (n = 11) of NOD-SCID–derived CD34+ cells (Fig. 1 C) (12). CD34+CD19− cells were further fractionated into Thy1+ or CD38− cells, as both cell types exhibit primitive functions. Thy1+ cells represented 10 ± 4% (n = 5) of fresh CD34+CD19− CB cells (Fig. 1 B), whereas Thy1+ (Fig. 1 D) and CD38− (Fig. 1 E) cells accounted only for 2.5 ± 1.8% and 2.3 ± 0.6% of CD34+CD19− cells from NOD-SCID mice (n = 5), respectively.
B, NK, and Gr/M Cell Differentiation Potentials of CD34+ Subsets from Fresh CB and the Marrow of NOD-SCID Mice Transplanted with Human CD34+ CB Cells.
We have previously shown that murine MS5 stromal cells support myeloid (30) and B cell differentiation (20) from CD34+ CB cells when studied in separate assays but also from single bipotent B/myeloid progenitors in a switch system (20). Because MS5 cells also promote the production of NK cells from CD34+ cells in the presence of human IL-15 (31), we reasoned that slight modifications of these culture conditions should allow the simultaneous development of all three lineages (B, NK, and Gr/M). To this end, we supplemented the medium with rhu-SCF, IL-2, IL-15 (three-cytokine condition), and, in later experiments, PEG-rhu-MGDF, Flt3-ligand, and IL-7 were also added (six-cytokine condition). PEG-rhuMGDF and Flt3-ligand were chosen to trigger active proliferation of immature progenitors that may not respond to lineage-specific cytokines (32), and IL-7 was chosen to minimize the loss of B and/or T cell potential (33).
Thus, CD34+CD19− and CD34+CD19−Thy1+ fresh CB cells (2–10,000 cells/well), when cocultured on MS5 cell feeder layers in three- or six-cytokine conditions, reproducibly generated CD19+ B, CD56+ NK, and CD15+/ MPO+ Gr/M cells in 2–3 wk (Table I). In the presence of six cytokines, up to 70% of cells recovered after 2 wk of culture were still CD34+ (not shown). Table I and Fig. 2 A–C also show that CD34+CD19− cells collected from the marrow of NOD-SCID mice (n = 4) and cultured for 3 wk in the presence of six cytokines generated 3.1 ± 0.7% CD19+ B cells, 9.5 ± 4.1% CD56+ NK cells, and 22 ± 13% CD15+ Gr/M cells (mean ± SEM of nucleated cells). Interestingly, all three cell types also exhibited almost nonoverlapping forward scatter versus side scatter profiles. Similar differentiation profiles and lineage yields were observed in three experiments initiated with CD34+CD19−CD38− cells (1 ± 0.4, 44 ± 3, and 28 ± 5% B, NK, and Gr/M cells, respectively) and CD34+CD19−Thy1+ cells (6.4 ± 6, 47 ± 9, and 20 ± 5% B, NK, and Gr/M cells, respectively) (Table I). Cells coexpressing CD1a and HLA-DR, most likely of dendritic origin, were detected in a few experiments in which these markers were analyzed (data not shown). As previously described, cultured CD19+ cells coexpressed CD38 and CD10 but lacked CD20, CD22, and sIgM, whereas NK cells were mature and functional, as shown by their spontaneous cytotoxic activity against K562 and Daudi cells (reference 31; data not shown).
Table I.
Phenotype of Human Cells Grown from CD34+CD19−, CD34+CD19−Thy1+, and CD34+CD19−CD38− Cells from Fresh CB and NOD-SCID Mice
| Cell fraction | No. of experiments | Cytokines* | Time in culture | Proportion of cells in the gate‡ (mean ± SEM) | ||
|---|---|---|---|---|---|---|
| CD19+ | CD56+ | CD15+ | ||||
| wk | ||||||
| Fresh CB | ||||||
| CD34+CD19− | 2 | 6 | 2 | 0.3; 0.2 | 1.4; 5 | 24; 36 |
| 1 | 6 | 3 | 0.4 | 45 | 34 | |
| CD34+CD19−Thy1+ | 2 | 3 | 2-3 | 5.7 ± 1.1 | 10 ± 6 | 24 ± 6 |
| NOD-SCID mice | ||||||
| CD34+CD19− | 4 | 3 | 2 | 19 ± 6.4 | 44 ± 15 | 6 ± 1 |
| 4 | 6 | 3 | 3.1 ± 0.7 | 9.5 ± 4.1 | 22 ± 13 | |
| CD34+CD19−Thy1+ | 3 | 6 | 4-6 | 6.4 ± 6 | 47 ± 9 | 20 ± 5 |
| CD34+CD19−CD38− | 3 | 6 | 4-6 | 1 ± 0.4 | 44 ± 3 | 28 ± 5 |
Figure 2.

Assessment of the Gr/M and lymphoid potentials of human CD34+CD19− cells from the marrow of NOD-SCID mice transplanted with CB CD34+ cells. 10,000 human CD34+CD19− cells obtained from the marrow of NOD-SCID mice 4 mo after transplantation with 105 human CB CD34+ cells were cultured on MS5 cells with six cytokines (A–C) or in chimeric FTOC (D–G). Cells cultured on MS5 were stained with CD15-FITC, CD19-PE, and CD56-PE-Cy5, and cells grown in FTOC were stained with CD4-PE and CD8-FITC. In F and G, cells were analyzed after three-color labeling (CD3-FITC, TCR-α/β-PE, and CD4-PE-Cy5). Labeled cells were analyzed on a FACScan™ (CellQuest software) in the morphological gates indicated in A and D, which included all viable nucleated cells. Positivity set by the quadrant limits was defined using isotype-control Ig. These results are from one representative experiment in which FTOCs and stromal cultures were initiated with human cells purified from NOD-SCID mice.
CD34+CD19− Human Cells Isolated from NOD-SCID Recipients Express T Lymphoid Potential in FTOC.
The culture conditions described above were not permissive to T cell differentiation, which requires the presence of thymic stromal elements. We have recently reported the successful use of NOD-SCID embryonic thymus to identify T cell potential of CB and adult marrow CD34+, CD34+CD38+, and CD34+CD38− cells (24). Here, we applied this strategy to demonstrate that human CD34+CD19− cells selected from the bone marrow of NOD-SCID mice generated CD4bright and double positive (DP) CD4+CD8+ T cells in NOD-SCID FTOC. As illustrated in Fig. 2 B–G, in 5/5 experiments, human CD4+ cells (which represented 69 ± 13% of nucleated cells collected from thymi) were produced, and 82 ± 5% of those were CD4bright, 46 ± 12% were DP CD4+CD8+, 31 ± 5% were CD4+TCR-α/β+ (n = 3), and 32 ± 7% were CD4+CD3+ (n = 3). As described for fresh CB cells, very few CD4−CD8+ cells were produced (22).
Taken together, these experiments demonstrated that CD34+CD19− human cells detected in NOD-SCID recipients 4 mo after intravenous injection of CD34+ CB cells had retained the ability to differentiate into B, NK, and Gr/M lineages as well as T cells in vitro, even though T and NK terminal maturation did not occur in vivo.
Single CD34+CD19−Thy1+ and CD34+CD19−CD38− Cells from Fresh CB and Transplanted NOD-SCID Mice Are Capable of Generating T, B, NK, and Gr/M Cells.
Before investigating the full lymphomyeloid differentiative potential of single cells, we first determined the frequency of single cells that produce high numbers of B, NK, and Gr/M cells when cocultured on MS5 cells with six cytokines.
In two experiments, 180 single CD34+CD19− cells from fresh CB were cultured in the presence of six cytokines and MS5 cell feeder layers. Cells from 55 wells were phenotyped and found to contain multiple combinations of B, NK, and Gr/M cells (Table II). Nine clones (16%) contained cells from all three lineages, and 54% of the clones yielded cells of only one lineage. When CD34+CD19− Thy1+ cells were used, the frequency of the three-lineage clones increased to 28% (28 clones analyzed) and the frequency of one-lineage clones decreased (21%), in agreement with the preferential expression of Thy1 by immature progenitors. Lineages were similarly distributed in clones grown from single CD34+CD19−Thy1+ or CD38− cells isolated from the marrow of transplanted NOD-SCID mice. Analysis, for instance, of cells from 240 wells, each seeded with a single CD34+CD19−Thy1+ (120 wells) or CD34+CD19−CD38− (120 wells) cell, indicated that >50% of the clones contained cells of two lineages and 15–33% cells of three lineages but only 13–23% cells of one lineage (Table II). In contrast and as noted above, 69% of the more mature CD34+CD19− cells generated cells of only one lineage.
Table II.
Lymphoid (B, NK) and Granulocytic Potentials of Single CD34+ Cells from Fresh CB and NOD-SCID Mice
| CD34+ fractions | No. of wells* | No. of wells with cells of the indicated lineage | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| one lineage‡ | two lineages | three lineages | |||||||||
| NK | B | M | Total % | NK/B | NK/M | B/M | Total % | NK/B/M | Total % | ||
| Fresh CB | |||||||||||
| CD19− | 180/55 | 8 | 2 | 20 | 54 | 13 | 2 | 1 | 29 | 9 | 16 |
| CD19−Thy1+ | 120/28 | 2 | 1 | 3 | 21 | 13 | 0 | 1 | 50 | 8 | 28 |
| NOD-SCID mice | |||||||||||
| CD19− | 600/90 | 40 | 1 | 21 | 69 | 14 | 5 | 2 | 23 | 8 | 9 |
| CD19−Thy1+ | 120/30 | 0 | 0 | 4 | 13 | 14 | 2 | 0 | 53 | 10 | 33 |
| CD19−CD38− | 120/34 | 3 | 1 | 4 | 23 | 19 | 1 | 1 | 62 | 5 | 15 |
Importantly, individual clones grown as described above in six cytokines on stromal feeders contained high numbers of nucleated cells (from 10,000 to 400,000 total cells per well at week 2–3), which made it possible to remove part of the clone to separately assay T cell differentiation in FTOC. Thus, in a second set of experiments, multiple wells were seeded with CD34+CD19−Thy1+ from fresh CB (420 wells) and with CD34+CD19−Thy1+ and CD34+ CD19−CD38− cells from the marrow of transplanted NOD-SCID mice (each population, 240 wells). After 2–3 wk, clones that had proliferated (150, 76, and 92, respectively) were divided to initiate NK-B-Gr/M and FTOC assays (Table III).
Table III.
Analysis of the Progeny of Single CD34+CD19− or CD34+CD19−Thy1+ or CD34+CD19−CD38− Subpopulations from Fresh CB and NOD-SCID Mice
| CD34+CD19− fractions | No. of wells* | No. of wells with cells of the indicated phenotype‡ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| one lineage | two lineages | ||||||||||
| NK | M | T | Total % | NK/B | NK/M | B/M | M/T | NK/T | Total % | ||
| Fresh CB Thy1+ | 420/150 | 30 | 16 | 3 | 33 | 25 | 15 | 1 | 1 | 4 | 31 |
| NOD-SCID mice Thy1+ | 240/76 | 29 | 15 | 0 | 58 | 11 | 11 | 1 | 0 | 0 | 30 |
| CD38− | 240/92 | 14 | 6 | 1 | 23 | 20 | 8 | 0 | 0 | 3 | 34 |
| CD34+CD19− fractions | No. of wells* | three lineages | four lineages | ||||||||
| NK/B/M | M/B/T | NK/M/T | NK/B/T | Total % | NK/B/T/M | Total % | |||||
| Fresh CB Thy1+ | 420/150 | 31 | 1 | 4 | 9 | 30 | 10 | 7 | |||
| NOD-SCID mice Thy1+ | 240/76 | 8 | 0 | 0 | 0 | 10 | 1 | 1.3 | |||
| CD38− | 240/92 | 19 | 0 | 4 | 4 | 29 | 13 | 14 |
The most remarkable result was that 10/150 clones grown from fresh CD34+CD19−Thy1+ CB cells and 14/ 68 clones grown from human CD34+CD19−Thy1+ (or CD38−) cells from NOD-SCID chimeras generated not only B, NK, and Gr/M cells but also T cells (CD4bright cells usually associated with DP CD4+CD8+ cells) in FTOC assays (Table III). As illustrated in Fig. 3, the proportions of lymphoid (CD19+, CD56+) and Gr/M (CD15+) cells varied between clones. However, the distribution of clones containing one, two, three, or four lineages in experiments initiated with either fresh CB cells or cells from NOD-SCID mice was almost identical (Table III). Interestingly, the distribution of lineages was not strictly identical in clones grown from CD34+CD19−Thy1+ and CD34+ CD19−CD38− NOD-SCID–derived human cells. The biological significance of this observation is unclear, and this result must be confirmed in experiments initiated with fresh CB cells of both phenotypes. In addition to totipotent clones, multiple combinations of lineages were detected: 30% of the clones (45/150 wells seeded with CD34+ CD19−Thy1+ fresh CB cells and 27/92 wells seeded with CD34+CD19−CD38− cells from transplanted NOD-SCID mice) generated combinations of three lineages. Among these three-lineage clones, 30% included T cells, and 4% combined B, NK, and T cells but no CD15+ cells and were most likely derived from lymphoid-restricted progenitors (Fig. 4 A). None of the clones contained B cells alone and <2% exclusively T cells (Table III). Both T (52/54) and B (151/154) cells were almost always combined with NK cells. This was the predominant phenotype in our culture conditions: 128/150 clones from fresh CB and 145/168 clones from marrow of transplanted NOD-SCID mice contained human NK cells.
Figure 3.
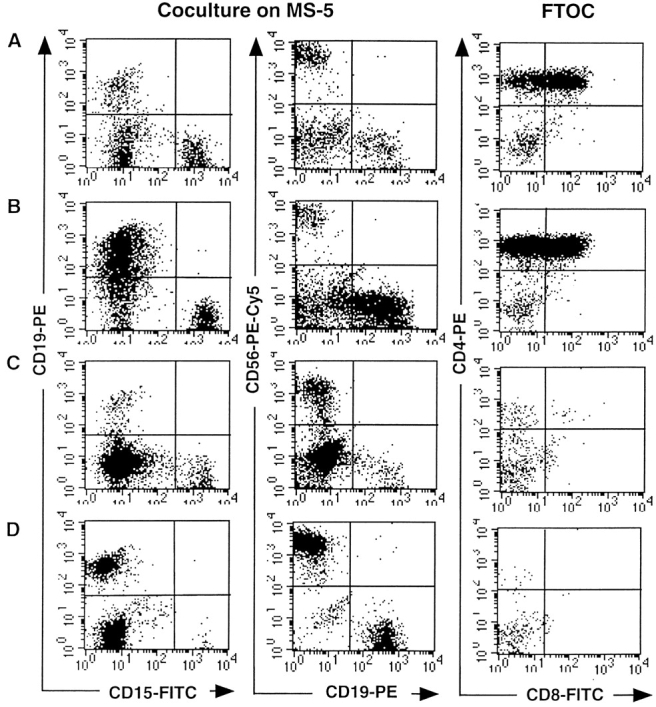
Production of T, B, NK, and Gr/M cells by single CD34+CD19−CD38− cells from the marrow of NOD-SCID mice transplanted with CB CD34+ cells. Analysis of the progeny of four individual clones is shown (A–D). Single CD34+CD19_−_CD38− cells were cultured on MS5 with a cocktail of six cytokines during 2 wk and the progeny separated to initiate secondary MS5 cocultures and FTOC. Cells were cultured for another 2 wk with MS5 and cytokines and an additional 4 wk in FTOC, after which time they were labeled with lineage-specific antibodies as described in the Fig. 2 legend. Analysis was performed in the gates defined in Fig. 2, A and D.
Figure 4.
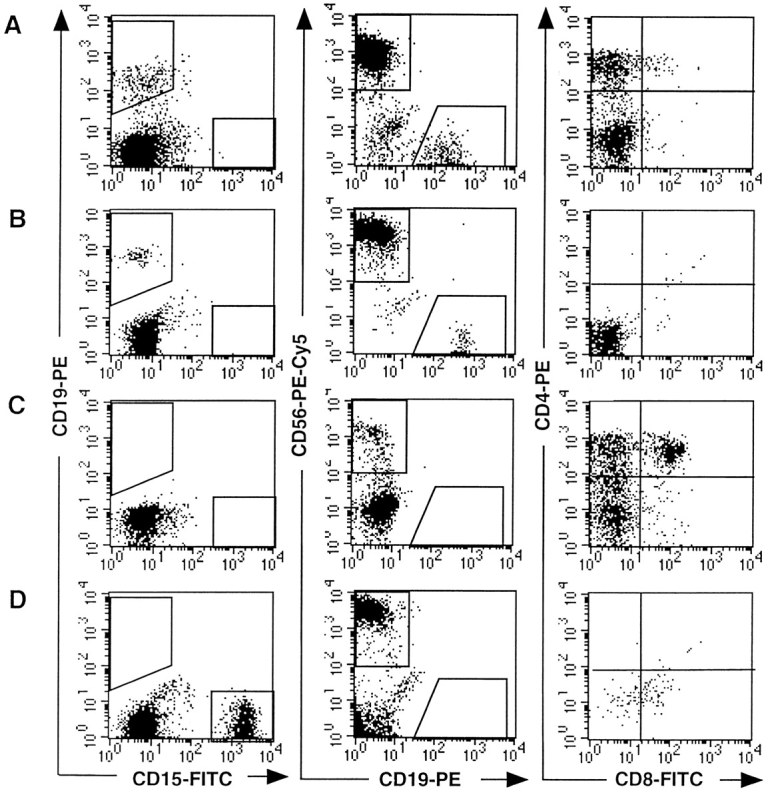
Combinations of cells from different lineages produced by single CD34+CD19−CD38− cells from CB or from the marrow of NOD-SCID mice transplanted with CB CD34+ cells. Single cells from fresh CB (A) and NOD-SCID bone marrow (B, C, and D) were cultured and their progeny analyzed as described for Fig. 3. The progeny of four clones with different differentiative capacities is shown: clone A (A) contained B, NK and T cells; clone B (B) contained B and NK cells; clone C (C) contained NK and T cells, and clone D (D) contained Gr/M cells and NK cells. Analysis was performed in the gates defined in Fig. 2, A and D.
Discussion
In this study we provide, for the first time in humans, direct evidence that a significant proportion of CD34+ CD19−Thy1+ (and/or CD38−) fresh CB cells are totipotent and that similar totipotent cells can be isolated from the marrow of NOD-SCID mice transplanted several months earlier with fresh human CD34+ CB cells. The ability of a single cell to generate T, B, and NK lymphocytes and Gr/M cells defines totipotency and was demonstrated in vitro by combining FTOCs and cocultures on competent murine stromal cell feeder layers.
A prerequisite for the success of these experiments was to first overcome previous limitations in the assay system for B and T lymphoid potential of human cells in vitro, and a decisive step was to use the murine stromal cell line MS5 (34). Feeder layers from MS5 and other cell stromal lines (17, 19, 35), in contrast to those from human stromal cells, support the differentiation of CD34+ cells into pro-B cells; although spontaneous terminal B cell maturation remains compromised, probably because T helper function is lacking, it can be induced (36). However, despite recent suggestions that extra-thymic differentiation may occur in the marrow environment (37) or in the gut and the unique finding of CD3+ human cells in the marrow of bgnuXid mice engrafted with CD34+ marrow cells (7), human T cells never developed in vitro in MS5 cocultures. The presence of an intact thymic structure remains essential, although recent results indicate that thymic stromal feeders can replace the intact thymus (27, 38). The observation that cultures of embryonic thymic lobes, initially used to follow mouse T cell development, could be successfully applied to the study of human T cell differentiation represents an improvement over the use of human embryonic thymus. Thus, we (24) and others (22, 23) recently described that FTOC initiated with thymic lobes from immunodeficient mice efficiently supported the first steps of human T cell differentiation up to the DP CD4+CD8+ stage and are sensitive enough to detect the production of CD4+CD8+ T cells from as few as 100 CD34+ CB cells (24).
To prove cell totipotency, it was essential to work at the clonal level, which imposed the need to initiate FTOC and cocultures on MS5 feeders with cells issued from the same clone. This required induction of the proliferation of input single cells to allow subdivision of the clone without loss of potentials. This was accomplished by adding early-acting proliferative cytokines, including IL-7, to avoid the loss of early T cell progenitors (33) and by plating the cells directly on MS5 cells, because we have shown that these stromal cells are important to retain the primitive potential of actively proliferating adult CD34+CD38− marrow long-term culture–initiating cells (39).
Our results showed that 6.7% of CD34+CD19−Thy1+ CB cells were totipotent, which leads to an estimate of one totipotent progenitor per 200 CD34+ CB cells. This number is surprisingly high and may even be underestimated, as we selected only actively proliferating clones and also because the hanging drop procedure that we used may limit the homing of T cell progenitors into thymic lobes. Although it is hazardous to draw any firm conclusion about stem cell hierarchy based on the distribution and combination of lineages observed in single clones grown in vitro, both were very similar to those reported from the analysis of lymphomyeloid clones grown in vitro from murine fetal liver (26) or embryonic splanchnopleura (40) and in vivo from recipients of genetically marked donor cells (1). Particularly interesting was the observation in our study that some clones, derived from fresh CB cells and the marrow of transplanted NOD-SCID mice, produced T, B, and NK but no Gr/M cells. Although we cannot exclude that other conditions could have unmasked additional myeloid potentials, erythroid or megakaryocytic, the total lack of CD15+ cells makes this hypothesis very unlikely. The parental progenitors most probably represented the human counterpart of the murine lymphoid-restricted progenitors identified among IL-7Rα+c-kit+Lin−Sca+ adult mouse marrow cells (28). As reported in the mouse (26), we also failed to detect bipotent T/B progenitors, an observation that contrasted with the high frequency, in our study, of clones with both NK and B cells. It is important to stress that NK cells were the most frequent cell type in our cultures and that their outgrowth might have interfered (in a positive or negative way) with the development/survival of other lineages. Indeed, there were very few if any clones containing only B or T cells. A possible explanation for the lack of B cell clones was the lack of signals released by T cells, which was not compensated for by the action of MS5 cells.
A second major observation was that human totipotent cells were detected in the marrow of NOD-SCID recipients several months after transplantation of CD34+ CB cells. Tracing individual stem cell fate and function in vivo is complicated in the NOD-SCID chimeras, as opposed to the syngenic murine situation, as terminal T and NK cell differentiation, in contrast to B cell differentiation, does not take place in vivo. However, human CD34+ cells isolated from NOD-SCID chimeras reproducibly produced CD4+ CD8+ T cells and NK cells in vitro, indicating that human cells have not lost their intrinsic T and NK potential after their engraftment in the NOD-SCID environment. A more likely hypothesis is that regulatory steps involved in the development of T and NK pathways do not take place in this xenogenic model. For T cells, the defect most likely involves molecules regulating cell trafficking, whereas the maturation of human NK cells was blocked because human cells do not respond to murine IL-2 and IL-15.
A close frequency of totipotent clones (7%) was found in experiments initiated with fresh CD34+CD19− Thy1+ CB cells and CD34+CD19−CD38− cells from transplanted NOD-SCID mice (14%). Calculations yielded an absolute number of ∼50 totipotent cells in four long bones of a NOD-SCID 4-mo posttransplant. Considering that 500 totipotent cells were present among the 105 CD34+ cells injected, this suggests loss rather than amplification of totipotent cells. However, totipotent clones may in fact actively proliferate in vivo. A strong argument in favor of this hypothesis is that, in our experience as well as in others' (8), <1% of the injected CD34+ human cells are detected in the marrow of NOD-SCID mice 48 h after their injection (range 0.1 to 4% in six experiments; our unpublished data). Therefore, it is conceivable that <10 totipotent cells eventually colonize the marrow of NOD-SCID mice and contribute to the reconstitution of all compartments, a hypothesis that is in agreement with murine studies (1, 4, 41, 42). Definite determination of the number of human clones contributing to the reconstitution of hematopoiesis in NOD-SCID mice will await results from transplantation experiments using genetically marked human cells. These studies will be facilitated by the availability of conditions that allow the expression of the full differentiative potentials of individual clones, as demonstrated in this study, but also by improved procedures that will allow efficient transduction of NOD-SCID-CRU competitive repopulating unit (43, 44).
Acknowledgments
We thank Annie Rouches and Patrice Ardouin for their invaluable contribution to the breeding and care of the NOD-SCID mice; Brigitte Izac for her excellent technical assistance; Philippe Rameau and André Katz for cell sorting; John Dick for providing the original NOD-SCID breeding pairs and for his constant support; Marina Cavazzana-Calvo, Jean Plum, and Magda de Smedt for their advice on the initial device of the FTOC system; and Anne-Lise Bennaceur-Griscelli and Cristina Tourino for stimulating discussions. We also thank Amgen-USA and Amgen-France for their gift of PEG-rhu-MGDF and rhu-SCF.
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale (INSERM), Electricité de France, Ligue Nationale Contre le Cancer, Association de la Recherche contre le Cancer (ARC 6532 to L. Coulombel), Institut Gustave Roussy, and Ministère de la Recherche et de la Technologie (to L. Coulombel). C. Robin was funded by fellowships from the French Ministère de la Recherche et de la Technologie and French Society of Hematology.
Abbreviations used in this paper
CB
cord blood
DP
double positive
FTOCs
fetal thymic organotypic cultures
Gr/M
granulomonocytic
MGDF
megakaryocyte growth and differentiation factor
NOD
nonobese diabetic
PEG
pegylated
rhu
recombinant human
SCF
stem cell factor
Footnotes
C. Robin and F. Pflumio contributed equally to this work.
This work was presented in part at the International Society for Experimental Hematology meeting, Vancouver, Canada, August 1998, and published in abstract form (Robin, C., F. Pflumio, C. Tourino, and L. Coulombel. 1998. Exp. Hematol. 26:693).
References
- 1.Jordan CT, Lemischka IR. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genes Dev. 1990;4:220–232. doi: 10.1101/gad.4.2.220. [DOI] [PubMed] [Google Scholar]
- 2.Keller G, Snodgrass R. Life span of multipotential hematopoietic stem cells in vivo. J Exp Med. 1990;171:1407–1418. doi: 10.1084/jem.171.5.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osawa M, Hanada KI, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 4.Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, Eaves CJ. Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proc Natl Acad Sci USA. 1990;87:8736–8740. doi: 10.1073/pnas.87.22.8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zijlmans JM, Visser JW, Laterveer L, Kleiverda K, Heemskerk DP, Kluin PM, Willemze R, Fibbe WE. The early phase of engraftment after murine blood cell transplantation is mediated by hematopoietic stem cells. Proc Natl Acad Sci USA. 1998;95:725–729. doi: 10.1073/pnas.95.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller CL, Eaves CJ. Expansion in vitro of adult murine hematopoietic stem cells with transplantable lympho-myeloid reconstituting ability. Proc Natl Acad Sci USA. 1997;94:13648–13653. doi: 10.1073/pnas.94.25.13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nolta JA, Dao MA, Wells S, Smorgorzewska EM, Kohn DB. Transduction of pluripotent human hematopoietic stem cells demonstrated by clonal analysis after engraftment in immune-deficient mice. Proc Natl Acad Sci USA. 1996;93:2414–2419. doi: 10.1073/pnas.93.6.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cashman J, Bockhold K, Hogge DE, Eaves AC, Eaves CJ. Sustained proliferation, multi-lineage differentiation and maintenance of primitive human haematopoietic cells in NOD/SCID mice transplanted with human cord blood. Br J Haematol. 1997;98:1026–1036. doi: 10.1046/j.1365-2141.1997.3233140.x. [DOI] [PubMed] [Google Scholar]
- 9.Hogan CJ, Shpall EJ, McNulty O, McNiece I, Dick JE, Shultz LD, Keller G. Engraftment and development of human CD34(+)-enriched cells from umbilical cord blood in NOD/LtSz-scid/scid mice. Blood. 1997;90:85–96. [PubMed] [Google Scholar]
- 10.Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science. 1992;255:1137–1141. doi: 10.1126/science.1372131. [DOI] [PubMed] [Google Scholar]
- 11.Nolta J, Hanley M, Kohn D. Sustained human hematopoiesis in immunodeficient mice by cotransplantation of marrow stroma expressing human interleukin-3: analysis of gene transduction of long-lived progenitors. Blood. 1994;83:3041–3051. [PubMed] [Google Scholar]
- 12.Pflumio F, Izac B, Katz A, Shultz LD, Vainchenker W, Coulombel L. Phenotype and function of human hematopoietic cells engrafting immune-deficient CB17- severe combined immunodeficiency mice and nonobese diabetic-severe combined immunodeficiency mice after transplantation of human cord blood mononuclear cells. Blood. 1996;88:3731–3740. [PubMed] [Google Scholar]
- 13.Conneally E, Cashman J, Petzer A, Eaves C. Expansion in vitro of transplantable human cord blood stem cells demonstrated using a quantitative assay of their lympho-myeloid repopulating activity in nonobese diabetic-scid/scid mice. Proc Natl Acad Sci USA. 1997;94:9836–9841. doi: 10.1073/pnas.94.18.9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang JC, Doedens M, Dick J. Primitive human hematopoietic cells are enriched in cord blood compared with adult bone marrow or mobilized peripheral blood as measured by the quantitative in vivo SCID-repopulating cell assay. Blood. 1997;89:3919–3924. [PubMed] [Google Scholar]
- 15.Yurasov S, Kollman TR, Kim A, Raker CA, Hachamovitch M, Wong-Staal F, Goldstein H. Severe combined immunodeficiency mice engrafted with human T cells, B cells and myeloid cells after transplantation with human fetal bone marrow or liver cells and implanted with human fetal thymus: a model for studying human gene therapy. Blood. 1997;89:1800–1810. [PubMed] [Google Scholar]
- 16.van der Loo JC, Hanenberg H, Cooper RJ, Luo FY, Lazaridis EN, Williams DA. Nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mouse as a model system to study the engraftment and mobilization of human peripheral blood stem cells. Blood. 1998;92:2556–2570. [PubMed] [Google Scholar]
- 17.Galy A, Travis M, Cen D, Chen B. Human T, B, natural killer and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3:459–473. doi: 10.1016/1074-7613(95)90175-2. [DOI] [PubMed] [Google Scholar]
- 18.Mrozek E, Anderson P, Caligiuri M. Role of interleukin 15 in the development of human CD56+ natural killer cells from CD34+hematopoietic progenitor cells. Blood. 1996;87:2632–2640. [PubMed] [Google Scholar]
- 19.Rawlings DJ, Quan S, Hao QL, Thiemann FT, Smogorzewska M, Witte ON, Crooks GM. Differentiation of human CD34+CD38−cord blood stem cells into B cell progenitors in vitro. Exp Hematol. 1997;25:66–72. [PubMed] [Google Scholar]
- 20.Berardi AC, Meffre E, Pflumio F, Katz A, Vainchenker W, Schiff C, Coulombel L. Individual CD34+ CD38lowCD19−CD10−progenitor cells from human cord blood generate B lymphocytes and granulocytes. Blood. 1997;89:3554–3564. [PubMed] [Google Scholar]
- 21.Merkenschlager M, Fisher A. Human postnatal thymocytes generate phenotypically immature CD3dim, CD5dim, CD1abrightprogeny in organ culture. J Immunol. 1992;148:1012–1015. [PubMed] [Google Scholar]
- 22.Plum J, De Smedt M, Defresne M-P, Leclercq G, Vandekerckhove B. Human CD34+fetal liver stem cells differentiate to T cells in a mouse thymic microenvironment. Blood. 1994;84:1587–1593. [PubMed] [Google Scholar]
- 23.Verhasselt B, De Smedt M, Verhelst R, Naessens E, Plum J. Retrovirally transduced CD34++human cord blood cells generate T cells expressing high levels of the retroviral encoded green fluorescent protein marker in vitro. Blood. 1998;91:431–440. [PubMed] [Google Scholar]
- 24.Robin C, Bennaceur-Griscelli A, Louache F, Vainchenker W, Coulombel L. Identification of T lymphoid progenitor cells in CD34+CD38low and CD34+ CD38+subsets of human cord blood and bone marrow cells using NOD-SCID fetal thymus organ cultures. Br J Haematol. 1999;104:809–819. doi: 10.1046/j.1365-2141.1999.01266.x. [DOI] [PubMed] [Google Scholar]
- 25.Barcena A, Galy A, Punnonen J, Muench M, Schols D, Grazia M, Roncarolo, de Vries J, Spits H. Lymphoid and myeloid differentiation of fetal liver CD34+lineage cells in human thymic organ culture. J Exp Med. 1994;180:123–132. doi: 10.1084/jem.180.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawamoto H, Ohmura K, Katsura Y. Direct evidence for the commitment of hematopoietic stem cells to T, B and myeloid lineages in murine fetal liver. Int Immunol. 1997;9:1011–1019. doi: 10.1093/intimm/9.7.1011. [DOI] [PubMed] [Google Scholar]
- 27.Rosenzweig M, Marks DF, Zhu H, Hempel D, Mansfield KG, Sehgal PK, Kalams S, Scadden DT, Johnson RP. In vitro T lymphopoiesis of human and rhesus CD34+progenitor cells. Blood. 1996;87:4040–4048. [PubMed] [Google Scholar]
- 28.Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 29.Jenkinson EJ, Anderson G. Fetal thymic organ cultures. Curr Opin Immunol. 1994;6:293–297. doi: 10.1016/0952-7915(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 30.Croisille L, Auffray I, Katz A, Izac B, Vainchenker W, Coulombel L. Hydrocortisone differentially affects the ability of murine stromal cells and human marrow-derived adherent cells to promote the differentiation of CD34++/CD38−long-term culture-initiating cells. Blood. 1994;84:4116–4124. [PubMed] [Google Scholar]
- 31.Carayol G, Robin C, Bourhis J-H, Chouaib S, Coulombel L, Caignard A. NK cells differentiated from bone marrow, cord blood and peripheral blood stem cells exhibit similar phenotype and functions. Eur J Immunol. 1998;28:1991–2002. doi: 10.1002/(SICI)1521-4141(199806)28:06<1991::AID-IMMU1991>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 32.Petzer AL, Zandstra PW, Piret JM, Eaves CJ. Differential cytokine effects on primitive (CD34+CD38−) human hematopoietic cells: novel responses to Flt3-ligand and thrombopoietin. J Exp Med. 1996;183:2551–2558. doi: 10.1084/jem.183.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akashi K, Kondo M, von Freeden-Jeffry U, Murray R, Weissman IL. Bcl-2 rescues T lymphopoiesis in Interleukin-7 receptor-deficient mice. Cell. 1997;89:1033–1041. doi: 10.1016/s0092-8674(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 34.Itoh K, Tezuka H, Sakoda H, Konno M, Nagata K, Uchiyama T, Uchino H, Mori KJ. Reproducible establishment of hemopoietic supportive stromal cell lines from murine bone marrow. Exp Hematol. 1989;17:145–153. [PubMed] [Google Scholar]
- 35.Hao QL, Smogorzewska EM, Barsky LW, Crooks GM. In vitro identification of single CD34+CD38−cells with both lymphoid and myeloid potential. Blood. 1998;91:4145–4151. [PubMed] [Google Scholar]
- 36.Fluckiger A, Sanz E, Garcia-Lioret M, Su T, Hao QL, Kato R, Quan S, de la Hera A, Crooks G, Witte ON, et al. In vitro reconstitution of human B-cell ontogeny: from CD34+multipotent progenitors to Ig-secreting cells. Blood. 1998;92:4509–4520. [PubMed] [Google Scholar]
- 37.García-Ojeda ME, Dejbakhsh-Jones S, Weissman IL, Strober S. An alternate pathway for T cell development supported by the bone marrow microenvironment: recapitulation of thymic maturation. J Exp Med. 1998;187:1813–1823. doi: 10.1084/jem.187.11.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner JP, Rosenzweig M, Marks DF, Harper D, Gaynor K, Fallon RJ, Wall DA, Johnson RP, Scadden DT. T-lymphopoietic capacity of cord blood- derived CD34+progenitor cells. Exp Hematol. 1998;56:991–999. [PubMed] [Google Scholar]
- 39.Bennaceur-Griscelli, A.L., C. Tourino, B. Izac, W. Vainchenker, and L. Coulombel. 1999. Murine stromal cells counteract the loss of LTC-IC potential induced by cytokines in CD34+CD38low/neg human bone marrow cells. Blood. In press. [PubMed]
- 40.Cumano A, Dieterlen-Lievre F, Godin I. Lymphoid potential, probed before circulation in mouse, is restricted to caudal intraembryonic splanchnopleura. Cell. 1996;86:907–916. doi: 10.1016/s0092-8674(00)80166-x. [DOI] [PubMed] [Google Scholar]
- 41.Snodgrass R, Keller G. Clonal fluctuation within the haematopoietic system of mice reconstituted with retrovirus-infected stem cells. EMBO (Eur Mol Biol Organ) J. 1987;6:3955–3960. doi: 10.1002/j.1460-2075.1987.tb02737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pawliuk R, Eaves C, Humphries RK. Evidence of both ontogeny and transplant dose-regulated expansion of hematopoietic stem cells in vivo. Blood. 1996;88:2852–2858. [PubMed] [Google Scholar]
- 43.Marandin A, Dubart A, Pflumio F, Cosset FL, Cordette V, Chapel-Fernandes S, Coulombel L, Vainchenker W, Louache F. Retrovirus-mediated gene transfer into human CD34+/CD38lowprimitive cells capable of reconstituting long-term cultures in vitro and non obese diabetic severe combined immunodeficient mice in vivo. Hum Gene Ther. 1998;9:1497–1511. doi: 10.1089/hum.1998.9.10-1497. [DOI] [PubMed] [Google Scholar]
- 44.Conneally E, Eaves CJ, Humphries RK. Efficient retroviral-mediated gene transfer to human cord blood stem cells with in vivo repopulating potential. Blood. 1998;91:3487–3493. [PubMed] [Google Scholar]