A Kinetic Threshold between Negative and Positive Selection Based on the Longevity of the T Cell Receptor–Ligand Complex (original) (raw)
Abstract
We have developed a unique in vivo system to determine the relationship between endogenous altered peptide ligands and the development of major histocompatibility complex class II– restricted T cells. Our studies use the 3.L2 T cell receptor (TCR) transgenic mouse, in which T cells are specific for Hb(64–76)/I-Ek and positively selected on I-Ek plus self-peptides. To this endogenous peptide repertoire, we have individually added one of six well-characterized 3.L2 ligands. This transgenic approach expands rather than constrains the repertoire of self-peptides. We find that a broad range of ligands produce negative selection of thymocytes in vivo. When compared with the in vitro TCR–ligand binding kinetics, we find that these negatively selecting ligands all have a half-life of 2 s or greater. Additionally, one of two ligands examined with no detectable binding to the 3.L2 TCR and no activity on mature 3.L2 T cells (Q72) enhances the positive selection of transgenic thymocytes in vivo. Together, these data establish a kinetic threshold between negative and positive selection based on the longevity of TCR–ligand complexes.
Keywords: T cell receptor, thymocyte, transgenic, selection, altered peptide ligand
Tcell development in the thymus depends upon the interaction between the α/β TCR and self-peptides bound to MHC molecules expressed on thymic APCs and on thymic stromal cells (1–6). This recognition is flexible, in that any given TCR has the capacity to interact at some level with a broad spectrum of related MHC/peptide ligands (7). For mature T cells, the functional correlates of these interactions are well described and include full activation, partial activation, and antagonism (8–11). For developing thymocytes, productive engagement of the TCR results either in continued maturation (positive selection) or in activation-induced cell death (negative selection). This apparent paradox, where TCR recognition of a single set of MHC ligands results in two very different functional outcomes, has led to the proposal that the fate of each developing T cell depends on the strength and timing of TCR–MHC interaction (12–15). In this hypothesis, “weak” interactions promote positive selection, while “strong” interactions lead to thymocyte activation and cell death. No specific interaction with ligand results in death by neglect (16).
T cell signaling is a complex series of events which begins with localization of the TCR to areas of contact between T cell and APC. This localization or clustering results in signal transduction, which begins by phosphorylation of the CD3 ζ chain (17). These events have been studied extensively in mature T cells, where evidence suggests that the potency of each ligand is determined by the half-life of the TCR–MHC complex (18–22). In this kinetic model of T cell activation, weak interactions are those with a short half-life. The short half-life limits formation of the contact cap, reduces T cell clustering, and produces a pattern of ζ phosphorylation distinct from that seen with more potent ligands. Conversely, a long half-life of the TCR–MHC complex allows for complete ζ chain phosphorylation, Zap-70 recruitment to the signaling complex, a rise in intracellular calcium, and T cell activation as measured by IL-2 production (23, 24).
When thymocyte development is used to measure ligand strength, the data is open to interpretation. Although many studies are at least consistent with a kinetic model of T cell selection (14, 25–28), some have suggested that integration of TCR signaling is based on TCR occupancy (12, 29–32). In an occupancy model, weak ligands have low equilibrium binding affinities (30). Ligands with low equilibrium binding affinity but at high concentration could occupy sufficient TCR for T cell signaling, resulting in positive selection (32). High-affinity ligands at low concentration could have the same effect (29). Thus, for each developing thymocyte there is predicted to be an occupancy threshold between positive and negative selection, which is determined by the product of ligand concentration and ligand equilibrium binding affinity (30, 33). One problem with this view is that T cells selected on low concentrations of a high-affinity ligand are not functional (27). Also, ligands with low equilibrium binding affinities often have fast off-rates, thereby blurring the distinction between kinetic and occupancy models (32). A clear understanding of how kinetic factors influence thymic selection and which factors are important has not been achieved.
These observations and others (34–40) support the conclusion that positive selection of both class I– and class II– restricted T cells is peptide specific. They suggest that certain kinetic parameters of the interaction between a TCR and its peptide/MHC ligand regulate the activation of mature T cells and determine the fate of developing thymocytes. For MHC class I–restricted T cells, this analysis has been facilitated through the use of fetal thymic organ culture (FTOC) by allowing investigators to examine a large number of ligands (12, 14, 25, 28). For class II–restricted T cells, the precise contribution of peptides to thymic selection has been difficult to dissect (41, 42). No comparable FTOC system exists for studying selection of MHC class II–restricted T cells. Instead, investigators have focused on experiments using a single peptide ligand attached to an MHC molecule, expressed at high concentration (32, 43–46). How either of these systems relates to in vivo biology has not been firmly established, and any strict correlation between the biological activity (binding kinetics) of the selecting ligand and the ligand's effect on T cell development remains to be determined.
In this report, we present results obtained with a novel system designed to determine how self-peptides influence the selection of class II–restricted T cells in vivo. We describe the functional consequences for a single antigen-specific T cell when an extended spectrum of well-characterized altered peptide ligands (APLs)1 of hemoglobin peptide Hb(64–76) are individually expressed in the thymi of TCR transgenic mice at physiologic levels. Thus, our approach differs significantly from other studies of class II–restricted T cells in that we have expanded rather than constrained the repertoire of self-peptides. Results demonstrate that a surprisingly broad range of ligands produce negative selection of transgenic T cells. Importantly, we have identified a single ligand, one with no effect on mature T cells, that enhances positive selection of thymocytes with the transgenic receptor. When viewed in the context of the equilibrium binding kinetics (47) and the activity of these ligands on mature T cells (48), these data define a kinetic threshold for negative selection of 3.L2tg T cells where the outcome is determined by the half-life of the TCR–ligand complex. They suggest that positive selection of MHC class II–restricted T cells involves specific interactions with ligands which have short half-lives. Taken together, our studies are most consistent with a kinetic model of thymic selection.
Materials and Methods
Transgenic Mice.
Lines of transgenic mice were generated that express a membrane form of hen egg-white lysozyme (mHEL) containing Hb(64–76) or one of five different APLs of Hb(64– 76) as an epitope tag. The self-peptide Hb(64–76) is derived from the minor d allele of the β chain of mouse Hb. When presented by I-Ek, this epitope is recognized by the 3.L2 T cell hybridoma and clone (49). Many of the APLs recognized by 3.L2 T cells involve substitutions at the 72 position of the Hb(64–76) peptide, the TCR contact residue in the middle of the MHC/peptide complex (P5 position) as determined by crystallography (50). The specific ligands and APLs studied in this report are Hb(64–76), Hb(64–76) N72T, Hb(64–76) N72I, Hb(64–76) N72A, Hb(64– 76) N72Q, and Hb(64–76) N72E. Henceforth these ligands are called N72(wt), T72, I72, A72, Q72, and E72.
The epitope tags were placed between amino acids 43 and 44 of mHEL, and the transgenic mice were generated as described previously (51). Expression of each transgenic construct was controlled by the MHC Eα promoter, limiting expression to all class II–positive cells (43, 52). Founders were obtained, characterized, and bred to the 3.L2tg mouse, which is specific for Hb(64–76)/I-Ek (53), and the 3A9 TCR transgenic mouse, which is specific for HEL(46–61)/I-Ak (54). Progeny were screened by PCR analysis of purified tail digest DNA, and all mice used in experiments were heterozygotes for TCR and mHEL/APL transgenes.
Peptides.
The peptides used in this study were synthesized, purified, and analyzed as described previously (48). The peptide sequences, in single letter amino acid code, are: GKKVITAFNEGLK [N72(wt)]; GKKVITAFQEGLK (Q72); and NTDGSTDYGILQINSR [HEL(46–61)].
T Cell Hybridoma Assays.
T cell hybridomas were used to detect the presence of stimulatory ligands on splenocytes from transgenic mice as described (51). In brief, increasing numbers of splenocytes were incubated with 105 hybridoma cells for 24 h. Stimulation of each hybridoma was then determined by measuring the level of IL-2 produced using the IL-2–dependent cell line CTLL-2 as described (55). The hybridomas used in these assays were 3.L2.12 (56), 3A9 (57), QC6.2, and QC85.5. The latter two hybridomas were generated by immunizing CE/J mice with the Q72 peptide, and each hybridoma was subcloned twice before use in this study. The QC6.2 hybridoma is specific for Q72, whereas the QC85.5 hybridoma is stimulated by Q72, A72, T72, and N72(wt).
Primary T Cell Proliferation.
Primary T cells were stimulated with Hb(64–76) peptide as described (58). In brief, 5 × 105 splenocytes/well were incubated with increasing amounts of peptide for 48 h, pulsed with 0.4 μCi of [3H]TdR for 18 h, and harvested. Proliferation was measured as cpm incorporated (mean of triplicate wells). Results from several experiments were then averaged to obtain the data shown in Fig. 4.
Figure 4.

The number of CAB+ cells in the periphery of APLtg × 3.L2tg mice reflects the effect of the APL on thymic selection. Three-color FACS® analysis was performed on splenocytes from APLtg × 3.L2tg mice and 3.L2tg littermate controls. Splenocytes were stained for CD4 (PE), CD8 (FITC), and the 3.L2 clonotypic receptor (CAB-biotin plus Tricolor-streptavidin), and 105 live cell events were collected per sample. Results were first examined as dot plots of CD4 versus CD8 (not shown), and the CD4+ region was selected for further analysis. The level of 3.L2 clonotypic TCR expression was determined in this population of cells, and the results were plotted as histograms of cell number versus log10 fluorescence. Representative histograms are shown for each APLtg × 3.L2tg (solid lines) and for 3.L2tg littermate controls (dotted lines).
Antibodies.
The antibodies used were PE-conjugated anti– mouse CD4 (PharMingen); FITC-conjugated anti–mouse CD8a (PharMingen); biotinylated 3.L2 clonotypic antibody (53); biotinylated 3A9 clonotypic antibody (51); biotinylated F10.6.6 (59); and biotinylated 14-4-4S (60). Cells stained with biotinylated antibodies were subsequently incubated with Tricolor-streptavidin (Caltag) or PE-streptavidin (Caltag).
Flow Cytometry.
Single-cell suspensions of thymocytes or splenocytes were stained in FACS buffer (PBS supplemented with 0.5% BSA and 0.1% sodium azide) using the following protocol. Aliquots of cells (106/sample in 100 μl FACS buffer) were placed in polypropylene culture tubes (12 × 75 mm; VWR) and incubated on ice for 1 h with the biotinylated or directly labeled antibodies. Cells were then washed once with 3 ml of FACS buffer and incubated for 30 min on ice with the streptavidin-fluorochrome conjugate where appropriate. Cells were washed again, fixed for 18–24 h in FACS buffer plus 1% paraformaldehyde, and analyzed on a FACScan® (Becton Dickinson) flow cytofluorometer using CELLQuest® (Becton Dickinson) software. Samples were gated on live cells, and 105 live cell events per sample were collected.
Single-label Immunocytochemistry.
Thymi from 8–16-wk-old transgenic mice and nontransgenic littermates were excised, embedded in OCT compound (Tissue-Tek), and frozen in liquid nitrogen. Thymic tissue was then cut into sagittal sections (4 μm thick), mounted on glass slides, and fixed in acetone for 10 min at room temperature (RT). Sections were stored at −70°C. Before immunolabeling, fixed tissue sections were allowed to reach RT and were washed three times in PBS. To block binding of antibody reagents to endogenous Fc receptors, either supernatant from the 2.4G2 hybridoma was applied (neat) for 15 min at RT or purified anti–mouse CD16/CD32 (PharMingen) diluted 1:200 (2.5 μg/ml) in PBS blocking buffer (PBS, pH 7.4, with 0.1% BSA, 5% normal mouse serum) was applied for 15 min at RT. Tissues were then washed three times in PBS. To block nonspecific binding of secondary streptavidin reagents to endogenous biotin, sections were incubated in avidin D and biotin blocking solutions (Vector Labs), respectively, for 15 min. Tissues were washed three times in PBS before and after each blocking application.
Biotin-conjugated primary antibodies were diluted in PBS blocking buffer and applied to tissues for overnight incubation in a humidified chamber at 4°C. To detect mHEL/APL transgene expression, biotinylated F10.6.6 was diluted 1:25 (33 μg/ml) and applied as above. For detection of MHC class II expression, biotinylated 14-4-4S was diluted 1:50 (10 μg/ml) and applied as above. Tissues were then washed three times in PBS. Cy3-conjugated streptavidin (Jackson ImmunoResearch Laboratories) was diluted in PBS blocking buffer 1:1,000 (1 μg/ml) and applied for 30 min at RT. After tissues were washed three times in PBS, coverslips were mounted to slides in PBS/glycerol (1:1). To control for irrelevant binding of F10.6.6, thymi from nontransgenic littermates were stained according to the above protocol. To control for nonspecific binding of Cy3-conjugated streptavidin, anti-TNP IgG1 (PharMingen) and anti-TNP IgG2 (PharMingen) were diluted 1:25 in PBS blocking buffer and applied as primary antibodies. Immunofluorescence was visualized on a Zeiss Axiophot microscope. Electronic images were captured by a Spot camera (Diagnostic Images, Inc.) and processed using Northern Eclipse version 2.0 software.
Results
APCs Express the Chimeric mHEL/APL Transgenes at Equivalent Levels.
These experiments are designed to study the functional consequences for T cells when endogenous APLs were expressed in the thymus and in the periphery. The system is based on the self-antigen Hb, and uses the 3.L2 TCR transgenic mouse (3.L2tg), which is specific for Hb(64–76)/I-Ek (53). The 3.L2tg mouse is ideally suited for these studies, as positive selection of the transgenic receptor is moderate in H2k mice, making it possible to observe enhanced positive as well as negative selection. To the 3.L2tg mouse, we have genetically introduced APLs of Hb(64–76). These APLs differ by as much as 150,000-fold in their relative activity towards mature 3.L2 T cells (48), and the relative activity correlates with the half-life of the TCR–ligand complex (47; Table I). Ligand levels in the APLtg mice are clearly physiologic, and peptide presentation is achieved through normal antigen processing pathways. Thus our approach should be distinguished from experiments using mice which express a single peptide covalently attached to the class II β chain (32, 43). In this latter approach, ligand levels are much higher than those achieved under normal circumstances, and the covalent linker may potentially influence binding of the peptide to the class II molecule.
Table I.
A Comparison of 3.L2 Ligands
| Ligand | Relative activity* | Half-life‡ | K D equilibrium§ | K D k off/k on |
|---|---|---|---|---|
| s | μM | μM | ||
| N72(wt) | 100 | 10.8 | 52 | 12.0 |
| T72 | 2 | 5.1 | 54 | 9.9 |
| I72 | 0.006 | 2.3–3.4 | 62.5 | 10–20 |
| A72 | 0.0007 | <2 | 170 | — |
| Q72 | none | — | — | — |
| E72 | none | — | — | — |
Our strategy for introducing these ligands into the 3.L2 TCR transgenic mouse was recently published for mice expressing the antagonist A72 (51). In brief, the APLs are introduced as an epitope tag contained within a carrier protein in transgenic mice. This carrier protein is a membrane form of HEL whose expression is controlled by an MHC Eα promoter. The Eα promoter was derived from the established expression vector pDOI-5, which has been shown to efficiently target expression of transgenes to all class II– positive cells in the thymus and the spleen (43, 52). As integral membrane proteins, all transgenes are also targeted to the class II antigen processing and presentation pathway (61, 62). The APLtg mice are then bred to the 3.L2tg mice, and the progeny are analyzed. Thus, the net effect of this approach is to add one specific ligand for 3.L2 T cells to the pool of endogenous ligands already present in the B6.AKR background. The specific Hb(64–76)–related ligands described in this report are N72(wt), the natural epitope with an Asn at position 72; T72, a weak agonist; I72, an antagonist; Q72, a “null” ligand with no effect on mature T cells; and E72, also a null ligand. Note that we also include new data from the A72tg mouse, as it is instructive to consider these data in the context of the spectrum of APLs described in this report.
Thymic expression of the mHEL/APL proteins and I-Ek from each transgenic line was assessed by immunofluorescence of frozen sections using mAbs. Results shown in Fig. 1, A–F, demonstrate that each line expresses the mHEL/APL protein in both the thymic cortex and medulla. Furthermore, the pattern of expression observed is identical to that seen for MHC class II (Fig. 1, G and H). Peripheral expression of the transgenes was determined by two-color FACS® analysis of splenocytes. The results shown in Fig. 2 demonstrate that each line expresses the mHEL/APL protein in all MHC class II–positive cells. This population represents ∼60% of splenocytes and is largely B220+ (not shown). Importantly, none of the transgenes appears to influence MHC class II expression in the thymus or in the periphery, as the data demonstrate that class II expression in these six transgenic lines is identical (Fig. 2, and data not shown). Within a given line, identical levels of mHEL/ APL expression are seen in splenocytes from 3–4-wk-old mice and in the 6–12-wk-old mice used in this study (data not shown). This implies that any potential differences in the timing of embryonic transgene expression could not influence T cell development in mature mice. However, when comparing one line to another, slight variability in the level of transgene expression was noted. Using both biotinylated and directly labeled bivalent F10.6.6, we estimate that at most there is a three- to fivefold difference in cell surface expression of the various chimeric proteins, with N72 the highest, Q72 the lowest, and ranked as follows: N72(wt) > T72 > E72 > I72 > A72 > Q72 (Figs. 1 and 2, and data not shown). These data demonstrate expression of the mHEL/APL proteins at similar levels and in all MHC class II–positive cells in both the thymus and the periphery.
Figure 1.
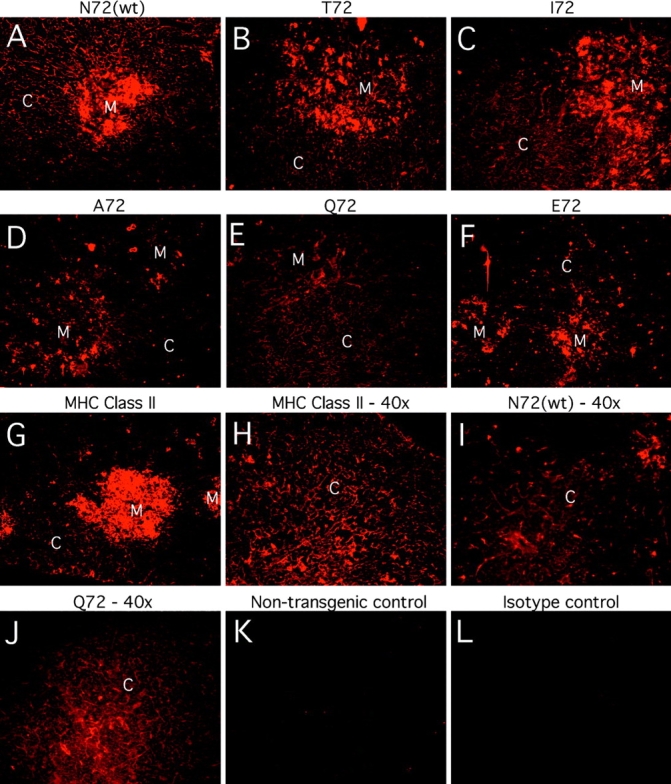
All mHEL/APL transgenes are expressed in both the thymic cortex and thymic medulla. Frozen thymic sections were stained for either an HEL epitope common to all transgenes (A–F, I, and J) or I-Ek (G and H) as described in Materials and Methods. The original magnification of all photographs is 20× unless otherwise specified. The cortex (C) and medulla (M) of each section are labeled. Note that mHEL/APL expression in the thymus did not influence MHC class II expression (shown only for the E72 and Q72 lines in G and H, respectively). Also, the relative intensity of fluorescence detected with the anti-HEL antibody matched that seen by FACS® analysis of splenocytes using the same primary reagent [N72(wt) > T72 > E72 > I72 > A72 > Q72]. A–F, K, and L are from a representative experiment in which all photographs shown were taken on the same camera settings (14-s exposure, gain of 4; original magnification: 20×). G–J were from another experiment: G is the E72 thymus stained for I-Ek (6-s exposure, gain of 4; original magnification: 20×); H is the Q72 thymic cortex stained for I-Ek (8-s exposure, gain of 2; original magnification: 40×); I is the N72(wt) thymic cortex stained for mHEL/APL expression (12-s exposure, gain of 8; original magnification: 40×); J is the Q72 thymic cortex stained for mHEL/ APL expression (14-s exposure, gain of 4; original magnification: 40×). The mHEL/APL-specific antibody does not bind nontransgenic thymus (K), and there is no nonspecific binding of the streptavidin-Cy3 reagent (L). This entire experiment was repeated four times with similar results.
Figure 2.

The mHEL/APL transgenes are expressed in all MHC class II– positive cells. Splenocytes from each transgenic line were stained for HEL epitopes common to all transgenes and I-Ek using mAbs. Dot plots from two-color FACS® analysis of stained cells from one of five similar experiments are shown (log10 fluorescence). All experiments demonstrate expression of the transgenes in MHC class II–positive splenocytes at comparable levels. Transgene expression using the Eα promoter does not influence endogenous class II expression.
The Effect of Most APLs on Developing Thymocytes Correlates with their Effect on Mature T Cells.
We tested the effect of each endogenous APL on 3.L2 T cell development by crossing 3.L2tg mice with APLtg mice, and then performing three-color FACS® analysis on thymocytes. All mice examined were heterozygotes for the TCR and/or mHEL/APL transgenes, approximately equally distributed between male and female, and ranged from 6 to 12 wk of age. Where possible, 3.L2tg-only littermates were used as positive controls; otherwise, age-matched 3.L2tg heterozygotes were selected. For each cross, we examined at least 8 mice, and in the case of Q72tg × 3.L2tg we examined 16 mice (Table II). CD4 single-positive (SP) cells were analyzed for their expression of the 3.L2 clonotypic receptor. For each experiment, the location of the quadrants, the CD4-SP region, and the marker delineating CD4-SP, 3.L2 clonotypehi antibody (CAB)hi population of cells was set using the 3.L2tg control. These same markers or gates were used to analyze the experimental population. The data are summarized in Table II. Note that the 3.L2tg phenotype is based on analysis of 36 thymi and 26 spleens.
Table II.
Summary of FACS® Analysis Data from mHEL/APLtg × 3.L2tg Mice
| Mouse | Thymus | Spleen | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Age | % CD4-SP | % DP | % DN | % CD8-SP | CD4+ gate: % CABhi | Size | % CD4+ | % CD8+ | CD4+ gate: % CABhi | Size | |
| wk | × 106 | × 106 | ||||||||||
| 3.L2tg | 36 | 8.5 | 10.2 | 70.7 | 13.9 | 3.8 | 58.6 ± 1.5 * | 108 ± 9* | 9.4 | 4.4 | 50.0 ± 2.4* | 159 ± 13* |
| N72tg × 3.L2tg | 8 | 10.4 | 6.2 | 62.4 | 24.8 | 6.7 | 0.4 ± 0.1 | 69 ± 6 | 5.0 | 4.4 | 2.7 ± 1.7 | 224 ± 15 |
| T72tg × 3.L2tg | 8 | 10.5 | 7.9 | 44.1 | 41.9 | 6.1 | 4.2 ± 0.6 | 42 ± 12 | 4.6 | 2.1 | 6.7 ± 1.8 | 130 ± 19 |
| I72tg × 3.L2tg | 10 | 8.4 | 6.9 | 57.2 | 27.4 | 8.5 | 1.8 ± 0.6 | 49 ± 6 | 5.5 | 3.8 | 3.0 ± 0.8 | 176 ± 32 |
| A72tg × 3.L2tg | 8 | 12.0 | 10.3 | 74.5 | 11.8 | 3.4 | 37.5 ± 5.3 | 123 ± 12 | 11.8 | 6.2 | 37.0 ± 4.2 | 148 ± 24 |
| Q72tg × 3.L2tg | 16 | 12.1 | 9.9 | 70.4 | 12.9 | 3.8 | 70.4 ± 2.3 | 81 ± 11 | 11.1 | 4.8 | 52.9 ± 2.4 | 131 ± 14 |
| E72tg × 3.L2tg | 9 | 7.0 | 9.7 | 74.7 | 12.8 | 2.6 | 57.8 ± 1.2 | 86 ± 6 | 8.0 | 3.4 | 44.5 ± 2.3 | 147 ± 28 |
| mHELtg × 3.L2tg | 9 | 11.4 | 9.7 | 71.1 | 16.4 | 2.8 | 59.3 ± 3.5 | 60 ± 6 | 8.6 | 4.8 | 45.3 ± 4.0 | 218 ± 16 |
For each cross, examples of CD4 versus CD8 dot plots and histograms showing CAB expression in the CD4-SP population are shown in Fig. 3. In the histograms, the 3.L2 control is shown as a dotted line, and the experimental condition as a heavy solid black line. The ligands are listed in order of their effect on mature T cells (48): N72(wt), the strongest agonist; T72, a weaker agonist; I72, an antagonist; A72, a weaker antagonist; Q72, a ligand with no effect on mature 3.L2 T cells; and E72, a ligand with no effect on mature T cells and with a negatively charged residue replacing the normal asparagine.
Figure 3.
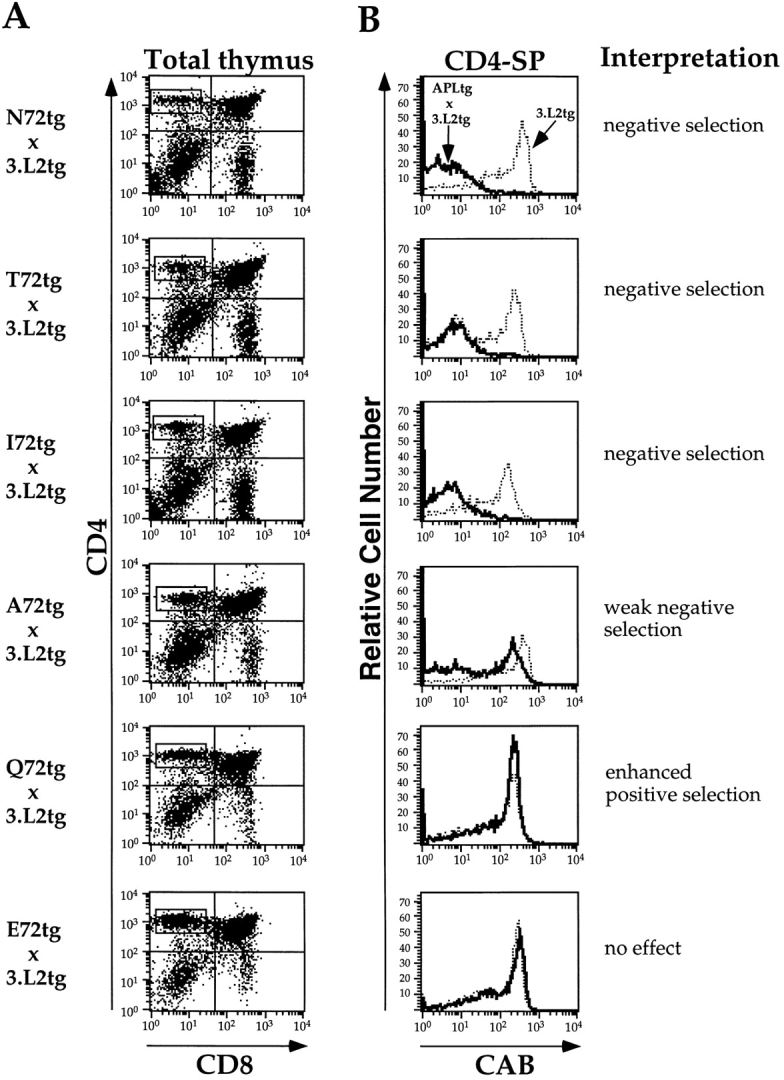
The effects of APLs on the thymic selection correlate with the relative activity of these ligands on mature T cells. (A) Three-color FACS® analysis of thymocytes derived from APLtg × 3.L2tg F1 mice was performed, with 105 live cell events collected per sample. The results were first examined as dot plots of CD4 (PE) versus CD8 (FITC), and the CD4-SP region was selected for further analysis. Representative thymi are depicted (log10 fluorescence), with the results from all mice summarized in Table II. (B) The CD4-SP gates in A were analyzed for expression of the 3.L2 clonotypic receptor (CAB-biotin plus Tricolor-streptavidin). The results are shown as histograms of cell number versus log10 fluorescence for both APLtg × 3.L2tg mice (solid lines) and 3.L2tg littermate controls (dotted lines). The ligands are listed in terms of their relative activity on mature T cells from strongest (top) to weakest (bottom) (reference 48). The interpretation, based on the cumulative observed effects for each APL, is listed beside the histogram.
As expected, we find that expressing the 3.L2 TCR agonist N72(wt) in the thymus of 3.L2tg mice results in complete negative selection of all thymocytes with the transgenic receptor (Fig. 3). Overall, the thymus size is about half that seen in 3.L2 controls, there are fewer CD4+CD8+ double-positive (DP) and CD4-SP thymocytes, and increased numbers of double-negative (DN) cells (Table II). There are no CD4-SP cells with the clonotypic receptor. The weaker agonist T72, a ligand that is 50-fold less active (48), has an identical effect when expressed in the thymus of 3.L2tg mice. Most surprisingly, thymic expression of the antagonist I72 also results in complete negative selection of specific T cells. The weaker antagonist A72 produces only modest negative selection, as described previously (51). We conclude that, for the 3.L2 TCR, the range of negatively selecting ligands in vivo is broad, and includes ligands that function as agonists and as antagonists of mature T cells.
The Ligand Q72 Has No Effect on Mature T Cells but Enhances Positive Selection.
Of significant interest, adding the ligand Q72 to the pool of endogenous ligands already present on the B6.AKR background results in an increase in the percentage of CD4-SP cells with high levels of the 3.L2 TCR over that seen in 3.L2tg control mice (Table II). This difference is 12% and is statistically significant, with P < 0.01. Also note that the percentage of thymocytes in the CD4-SP quadrants of these two groups is the same, and that the overall size of thymi in the two groups is also approximately the same. Thus, this increase in the proportion of CD4-SP, CABhi cells is likely to represent an increase in the absolute number of cells with the 3.L2 TCR. When Q72 is replaced by E72, this effect is not observed and 3.L2 × E72 mice are identical to 3.L2 mice, both in terms of the number of CD4-SP thymocytes and the percentage of CD4-SP thymocytes with high levels of the clonotypic receptor. It is important to note that the Q for N substitution at position 72 conserves charge while lengthening the amino acid side chain by one methyl group. This is in contrast to the E for N substitution, which both lengthens the side chain and replaces the polar amide with a negatively charged carboxyl group. Taken together, these data suggest that ligands capable of positively selecting the 3.L2 TCR are weak ligands like Q72, with no activity on mature T cells but capable of forming specific interactions.
To further control for the effect of expressing a transgenic membrane protein in all APCs, we also crossed 3.L2tg mice to a line of mice which express mHEL only, that is, they lack the APL epitope tag but use the same Eα promoter. We find that mice expressing the 3.L2 TCR and mHEL are identical to 3.L2 mice (Table II). For example, in these mice there are 9.7% CD4-SP thymocytes. Of these CD4-SP cells, 59.3% are CABhi. This experiment provides evidence that the effects we have observed are specific for the epitope tags, and not a result of the HEL components of the transgenes interfering with or altering processing and presentation of endogenous epitopes.
The Peripheral Phenotype Is Generally Predicted by the Number of CD4-SP Thymocytes with High Levels of the Clonotypic Receptor.
Splenocytes from the mice analyzed above were also examined by three-color FACS®. The data demonstrate that when considering naive T cells, the peripheral phenotype is largely determined by selection events occurring in the thymus. For example, the percentage of CD4+ splenocytes in N72tg × 3.L2tg mice is roughly one half that seen in 3.L2 controls (Table II). This decrease represents the loss of all cells with high levels of 3.L2 TCR (Fig. 4, top left). This same phenotype is also seen in T72tg × 3.L2tg and I72 × 3.L2tg mice. In A72tg × 3.L2tg mice, a reduced number of CD4+CAB+ cells is seen. In the case of Q72tg × 3.L2tg mice, a small increase in the percentage of CD4+ CAB+ splenocytes is seen compared with 3.L2 controls. This increase is not statistically significant. In E72tg × 3.L2tg mice, a slight reduction in the percentage of CD4+CAB+ splenocytes is seen. Finally, there is no statistically significant difference between mHEL-only × 3.L2tg mice and 3.L2tg controls (Table II). Again, this implies that epitopes from the mHEL portion of the constructs have not influenced the results.
Interpretation of the data in negatively selecting mice is relatively straightforward. Nearly all cells with the 3.L2 clonotypic receptor are eliminated in the thymus, and no cells with high levels of this receptor are found in the spleen. For the weaker ligands, interpretation of the data is more complicated. There are many determinants, other than genetic factors, that influence the size of the spleen and the relative size of the CD4+ and CD8+ compartments. In previous studies with the A72tg × 3.L2tg mice, we found that CD4+CAB+ cells in the periphery were naive. Yet antigen-induced expansion of any particular population of splenocytes could clearly influence the relative numbers of those cells. In this set of experiments, we did not routinely examine the CD4+CAB+ or the CD4+ CAB− populations of cells for activation markers, making the potential contribution of such an event difficult to determine. Also, we have not examined what influence, if any, these ligands have on the survival of 3.L2tg T cells. These caveats notwithstanding, the peripheral phenotypes are generally consistent with those observed in the thymus, in terms of the size of the CD4+ compartment and the percentage of CD4+CAB+ cells.
The Proliferative Response of Naive Splenocytes Reflects the Selecting Background.
We wished to determine the functional state of naive T cells selected on the various APL backgrounds. For these experiments, we added increasing amounts of Hb(64–76) peptide to splenocytes as described (51). Each curve represents the average cpm incorporated at each antigen dose for all experiments performed. As shown in Fig. 5 (top), splenocytes from the negatively selecting backgrounds are tolerant. They proliferate only weakly to high concentrations of Hb(64–76) peptide. For N72 × 3.L2 mice, a response is first measurable at 1.0 μM, compared with 0.003 μM for 3.L2 controls. Overall, these data demonstrate a 500-fold shift in the dose–response curve. T72tg × 3.L2tg and I72tg × 3.L2tg mice are also tolerant, with 500- and 100-fold shifts in their response curves, respectively.
Figure 5.

The proliferative responses of naive splenocytes from APLtg × 3.L2tg mice are consistent with the observed effects of these ligands on T cell development. They also demonstrate that T cells selected on the Q72 background are functional. The proliferative responses were measured by tritiated thymidine incorporation. Splenocytes (5 × 105/well) were added along with the indicated concentrations of Hb(64– 76) peptide. Cells were incubated for 48 h, pulsed with tritiated thymidine for 18 h, harvested, and counted. Within a given experiment, triplicate wells were performed at each peptide concentration, and the mean cpm was determined. Each experiment was repeated several times. Symbols are as follows: •, N72tg (n = 5); ▪, T72tg (n = 9); ▴, I72tg (n = 11); ⋄, A72tg (n = 8); ▿, Q72tg (n = 16); ♦, 3.L2tg (n = 20); and □, B6.AKR (n = 5). The data shown represent the average of these experiments, with the error calculated as SEM. The 3.L2tg and B6.AKR controls are shown in each panel for comparison.
The response of A72tg × 3.L2tg splenocytes to Hb(64–76) peptide was recently published (51). In that report, we found that the presence of the endogenous antagonist A72 inhibited the proliferative response of 3.L2tg T cells. Using an adoptive transfer approach, we have also observed peripheral antagonism with A72 in vivo (63). The reduced response shown in Fig. 5 (bottom left) reflects both this peripheral antagonism and the reduced numbers of CD4+ CAB+ cells present when all experiments are averaged. The response of Q72tg × 3.L2tg T cells is identical to 3.L2 controls at low and intermediate antigen doses. At doses above 1.0 μM, the Q72tg × 3.L2tg response is greater than that seen in 3.L2tg controls, reflecting greater numbers of specific T cells proliferating. The response observed in E72tg × 3.L2tg mice is identical to 3.L2 controls, within the error of the experiments.
We conclude from the above that 3.L2tg mice selected on the endogenous ligands N72, T72, and I72 are tolerant to Hb(64–76). A72 produces a degree of tolerance by peripheral antagonism as described (51). Expression of Q72 in the thymus and spleen of 3.L2tg mice results in functional T cells and a proliferative response slightly greater than control mice. Finally, E72 has no effect on the response of naive 3.L2 T cells to Hb(64–76) peptide.
APCs in the Spleen and Thymus Process and Present Both Hb-related APL and HEL Epitopes from the Chimeric Proteins.
It was possible that differential processing and presentation of the Hb-related APL epitopes in each founder line could influence our interpretation by allowing us to either underestimate or overestimate the effect of any particular ligand. Many of the ligands examined are not agonists for 3.L2tg T cells, and there is no single assay available to detect presentation of these APL epitopes. Instead, we have used a combination of T cell hybridomas and TCR transgenic mice to provide convincing evidence that all chimeric proteins are equivalently processed and presented in both the thymus and the spleen of APLtg mice.
Processing and presentation of the relevant epitopes in each chimeric protein were demonstrated by hybridoma stimulation assays using splenocytes as APCs. The 3.L2.12 hybridoma is specific for Hb(64–76), and is weakly stimulated (50-fold reduction) by T72 (48). As expected, as few as 3 × 103 N72tg splenocytes stimulate the 3.L2.12 hybridoma (Fig. 6, top left). At higher numbers of splenocytes per well (105–106), T72tg splenocytes will also stimulate the 3.L2.12 hybridoma, although this stimulation is reduced 35–50-fold compared with that achieved with N72tg splenocytes and is not visible in the scale of the graph. In a similar fashion, the hybridoma QC85.5 is stimulated most strongly by the Q72 peptide, but will also produce IL-2 when stimulated by N72, T72, and A72. When splenocytes from each transgenic mouse line were used as APCs, the response observed was consistent with this observation, confirming the presence of these ligands (Fig. 6, bottom right). Again, the response to N72tg and A72tg splenocytes, while fivefold above background, is not visible in the scale of the Q72tg response. The presence of the Q72 ligand was also demonstrated by using Q72tg splenocytes to stimulate the QC6.2 hybridoma (Fig. 6, bottom left), which is specific for Q72 and no other APL of Hb (64–76). Finally, APCs from all transgenic lines, regardless of which specific APL of Hb(64–76) they express, should also process and present epitopes from the common HEL portion of the chimeric protein. This expectation was tested with the 3A9 T cell hybridoma, which is specific for HEL(46–61)/I-Ak. We find that splenocytes from all transgenic lines stimulate the 3A9 hybridoma, and that the induced responses are equivalent (Fig. 6, top right).
Figure 6.

Both Hb-related APL and HEL(46–61) epitopes from the chimeric proteins are processed and presented. Splenocytes from each transgenic line were used as APCs in hybridoma proliferation assays to demonstrate properly processed and presented APL and HEL epitopes. In brief, increasing numbers of splenocytes per well (3 × 102 to 1 × 106) were added to a fixed number of T cell hybridoma cells per well (105). The cells were incubated for 24 h, after which 100 ml of supernatant was removed and assayed for IL-2 content by CTLL-2 assay. Results are plotted as the cpm incorporated in the CTLL-2 assay versus the number of splenocytes per well in the original culture. Each datapoint is the mean of triplicate wells. Considering the spectrum of APLs used in this report, 3.L2.12 hybridoma is specific for Hb(64– 76)/I-Ek and stimulated weakly by T72; QC6.2 is specific for Q72/I-Ek; QC85.5 is specific for Q72/I-Ek and stimulated weakly by T72, N72(wt), and A72; and 3.A9 is specific for HEL(46–61)/I-Ak. Most weak responses are not visible in the context of the scale used for the predominant response. This experiment was performed five times with similar results, all of which clearly demonstrate processing and presentation of APL and HEL epitopes from the transgenic proteins. Symbols are as follows: •, N72tg; ▪, T72tg; ▴, I72tg; ⋄, A72tg; ▿, Q72tg; ○, E72tg; and □, the B6.AKR control.
Thymic expression, processing, and presentation of mHEL/ APL transgenic proteins were demonstrated by breeding each line of APLtg mice to 3A9 TCR transgenic mice. T cells in 3A9 TCR transgenic mice are specific for HEL(46–61)/ I-Ak, and therefore should be negatively selected in the presence of this strong agonist ligand. As shown in Table III and Fig. 7, this is indeed the case. When thymocytes from mHEL/APL+/−, 3A9+/− mice were examined by FACS® analysis for CD4, CD8, and the 3A9 clonotypic receptor, there were virtually no DP cells. The number of CD4-SP (CD4+CD8−) cells was reduced by ∼75%, and in this latter population no cells expressed the 3A9 clonotypic TCR (shown for 3A9 × Q72tg; Fig. 7 A, right). Consistent with this observation, the size of thymi in 3A9 × mHEL/APL mice was reduced by ∼80% relative to 3A9 littermate controls. In these mice, no splenocytes were found with the 3A9 clonotypic receptor, and naive splenocytes failed to proliferate to the HEL(46–61) peptide (not shown). We conclude 3A9 × mHEL/APL mice are tolerant to HEL(46–61), and the mechanism for this tolerance is negative selection, demonstrating the presence of properly processed and presented HEL(46–61) epitopes in the thymi of all mHEL/APL mice.
Table III.
Summary of FACS® Analysis Data from mHEL/APLtg × 3A9 Mice
| Mouse | Thymus | |||||||
|---|---|---|---|---|---|---|---|---|
| n | Age | % CD4-SP | % DP | % DN | % CD8-SP | CD4+ gate: % clonotypehi | Size | |
| wk | × 106 | |||||||
| 3A9 | 7 | 7.9 | 35.8 | 46.7 | 13.1 | 4.3 | 81.3 ± 3.2* | 155 ± 29* |
| N72tg × 3A9 | 3 | 8.0 | 10.8 | 2.2 | 82.4 | 4.6 | <2.0‡ | 17 ± 1 |
| T72tg × 3A9 | 3 | 5.0 | 2.8 | 0.5 | 94.3 | 2.2 | <2.0‡ | 30 ± 2 |
| I72tg × 3A9 | 2 | 9.0 | 16.2 | 1.8 | 80.2 | 1.8 | <2.0‡ | 18 ± 1 |
| A72tg × 3A9 | 3 | 11.0 | 10.3 | 0.9 | 81.1 | 2.7 | <2.0‡ | 21 ± 1 |
| Q72tg × 3A9 | 3 | 5.5 | 4.5 | 0.8 | 92.5 | 2.2 | <2.0‡ | 18 ± 5 |
| E72tg × 3A9 | 2 | 7.0 | 8.7 | 1.4 | 88.0 | 2.0 | <2.0‡ | 10 ± 1 |
Figure 7.

Expression of the mHEL/APL transgenic proteins in the thymus results in complete negative selection of 3A9 transgenic T cells. (A) Three-color FACS® analysis was performed on the mice described in Table III. The left panel shows a dot plot of CD4 (PE) versus CD8 (FITC) for a representative control 3A9 TCR transgenic mouse (log10 fluorescence). From this plot, the CD4-SP region was selected for further analysis of 3A9 clonotypic TCR expression. Shown in the right panel are representative histograms derived from the CD4-SP region of the 3A9 control and the Q72tg × 3A9 mouse shown in B. The histogram plots the log10 fluorescence (biotinylated 3A9 clonotypic antibody plus Tricolor-streptavidin) and the number of cells with the 3A9 TCR in these populations. The results clearly demonstrate the complete loss of all CD4-SP, 3A9 TCRhi thymocytes in Q72tg × 3A9 mice. Identical results were obtained with all other APLtg × 3A9 mice (not shown). (B) Three-color FACS® analysis was performed on the APLtg × 3A9 mice described in Table III. Representative dot plots of CD4 (PE) versus CD8 (FITC) are shown. Compared with the 3A9 control in Fig. 7 A, all mice show a characteristic loss of DP and CD4-SP thymocytes consistent with negative selection. Thus, these data directly demonstrate that all transgenes are expressed, processed, and presented by thymic APCs.
In summary, these data clearly show that the APL portion of each chimeric transgenic protein is processed and presented by APCs in vivo. For those hybridomas stimulated by more than one APL, the relative magnitude of the response observed with splenocytes as APCs compares favorably with the response observed with purified peptide in a standard hybridoma proliferation assay (48, and data not shown). When considered together, these data strongly suggest that each transgenic line expresses similar numbers of I-Ek/APL complexes on APCs in both the thymus and the periphery.
Discussion
In this study, we have determined the effect of a broad spectrum of APLs on the selection of an MHC class II– restricted, antigen-specific transgenic T cell in vivo. We find a strong correlation between the effect of the ligand on thymocyte selection and the relative biological activity of the ligand. Specifically, all ligands with measurable relative activities in vitro produce some level of negative selection in vivo. When combined with our previous observation that the relative biological activity is kinetically related to half-life of the TCR–ligand complex (47), the data demonstrate that all ligands examined with a half-life >2 s in vitro produce complete negative selection in vivo. Thus, these results establish a kinetic threshold for the negative selection of an MHC class II–restricted TCR. Negatively selecting ligands include both weak agonists and antagonists of mature T cells. Our results also identify one ligand (Q72) that appears to enhance the positive selection of specific thymocytes. Q72 has no effect on mature 3.L2 T cells, and no binding can be detected in BIAcore™ experiments. However, when expressed in the thymus of 3.L2tg mice, this ligand results in increased numbers of CD4-SP thymocytes with high levels of the 3.L2 clonotypic receptor. E72, another ligand with no effect on mature 3.L2 T cells, has no effect on 3.L2tg T cell development. Taken together, these results clearly illustrate an ordered progression from negative selection to positive selection to no effect, as the potency of the ligand is decreased. When combined with the affinity measurements of ligand binding, they suggest that there are kinetic thresholds for negative and positive selection which are related to the half-life of the TCR–ligand complex. Specific interactions between TCR and ligand of sufficient longevity will result in negative selection, whereas weaker (shorter) interactions may be sufficient for positive selection. The results also suggest that positive selection involves specific interactions between TCR and ligand, as only one of the two weak or null ligands examined increased the percentage of specific T cells.
For the ligands used in this study, the correlation between the biological activity on mature T cells and the half-life of the TCR–ligand complexes in vitro is compelling. Accordingly, we have chosen to interpret the in vivo selection data using a kinetic model of thymocyte selection, as this model best fits all of the available data (47). Other interpretations are possible, particularly when using negative selection as the sole parameter to be correlated with the kinetic measurements. Specifically, on the basis of negative selection alone, one could use an occupancy model to interpret the data. In this model, negative selection of 3.L2tg T cells would result for all ligands that bind the TCR with an equilibrium binding affinity of 170 μM or greater, the limit of detectable binding in our experiments.
Recent data from a single peptide/MHC class II complex system suggest that a substantial increase in the concentration of a positively selecting ligand can result in negative selection (39). We have no direct information on how the small differences in the level of expression of endogenous membrane proteins in this study might influence the number of available peptide/MHC complexes derived from these proteins. Data using an mAb to detect specific MHC/ peptide complexes on the cell surface have suggested that only a fraction of the class II molecules on an APC may be available for loading with a single epitope. These experiments also suggested that there may be an upper limit to the fraction of class II that can be loaded from endogenously expressed membrane proteins (64). The 3A9 T cell hybridoma assays described above demonstrate that, for at least this particular functional assessment, the consequences of slight differences in expression are minimal. One interpretation is that we have achieved maximal or near-maximal levels of the appropriate complexes using our approach of continuously delivering the transgenic membrane protein to the class II antigen processing pathway in all APCs.
Furthermore, in a kinetic model, outcome is determined primarily by the TCR dissociation rate, which does not depend on ligand concentration. This important distinction makes it unlikely that the three- to fivefold difference in expression of the APL transgenes would have any effect on negative selection, where the association between APL and TCR–ligand half-life is strongest. Additionally, data from our laboratory using an inducible system to control expression of N72(wt) support this conclusion (not shown). Therefore, for example, the significant differences between the effects of I72 and A72 on negative selection cannot be explained by small differences in ligand concentration.
The relationship between TCR–ligand kinetics and positive selection remains speculative, as the half-lives of the relevant complexes cannot be accurately measured with current technology. Furthermore, practical considerations in this study have limited our investigations to one level of expression for each ligand; thus, the influence of ligand density on positive selection remains to be determined. It is possible, for example, that the A72 ligand expressed at lower levels could give a result similar to that seen with Q72, as predicted by an occupancy model of thymic selection. One way to integrate occupancy and kinetic models is to postulate that signaling in both positive and negative selection requires a certain number of complexes with an appropriate half-life. Once the occupancy threshold is exceeded, the relevant kinetic parameter becomes the stability of the TCR–ligand complex. Short-lived complexes could result in contact caps with a different composition of signaling molecules than complexes with longer half-lives (65, 66), thereby altering subsequent events in the signaling cascade. Differential, ordered phosphorylation of the TCR ζ chain has been observed in mature T cell signaling (24), and it is not unreasonable to postulate a similar process in developing thymocytes. Thus, positive and negative selection may involve distinct signaling pathways (67).
It is important to note that adding one specific ligand to the pool of endogenous ligands is not a definitive test of positive selection per se. On any particular background, positive selection may already be maximized, so that the only additive effect that could be observed would be negative selection. We feel that, as indicated by the data presented here and elsewhere (53), this is not the case for 3.L2tg T cells. Therefore, the observed transition from negative selection to positive selection to no effect matches the predicted outcome as the potency of the ligand is decreased.
In summary, we have presented data examining the role of APLs in thymic selection of 3.L2 T cells in vivo. Our system individually adds one specific ligand at one fixed concentration to MHC class II–positive cells in the thymus and spleen. Under these conditions, we find that all ligands with measurable effects on mature T cells produce at least some level of negative selection. We also find that one particular null ligand, Q72, enhances the development of 3.L2tg T cells. These observations lead us to conclude that there is a direct correlation between the biological activity of the APLs of 3.L2tg T cells and their effects on thymic selection. We have previously demonstrated that these biological effects reflect the stability of the interaction between 3.L2tg T cells and each MHC/peptide complex (47). The logical conclusion is that there are kinetic thresholds for positive and negative selection related to the half-life of the TCR–ligand complex.
Acknowledgments
This work was supported by grants from the National Institutes of Health (AR01998 and AI24157). C. Williams is a Scholar of the Child Health Research Center of Excellence in Developmental Biology at Washington University School of Medicine (HD33688).
Abbreviations used in this paper
APL
altered peptide ligand
APLtg
transgenic mouse expressing mHEL/APL in all class II–positive cells
CAB
3.L2 clonotypic antibody
DN
double-negative (CD4−CD8−)
DP
double-positive (CD4+CD8+)
Hb
hemoglobin
HEL
hen egg-white lysozyme
3.L2tg
3.L2 TCR transgenic mouse
mHEL/APL
chimeric membrane protein containing HEL and one of the APLs listed below
N72(wt)
the native Hb(64–76) peptide with asparagine at position 72
RT
room temperature
SP
single positive
T72
I72, A72, Q72, and E72, APLs of Hb(64–76) with position 72 substitutions as indicated
Footnotes
Many colleagues have provided reagents and helpful discussions. In particular, we wish to thank Dr. Emil Unanue, Dr. Chris Nelson, and Daniel Peterson for the 3A9 clonotypic antibody, the mHEL construct, and the mHEL transgenic mouse; Dr. Mark Davis for the 3A9 TCR transgenic mice; Drs. Diane Mathis and Christophe Benoist for the Eα promoter construct; Kathy Frederick, Darren Kreamalmeyer, and Donna Thompson for help breeding the mice; David Donermeyer and Stephen Horvath for help screening the mice; Dr. Peiqing Qian and Dr. Karine Vidal for assistance with the immunofluorescence; and Jerri Smith for her assistance in preparation of the manuscript.
References
- 1.Kappler JW, Noehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 2.MacDonald HR, Schneider R, Lees RK, Howe RC, Acha-Orbea H, Festenstein H, Zinkernagel RM, Hengartner H. T-cell receptor Vβ use predicts reactivity and tolerance to Mlsa-encoded antigens. Nature. 1988;332:40–45. doi: 10.1038/332040a0. [DOI] [PubMed] [Google Scholar]
- 3.Kisielow P, Blüthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+thymocytes. Nature. 1988;333:742–746. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- 4.Sha WC, Nelson CA, Newberry RD, Kranz DM, Russell JH, Loh DY. Positive and negative selection of an antigen receptor on T cells in transgenic mice. Nature. 1988;336:73–79. doi: 10.1038/336073a0. [DOI] [PubMed] [Google Scholar]
- 5.Bevan MJ. In a radiation chimera host H-2 antigens determine the immune responsiveness of donor cytotoxic cells. Nature. 1977;269:417–419. doi: 10.4049/jimmunol.176.1.677. [DOI] [PubMed] [Google Scholar]
- 6.Zinkernagel RM, Callahan GN, Cooper AAS, Klein PA, Klein J. On the thymus in the differentiation of “H-2 self-recognition” by the cells: evidence for dual recognition? . J Exp Med. 1978;147:882–896. doi: 10.1084/jem.147.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniel C, Horvath S, Allen PM. A basis for alloreactivity: MHC helical residues broaden peptide recognition by the TCR. Immunity. 1998;8:543–552. doi: 10.1016/s1074-7613(00)80559-2. [DOI] [PubMed] [Google Scholar]
- 8.Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science. 1991;252:1308–1310. doi: 10.1126/science.1833816. [DOI] [PubMed] [Google Scholar]
- 9.De Magistris MT, Alexander J, Coggeshall M, Altman A, Gaeta FCA, Grey HM, Sette A. Antigen analog-major histocompatibility complexes act as antagonists of the T cell receptor. Cell. 1992;68:625–634. doi: 10.1016/0092-8674(92)90139-4. [DOI] [PubMed] [Google Scholar]
- 10.Evavold BD, Sloan-Lancaster J, Wilson KJ, Rothbard JB, Allen PM. Specific T cell recognition of minimally homologous peptides: evidence for multiple endogenous ligands. Immunity. 1995;2:655–663. doi: 10.1016/1074-7613(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 11.Evavold BD, Sloan-Lancaster J, Allen PM. Tickling the TCR: selective T cell functions stimulated by altered peptide ligands. Immunol Today. 1993;14:602–609. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- 12.Ashton-Rickardt PG, Bandeira A, Delaney JR, Van Kaer L, Pircher H-P, Zinkernagel RM, Tonegawa S. Evidence for a differential avidity model of T cell selection in the thymus. Cell. 1994;76:651–663. doi: 10.1016/0092-8674(94)90505-3. [DOI] [PubMed] [Google Scholar]
- 13.Ashton-Rickardt PG, Tonegawa S. A differential-avidity model for T-cell selection. Immunol Today. 1994;15:362–366. doi: 10.1016/0167-5699(94)90174-0. [DOI] [PubMed] [Google Scholar]
- 14.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 15.Sebzda E, Kündig TM, Thomson CT, Aoki K, Mak S-Y, Mayer JP, Zamborelli T, Nathenson SG, Ohashi PS. Mature T cell reactivity altered by peptide agonist that induces positive selection. J Exp Med. 1996;183:1093–1104. doi: 10.1084/jem.183.3.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bevan MJ. In thymic selection, peptide diversity gives and takes away. Immunity. 1997;7:175–178. doi: 10.1016/s1074-7613(00)80520-8. [DOI] [PubMed] [Google Scholar]
- 17.Shaw AS, Dustin ML. Making the T cell receptor go the distance: a topological view of T cell activation. Immunity. 1997;6:361–369. doi: 10.1016/s1074-7613(00)80279-4. [DOI] [PubMed] [Google Scholar]
- 18.Matsui K, Boniface JJ, Steffner P, Reay PA, Davis MM. Kinetics of T-cell receptor binding to peptide/ I-Ekcomplexes: correlation of the dissociation rate with T-cell responsiveness. Proc Natl Acad Sci USA. 1994;91:12862–12866. doi: 10.1073/pnas.91.26.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rabinowitz JD, Beeson C, Lyons DS, Davis MM, McConnell HM. Kinetic discrimination in T-cell activation. Proc Natl Acad Sci USA. 1996;93:1401–1405. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien Y-H, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 22.Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y-H. Ligand recognition by αβ T cell receptors. Annu Rev Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 23.Boniface JJ, Rabinowitz JD, Wülfing C, Hampl J, Reich Z, Altman JD, Kantor RM, Beeson C, McConnell HM, Davis MM. Initiation of signal transduction through the T cell receptor requires the peptide multivalent engagement of MHC ligands. Immunity. 1998;9:459–466. doi: 10.1016/s1074-7613(00)80629-9. [DOI] [PubMed] [Google Scholar]
- 24.Kersh EN, Shaw AS, Allen PM. Fidelity of T cell activation through multistep T cell receptor ζ phosphorylation. Science. 1998;281:572–575. doi: 10.1126/science.281.5376.572. [DOI] [PubMed] [Google Scholar]
- 25.Hogquist KA, Gavin MA, Bevan MJ. Positive selection of CD8+T cells induced by major histocompatibility complex binding peptides in fetal thymic organ culture. J Exp Med. 1993;177:1469–1473. doi: 10.1084/jem.177.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jameson SC, Hogquist KA, Bevan MJ. Specificity and flexibility in thymic selection. Nature. 1994;369:750–752. doi: 10.1038/369750a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogquist KA, Jameson SC, Bevan MJ. Strong agonist ligands for the T cell receptor do not mediate positive selection of functional CD8+T cells. Immunity. 1995;3:79–86. doi: 10.1016/1074-7613(95)90160-4. [DOI] [PubMed] [Google Scholar]
- 28.Hogquist KA, Tomlinson AJ, Kieper WC, McGargill MA, Hart MC, Naylor S, Jameson SC. Identification of a naturally occurring ligand for thymic positive selection. Immunity. 1997;6:389–399. doi: 10.1016/s1074-7613(00)80282-4. [DOI] [PubMed] [Google Scholar]
- 29.Sebzda E, Wallace VA, Mayer J, Yeung RSM, Mak TW, Ohashi PS. Positive and negative thymocyte selection induced by different concentrations of a single peptide. Science. 1994;263:1615–1618. doi: 10.1126/science.8128249. [DOI] [PubMed] [Google Scholar]
- 30.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NRJ. T-cell- receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 31.Schodin BA, Tsomides TJ, Kranz DM. Correlation between the number of T cell receptors required for T cell activation and TCR-ligand affinity. Immunity. 1996;5:137–146. doi: 10.1016/s1074-7613(00)80490-2. [DOI] [PubMed] [Google Scholar]
- 32.Liu C-P, Crawford F, Marrack P, Kappler J. T cell positive selection by a high density, low affinity ligand. Proc Natl Acad Sci USA. 1998;95:4522–4526. doi: 10.1073/pnas.95.8.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sykulev Y, Cohen RJ, Eisen HN. The law of mass action governs antigen-stimulated cytolytic activity of CD8+cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1995;92:11990–11992. doi: 10.1073/pnas.92.26.11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. β2-microglobulin deficient mice lack CD4−8+cytolytic T cells. Nature. 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- 35.Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in β2M, MHC class I proteins, and CD8+T cells. Science. 1990;248:1227–1230. doi: 10.1126/science.2112266. [DOI] [PubMed] [Google Scholar]
- 36.Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4−8+T cells. Cell. 1992;71:1205–1214. doi: 10.1016/s0092-8674(05)80068-6. [DOI] [PubMed] [Google Scholar]
- 37.Ashton-Rickardt PG, Van Kaer L, Schumacher TNM, Ploegh HL, Tonegawa S. Peptide contributes to the specificity of positive selection of CD8+T cells in thymus. Cell. 1993;73:1041–1049. doi: 10.1016/0092-8674(93)90281-t. [DOI] [PubMed] [Google Scholar]
- 38.Sant'Angelo DB, Waterbury PG, Cohen BE, Martin WD, Van Kaer L, Hayday AC, Janeway CA., Jr The imprint of intrathymic self-peptides on the mature T cell receptor repertoire. Immunity. 1997;7:517–524. doi: 10.1016/s1074-7613(00)80373-8. [DOI] [PubMed] [Google Scholar]
- 39.Fukui Y, Ishimoto T, Utsuyama M, Gyotoku T, Koga T, Nakao K, Hirokawa K, Katsuki M, Sasazuki T. Positive and negative CD4+thymocyte selection by a single MHC class II/peptide ligand affected by its expression level in the thymus. Immunity. 1997;6:401–410. doi: 10.1016/s1074-7613(00)80283-6. [DOI] [PubMed] [Google Scholar]
- 40.Sant'Angelo DB, Lucas B, Waterbury PG, Cohen B, Brabb T, Goverman J, Germain RN, Janeway CA., Jr A molecular map of T cell development. Immunity. 1998;9:179–186. doi: 10.1016/s1074-7613(00)80600-7. [DOI] [PubMed] [Google Scholar]
- 41.Spain LM, Jorgensen JL, Davis MM, Berg LJ. A peptide antigen antagonist prevents the differentiation of T cell receptor transgenic thymocytes. J Immunol. 1994;152:1709–1717. [PubMed] [Google Scholar]
- 42.Page DM, Alexander J, Snoke K, Appella E, Sette A, Hedrick SM, Grey HM. Negative selection of CD4+CD8+thymocytes by T-cell receptor peptide antagonists. Proc Natl Acad Sci USA. 1994;91:4057–4061. doi: 10.1073/pnas.91.9.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ignatowicz L, Kappler J, Marrack P. The repertoire of T cells shaped by a single MHC/peptide ligand. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]
- 44.Miyazaki T, Wolf P, Tourne S, Waltzinger C, Dierich A, Barois N, Ploegh H, Benoist C, Mathis D. Mice lacking H2-M complexes, enigmatic elements of the MHC class II peptide-loading pathway. Cell. 1996;84:531–541. doi: 10.1016/s0092-8674(00)81029-6. [DOI] [PubMed] [Google Scholar]
- 45.Martin WD, Hicks GG, Mendiratta SK, Leva HI, Ruley HE, Van Kaer L. H2-Mmutant mice are defective in the peptide loading of class II molecules, antigen presentation, and T cell repertoire selection. Cell. 1996;84:543–550. doi: 10.1016/s0092-8674(00)81030-2. [DOI] [PubMed] [Google Scholar]
- 46.Fung-Leung W-P, Surh CD, Liljedahl M, Pang J, Leturcq D, Peterson PA, Webb SR, Karlsson L. Antigen presentation and T cell development in H2-M-deficient mice. Science. 1996;271:1278–1281. doi: 10.1126/science.271.5253.1278. [DOI] [PubMed] [Google Scholar]
- 47.Kersh, G.J., E.N. Kersh, D.H. Fremont, and P.M. Allen. 1998. High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. _Immunit_y. 9:817–826. [DOI] [PubMed]
- 48.Kersh GJ, Allen PM. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J Exp Med. 1996;184:1259–1268. doi: 10.1084/jem.184.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evavold BD, Williams SG, Hsu BL, Buus S, Allen PM. Complete dissection of the Hb(64-76) determinant using Th1, Th2 clones, and T cell hybridomas. J Immunol. 1992;148:347–353. [PubMed] [Google Scholar]
- 50.Fremont DH, Hendrickson WA, Marrack P, Kappler J. Structures of an MHC class II molecule with covalently bound single peptides. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
- 51.Williams CB, Vidal K, Donermeyer DL, Peterson DA, White JM, Allen PM. In vivo expression of a T cell receptor antagonist: T cells escape central tolerance but are antagonized in the periphery. J Immunol. 1998;161:128–137. [PubMed] [Google Scholar]
- 52.Kouskoff V, Fehling H-J, Lemeur M, Benoist C, Mathis D. A vector driving the expression of foreign cDNAs in the MHC class II-positive cells of transgenic mice. J Immunol Methods. 1993;166:287–291. doi: 10.1016/0022-1759(93)90370-m. [DOI] [PubMed] [Google Scholar]
- 53.Kersh GJ, Donermeyer DL, Frederick KE, White JM, Hsu BL, Allen PM. TCR transgenic mice in which usage of transgenic α- and β-chains is highly dependent on the level of selecting ligand. J Immunol. 1998;161:585–593. [PubMed] [Google Scholar]
- 54.Ho WY, Cooke MP, Goodnow CC, Davis MM. Resting and anergic B cells are defective in CD28- dependent costimulation of naive CD4+T cells. J Exp Med. 1994;179:1539–1549. doi: 10.1084/jem.179.5.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evavold BD, Sloan-Lancaster J, Allen PM. Antagonism of superantigen-stimulated helper T-cell clones and hybridomas by altered peptide ligand. Proc Natl Acad Sci USA. 1994;91:2300–2304. doi: 10.1073/pnas.91.6.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evavold BD, Sloan-Lancaster J, Hsu BL, Allen PM. Separation of T helper 1 clone cytolysis from proliferation and lymphokine production using analog peptides. J Immunol. 1993;150:3131–3140. [PubMed] [Google Scholar]
- 57.Johnson NA, Carland F, Heilig JS, Allen PM, Glimcher LH. T cell receptor gene segment usage in a panel of hen egg-white lysozyme (HEL)-specific, I-Ak- restricted T helper hybridomas. J Immunol. 1989;142:3298–3304. [PubMed] [Google Scholar]
- 58.Vidal K, Hsu BL, Williams CB, Allen PM. Endogenous altered peptide ligands can affect peripheral T cell responses. J Exp Med. 1996;183:1311–1321. doi: 10.1084/jem.183.4.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fischmann T, Souchon H, Riottot M-M, Tello D, Poljak RJ. Crystallization and preliminary X-ray diffraction studies of two new antigen-antibody (Lysozyme-Fab) complexes. J Mol Biol. 1988;203:527–529. doi: 10.1016/0022-2836(88)90022-8. [DOI] [PubMed] [Google Scholar]
- 60.Ozato K, Mayer N, Sachs DH. Hybridomas cell lines secreting monoclonal antibodies to mouse H-2 and Ia antigens. J Immunol. 1980;124:533–540. [PubMed] [Google Scholar]
- 61.Rudensky AY, Preston-Hurlburt P, Hong S-C, Barlow A, Janeway CA., Jr Sequence analysis of peptides bound to MHC class II molecules. Nature. 1991;353:622–627. [PubMed] [Google Scholar]
- 62.Brooks A, Hartley S, Kjer-Nielsen L, Perera J, Goodnow CC, Basten A, McCluskey J. Class II- restricted presentation of an endogenously derived immunodominant T-cell determinant of hen egg lysozyme. Proc Natl Acad Sci USA. 1991;88:3290–3294. doi: 10.1073/pnas.88.8.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Basu D, Williams CB, Allen PM. In vivoantagonism of a T cell response by an endogenously expressed ligand. Proc Natl Acad Sci USA. 1998;95:14332–14336. doi: 10.1073/pnas.95.24.14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dadaglio G, Nelson CA, Deck MB, Petzold SJ, Unanue ER. Characterization and quantitation of peptide-MHC complexes produced from hen egg-white lysozyme using a monoclonal antibody. Immunity. 1997;6:727–738. doi: 10.1016/s1074-7613(00)80448-3. [DOI] [PubMed] [Google Scholar]
- 65.Leitenberg D, Boutin Y, Constant S, Bottomly K. CD4 regulation of TCR signaling and T cell differentiation following stimulation with peptides of different affinities for the TCR. J Immunol. 1998;161:1194–1203. [PubMed] [Google Scholar]
- 66.Dustin ML, Olszowy MW, Holdorf AD, Li J, Bromley S, Desai N, Widder P, Rosenberger F, van der Merwe PA, Allen P, Shaw AS. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contracts. Cell. 1998;94:667–677. doi: 10.1016/s0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- 67.Alberola-Ila J, Hogquist KA, Swan KA, Bevan MJ, Perlmutter RM. Positive and negative selection invoke distinct signaling pathways. J Exp Med. 1996;184:9–18. doi: 10.1084/jem.184.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]