Pivotal Role of KARAP/DAP12 Adaptor Molecule in the Natural Killer Cell–mediated Resistance to Murine Cytomegalovirus Infection (original) (raw)
Abstract
Natural killer (NK) cells are major contributors to early defense against infections. Their effector functions are controlled by a balance between activating and inhibiting signals. To date, however, the involvement of NK cell activating receptors and signaling pathways in the defense against pathogens has not been extensively investigated. In mice, several NK cell activating receptors are coexpressed with and function through the immunoreceptor tyrosine-based activation motif (ITAM)-bearing molecule KARAP/DAP12. Here, we have analyzed the role of KARAP/DAP12 in the early antiviral response to murine cytomegalovirus (MCMV). In KARAP/DAP12 mutant mice bearing a nonfunctional ITAM, we found a considerable increase in viral titers in the spleen (30–40-fold) and in the liver (2–5-fold). These effects were attributed to NK cells. The formation of hepatic inflammatory foci appeared similar in wild-type and mutant mice, but the latter more frequently developed severe hepatitis with large areas of focal necrosis. Moreover, the percentage of hepatic NK cells producing interferon γ was reduced by 56 ± 22% in the absence of a functional KARAP/DAP12. This is the first study that shows a crucial role for a particular activating signaling pathway, in this case the one induced through KARAP/DAP12, in the NK cell–mediated resistance to an infection. Our results are discussed in relation to recent reports demonstrating that innate resistance to MCMV requires the presence of NK cells expressing the KARAP/DAP12-associated receptor Ly49H.
Keywords: animal models, activating receptors, signal transduction, pathogens, cytokines
Introduction
NK cells form a lymphocyte population that participates in early innate immune responses (1, 2). Without need of prior sensitization, NK cells contribute to the defense against a variety of viral and bacterial infections as well as against transformed or allogeneic cells (3, 4). Acute infection of murine cytomegalovirus (MCMV)* represents a useful model for studies of the protective effects of NK cells against viruses (4). Studies based on mice genetically deficient in NK cell functions or on NK cell depletion have shown that the absence of NK cells renders mice more sensitive to MCMV, with increased MCMV synthesis and MCMV-induced pathology (5–8). Furthermore, adoptive transfer of purified culture-derived NK cells enhanced survival and reduced MCMV replication in irradiated adult mice as well as in suckling mice, which have a low NK cell activity and are very sensitive to MCMV (9, 10).
However, the mechanisms by which NK cells counteract viral infections in vivo have been only partly defined. NK cell effector mechanisms involved in resistance to MCMV are organ dependent. In the liver, the antiviral effects are predominantly mediated through IFN-γ production (11–13). Upon MCMV infection, the β-chemo-kine macrophage inflammatory protein 1α (MIP-1α) promotes recruitment of NK cells and formation of inflammatory foci in the infected liver (14). Within the foci, NK cells encompass infected cytomegalic cells while producing local high levels of IFN-γ that controls MCMV replication (11, 14, 15). IFN-γ production is induced also in the serum and in the spleen, but with a mechanism that is independent of MIP-1α (15).
In the spleen, NK cells uphold MCMV resistance mainly via a perforin-dependent mechanism, suggesting that cell-mediated cytotoxicity is crucial (12). In addition, a specific locus, Cmv-1, appears to be strongly related to the control of viral replication in the spleen (16). Cmv-1 maps in the NK-complex on mouse chromosome 6, a region encoding several NK cell receptors (17). While this work was in progress, two independent groups have reported that the Cmv-1 locus may correspond to the NK cell activating receptor Ly49H (18, 19). These groups and an additional one (20) also showed that depletion of the Ly49H+ NK cell subset rendered mice more susceptible to MCMV.
A balance between inhibiting and activating signals regulates the different effector mechanisms of NK cells (21, 22). Triggering signals can be transduced from the activating receptors into the cell via immunoreceptor tyrosine-based activation motifs (ITAMs) coupled to downstream protein kinases. Activating receptors lack intrinsic ITAMs and they instead act by forming noncovalent complexes with adaptor molecules carrying the ITAMs (23). After cross-linking of inhibitory receptors, signals are transduced inside the cell through immunoreceptor tyrosine-based inhibition motifs (ITIMs) present on the intracellular part of the receptors and coupled to downstream protein phosphatases (24). These phosphatases dampen cell activation by dephosphorylating several effector/adaptor molecules involved in the early activation signaling events (22, 24).
KARAP/DAP12 is an ITAM-bearing adaptor protein known to associate with several human and mouse receptors expressed on NK cells (i.e., Ly49D, Ly49H, KIR2DS, CD94-NKG2C, and NKp44) and on myeloid cells (i.e., MDL-1, SIRPβ, TREM-1, and TREM-2; for a review, see reference 25). KARAP/DAP12 is expressed as a disulfide-linked homodimer, each subunit bearing one ITAM within its intracellular region (26). Upon cross-linking of these receptor complexes, it is crucial that both tyrosine residues within each of the ITAM are phosphorylated. They then act as a docking site for Syk or ZAP-70 protein tyrosine kinases (26–28), which elicits a cascade of protein tyrosine phosphorylation events that in NK cells results in the activation of cell-mediated cytotoxicity and cytokine production (21, 22).
Recently, two groups have independently generated mice deficient in KARAP/DAP12 (29, 30). While the mice generated by Bakker et al. lack the expression of KARAP/DAP12 and its associated receptors (30), the mice used in the present study are KARAP/DAP12 loss-of-function mice. In this case, the adaptor molecule is expressed but the integrity of the ITAM sequence has been disrupted and the signal transduction is therefore abrogated (29). In spite of this mutation, the frequency of NK cells expressing the KARAP/DAP12-associated activating receptors is comparable to wild-type mice, though the expression levels of these receptors appear to be decreased (29). There is also evidence for unaffected expression of inhibitory receptors (29, 30). Furthermore, these mice show normal NK cell development and normal numbers of lymphoid and myeloid subsets, though a restriction of the NK cell–mediated tumor killing was noticed together with a resistance to antigen sensitization and an accumulation of dendritic cells in muco-cutaneous epithelia (29).
Among the NK cell receptors not associated with KARAP/DAP12, some are instead associated with CD3ς and/or FcRγ, while a third category transmits activating signals via the adaptor molecule DAP10 (23). At present, little is known regarding the role of all these different activating receptors in distinct defense situations, including antiviral activity of NK cells. Are certain activating receptors mandatory in some situations, or is activation always provided by parallel and redundant molecular interactions? In this study, KARAP/DAP12 loss-of-function mutant mice were infected with MCMV in order to investigate the importance of KARAP/DAP12-dependent pathways in MCMV resistance.
Materials and Methods
Mice.
KARAP/DAP12 loss-of-function −/− mice have been described previously (29). The mice used in this study were however not initially mated with C57BL/6 Cre transgenic mice and they therefore carried the loxP-neo-loxP cassette (29). KARAP/DAP12−/− mice were backcrossed into C57BL/6 mice six or seven times and all the animals used in the experiments were derived from the offspring of KARAP/DAP12+/− × KARAP/DAP12+/− breeding pairs. These mice were conventionally housed and they were used between 6 and 13 wk of age. The mice used in the experiments were of either sex and they were −/−, +/−, and +/+ littermates, unless otherwise stated. C57/BL6 mice were initially purchased from The Jackson Laboratory. For production of MCMV viral stock, Swiss Webster outbred CD-1 mice were obtained from Charles River Laboratories. Experiments were performed in accordance with institutional guidelines for animal care and use.
Virus and Viral Titer Determination.
The Smith strain of MCMV was obtained from the American Type Culture Collection (catalog no. VR-1399). Subsequently, it was propagated in vivo in salivary glands of CD-1 mice, as described previously (11, 31). The preparation used in this study was from a tertiary passage through CD-1 mice. Infected salivary glands homogenates were titrated on NIH3T3 fibroblasts, a gift of Drs. G. Pien and C. Biron (Brown University, Providence, RI). In all experiments described, mice were inoculated intraperitoneally with 105 PFU of MCMV and killed 2 or 3 d after. Viral titers were determined by plaque assays as described previously (11). In brief, spleen, liver, and both lungs were harvested after infection and livers were weighed individually. After homogenization of the organs in 199 1× medium supplemented with 3% heat-inactivated fetal calf serum, the samples were centrifuged twice and the supernatants were stored in −80°C. On the day of the experiment, the homogenates were thawed and duplicate log dilution samples were titrated on NIH3T3 fibroblasts monolayers. After 1 h of incubation at 37°C, a mixture of 0.5% low melt agarose (FMC) diluted in DMEM medium (Life Technologies) was added to each well and the cells were incubated at 37°C for additional 6–7 d. The cells were fixed by using 1–5% buffered formalin and stained with 0.1% crystal violet. To calculate viral titers for one individual mouse, the average of the raw PFU data derived from duplicate dilution samples was calculated and then expressed in log10. When data from several experiments and mice are shown, the numbers presented are the arithmetic averages of the log10 data, derived from each individual mouse tested (± SD).
Depletion of NK Cells In Vivo.
Mice were depleted of NK cells by a single intraperitoneal injection of 100 μg of column-purified anti-NK1.1 mAb PK136 2 d before infection with MCMV. As controls, mice were injected with PBS. As shown previously for C57BL/6 mice, PBS injection results in MCMV titers undistinguishable from inoculations with mAbs of the same isotype as the PK136 mAb (19, 32).
Histology and Evaluation of Liver Disease.
Livers were isolated, fixed in 10% buffered formalin, and paraffin embedded. Tissue sections (5 μm) were stained with hematoxylin and eosin (H&E) and analyzed microscopically. Inflammatory foci were defined as discrete clusters of at least 10 individual, small, nucleated cells visible throughout the liver (11, 14, 33). Numbers of inflammatory foci were determined by counting clusters of cells in five randomly selected fields of 3.14 mm2 each, at a magnification of ×10 low power fields. Necrotic areas were histologically identified as large, subcapsular, eosinophilic areas of necrotic hepatocytes.
Isolation of Liver Leukocytes and Intracellular IFN-γ Assay.
Liver leukocytes were isolated by flushing blood from the liver by injecting PBS through the portal vein, crushing the liver and harvesting the leukocytes layer from a Percoll gradient. Leukocytes were subsequently stained for the intracellular accumulation of IFN-γ using the reagents and protocol from BD PharMingen. Cells were incubated in RPMI medium (Life Technologies) containing GolgiStop (BD PharMingen) for 4 h at 37°C in Falcon U-bottomed 96-well plates. As a positive control, in some wells cells were incubated with 50 ng/ml PMA and 500 ng/ml ionomycin (Sigma-Aldrich; data not shown). Cells were then washed and incubated for 20 min at 4°C with the purified anti-CD16 2.4G2 mAb to block unspecific staining. Samples were washed and incubated with a combination of FITC-conjugated anti-NK1.1 mAb or DX5 mAb (data not shown) (34) and PerCP-conjugated anti-CD3 mAb (both from BD PharMingen). After 30 min of incubation at 4°C, cells were washed and permeabilized using 100 μl of cytofix/cytoperm solution for 20 min at 4°C. Samples were washed twice using perm/wash solution and stained with rat IgG1-PE mAb to mouse IFN-γ or with a control rat IgG1-PE for 30 min at 4°C (BD PharMingen). Cells were then washed in perm/wash solution and analyzed on a FACStar™ Plus (Becton Dickinson). At least 10,000 events were acquired in the DX5+ gate in order to ensure a sizable NK cell population.
Ly49H Staining.
Freshly isolated splenocytes, peripheral blood, and liver leukocytes were incubated for 20 min at 4°C with the purified anti-CD16 2.4G2 mAb to block unspecific staining. Samples were washed and incubated with a combination of the FITC-conjugated anti-Ly49H mAb 3D10 (provided by Dr. W. Yokoyama, Howard Hughes Medical Institute, St. Louis, MO), the PE-conjugated anti-NK1.1 mAb, and the PerCP-conjugated anti-CD3 mAb (both from BD PharMingen). The cells were then washed and analyzed on a FACStar™ Plus (Becton Dickinson).
Statistical Analysis.
Data were analyzed by the unpaired Student's t test.
Results
MCMV Titers in KARAP/DAP12 Loss-of-Function Mutant Mice.
To evaluate the role of KARAP/DAP12 in the early control of MCMV replication and spread by NK cells, KARAP/DAP12 loss-of-function (−/−) and control (+/− and +/+) mice were infected with MCMV and viral titers were measured 3 d after infection in the spleen, liver, and lungs (Fig. 1) . Replication of MCMV in the spleen of KARAP/DAP12−/− mice was in all experiments performed remarkably elevated (30–40-fold) compared with +/− and +/+ mice (Fig. 1). In a total of five experiments performed, the average in viral titers (± SD) was 4.21 ± 0.42 in −/−, 2.96 ± 0.59 in +/−, and 2.72 ± 0.3 in +/+ mice. In the liver of KARAP/DAP12−/− mice, the increase in viral load was 2–5 times higher compared with +/+ controls, whereas the difference between −/− and +/− mice ranged between 1.5 and 3-fold increase (Fig. 1). In a total of five experiments performed, the average in viral titers (± SD) was 4.53 ± 0.27 in −/−, 4.22 ± 0.34 in +/−, and 4.13 ± 0.32 in +/+ mice. Interestingly, in both spleen and liver there was a slight, though not statistically significant, difference in viral titers between +/− and +/+ mice (Fig. 1). We could not detect any significant difference in viral titers in the lungs of −/− compared with either +/− or +/+ control mice. However, a tendency to increased viral titers in −/− mice was observed also in this organ, with the average in viral titers (± SD) being 2.32 ± 0.6 in −/−, 1.89 ± 0.56 in +/−, and 1.77 ± 0.65 in +/+ mice. Altogether, these results indicate that the absence of a functional KARAP/DAP12 molecule results in an increase of viral titers, which was statistically significant in the spleen and liver.
Figure 1.

MCMV replication in KARAP/DAP12 +/+, +/−, or −/− loss-of-function mice. 3 d after infection with 105 PFU of MCMV, organs were harvested, homogenized, and viral titers were measured as described in Materials and Methods. Each point represents the average titer determined for an individual mouse. Horizontal bars depict mean viral titers for each group. The limit of detection of the assay is shown (DL) and for mice with titers below this limit the minimum number of detectable PFU was assumed to determine the mean. This assumption overestimates the mean of the group having titers below detectable levels. This figure combines five independent experiments, each including spleen, liver, and lungs data obtained from the same mice. A total of 11 −/−, 10 +/−, and 6 +/+ mice were tested. P values are as follows: P < 0.00001 (***); P < 0.001 (**); P < 0.05 (*).
Depletion of NK1.1+ Cells in KARAP/DAP12 Loss-of-Function Mutant Mice.
To evaluate whether NK cell dependency and KARAP/DAP12 dependency were strictly parallel in the control of MCMV replication, KARAP/DAP12 −/−, +/+, or C57BL/6 mice were injected with an anti-NK1.1 antibody 2 d before MCMV infection. Fig. 2 shows that in the spleen of −/− mice, depletion of NK1.1+ cells affected viral titers only marginally, with no statistically significant difference between PBS and anti-NK1.1 treated −/− mice. This suggests that KARAP/DAP12 is required for the NK cell–mediated control of MCMV replication. This is also supported by a comparison between PBS-treated −/− mice and NK1.1-treated +/+ mice that showed similar viral titers. Moreover, the comparison between NK1.1-treated +/+ and −/− mice suggests that there is no major direct contribution to MCMV resistance by KARAP/DAP12-associated receptors expressed on other cells than the ones expressing NK1.1. In the liver, the effect of anti-NK1.1 treatment on +/+ mice appeared to be stronger than the impact of a defective KARAP/DAP12 gene (P = 0.016 between PBS-treated −/− and anti-NK1.1-treated +/+ mice). In addition, there was a 10-fold increase in viral titers after NK1.1 treatment also in −/− mice (P = 0.0022 between PBS and anti-NK1.1–treated −/− mice). One possible interpretation of these results is that other NK cell–mediated mechanisms, independent of KARAP/DAP12, may play a more prominent role in the control of MCMV infection in the liver. Finally, treatment of −/− and +/+ mice with the anti-NK1.1 mAb, resulted in similar liver viral titers. This suggests that in the liver, as well as in the spleen, KARAP/DAP12-associated receptors expressed on NK1.1-negative cells are not playing a crucial direct role in MCMV infection, at least at an early stage after infection.
Figure 2.

Effect of NK1.1 mAb treatment on the replication of MCMV in KARAP/DAP12 −/− and +/+ mice. Age-matched −/−, +/+, or C57BL/6 mice were either injected with anti-NK1.1 mAb PK136 in PBS or with PBS alone 2 d before MCMV infection. 3 d after infection, spleens and livers were harvested and viral titers determined. Each point represents the average titer determined for an individual mouse. Horizontal bars depict mean values of viral titers for each group. Values (± SD) in the spleen were as follows: −/− PBS, 4.11 ± 0.71; −/− NK1.1, 4.69 ± 0.56; +/+ PBS, 2.56 ± 0.38; +/+ NK1.1, 4.42 ± 0.47. Values (± SD) in the liver were as follows: −/− PBS, 3.97 ± 0.63; −/− NK1.1, 5.01 ± 0.38; +/+ PBS, 3.53 ± 0.37; +/+ NK1.1, 4.91 ± 0.55. This figure combines four independent experiments, each including spleen and liver data obtained from the same mice.
Characterization of MCMV-induced Hepatitis in KARAP/DAP12 Loss-of-Function Mice.
Virus-induced liver disease can also be used to estimate the degree of MCMV replication in this organ. A mononuclear cell infiltrate, specific cytological alterations and areas of necrosis characterize the MCMV-induced hepatitis (11, 33, 35). Histologically, the cellular inflammation has the form of discrete foci or clusters of cells present throughout the liver. It has been shown that foci formation requires the chemokine MIP-1α and requires NK cells, and that the antiviral response colocalizes NK cells and IFN-γ production at sites of viral infection (14). To test the hypothesis that the higher viral titers observed in the livers of KARAP/DAP12 loss-of-function mice were due to an impaired recruitment of inflammatory cells and/or formation of inflammatory foci, tissue sections from −/−, +/−, +/+, and C57BL/6 mice were prepared and H&E stained for the histomorphological evaluation. Similar to the observations previously reported for C57BL/6 mice (14, 35), MCMV-infected livers from KARAP/DAP12+/+ mice were characterized by discrete inflammatory foci, generally localized between portal triads and central veins (Fig. 3 A). A similar pattern was observed in −/− mice (Fig. 3 B). Cytomegalic cells and nuclear inclusions were often seen associated with inflammatory foci in +/+, +/−, as well as in −/− mice (Fig. 3 C). Contrary to what we expected from the hypothesis, there was only a marginal difference in the number of inflammatory foci in −/− (64 ± 22) compared with control mice (80 ± 25 in +/+ and 88 ± 13 in +/− mice). The major difference observed in the MCMV-induced hepatitis was instead the more frequent formation in −/− mice of large areas of focal necrosis, both at the liver surface and inside the liver parenchyma (Fig. 3, D and E). Sometimes, necrotic areas were surrounded by residual inflammatory cells (Fig. 3 F). Table I shows a summary of the morphological observations. On the basis of hepatic necrosis, we could identify three groups of mice, each with different level of MCMV-induced hepatitis. These levels were defined by the presence of large areas of focal necrosis (severe hepatitis), by the presence of small and isolated areas of focal necrosis (moderate hepatitis), or by the absence of necrosis (minimal hepatitis). Specific signs of MCMV infection and inflammatory foci were present in all animals. However, we observed large areas of necrosis in 6/10 −/− mice (60%) compared with only 1/15 control mice (7%) (0/6 +/+ mice and 1/9 +/− mice). In general and as expected, there was a direct correlation between the severity of liver damage and the viral titers (Table I, and data not shown). In fact, mice with a severe hepatitis had statistically significant higher titers compared with mice with a moderate (P = 0.002) or minimal hepatitis (P = 0.0007). Taken together, these results indicate that the impaired KARAP/DAP12 function did not lead to a major defect in the formation of inflammatory foci, but rather impaired the ability of the inflammatory cells to limit virus-induced liver disease.
Figure 3.
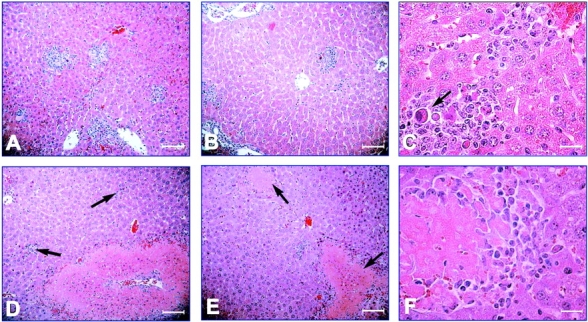
H&E staining of liver sections derived from KARAP/DAP12 loss-of-function mutant mice. Livers were harvested and tissue sections prepared from day 3 MCMV-infected mice as described in Materials and Methods. Inflammatory foci in +/+ (A) and −/− (B) were comparable in number (see Table I) and appearance, with inflammatory cells surrounding hepatocytes with MCMV-specific cytomorphological alterations (large arrow in C, from a liver of a −/− mouse). In 6/10 −/− mice (but in none out of six +/+ mice, see Table I), large areas of necrosis (shown by arrows in E) were present despite inflammatory foci (shown by arrows in D). Further amplification revealed necrotic areas surrounded by inflammatory mononuclear cells in these −/− mice (F). A, B, D, E: ×132; scale bars, 75.7 μm. C, F: ×270; scale bars, 37 μm).
Table I.
MCMV-induced Hepatitis in KARAP/DAP12 Loss-of-Function Mice
| Severea | Moderateb | Minimalc | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No.of mice | Viraltiters | Inflammatoryfoci/area liver | No.of mice | Viraltiters | Inflammatoryfoci/area liver | No.of mice | Viraltiters | Inflammatoryfoci/area liver | |
| −/− | 6 | 4.59 ± 0.21 | 66 ± 14 | 0 | — | — | 4 | 4.36 ± 0.24 | 61 ± 32 |
| +/− | 1 | 4.61 ± 0 | 100 ± 0 | 3 | 3.90 ± 0.29 | 81 ± 9 | 5 | 4.16 ± 0.28 | 90 ± 16 |
| +/+ | 0 | — | — | 0 | — | — | 6d | 3.90 ± 0.17 | 80 ± 25 |
Liver Lymphocytes Derived from KARAP/DAP12 Loss-of-Function Mice Show an Impaired IFN-γ Production after MCMV Infection.
To investigate if the increase in viral titers and the severity of liver damage in mutant mice were due to a decreased capability of NK cells to produce IFN-γ, we isolated liver lymphocytes from −/−, +/−, +/+, and C57BL/6 mice 2 d after MCMV infection, when a peak in the IFN-γ production occurs (13, 36). The cells were triple-stained with NK1.1, anti-CD3, and anti-IFN-γ mAbs and analyzed by flow cytometry. As shown in Fig. 4 A, among all liver lymphocytes isolated from C57BL/6 mice, the average percentage (± SD) of IFN-γ1 cells was 24 ± 7. This percentage was strongly decreased in −/− mice (9 ± 5) (P = 0.0004) and partially decreased in +/− mice (17 ± 9). Almost all of these IFN-γ producing cells were NK1.1+. Thus, while in C57BL/6 mice the average percentage of NK1.1+IFN-γ1 cells was 22 ± 6%, this percentage drastically decreased to 8 ± 4% in −/− mice. In +/− mice, we saw only a partial decrease in the NK1.1+IFN-γ1 population (16 ± 9%). When NK cells were analyzed in detail, by gating on the CD3−NK1.1+ population (Fig. 4 B), we observed that in C57BL/6 mice 36 ± 4% of NK cells were producing IFN-γ, while in −/− mice the corresponding proportion was only 16 ± 8% (P = 0.00017). In +/− mice 24 ± 12% (P = 0.039) of the NK cells produced IFN-γ. Thus, the percentage of IFN-γ producing liver NK cells isolated from the KARAP/DAP12 loss-of-function mice 2 d after MCMV infection showed a reduction of 56 ± 22 compared with C57BL/6 mice. Similar results were obtained when cells were stained with DX5 mAb, instead of anti-NK1.1 mAb (data not shown).
Figure 4.

Decreased IFN-γ production by liver NK cells from KARAP/DAP12 loss-of-function mice. Groups of C57BL/6, +/−, and −/− mice were MCMV-infected and on day 2 after infection, liver leukocytes were isolated, stained for CD3, NK1.1, and IFN-γ expression, and analyzed by flow cytometry. (A) Representative density plots obtained from cells gated in the lymphocyte population are shown. The numbers in the quadrants indicate the percentage of liver lymphocytes expressing NK1.1 and/or IFN-γ. (B) The same cells shown in A were instead gated on CD3−NK1.1+ NK cells. The percentage of NK cells producing IFN-γ is shown (thick solid line). The thin solid line represents histograms of cells stained with isotype control mAb. The results shown are derived from one mouse for each group and are representative of a total of six C57BL/6, six +/−, and seven −/− mice tested in two independent experiments, with similar results.
Expression of Ly49H on NK Cells in KARAP/DAP12 Loss-of-Function Mice.
While this work was in progress, three groups have independently shown that the NK cell–mediated MCMV resistance is dependent on cells expressing the KARAP/DAP12-associated receptor Ly49H (18–20). Thus, we wanted to investigate the expression of Ly49H on NK cells derived from the KARAP/DAP12 mutant mice used in our study. Splenocytes, liver, and peripheral blood leukocytes from −/−, +/−, and +/+ mice were isolated and stained with the anti-Ly49H mAb 3D10, together with anti-NK1.1 and anti-CD3 mAbs, and analyzed by flow cytometry. Analysis of NK1.1+CD3− peripheral blood lymphocytes showed that the percentage of Ly49H+ NK cells was 47 ± 2 in −/−, 57 ± 3 in +/−, and 53 ± 6 in +/+ mice (Fig. 5) . The median of fluorescence intensity (MFI) of Ly49H staining was reduced at a greater extent. MFI values were 32 ± 0 in −/−, 79 ± 2 in +/−, and 88 ± 2 in +/+ mice (Fig. 5). Analysis of splenocytes and liver lymphocytes resulted in a similar pattern, with the greatest reduction observed on MFI values (data not shown). Ly49H expression was reduced at a similar extent also in KARAP/DAP12 mutant mice were the loxP-neo-loxP cassette has been deleted (reference 29, and data not shown).
Figure 5.

Expression of Ly49H on NK cells from KARAP/DAP12 loss-of-function mice. Peripheral blood leukocytes were isolated and triple-stained with anti-Ly49H mAb 3D10, anti-NK1.1, and anti-CD3 mAbs, and analyzed by flow cytometry. The results shown are derived from one representative mouse per group. The histograms are obtained from cells gated on CD3−NK1.1+ NK cells. Dotted line represents negative control.
Discussion
Our work demonstrates for the first time that the pathways dependent on the ITAM-bearing molecule KARAP/DAP12 are crucial for the NK cell–mediated resistance to MCMV infection in vivo, and that there is no redundancy in these pathways. Numerous studies have shown a clear role for NK cell inhibitory and activating receptors in bone marrow graft rejection and tumor resistance, but less is known about the role of these receptors in the regulation of anti-viral functions. On murine NK cells, all MHC class I–specific activating receptors known so far (i.e., Ly49H, Ly49D, Ly49P, and CD94/NKG2C molecules) are associated with KARAP/DAP12 dimers (25). Therefore, our KARAP/DAP12 loss-of-function mice serve as a unique tool when studying regulation of NK cell functions by this subset of activating receptors. Viral titers increased up to 40-fold in the spleen and fivefold in the liver of these mutant mice. This difference strongly suggests KARAP/DAP12-dependent pathways to be critical for the control of MCMV replication in these organs. We observed higher viral titers also in the lungs of the mutant mice. The increase was not statistically significant, but similar to what has been previously observed in NK cell–depleted C57BL/6 mice (6), suggesting that, also in the lungs, the control of MCMV infection and spread is influenced by KARAP/DAP12.
In the liver, MCMV induces distinct patterns of disease at early times after infection. Inflammatory foci are observed within the liver parenchyma and necrotic foci are visible in some strains of mice (11, 33, 35). Interestingly, it appears from our experiments that the ability of NK cells to be recruited to the infected liver (most likely through a MIP-1α–dependent mechanism [14]), as well as their capability to cluster and form inflammatory foci was not greatly affected by the absence of a functional KARAP/DAP12 molecule. This indicates that KARAP/DAP12-associated receptors are not crucial for these early events. Our results rather suggest that the KARAP/DAP12 deficiency instead impaired the capacity of NK cells to limit replication and spread of the virus in the initial foci. This more frequently led to large areas of focal necrosis. The main mechanism for NK cell–mediated control of MCMV replication is known to be dependent mostly on IFN-γ production (11, 12). Orange et al. have shown that the number of both the inflammatory and necrotic foci increases after neutralization of early NK cell–dependent IFN-γ production (11). It has recently been reported that, after MCMV infection in C57BL/6 mice, ∼40% of the NK1.1+CD3− liver NK cells spontaneously produced IFN-γ (see also Fig. 4 B in this study) and that the majority of them were Ly49H+ (20). Our results from the triple staining of liver-derived lymphocytes show an average of 56 ± 22% decrease in the percentage of IFN-γ producing NK cells within the livers of the −/− mice, suggesting that MCMV-induced IFN-γ production is partly dependent on KARAP/DAP12-dependent pathways in NK cells. It is therefore likely that the observed development of a more severe hepatitis was due to lower IFN-γ production. We consistently observed a difference in hepatic viral titers between untreated −/− mice and −/− mice treated with anti-NK1.1 mAb. Additional NK cell–dependent mechanisms, which can mediate the clearance of MCMV in the liver, independently of KARAP/DAP12 expression and signaling, can therefore not be excluded. In fact, it has been previously shown that IL-12–dependent production of IFN-γ by NK cells is important for an efficient anti-MCMV response (11, 13, 36). Thus, it is possible that in order to achieve an optimal level of IFN-γ in the liver, both IL-12–dependent and KARAP/DAP12-dependent signals are required. It remains open whether IL-12–induced IFN-γ production occurs through a completely independent pathway or whether IL-12 is required for an optimal IFN-γ release triggered via KARAP/DAP12 associated receptors. On the basis of all the observations discussed above, we favor the hypothesis that at early times after MCMV infection in KARAP/DAP12 loss-of-function mice, NK cells (possibly Ly49H+) migrate into the liver and form inflammatory foci. However, their impaired capability to signal via KARAP/DAP12 for production of IFN-γ leads to higher viral titers, to a more severe hepatic necrosis and most likely to a more generalized infection.
In the spleen, the immune response to MCMV infection has been shown to be dependent mainly on perforin, most likely through cell-mediated cytotoxicity, and on the Cmv-1 locus (12, 16). The fact that treatment of KARAP/DAP12−/− mice with anti-NK1.1 mAb only resulted in a marginal and not statistically significant effect on splenic viral titers indicates that KARAP/DAP12-associated receptors expressed on NK1.1+ cells are the major players in early MCMV resistance in the spleen. An additional observation in our study was that depletion of NK1.1+ cells from −/− and +/+ mice resulted in similar viral titers, both in the spleen and in the liver. We cannot exclude that KARAP/DAP12-associated receptors expressed on NK1.1− cells (i.e., dendritic cells, monocytes) play a role in MCMV resistance, but these data suggest that their contribution, if any, is indirect and less important. Our results agree with and extend the work of three independent groups, published during the course of this study (18–20, 37). These investigators all concluded that the NK cell–mediated MCMV resistance is dependent on cells expressing the activating receptor Ly49H. While indicating that Ly49H is the key receptor in MCMV resistance in the spleen, Brown et al. and Lee et al. also suggested that this receptor may correspond to the Cmv-1 locus, that controls MCMV replication in the spleen (18, 19). Thus, our study together with recent and previous reports indicates Ly49H/KARAP(DAP12)-mediated cell cytotoxicity as the main mechanism for MCMV clearance in the spleen.
The studies recently published on Ly49H and MCMV are based on mice deficient in Ly49H expression, either as a result of genetic modifications (18, 19) or after in vivo depletion of Ly49H+ cells using mAbs (19, 20). These reports clearly demonstrated that MCMV resistance depends on the presence of Ly49H-expressing cells. If then the Ly49H receptor has a triggering function, one would expect NK cell–mediated MCMV resistance to be impaired to the same extent in a mouse where the signaling through the only known Ly49H-associated adaptor molecule, KARAP/DAP12, is abrogated and in a mouse depleted of Ly49H+ cells (29, 30). This was tested and confirmed in our model. Even if the role of other KARAP/DAP12 associated molecules cannot be completely excluded, the simplest and most straightforward interpretation of the combined evidence from our and the previous studies is that Ly49H is directly involved in resistance to MCMV through its cell activation capacity via KARAP/DAP12. The quantitative effect on MCMV resistance of Ly49H gene deficiency, Ly49H+ cell depletion, and KARAP/DAP12 loss-of-function appears similar, with one possible exception. The viral titers in the spleen were higher in mice lacking Ly49H as a result of a genetic modification (103 to 104 fold) (18, 19) compared with our mice (30–40-fold; present study) and to mice depleted of Ly49H+ NK cells (20–103-fold) (19, 20), at least at an early time after infection. The reason for this difference is at present unknown and may simply be technical, i.e. related to the different recombinant strains of mice or to different amounts of virus used in the experiments. The increase in viral titers in the liver of deficient mice was instead about fivefold in all models including ours (19, 20).
In light of these recent findings regarding the role of Ly49H in resistance to MCMV infection (18–20, 37), we analyzed Ly49H expression in the KARAP/DAP12 mutant mice. Similar to the mice where the lox-P flanked neo cassette at the site of mutation is deleted (29), we found that Ly49H was expressed on NK cells from blood, liver, and spleen of our mutant mice, though at a lower level compared with control mice. Thus, the reduction is due to the disruption of the ITAM, and not to the presence of the loxP-neo-loxP cassette in the cytoplasmic tail of KARAP/DAP12. We therefore believe that a functional ITAM is required for normal expression of Ly49H, either because it is required for optimal transport to the cell surface or because Ly49H/KARAP(DAP12) complex turnover is affected by the absence of signaling through KARAP/DAP12. It could be argued that the reduction in Ly49H expression might result in susceptibility to MCMV infection, independently of signaling through this receptor. However, if MCMV resistance is dependent only on Ly49H expression and not signaling through KARAP/DAP12, the partial reduction in Ly49H expression observed in our −/− mice would have resulted in only a partial susceptibility to MCMV. Our results instead clearly show that in our mutant mice viral titers were as high as in mice where Ly49H+ cells were depleted (19, 20). Thus, although we cannot formally exclude the possibility that the density of Ly49H molecules on the cell surface has to reach a certain threshold level in order to enable adequate MCMV resistance, our results suggest that the signaling pathways activated by and**/**or dependent on Ly49H/KARAP(DAP12) are necessary for the clearance of MCMV and that there is no redundancy in these pathways.
The questions of when, where and by what KARAP/DAP12-dependent activating signals are triggered remain to be studied. We believe that the answers will be crucial for a better understanding of the mechanisms responsible of the NK cell–dependent control of MCMV in vivo. Such data may also lead to novel and alternative therapeutic strategies that could be transferred to human CMV-induced diseases.
In the last years the discussion of specificity of NK cell cytotoxicity has emphasized “missing self recognition” (38). This concept is based on the idea that triggering is mediated via multiple, overlapping and redundant receptors recognizing most cells, while the crucial step in discrimination between normal and infected or otherwise abnormal cells occurs via inhibitory, MHC class I–specific receptors. However, most evidence argue against the possibility that NK cells can discriminate between infected and uninfected cells only on the basis of virus-induced MHC class I downmodulation (39–42). Perhaps the virus has evolved additional mechanisms to escape missing self recognition by NK cells, and NK cell–mediated resistance therefore is heavily dependent on the recognition of a particular triggering ligand. In fact, the evidence from recent and the present studies all point out to one distinct activating receptor (Ly49H) and signaling adaptor molecule (KARAP/DAP12), arguing against NK cell activation via numerous redundant receptors in resistance to CMV infection.
Acknowledgments
The authors wish to thank Maj-Britt Alter and Margerita Hagelin for excellent assistance in the animal facility, Ros-Mari Johansson for assistance with the histology, and Dr. Uwe Roblick for the help with the pictures. We also thank Drs. Christine A. Biron and Gary C. Pien for the NIH3T3 cell line and for advice, and Dr. Wayne Yokoyama for the anti-Ly49H mAb.
This work was supported by the Swedish Cancer Society, the Tobias Foundation, Karolinska Institute, Wenner-Gren, and “C.M. Lerici” Foundations (C. Cerboni); by institutional grants from Institut National de la Sante et de la Recherche Medicale, Centre National de la Recherche Scientifique, Ministère de l'Enseignement Supérieur et de la Recherche, and specific grants from Ligue Nationale contre le Cancer (Equipe labelisée La Ligue), from Association pour la Recherche contre le Cancer (E. Vivier), from Fondation pour la Recherche Médicale (E. Tomasello).
Footnotes
*
Abbreviations used in this paper: ITAM, immunoreceptor tyrosine-based activatory motif; MCMV, murine CMV; MFI, median of fluorescence intensity; MIP-1α, macrophage inflammatory protein-1α.
References
- 1.Trinchieri, G. 1989. Biology of natural killer cells. Adv. Immunol. 47:187–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yokoyama, W.M. 1999. Natural killer cells. Fundamental Immunology. W.E. Paul, editor. Lippincott-Raven, Philadelphia. 575–603.
- 3.Scott, P., and G. Trinchieri. 1995. The role of natural killer cells in host-parasite interactions. Curr. Opin. Immunol. 7:34–40. [DOI] [PubMed] [Google Scholar]
- 4.Biron, C.A., K.B. Nguyen, G.C. Pien, L.P. Cousens, and T.P. Salazar-Mather. 1999. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17:189–220. [DOI] [PubMed] [Google Scholar]
- 5.Shellam, G.R., J.E. Allan, J.M. Papadimitriou, and G.J. Bancroft. 1981. Increased susceptibility to cytomegalovirus infection in beige mutant mice. Proc. Natl. Acad. Sci. USA. 78:5104–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bukowski, J.F., B.A. Woda, and R.M. Welsh. 1984. Pathogenesis of murine cytomegalovirus infection in natural killer cell-depleted mice. J. Virol. 52:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shanley, J.D. 1990. In vivo administration of monoclonal antibody to the NK 1.1 antigen of natural killer cells: effect on acute murine cytomegalovirus infection. J. Med. Virol. 30:58–60. [DOI] [PubMed] [Google Scholar]
- 8.Welsh, R.M., C.L. O'Donnell, and L.D. Shultz. 1994. Antiviral activity of NK 1.1+ natural killer cells in C57BL/6 scid mice infected with murine cytomegalovirus. Nat. Immunol. 13:239–245. [PubMed] [Google Scholar]
- 9.Bukowski, J.F., J.F. Warner, G. Dennert, and R.M. Welsh. 1985. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J. Exp. Med. 161:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bukowski, J.F., H. Yang, and R.M. Welsh. 1988. Antiviral effect of lymphokine-activated killer cells: characterization of effector cells mediating prophylaxis. J. Virol. 62:3642–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orange, J.S., B. Wang, C. Terhorst, and C.A. Biron. 1995. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 182:1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tay, C.H., and R.M. Welsh. 1997. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J. Virol. 71:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orange, J.S., and C.A. Biron. 1996. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 156:1138–1142. [PubMed] [Google Scholar]
- 14.Salazar-Mather, T.P., J.S. Orange, and C.A. Biron. 1998. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP-1alpha)-dependent pathways. J. Exp. Med. 187:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salazar-Mather, T.P., T.A. Hamilton, and C.A. Biron. 2000. A chemokine-to-cytokine-to-chemokine cascade critical in antiviral defense. J. Clin. Invest. 105:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scalzo, A.A., N.A. Fitzgerald, A. Simmons, A.B. La Vista, and G.R. Shellam. 1990. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J. Exp. Med. 171:1469–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scalzo, A.A., P.A. Lyons, N.A. Fitzgerald, C.A. Forbes, W.M. Yokoyama, and G.R. Shellam. 1995. Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics. 27:435–441. [DOI] [PubMed] [Google Scholar]
- 18.Lee, S.H., S. Girard, D. Macina, M. Busa, A. Zafer, A. Belouchi, P. Gros, and S.M. Vidal. 2001. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat. Genet. 28:42–45. [DOI] [PubMed] [Google Scholar]
- 19.Brown, M.G., A.O. Dokun, J.W. Heusel, H.R. Smith, D.L. Beckman, E.A. Blattenberger, C.E. Dubbelde, L.R. Stone, A.A. Scalzo, and W.M. Yokoyama. 2001. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 292:934–937. [DOI] [PubMed] [Google Scholar]
- 20.Daniels, K.A., G. Devora, W.C. Lai, C.L. O'Donnell, M. Bennett, and R.M. Welsh. 2001. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to ly49h. J. Exp. Med. 194:29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bakker, A.B., J. Wu, J.H. Phillips, and L.L. Lanier. 2000. NK cell activation: distinct stimulatory pathways counterbalancing inhibitory signals. Hum. Immunol. 61:18–27. [DOI] [PubMed] [Google Scholar]
- 22.Blery, M., L. Olcese, and E. Vivier. 2000. Early signaling via inhibitory and activating NK receptors. Hum. Immunol. 61:51–64. [DOI] [PubMed] [Google Scholar]
- 23.Lanier, L.L. 2001. On guard—activating NK cell receptors. Nat. Immunol. 2:23–27. [DOI] [PubMed] [Google Scholar]
- 24.Vivier, E., and M. Daeron. 1997. Immunoreceptor tyrosine-based inhibition motifs. Immunol. Today. 18:286–291. [DOI] [PubMed] [Google Scholar]
- 25.Lanier, L.L., and A.B. Bakker. 2000. The ITAM-bearing transmembrane adaptor DAP12 in lymphoid and myeloid cell function. Immunol. Today. 21:611–614. [DOI] [PubMed] [Google Scholar]
- 26.Lanier, L.L., B.C. Corliss, J. Wu, C. Leong, and J.H. Phillips. 1998. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 391:703–707. [DOI] [PubMed] [Google Scholar]
- 27.Tomasello, E., L. Olcese, F. Vely, C. Geourgeon, M. Blery, A. Moqrich, D. Gautheret, M. Djabali, M.G. Mattei, and E. Vivier. 1998. Gene structure, expression pattern, and biological activity of mouse killer cell activating receptor-associated protein (KARAP)/DAP-12. J. Biol. Chem. 273:34115–34119. [DOI] [PubMed] [Google Scholar]
- 28.McVicar, D.W., L.S. Taylor, P. Gosselin, J. Willette-Brown, A.I. Mikhael, R.L. Geahlen, M.C. Nakamura, P. Linnemeyer, W.E. Seaman, S.K. Anderson, et al. 1998. DAP12-mediated signal transduction in natural killer cells. A dominant role for the Syk protein-tyrosine kinase. J. Biol. Chem. 273:32934–32942. [DOI] [PubMed] [Google Scholar]
- 29.Tomasello, E., P.O. Desmoulins, K. Chemin, S. Guia, H. Cremer, J. Ortaldo, P. Love, D. Kaiserlian, and E. Vivier. 2000. Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity. 13:355–364. [DOI] [PubMed] [Google Scholar]
- 30.Bakker, A.B., R.M. Hoek, A. Cerwenka, B. Blom, L. Lucian, T. McNeil, R. Murray, L.H. Phillips, J.D. Sedgwick, and L.L. Lanier. 2000. DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity. 13:345–353. [DOI] [PubMed] [Google Scholar]
- 31.Selgrade, M.K., J.G. Nedrud, A.M. Collier, and D.E. Gardner. 1981. Effects of cell source, mouse strain, and immunosuppressive treatment on production of virulent and attenuated murine cytomegalovirus. Infect. Immun. 33:840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tay, C.H., L.Y. Yu, V. Kumar, L. Mason, J.R. Ortaldo, and R.M. Welsh. 1999. The role of LY49 NK cell subsets in the regulation of murine cytomegalovirus infections. J. Immunol. 162:718–726. [PubMed] [Google Scholar]
- 33.Orange, J.S., T.P. Salazar-Mather, S.M. Opal, and C.A. Biron. 1997. Mechanisms for virus-induced liver disease: tumor necrosis factor-mediated pathology independent of natural killer and T cells during murine cytomegalovirus infection. J. Virol. 71:9248–9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arase, H., T. Saito, J.H. Phillips, and L.L. Lanier. 2001. Cutting edge: the mouse NK cell-associated antigen recognized by DX5 monoclonal antibody is CD49b (alpha(2) integrin, very late antigen-2). J. Immunol. 167:1141–1144. [DOI] [PubMed] [Google Scholar]
- 35.Bukowski, J.F., B.A. Woda, S. Habu, K. Okumura, and R.M. Welsh. 1983. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J. Immunol. 131:1531–1538. [PubMed] [Google Scholar]
- 36.Pien, G.C., A.R. Satoskar, K. Takeda, S. Akira, and C.A. Biron. 2000. Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J. Immunol. 165:4787–4791. [DOI] [PubMed] [Google Scholar]
- 37.Dokun, A.O., S. Kim, H.R. Smith, H.S. Kang, D.T. Chu, and W.M. Yokoyama. 2001. Specific and nonspecific NK cell activation during virus infection. Nat. Immunol. 2:951–956. [DOI] [PubMed] [Google Scholar]
- 38.Ljunggren, H.G., and K. Karre. 1990. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today. 11:237–244. [DOI] [PubMed] [Google Scholar]
- 39.Tay, C.H., R.M. Welsh, and R.R. Brutkiewicz. 1995. NK cell response to viral infections in beta 2-microglobulin-deficient mice. J. Immunol. 154:780–789. [PubMed] [Google Scholar]
- 40.Polic, B., S. Jonjic, I. Pavic, I. Crnkovic, I. Zorica, H. Hengel, P. Lucin, and U.H. Koszinowski. 1996. Lack of MHC class I complex expression has no effect on spread and control of cytomegalovirus infection in vivo. J. Gen. Virol. 77:217–225. [DOI] [PubMed] [Google Scholar]
- 41.Ploegh, H.L. 1998. Viral strategies of immune evasion. Science. 280:248–253. [DOI] [PubMed] [Google Scholar]
- 42.Alcami, A., and U.H. Koszinowski. 2000. Viral mechanisms of immune evasion. Immunol. Today. 21:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]