Human Cytomegalovirus Glycoprotein UL16 Causes Intracellular Sequestration of NKG2D Ligands, Protecting Against Natural Killer Cell Cytotoxicity (original) (raw)
Abstract
The activating receptor, NKG2D, is expressed on a variety of immune effector cells and recognizes divergent families of major histocompatibility complex (MHC) class I–related ligands, including the MIC and ULBP proteins. Infection, stress, or transformation can induce NKG2D ligand expression, resulting in effector cell activation and killing of the ligand-expressing target cell. The human cytomegalovirus (HCMV) membrane glycoprotein, UL16, binds to three of the five known ligands for human NKG2D. UL16 is retained in the endoplasmic reticulum and cis-Golgi apparatus of cells and causes MICB to be similarly retained and stabilized within cells. Coexpression of UL16 markedly reduces cell surface levels of MICB, ULBP1, and ULBP2, and decreases susceptibility to natural killer cell–mediated cytotoxicity. Domain swapping experiments demonstrate that the transmembrane and cytoplasmic domains of UL16 are important for intracellular retention of UL16, whereas the ectodomain of UL16 participates in down-regulation of NKG2D ligands. The intracellular sequestration of NKG2D ligands by UL16 represents a novel HCMV immune evasion mechanism to add to the well-documented viral strategies directed against antigen presentation by classical MHC molecules.
Keywords: ULBP, MIC, NKG2D, UL16, HCMV
Introduction
Human cytomegalovirus is well adapted, establishing a lifelong, usually benign, relationship with most hosts. Typically, primary infections occur in childhood resulting in mild or inapparent disease followed by asymptomatic, lifetime persistence of the virus with intermittent low level shedding of infectious particles. However, in very young children and immunocompromised individuals, human CMV (HCMV)*replicates to relatively high levels in several different organs, frequently resulting in morbidity and mortality (1). After reactivation from latency, HCMV faces robust, fully primed host immunity. To counter this, the virus uses a repertoire of immune evasion strategies that can open “a window of opportunity” allowing virus replication for a time or in a specific cell type. There are four membrane glycoproteins encoded in the US region of the HCMV genome (US2, US3, US6, and US11) that can inhibit the MHC class I antigen presentation pathway by independent mechanisms. These include prevention of cell surface expression of MHC class I by retention in the ER, increased degradation of MHC class I, and prevention of the transport of peptides into the ER by the TAP transporter (2, 3). In addition, two of these, US2 and US3, can block MHC class II–mediated presentation to CD4+ T cells (4–6).
Decreased antigen presentation by virus-infected cells would be expected to protect from T cell–mediated recognition, but low levels of MHC class I expression might also predispose these cells to lysis by NK cells, due to decreased engagement of NK cell MHC class I–specific inhibitory receptors. The importance of NK cells in controlling infection by both HCMV and mouse CMV (MCMV) has been documented (7, 8), but much less is known about HCMV immune evasion mechanisms directed at NK cells. Although HCMV-encoded proteins UL18 and UL40 have been proposed to inhibit NK function by different mechanisms, our understanding of how HCMV avoids immunosurveillance by NK cells is clearly incomplete (9–11).
Recent studies have revealed that NK cells and other leukocytes express a variety of inhibitory and activating receptors for classical and nonclassical MHC class I antigens and other ligands. The balance between engagement of these opposing classes of receptors is believed to control leukocyte activation (12–14). One of the activating receptors, the C-type lectin-like molecule, NKG2D, has attracted particular attention as an important mediator of innate and adaptive immune responses (15). NKG2D is expressed on NK cells, CD8+ T cells, some γδ T cells, some NK-T cells, and activated macrophages (16–22). NKG2D associates with the membrane-bound signaling adaptor protein, DAP10, to transduce a potent activating signal that can stimulate cytotoxicity, proliferation, and the production of cytokines, chemokines, and nitric oxide (16–19, 23–27). Very recently, NKG2D was also shown to associate with the DAP12/KARAP signaling adaptor in mouse NK cells and macrophages (28, 29).
Ligands for NKG2D belong to distinct and divergent families of nonclassical MHC class I–like molecules. Human MICA and MICB are encoded by closely related, polymorphic genes that map to the MHC (30, 31). Like classical MHC class I antigens, they contain α1, α2, and α3 extracellular domains and are type 1 transmembrane proteins. Human ULBP1, ULBP2, and ULBP3 genes map outside the MHC on chromosome 6q25, and the encoded proteins contain only α1 and α2 domains, and are glycosylphosphatidylinositol-linked to the cell surface (24). There are no known mouse equivalents to the MIC genes, but several mouse ligands for NKG2D have been described (18, 19, 32). All contain α1 and α2 domains, like ULBPs, but with low sequence identity. The Rae1 proteins are glycosylphosphatidylinositol-linked, whereas H60 and MULT-1 are type 1 membrane proteins.
Studies on the regulation of NKG2D ligand expression have shown increased MIC expression after heat shock, oxidative stress, transformation, and infection by certain viruses and bacteria (27, 33–38). NKG2D ligand expression is commonly found on tumor-derived cell lines (18, 19, 24, 39, 40), and both H60 and Rae1 are induced in mouse skin by carcinogen treatment (41). Studies using target cells transfected with NKG2D ligands have shown that their expression stimulates NK cytotoxicity even in the presence of normal levels of classical MHC class I antigens on the target cells (16, 24). These findings support a general model in which increased NKG2D ligand expression provides a “danger” or “damage” signal to immune effector cells that allows them to kill stressed, transformed, or infected cells (15). Thus, the NKG2D/NKG2D ligand system potentially represents a new type of immune surveillance mechanism that can operate against cells with normal MHC class I expression.
ULBP1 was initially discovered as a protein that bound to the HCMV-encoded membrane glycoprotein, UL16. This viral protein also binds to ULBP2 and MICB, but not to ULBP3 nor MICA, suggesting that it might be involved in subversion of the NKG2D system. Two possible mechanisms by which this could occur in the context of a CMV-infected cell were proposed. First, cell surface UL16 could bind to cell surface NKG2D ligands and thereby prevent NKG2D–NKG2D ligand interaction, or second, UL16 and NKG2D ligands could interact intracellularly to alter the trafficking of NKG2D ligands and prevent their expression on the cell surface (24, 42). Here, we present evidence that favors the second hypothesis, provides insight into the mechanisms involved, and supports a role for UL16 as an HCMV-encoded immunoevasion protein.
Materials and Methods
Cell Lines and Purification of Cells.
Daudi cells, transduced to express MHC class I antigens and ULBP1, have been described (24). EL4 (American Type Culture Collection TIB-39) are a murine T lymphoma cell line. His16 cells, a derivative of U373 human glioma cells, have been described (4). U373-MICBNeo15 (MBN15) were derived by transfecting U373 cells with pDC409-MicB7 and pSV2-Neo, and selection in media containing 100 μg/ml G418 sulfate (GIBCO BRL).
For data shown in Fig. 1, short-term cultured primary human NK cells were obtained as previously described (43, 44). For data shown in Figs. 8 and 9, freshly isolated primary human NK cells were obtained from peripheral blood by negative selection using the Rosette Sep human NK cell enrichment cocktail kit (StemCell Technologies Inc.) according to the manufacturer's specifications.
Figure 1.

Soluble UL16 does not block ULBP-mediated cytotoxicity. Daudi cells expressing MHC class I and ULBP1 were used as targets in cytotoxicity assays using human NK cells, at the indicated effector/target ratios. Class I+/ULBP1+ Daudi targets were preincubated with the anti-ULBP1 Fab, M291, a control Fab, the UL16LZ protein, or a control LZ protein (RANK ligand-LZ), before the addition of effector cells. Individual data points are calculated from the averages of triplicate samples. The results shown are representative of three experiments using three separate donors.
Figure 8.

Enhanced cytotoxicity of human and mouse NK cells against EL4 cells transduced with ULBPs or MICB. (A) EL4 cells expressing ULBP1, ULBP2, ULBP3, or MICB were tested as targets in cytotoxicity assays using mouse NK cells as effectors. Individual data points are calculated from the averages of triplicate samples. The results shown are representative of three separate experiments. (B) EL4 cells expressing ULBP2, ULBP3, or MICB were tested as targets in cytotoxicity assays using human NK cells as effectors. The results shown are from a single donor and are representative of three experiments using three separate donors.
Figure 9.

Cell surface expression of ULBP1, ULBP2, or MICB correlates with sensitivity to NK lysis. EL4 cells were transduced with amphotropic retroviruses encoding ULBP1 (A), ULBP2 (B), ULBP3 (C), or MICB (D). The ULBP- or MICB-expressing EL4 populations were further transduced with VSVG-pseudotyped retroviruses encoding both UL16 and GFP. Transduced cells were separated into two cell populations: those expressing GFP and UL16 (UL16+) and those not expressing GFP and UL16 (UL16−). Cell populations were used as targets in cytotoxicity assays using mouse NK cells (A–C) or human NK cells (D). The results shown are representative of three separate experiments.
Murine NK cells were expanded from splenocytes of C57BL/6 SCID or RAG2−/− mice (The Jackson Laboratory) by growth for 4 d in 200 ng/ml rhuIL-15 (Immunex). Cultures containing ≥90% NK cells, as analyzed on day 3, were used in cytotoxicity assays.
Adenovirus (Ad) Constructs.
Replication-defective (E1−) Ad vectors expressing HCMV UL16, and the cellular proteins MICB, MICA, and ULBP2 were constructed and propagated as previously described (45). For glycoprotein expression, His16 or MBN15 cells were coinfected with Ad vectors AdtetUL16, AdtetMICB, AdtetMICA, or AdtetULBP2, and a second vector, Adtet-transactivator (Adtet-Trans; using 20% of the amount of other Ad vectors), which expresses a transactivator protein that activates the promoter without the need for tetracycline. AdtetUS9 (45) was used as a control Ad vector.
Flow Cytometric Analysis.
The following monoclonal antibodies were used for flow cytometric analysis: M90, anti-hCD40L used as a mouse IgG1 isotype control; M230, mouse IgG1 anti-UL16; M291 and M295, mouse IgG1 anti-ULBP1; M311, mouse IgG1 anti-ULBP2; M550, mouse IgG1 anti-ULBP3; M673 mouse IgG1 anti-MICA; M360, mouse IgG1 anti-MICB; M2, rat IgG2a anti–muIL-4R; and M149, rat IgG2a anti–muIL-15 used as a rat IgG2a isotype control.
Specific binding was detected with either a PE-conjugated F(ab′)2 fragment goat anti–mouse IgG (Jackson ImmunoResearch Laboratories), a PE-conjugated F(ab′)2 fragment goat anti–rat IgG (Jackson ImmunoResearch Laboratories), or FITC-conjugated goat anti–mouse IgG. After staining, cells were analyzed on a Becton Dickinson FACScan™ or FACSCalibur®.
Confocal Immunofluorescence Microscopy.
MBN15 or His16 cells were seeded onto 22-mm diameter glass coverslips overnight and infected with Ad vectors for 12 h before fixing in PBS 4% paraformaldehyde. Cycloheximide chase experiments were performed as described above, with an additional 4 h of treatment with 100 μg/ml cycloheximide. After permeabilization for 15 min with PBS 0.2% Triton X-100 and blocking for 1 h with PBS 2% goat serum, 1% fish gelatin, and 0.02% Tween 20, the cells were incubated with primary antibodies diluted in blocking buffer for 1 h. Antibodies to calreticulin and GM130 were obtained from Transduction Laboratories. Rabbit antiserum to a COOH-terminal peptide of UL16 (RLRIRLPHRYQRLRTED) was generated by immunizing rabbits with peptides conjugated onto keyhole limpet hemocyanin according to standard protocols. Cells were washed extensively with PBS containing 0.02% Tween 20 and incubated with goat anti–mouse IgG Alexa 488 and goat anti–rabbit IgG Alexa 594 secondary fluorescent antibodies (Molecular Probes) for 1 h, washed, and mounted with Prolong anti-fade agent (Molecular Probes). Cell staining was visualized on a Bio-Rad 1024 ES laser scanning confocal system attached to a Nikon Eclipse TE300 fluorescence microscope.
Labeling and Immunoprecipitation of Glycoproteins.
MBN15 or Ad-infected His16 cells were radiolabeled with 150–250 μCi/ml [35S]methionine-cysteine (PerkinElmer) 20 h after infection for the times indicated in the legends to the figures. After washing, the label was chased by culture in media containing 20-fold excess methionine-cysteine. Cell extracts were made using NP-40/deoxycholate lysis buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mg/ml BSA, and a cocktail of protease inhibitors). For immunoprecipitation, primary antibodies were rabbit anti–UL16 peptide serum, mouse anti–MICB, M360, or mouse anti–MICA, 3H5 (provided by T. Spies, Fred Hutchinson Cancer Institute, Seattle, WA), and immune complexes were collected with either protein A or protein G agarose beads (GIBCO BRL). Endoglycosidase H (endoH)f analyses were performed with enzyme preparations and protocols supplied by New England Biolabs, Inc. The protein samples were subjected to SDS-PAGE using 12% gels followed by autoradiography or PhosphorImager analysis.
Retroviral Vectors and Transduction.
EL4 cells were transduced with amphotropic retroviruses generated by insertion of cDNAs encoding ULBP1, ULBP2, ULBP3, or MICB into the LZRSpBMN-Z vector (46) followed by transfection into the Phoenix packaging line, or with vesicular stomatitis virus G protein (VSVG)-pseudotyped retroviruses (47) generated using the pBMN-internal ribosome entry site (IRES)-green fluorescent protein (GFP) vector in the 293GP packaging line (vectors and cells were provided by the Nolan Lab, Stanford University, Palo Alto, CA).
Construction and Expression of UL16/IL-4R and IL-4R/UL16 Chimeric Protein Retroviral Constructs.
The UL16/IL-4R chimera contains the extracellular domain of UL16 through residue 187, with a HindIII site 5′ and a BglII site 3′, fused to the transmembrane domain of the truncated murine IL-4R (IL-4R; reference 48) from residues 232 up to and including 258, with a BglII site 5′ and a NotI site 3′. This was then subcloned into the LZRSpBMN-Z vector, excised with SrfI, which cuts within the Psi sequence, and NotI, and subcloned into the pBMN-IRES-GFP vector.
The IL-4R/UL16 chimera contains the extracellular domain of the IL-4R through residue 231, with a HindIII site 5′ and a BglII site 3′, fused to the transmembrane and cytoplasmic domains of UL16 from residues 188 up to and including 230, with a BglII site 5′ and a NotI site 3′. This was then subcloned into the pBMN-IRES-GFP vector as described above. The sequences of the oligonucleotide primers used to amplify the selected sequences by PCR are available upon request.
Immunoblotting.
Proteins (15 μg of total EL4 lysate or 2 μg of CV-1 lysate) were treated with 2 μl recombinant _N_-glycanase (Glyko), separated on 1-mm thick 4–20% Tris glycine gels, and transferred to nitrocellulose filters. UL16 was detected with mAb M230 (1 μg/ml in PBS containing 2.5% BSA), followed by horseradish peroxidase–conjugated goat anti–mouse IgG (1:4,000 in PBST). Immunoreactive bands were visualized by enhanced chemiluminescence (Amersham Biosciences). Blots were stripped and reprobed with 0.5 μg/ml rabbit anti–STAT5a (Upstate Biotechnology) and horseradish peroxidase–conjugated goat anti–rabbit IgG (1:2,500 in PBST).
Cytotoxicity Assay.
Cytotoxicity assays were performed in the presence of Fab fragments of specific antibodies or leucine zipper (LZ) fusion proteins. UL16LZ has been described (24). 5 × 105 51Cr-labeled targets were incubated with 50 μg of the Fab fragments or LZ proteins for 20 min at 37°C, washed with media, and plated at 104 cells/well. Effectors were added and assays were performed as previously described (24).
Results
UL16 Is Not an Effective Competitor of NKG2D–NKG2D Ligand Interaction.
Previous experiments had shown that soluble UL16 prevented binding of ULBP1 to cell surface–expressed NKG2D, suggesting that UL16 could be a biological antagonist of NKG2D ligands (24). To test this hypothesis, soluble recombinant UL16 was added to a cytotoxicity assay that measured the ability of human NK cells to kill Daudi cells that coexpressed MHC class I and ULBP1. Previous work had shown that cytotoxicity in this system was dependent on ULBP1 expression and that killing could be blocked by anti-ULBP1 Fab (24) or anti-NKG2D Fab (unpublished data). In contrast, no diminution of killing was seen with the addition of soluble UL16 (UL16LZ, Fig. 1) . In other studies, we were unable to detect binding of UL16 to ULBP1, ULBP2, or MICB proteins using Biacore technology, under conditions where binding of these ligands to NKG2D was readily measured (unpublished data). The relatively weak binding of soluble UL16 to NKG2D ligands, compared with the relatively strong binding of NKG2D to NKG2D ligands, make it unlikely that UL16 could be an effective direct competitor for NKG2D–NKG2D ligand interactions.
UL16 Accumulates in the ER and cis-Golgi Apparatus.
The UL16 coding sequence predicts a type 1 membrane glycoprotein that could act to bind NKG2D ligands either in cytoplasmic membranes or on the cell surface. To examine the subcellular localization of UL16, we constructed a replication-defective Ad vector, AdtetUL16, that expresses UL16. His16 human glioma cells were infected with AdtetUL16, radiolabeled in a pulse-chase format, and UL16 was immunoprecipitated from cell extracts. Samples were treated with endoH, which removes high mannose oligosaccharides characteristic of glycoproteins in the ER and cis-Golgi. After a short pulse labeling, UL16 was largely or entirely sensitive to endoH, shifting to a faster migrating form without _N_-linked oligosaccharides (Fig. 2 A). Although glycoproteins that move out through the Golgi apparatus and to the cell surface become endoH resistant, UL16 remained predominantly endoH sensitive for the entire 8-h chase period.
Figure 2.
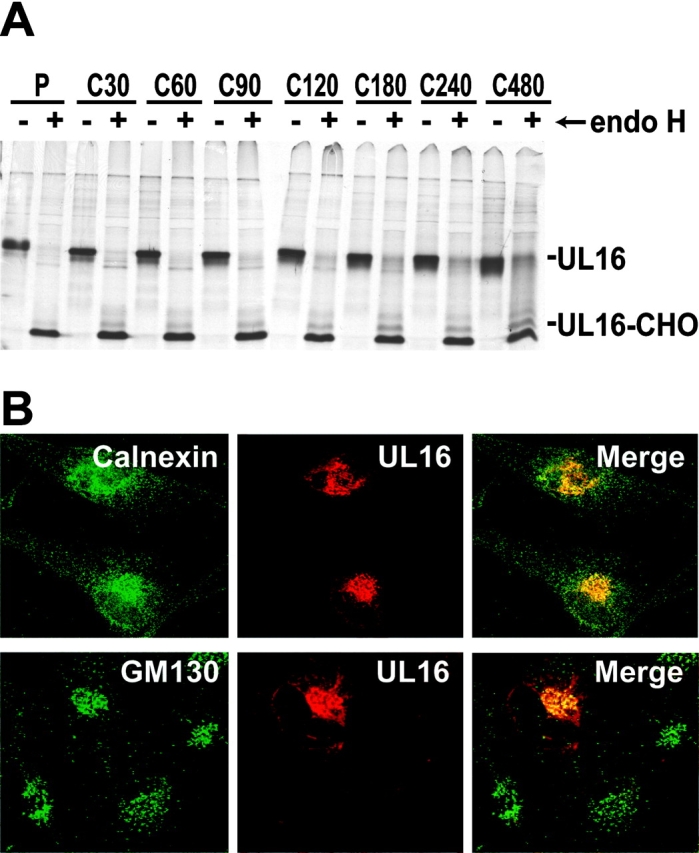
UL16 is a stable, highly glycosylated protein that localizes to the ER/cis-Golgi regions. (A) His16 cells were infected with AdtetUL16 and Adtet-trans (50 and 10 PFU/ml, respectively) for 20 h. Infected cells were labeled with [35S]methionine, a 10-min pulse (labeled P), and chases for the indicated times (in minutes, labeled C). UL16 was immunoprecipitated from cell lysates with rabbit anti–UL16-C-terminal peptide sera. For endoH treatment, samples were divided in half and not treated (−) or treated (+) with endoH. Immunoprecipitates were analyzed by SDS-PAGE and autoradiography. Bands representing UL16 or UL16 with _N_-linked oligosaccharides removed (UL16-CHO) are indicated. Preimmune rabbit serum did not precipitate these bands (unpublished data). (B) His16 cells were infected with AdtetUL16 and Adtet-trans (25 and 5 PFU/cell, respectively). After 12 h the cells were fixed, permeabilized, and incubated simultaneously with rabbit anti–UL16-C (red) and mouse antibodies to the cellular markers calnexin (green) and gm130 (green) as indicated. Primary antibodies were visualized with secondary fluorescent antibodies using a scanning laser confocal microscope. No staining was seen with preimmune rabbit serum (unpublished data).
Confocal immunofluorescence microscopy indicated that UL16 was found in perinuclear cytoplasmic membranes and rarely on the surfaces of cells (Fig. 2 B). UL16 extensively colocalized with the ER proteins, calnexin (Fig. 2 B), and calreticulin (unpublished data). There was also colocalization with two markers found predominately in the cis-Golgi apparatus, GM130 (Fig. 2 B) and p115 (unpublished data). The amount of UL16 that colocalized with these two Golgi apparatus markers was always less than with ER markers. We concluded that UL16 largely accumulates in the ER and to a lesser extent in the cis-Golgi apparatus, with little of the glycoprotein reaching the cell surface.
UL16 Prevents Cell Surface Expression of NKG2D Ligands.
To test the hypothesis that UL16 might function to alter the intracellular trafficking or sorting of NKG2D ligands, MICB and UL16, or MICB and a control protein US9, were coexpressed in His16 cells using Ad vectors. Cell surface expression of MICB was reduced by ∼90% when UL16 was coexpressed compared with cells infected with the control Ad vector (Fig. 3, A and B) . Two other NKG2D ligands, ULBP2, which is known to bind UL16, and MICA that does not bind UL16 (24), were also expressed using Ad vectors. UL16 reduced cell surface expression of ULBP2, but not MICA (Fig. 3 C). Therefore, there is a correlation between UL16 binding of NKG2D ligands and down-regulation of their cell surface expression.
Figure 3.

UL16 reduces cell surface expression of NKG2D ligands. (A and B) His16 cells were infected with 50 PFU/cell AdtetMICB and coinfected with AdtetUL16 and Adtet-trans (30 and 6 PFU/cell, respectively) to induce UL16 expression, or with Adtet-trans alone as a negative control (36 PFU/cell) for 20 h. Cells were stained with anti-MICB M360 and analyzed by flow cytometry. In A: thin black line, AdtetUL16-infected cells; thick gray line, Adtet-trans infected cells; dotted line, secondary antibody control. B quantitates cell surface expression of MICB on cells infected with Adtet-trans versus AdtetUL16. (C) His16 cells were coinfected with AdtetMICA, AdtetMICB, or AdtetULBP2 and Adtet-trans (50 and 10 PFU/cell, respectively). The cells were also infected with AdtetUL16 and Adtet-trans (50 and 10 PFU/cell, respectively) or as negative control with AdtetUS9 and Adtet-trans (also 50 and 10 PFU/cell, respectively) for 20 h. Cells were stained with anti-MICA M673, anti-MICB M360, and anti-ULBP2 M311. The mean fluorescence in each sample was normalized to the values obtained when cells were transduced with each of the NKG2D ligands alone. These results are representative of three separate experiments.
UL16 Causes MICB, but Not MICA, to be Retained in the ER and Golgi Apparatus and Stabilizes MICB.
To further characterize the effects of UL16 on MICB, a stably transfected cell line, MBN15, was produced that expresses MICB. MICB was immunoprecipitated from radiolabeled MBN15 cells in a pulse-chase format, and subsequently treated or not treated with endoH. In uninfected cells or cells infected with a control Ad vector, MICB was found largely as a single 41-kD protein species that was entirely endoH sensitive after the 10-min pulse labeling period. However, after the 60-min chase period a large fraction of the MICB displayed an increased apparent molecular weight of ∼44–52 kD, and these mature species were resistant to endoH (Fig. 4 A, top). By contrast, in cells infected with AdtetUL16, MICB remained largely in the faster migrating immature form that was endoH sensitive. To characterize the effects of UL16 on MICA, His16 cells were coinfected with AdtetMICA and AdtetUL16, or AdtetMICA and AdtetUS9. Similarly to MICB, MICA displayed an apparent molecular weight of ∼41 kD after a short pulse labeling period and this species was endoH sensitive (Fig. 4 A, bottom). The electrophoretic mobility of MICA decreased after the 60-min chase period and the glycoprotein was endoH resistant. Processing of MICA was not affected by UL16. We conclude that UL16 inhibits the processing of MICB to mature, endoH-resistant forms of the glycoprotein, but does not affect trafficking of MICA. These results are consistent with the hypothesis that in cells expressing UL16, most of the MICB leaves the ER slowly, or not at all, and does not reach the trans-Golgi apparatus.
Figure 4.

UL16 causes intracellular retention and stabilization of MICB. (A) MICB-transfected MBN15 cells (top) or His16 cells expressing MICA after infection with AdtetMICA and Adtet-trans (50 and 10 PFU/cell, respectively) were coinfected with AdtetUL16 (100 PFU/cell) or AdtetUS9 (100 PFU/cell) and Adtet-trans (20 PFU/cell) for 20 h or were left uninfected. Infected cells were labeled for 10 min with [35S]methionine and the label was chased for 60 min. MICB and MICA were immunoprecipitated from cell extracts with mouse monoclonal antibodies M360 and 3H5, respectively, treated with endoH where indicated, and analyzed by SDS-PAGE and phosphorimager. The positions of molecular weight markers of 45 and 31 kD and of several species representing mature MICB and MICA, as well as MICB and MICA with _N_-linked oligosaccharides removed (MICB-CHO, MICA-CHO), are indicated. MICA- and MICB-specific bands were identified by comparison with immunoprecipitates using isotype control antibodies and untransfected or uninfected cells (unpublished data). (B) MBN15 cells infected with AdtetUS9 or AdtetUL16 as described above were labeled for 10 min with [35S]methionine (labeled P) and the label was chased for the indicated times (in min, labeled C). MICB was immunoprecipitated as described above.
UL16 was found in perinuclear membranes, colocalizing with ER and cis-Golgi markers. To characterize the subcellular distribution of MICB in cells expressing UL16, MBN15 cells were infected with AdtetUL16 or a control Ad vector and stained with anti-MICB and anti-UL16 antibodies. Confocal microscopy indicated that UL16 extensively colocalized with MICB in a perinuclear region of the cells (Fig. 5, A–C) . In cells infected with the control Ad vector, MICB was distributed more uniformly throughout the cytoplasm and a large fraction of the protein was on the cell surface (Fig. 5, D–F). Therefore, UL16 caused MICB to be retained in perinuclear cytoplasmic membranes of cells, consistent with the endoH analyses.
Figure 5.
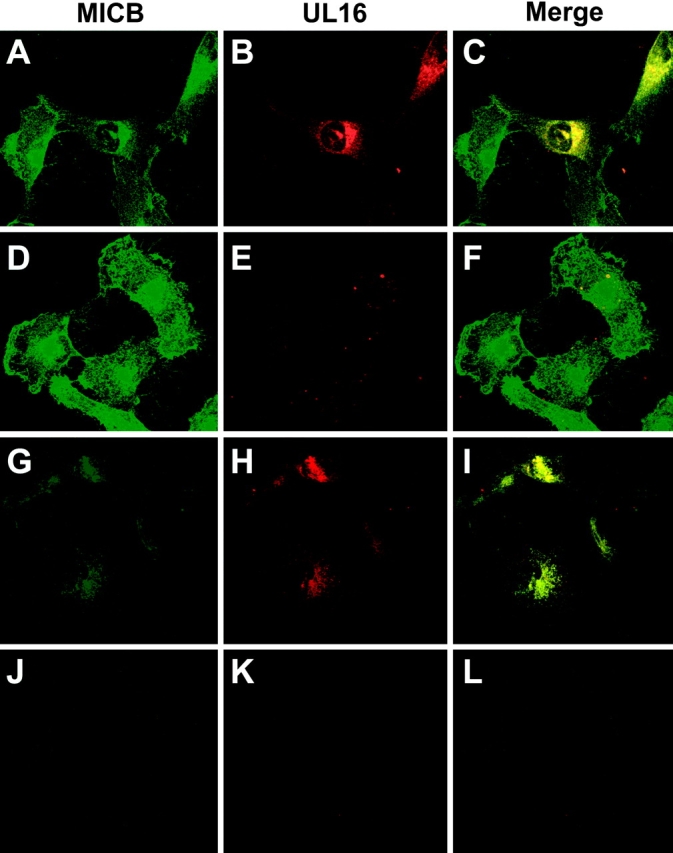
MICB colocalizes with UL16 and is retained/stabilized by the interaction. MBN15 cells expressing MICB were infected with either AdtetUL16 (25 PFU/cell) in A, B, C, G, H, and I or AdtetUS9 (25 PFU/cell) in D, E, F, J, K, and L and Adtet-trans (5 PFU/cell; in all cases) for 12 h. The infected cells were fixed immediately (A–F) or incubated for 4 h with 100 μg/ml cycloheximide and then fixed (G–L). The cells were permeabilized and incubated simultaneously with anti-MICB M360 (green) and rabbit anti–UL16-C sera (red), followed by secondary fluorescent antibodies and visualized by confocal microscopy. No staining was seen with preimmune rabbit serum or isotype control monoclonal antibody (unpublished data).
Based on pulse-chase and endoH digestion experiments, MICB traffics quickly from the ER through the Golgi apparatus to the cell surface and UL16 blocks this transport. The confocal experiments in Fig. 5, A–F, measure steady state levels of MICB. To trace the fate of MICB molecules produced after expression of UL16, MBN15 cells infected with AdtetUL16 or a control Ad vector were treated with cycloheximide to block protein synthesis. After 4 h of cycloheximide treatment, confocal microscopy of UL16-expressing cells revealed MICB in a perinuclear location colocalized with UL16 (Fig. 5, G–I). In contrast, MICB was largely undetectable after 4 h of cycloheximide treatment in cells infected with a control Ad vector (Fig. 5, J–L). To examine turnover of MICB directly, MBN15 cells were infected with AdtetUL16 or a control Ad vector, radiolabeled, and MICB was immunoprecipitated after longer chase periods. After 4 h, there was a marked loss of MICB in cells infected with the control Ad vector. PhosphorImager analysis of the 4-h chase compared with pulse samples indicated that ∼90% of the MICB protein was lost. By contrast, MICB was more stable in cells coexpressing UL16 (Fig. 4 B), with only an 11% loss of MICB during the 4-h chase period. Much of the MICB observed after the 4-h chase remained sensitive to endoH and was stabilized by the presence of UL16. It appears that in the absence of UL16, MICB reaches the cell surface relatively quickly, within 60–90 min, and is then rapidly turned over. The presence of UL16 prevents transport of MICB to the cell surface and reduces turnover.
Down-regulation of Cell Surface Expression of NKG2D Ligands by UL16.
To examine in more detail the consequences of coexpression of UL16 and NKG2D ligands on susceptibility to NK cytotoxicity, we wished to use stable transduction of a cell type that lacked expression of endogenous NKG2D ligands. His16 cells express several human NKG2D ligands and were efficiently killed by NK cells (unpublished data), but EL4 mouse thymoma cells have been shown to lack detectable expression of NKG2D ligands (18, 19, unpublished data) and were chosen for these experiments. EL4 cells were separately transduced with retroviral vectors expressing ULBP1, ULBP2, ULBP3, and MICB. Cell populations expressing high levels of the human NKG2D ligands were derived by cell sorting using specific antibodies to the ligands. UL16 was transduced into each of these EL4 cell populations using a bicistronic retroviral vector in which the UL16 cDNA was followed by an IRES and cDNA encoding GFP. GFP-expressing cells were enriched by cell sorting and analyzed by flow cytometry for NKG2D ligand and GFP expression. Fig. 6 A shows that the NKG2D ligand-transduced cells express uniformly high levels of MICB and the ULBPs, but that transduction by the UL16-IRES-GFP retrovirus followed by a single round of sorting for GFP-expressing cells resulted in decreased levels of cell surface MICB, ULBP1, and ULBP2 in a substantial fraction of cells. In contrast, surface expression of ULBP3, which does not bind to UL16 (24), was unchanged by the presence of UL16.
Figure 6.
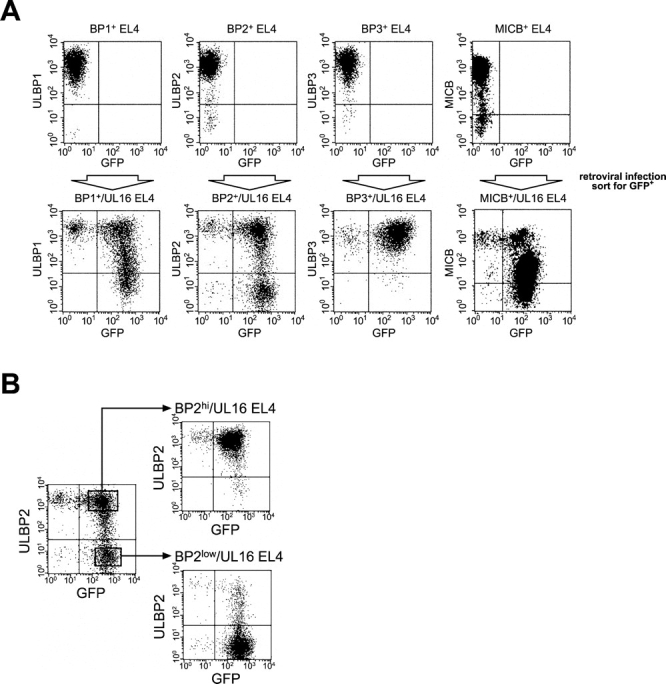

UL16 decreases cell surface expression of ULBP1, ULBP2, and MICB, but not ULBP3, in EL4 cells. (A) EL4 cells expressing the ULBPs or MICB were further transduced with retroviruses encoding both UL16 and GFP. Transduced cells were enriched by one round of fluorescence-activated cell sorting for cells expressing GFP, stained with specific monoclonal antibodies, and analyzed by flow cytometry. (B) EL4 cells transduced with ULBP2 and UL16 (ULBP2+/UL16 EL4) were further sorted by flow cytometry to obtain populations expressing high (BP2hi/UL16 EL4) or low (BP2low/UL16 EL4) cell surface levels of ULBP2. (C) Cell lysates were prepared from ULBP2+ EL4 cells, ULBP2+/UL16 EL4 cells, ULBP2hi/UL16 EL4 cells, ULBP2low/UL16 EL4 cells, or from CV-1 cells transiently transfected with an expression vector alone (vector) or with an expression vector containing the gene for UL16. Cell lysates were treated with _N_-glycanase and analyzed by immunoblotting with anti-UL16 M230. Blots were stripped and reprobed with anti-STAT5 to demonstrate equal protein loading.
To determine if the down-regulation of cell surface ULBP2 was directly correlated with UL16 expression, the heterogeneous population of UL16-IRES-GFP–transduced cells was further sorted by flow cytometry into those expressing higher cell surface levels of ULBP2 (BP2hi/UL16) and those that expressed lower or undetectable levels (BP2low/UL16; Fig. 6 B). UL16 expression in these populations was assessed by Western blot with a monoclonal antibody specific to UL16. Fig. 6 C shows that there was an inverse correlation between UL16 expression and cell surface ULBP2 expression. These findings confirm that UL16 is able to cause down-regulation of cell surface expression for all the NKG2D ligands to which it can bind.
UL16 Transmembrane and Cytoplasmic Domains Contribute to Intracellular Retention of UL16, Yet the Ectodomain Can Reduce Cell Surface Expression of ULBP2.
To examine which sequences within UL16 were responsible for intracellular retention, we constructed chimeric proteins between UL16 and a truncated mouse IL-4R that has a very short cytoplasmic domain and is readily expressed on the cell surface (48). These chimeric proteins were expressed using retroviral vectors in which the UL16/IL-4R chimeric cDNA was followed by an IRES and GFP. After transduction of EL4 cells or EL4 cells expressing ULBP2, transduced cells were enriched by sorting for GFP expression. As shown in Fig. 7, A and C , full-length UL16 transduction gives no detectable cell surface expression of UL16 despite abundant intracellular staining. When the extracellular domain of the IL-4R was fused to the UL16 transmembrane and cytoplasmic domains (IL-4R/UL16), there was no detectable surface expression of the chimeric protein, but it was readily detected intracellularly (Fig. 7, B and C). In contrast, replacement of the UL16 transmembrane and cytoplasmic domains by those of the IL-4R (UL16/IL-4R) allows cell surface expression of the UL16 ectodomain in a considerable fraction of transduced cells (Fig. 7 C). These results demonstrate that the UL16 transmembrane and cytoplasmic domains of UL16 contribute to intracellular retention of UL16 and can also dictate intracellular localization when fused to ectodomains of proteins that are normally located on the cell surface.
Figure 7.
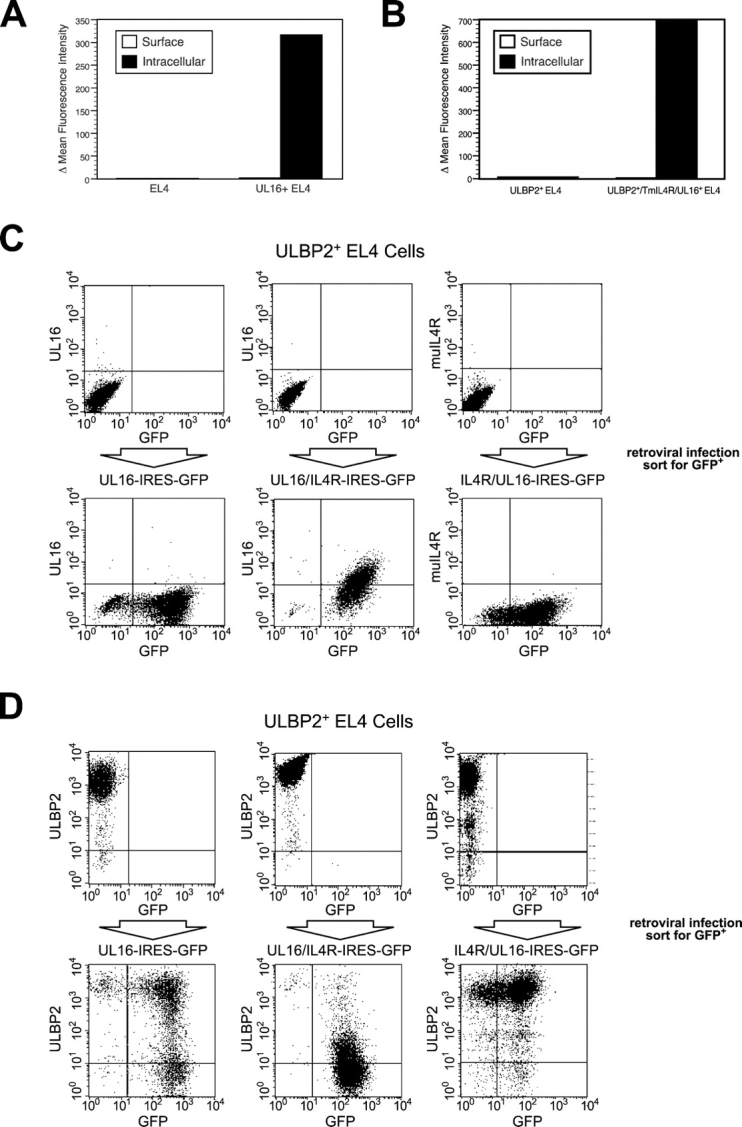
The UL16 transmembrane and cytoplasmic domains are involved in the intracellular retention of UL16, whereas the UL16 ectodomain is required for intracellular retention of ULBP2. (A) EL4 cells or EL4 cells transduced with a VSVG-pseudotyped retrovirus encoding UL16 and GFP were incubated in the presence or absence of 0.1% saponin to permeabilize cells. Cells were stained with anti-UL16 M230 or an isotype control antibody, fixed with 2% paraformaldehyde, and analyzed via flow cytometry. The histograms depict the mean fluorescence intensity (MFI) of cells stained with M230 minus the MFI of the cells stained with the control antibody. The results shown are representative of three separate experiments. (B) EL4 cells or EL4 cells transduced with a VSVG-pseudotyped retrovirus encoding the IL-4R/UL16 chimeric protein and GFP were stained as described above with anti-murine IL-4R M2 or an isotype control antibody. The histograms shown depict the MFI of cells stained with M2 minus the MFI of the cells stained with the isotype-matched control antibody. The results shown are representative of three separate experiments. (C) ULBP2+ EL4 cells were transduced with VSVG-pseudotyped retroviruses encoding UL16 and GFP, the UL16/IL-4R chimera and GFP, and the IL-4R/UL16 chimera and GFP. Transduced cells were enriched by fluorescence-activated cell sorting for cells expressing GFP, stained with anti-UL16 or with anti–IL-4R M2, and analyzed by flow cytometry. (D) ULBP2+ EL4 cells were transduced with VSVG-pseudotyped retroviruses encoding UL16 and GFP, the UL16/IL-4R chimera and GFP, and the IL-4R/UL16 chimera and GFP. Transduced cells were enriched by fluorescence-activated cell sorting of cells expressing GFP, stained with anti-ULBP2 M311, and analyzed by flow cytometry.
The retrovirus vectors encoding the chimeric constructs were then transduced into EL4 cells expressing ULBP2 and sorted for GFP expression (Fig. 7 D). Surprisingly, UL16/IL-4R substantially reduced cell surface expression of ULBP2 to an extent that was comparable to full-length UL16. The IL-4R/UL16 construct had no effect on ULBP2 surface expression (Fig. 7 D). Thus, the UL16 ectodomain, in the absence of UL16 transmembrane and cytoplasmic domains, was able to down-regulate NKG2D ligand surface expression.
Functional Cross-Reaction of Human NKG2D Ligands with Mouse NKG2D.
Previous work had shown that human NK cells could recognize ULBP and MIC when they were expressed in human target cells (24). To determine whether human or mouse NK cells would be able to mediate efficient NKG2D-mediated cytotoxicity against human NKG2D ligands expressed in mouse target cells, we used the EL4 lines expressing human NKG2D ligands as targets in cytotoxicity assays. MICB, ULBP1, and ULBP2-expressing EL4 cells were killed efficiently by IL-15–activated mouse NK cells compared with the parental EL4 cells, whereas ULBP3-expressing EL4 cells were killed at the same level as the parental EL4 cells (Fig. 8 A). The cytotoxicity results correlated completely with binding experiments showing that ULBP1, ULBP2, and MICB, but not ULBP3, could bind to recombinantly expressed mouse NKG2D (unpublished data). The enhanced killing was completely blocked by a monoclonal antibody directed against mouse NKG2D, demonstrating specificity (unpublished data). When human NK cells were tested against the same panel of mouse targets, all the transductants were killed more efficiently than EL4 cells and killing was blocked by a monoclonal antibody directed against human NKG2D (Fig. 8 B and unpublished data). These experiments establish that both mouse and human NK cells can recognize the human NKG2D ligands expressed in mouse lymphocytes.
UL16 Expression Increases Resistance to NKG2D Ligand–mediated NK Cytotoxicity.
The preceding experiments established that UL16 was able to interact intracellularly with NKG2D ligands, altering their trafficking within the cell and decreasing their cell surface expression. To determine how this might affect recognition of the cells by NKG2D-expressing immune effector cells, EL4 cells expressing the NKG2D ligands, ULBP1, ULBP2, and MICB, were transduced with the UL16-IRES-GFP retroviral vector, sorted into populations expressing GFP (designated UL16+), or not expressing GFP (designated UL16−), and examined for their ability to act as targets for NK cytotoxicity. As shown in Fig. 9 , cells expressing these NKG2D ligands were efficiently killed by mouse NK cells (Fig. 9, A and B, ULBP1 and ULBP2) or human NK cells (Fig. 9 D, MICB) compared with EL4 cells. NKG2D ligand–expressing cells transduced with UL16-IRES-GFP and sorted for GFP expression were killed much less efficiently than the GFP− cells. Expression of UL16 in EL4 cells or EL4 cells expressing ULBP3 did not change their sensitivity to killing by mouse or human NK cells (Fig. 9 C and unpublished data). The correlation between increased UL16 expression, decreased cell surface NKG2D ligand expression, and decreased killing by NK cells, together with the intracellular colocalization of UL16 and the NKG2D ligands, establishes a mechanism by which UL16 would be able to protect HCMV-infected cells from immune surveillance by preventing the cell surface expression of the NKG2D ligands.
Discussion
The central findings described here are that UL16 causes the NKG2D ligands, ULBP1, ULBP2, and MICB, to be retained in the ER and cis-Golgi apparatus of cells so that these molecules do not reach the cell surface, resulting in increased resistance to NK cell cytotoxicity. These data are consistent with a role of UL16 as a viral immunomodulatory protein that enables HCMV-infected cells to avoid recognition by immune effector cells that express NKG2D. Although our data examine only NK cells, it is likely that decreased cell surface expression of NKG2D ligands would increase resistance to killing of HCMV-infected cells by CD8+ or γδ T cells, given the demonstrated role of NKG2D ligands in stimulating cytotoxicity by these effector cells (27, 35).
The predicted protein sequence of UL16 suggests a type 1 membrane glycoprotein. When expressed in cells, the majority of UL16 was found in perinuclear membranes that also stained with antibodies specific to ER and cis-Golgi proteins. Little or no UL16 was found on the cell surface, either by cell surface staining or confocal microscopy. UL16 oligosaccharides were not processed from high mannose to complex oligosaccharides, a process that is completed in the trans-Golgi, supporting the hypothesis that UL16 does not traffic extensively beyond the cis- or medial-Golgi compartments and is largely retained in the ER. Alternatively, there might be transport from the ER and retrieval from the ER Golgi intermediate compartment or cis-Golgi apparatus back to the ER.
Efficient intracellular retention of UL16 required its own transmembrane and cytoplasmic domains. In their absence some of the UL16 reached the cell surface. These domains can function to retain the extracellular domain of the mIL-4R within the cell, indicating that they contribute substantially to ER localization. The amino acids within these domains that mediate retention are not yet defined. Inspection of the UL16 cytoplasmic amino acid sequence does not reveal known endoplasmic reticulum retention motifs such as di-lysine residues, or KDEL motifs. Arginine-rich sequences have been shown to function as endoplasmic reticulum retention motifs (49) and the short cytoplasmic domain of UL16 is rich in clustered arginine residues. Notably, most arginines are followed by a hydrophobic amino acid (RIPQRLCQRLRIRLPHRYQ RLRTED). Additionally, a YQRL sequence is present that matches a consensus tyrosine-based sorting signal, Yxxφ, where φ is a hydrophobic amino acid. Such tyrosine-based signals are used for protein sorting between many different subcellular compartments, depending on the context of the sequence and which adaptor protein complexes are bound (50). Mutational analysis will be required to determine which residues contribute to the intracellular trafficking of UL16.
UL16 expression in cells caused MICB and other NKG2D ligands to accumulate in cytoplasmic membranes. In the case of MICB, this accumulation was largely or exclusively in perinuclear membranes. Consistent with the notion that MICB accumulated in the ER/cis-Golgi, we found extensive colocalization of UL16 with MICB. UL16 expression also dramatically stabilized MICB within the cell. Confocal microscopy of cells expressing UL16, and treated with cycloheximide for 4 h, detected MICB in a perinuclear location. However, in the absence of UL16, MICB was not detected after 4 h of protein synthesis inhibition. Pulse-chase analysis also confirmed the rapid turnover of MICB and its stabilization by UL16. The precise molecular mechanisms responsible for these changes are not yet known, but it is possible to speculate as to how this might occur. The intracellular half-life of UL16 is long, as we observed little loss of the protein during an 8-h chase period. It is possible that binding of UL16 to MICB forms a relatively stable intracellular complex in the ER and cis-Golgi apparatus. In the absence of UL16, MICB moves rapidly to the Golgi apparatus and then to the cell surface, largely acquiring endoH resistance within 60 min. Turnover from the cell surface is likely to involve endocytosis and delivery to lysosomes for degradation, as well as enzymatic shedding, which has been described for MICA and ULBP2 (37, 51, 52). Intracellular retention of MICB by UL16 would preclude this turnover.
The cytoplasmic and transmembrane domains of UL16 clearly play a role in intracellular traffic and intracellular retention. When these domains were replaced with those of the IL-4R (IL-4R/UL16), there was detectable expression of the UL16 ectodomain on the cell surface. However, when the UL16 ectodomain was fused with the IL-4R transmembrane and cytoplasmic domains (UL16/IL-4R), the chimeric protein retained the ability to reduce cell surface expression of NKG2D ligands. To explain these findings, one hypothesis is that a proportion of the UL16/IL-4R chimera is still retained in the cytoplasm, by mechanisms involving the ectodomain of UL16, and is able to bind NKG2D ligands and retain or missort these complexes within cells. This model suggests that some of the sorting signals reducing UL16 surface expression would be present in the ectodomain. Alternatively, UL16 may function in a similar fashion as has been proposed for the HCMV US3 and the murine CMV m152 proteins that appear to interact transiently with MHC class I and II proteins (5, 53–55). This transient interaction might alter the posttranslational processing of MHC proteins, causing them to be mis-sorted either to lysosomes or into other cellular compartments, but not allowing transport to the cell surface. Additional studies are required to define more precisely the molecular mechanisms by which UL16 affects intracellular trafficking of NKG2D ligands.
Although UL16 is able to target ULBP1, ULBP2, and MICB, it is noteworthy that it does not affect the closely related MICA and ULBP3 molecules. MICA has been shown to be induced by HCMV infection in vitro and stimulate T cell–mediated recognition of the infected cells (27), so one could speculate that another, as yet unidentified, HCMV protein might target these other NKG2D ligands under certain circumstances. Alternatively, the absence of HCMV-encoded antagonists for MICA and ULBP3 might reflect an advantage to the host in the evolutionary arms race with the virus. Similarly, m152 was recently shown to down-regulate the mouse NKG2D ligand, H60, but not the other mouse NKG2D ligands (56). It is not clear whether the extensive polymorphism in the NKG2D ligands, coupled with sequence variation between strains of CMV, might alter the spectrum of NKG2D ligands affected by UL16 or m152.
How might the interaction of UL16 and NKG2D ligands play out in a virus infection in vivo? HCMV replicates slowly compared with many viruses and does not cause rapid cell destruction. Strong and long-lasting host immune responses are generated to CMV antigens during lifelong persistent infections that frequently involve many cycles of latency and reactivation. Many studies have described the inhibition of MHC class I and II antigen presentation by the HCMV US2, US3, US6, and US11 proteins, and have suggested that this contributes to viral persistence. The discovery that NKG2D ligands are up-regulated by cells undergoing infection, stress, or transformation, and that their recognition by NKG2D receptor-expressing cells can mediate destruction of these targets in the absence of any changes in classical MHC-mediated antigen presentation, provides a new paradigm for immunosurveillance. In this model, the pathogen is recognized indirectly by immune effector cells, via changes in cellular phenotype rather than by direct recognition of its encoded antigens. The data presented here suggest that once again the virus is “one step ahead of the game” (57), and that HCMV expresses UL16 in order to prevent cell surface expression of NKG2D ligands and their recognition by NKG2D-expressing immune effector cells. Strategies that enable the virus-infected cell to avoid immune recognition would be especially useful during the reactivation from latency in the face of a primed and robust cellular immune response. The need to evade preexisting humoral immunity might also explain why UL16 and other HCMV-encoded immunomodulatory glycoproteins are retained and act intracellularly. However, it is also possible that UL16 might act during the primary stages of HCMV infection, before the onset of an adaptive immune response, to inhibit recognition by innate immune effector cells and “buy time” for the virus to achieve a latent state.
In studying HCMV immune evasion mechanisms, there are difficulties in extrapolating in vitro molecular and cellular experimental findings to in vivo relevance. HCMV is highly species specific and therefore no in vivo model systems are available. The cell types, viral strains, and infection protocols commonly used to study HCMV in vitro do not always replicate the natural history of HCMV infection in vivo. These difficulties prevent a precise definition of the relative importance of proposed HCMV-encoded immune evasion proteins, how they may work in concert with each other, or even the types of infected cells in which they perform in vivo. Nevertheless, MCMV provides an experimental model in which sophisticated manipulation of the viral genome is now possible so that the roles of proposed immunomodulatory proteins can be tested in the context of an in vivo infection (58). Such studies have demonstrated the in vivo relevance of MCMV proteins that down-regulate surface expression of classical MHC class I antigens and NKG2D ligands (56, 59), so there is a solid rationale to believe that UL16-mediated intracellular retention of NKG2D ligands is an important component of the immune evasion arsenal of HCMV.
Acknowledgments
The authors thank Stacey Culp for purification of antibodies, the Amgen flow cytometry group, Aurelie Snyder for assistance with confocal microscopy, Gary Carlton and Tiffani Howard for preparation of the figures, Corinne Harmon for editorial assistance, and Melanie Spriggs for comments on the manuscript.
The work in the laboratory of D.C. Johnson was supported by a National Institutes of Health grant from the National Eye Institute (EY11245).
C. Dunn and N.J. Chalupny contributed equally to this work.
P.V. Sivakumar's present address is Zymogenetics, 1201 Eastlake Ave. E., Seattle, WA 98102.
Footnotes
*
Abbreviations used in this paper: Ad, adenovirus; endoH, endoglycosidase H; GFP, green fluorescent protein; HCMV, human CMV; IRES, internal ribosome entry site; LZ, leucine zipper; MCMV, mouse CMV; MFI, mean fluorescence intensity; VSVG, vesicular stomatitis virus G protein.
References
- 1.Britt, W., and C. Alford. 1996. Cytomegalovirus. Field's Virology. B.N. Field, D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven, Philadelphia, PA. 2493–2524.
- 2.Tortorella, D., B.E. Gewurz, M.H. Furman, D.J. Schust, and H.L. Ploegh. 2000. Viral subversion of the immune system. Annu. Rev. Immunol. 18:861–926. [DOI] [PubMed] [Google Scholar]
- 3.McFadden, G., and D.C. Johnson. 2002. Viral immune evasion. Immunology of Infectious Diseases. S.H.E. Kaufmann, A. Sher, and R. Ahmed, editors. American Society for Microbiology, Washington, D.C. 357–371.
- 4.Tomazin, R., J. Boname, N.R. Hegde, D.M. Lewinsohn, Y. Altschuler, T.R. Jones, P. Cresswell, J.A. Nelson, S.R. Riddell, and D.C. Johnson. 1999. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat. Med. 5:1039–1043. [DOI] [PubMed] [Google Scholar]
- 5.Hegde, N.R., R.A. Tomazin, T.W. Wisner, C. Dunn, J.M. Boname, D.M. Lewinsohn, and D.C. Johnson. 2002. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J. Virol. 76:10929–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson, D.C., and N.R. Hegde. 2002. Inhibition of the MHC class II antigen presentation pathway by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 269:101–115. [DOI] [PubMed] [Google Scholar]
- 7.Biron, C.A., K.S. Byron, and J.L. Sullivan. 1989. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 320:1731–1735. [DOI] [PubMed] [Google Scholar]
- 8.Farrell, H.E., M.A. Degli-Esposti, and N.J. Davis-Poynter. 1999. Cytomegalovirus evasion of natural killer cell responses. Immunol. Rev. 168:187–197. [DOI] [PubMed] [Google Scholar]
- 9.Cosman, D., N. Fanger, and L. Borges. 1999. Human cytomegalovirus, MHC class I and inhibitory signalling receptors: more questions than answers. Immunol. Rev. 168:177–185. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Botet, M., M. Llano, and M. Ortega. 2001. Human cytomegalovirus and natural killer-mediated surveillance of HLA class I expression: a paradigm of host-pathogen adaptation. Immunol. Rev. 181:193–202. [DOI] [PubMed] [Google Scholar]
- 11.Braud, V.M., P. Tomasec, and G.W. Wilkinson. 2002. Viral evasion of natural killer cells during human cytomegalovirus infection. Curr. Top. Microbiol. Immunol. 269:117–129. [DOI] [PubMed] [Google Scholar]
- 12.Lanier, L.L. 1998. NK cell receptors. Annu. Rev. Immunol. 16:359–393. [DOI] [PubMed] [Google Scholar]
- 13.Trowsdale, J. 2001. Genetic and functional relationships between MHC and NK receptor genes. Immunity. 15:363–374. [DOI] [PubMed] [Google Scholar]
- 14.Veillette, A., S. Latour, and D. Davidson. 2002. Negative regulation of immunoreceptor signaling. Annu. Rev. Immunol. 20:669–707. [DOI] [PubMed] [Google Scholar]
- 15.Vivier, E., E. Tomasello, and P. Paul. 2002. Lymphocyte activation via NKG2D: towards a new paradigm in immune recognition? Curr. Opin. Immunol. 14:306–311. [DOI] [PubMed] [Google Scholar]
- 16.Bauer, S., V. Groh, J. Wu, A. Steinle, J.H. Phillips, L.L. Lanier, and T. Spies. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729. [DOI] [PubMed] [Google Scholar]
- 17.Wu, J., Y. Song, A.B. Bakker, S. Bauer, T. Spies, L.L. Lanier, and J.H. Phillips. 1999. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 285:730–732. [DOI] [PubMed] [Google Scholar]
- 18.Diefenbach, A., A.M. Jamieson, S.D. Liu, N. Shastri, and D.H. Raulet. 2000. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1:119–126. [DOI] [PubMed] [Google Scholar]
- 19.Cerwenka, A., A.B. Bakker, T. McClanahan, J. Wagner, J. Wu, J.H. Phillips, and L.L. Lanier. 2000. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 12:721–727. [DOI] [PubMed] [Google Scholar]
- 20.Ho, E.L., L.N. Carayannopoulos, J. Poursine-Laurent, J. Kinder, B. Plougastel, H.R. Smith, and W.M. Yokoyama. 2002. Costimulation of multiple NK cell activation receptors by NKG2D. J. Immunol. 169:3667–3675. [DOI] [PubMed] [Google Scholar]
- 21.Gumperz, J.E., S. Miyake, T. Yamamura, and M.B. Brenner. 2002. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J. Exp. Med. 195:625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamieson, A.M., A. Diefenbach, C.W. McMahon, N. Xiong, J.R. Carlyle, and D.H. Raulet. 2002. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 17:19–29. [DOI] [PubMed] [Google Scholar]
- 23.Kubin, M., L. Cassiano, J. Chalupny, W. Chin, D. Cosman, W. Fanslow, J. Mullberg, A.M. Rousseau, D. Ulrich, and R. Armitage. 2001. ULBP1, 2, 3: novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur. J. Immunol. 31:1428–1437. [DOI] [PubMed] [Google Scholar]
- 24.Cosman, D., J. Mullberg, C.L. Sutherland, W. Chin, R. Armitage, W. Fanslow, M. Kubin, and N.J. Chalupny. 2001. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 14:123–133. [DOI] [PubMed] [Google Scholar]
- 25.Sutherland, C.L., N.J. Chalupny, K. Schooley, T. VandenBos, M. Kubin, and D. Cosman. 2002. UL16-binding proteins, novel MHC class I-related proteins, bind to NKG2D and activate multiple signaling pathways in primary NK cells. J. Immunol. 168:671–679. [DOI] [PubMed] [Google Scholar]
- 26.Wu, J., H. Cherwinski, T. Spies, J.H. Phillips, and L.L. Lanier. 2000. DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J. Exp. Med. 192:1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groh, V., R. Rhinehart, J. Randolph-Habecker, M.S. Topp, S.R. Riddell, and T. Spies. 2001. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2:255–260. [DOI] [PubMed] [Google Scholar]
- 28.Diefenbach, A., E. Tomasello, M. Lucas, A.M. Jamieson, J.K. Hsia, E. Vivier, and D.H. Raulet. 2002. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3:1142–1149. [DOI] [PubMed] [Google Scholar]
- 29.Gilfillan, S., E.L. Ho, M. Cella, W.M. Yokoyama, and M. Colonna. 2002. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 3:1150–1155. [DOI] [PubMed] [Google Scholar]
- 30.Bahram, S., M. Bresnahan, D.E. Geraghty, and T. Spies. 1994. A second lineage of mammalian major histocompatibility complex class I genes. Proc. Natl. Acad. Sci. USA. 91:6259–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leelayuwat, C., D.C. Townend, M.A. Degli-Esposti, L.J. Abraham, and R.L. Dawkins. 1994. A new polymorphic and multicopy MHC gene family related to nonmammalian class I. Immunogenetics. 40:339–351. [DOI] [PubMed] [Google Scholar]
- 32.Carayannopoulos, L.N., O.V. Naidenko, D.H. Fremont, and W.M. Yokoyama. 2002. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169:4079–4083. [DOI] [PubMed] [Google Scholar]
- 33.Groh, V., S. Bahram, S. Bauer, A. Herman, M. Beauchamp, and T. Spies. 1996. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. USA. 93:12445–12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groh, V., A. Steinle, S. Bauer, and T. Spies. 1998. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science. 279:1737–1740. [DOI] [PubMed] [Google Scholar]
- 35.Groh, V., R. Rhinehart, H. Secrist, S. Bauer, K.H. Grabstein, and T. Spies. 1999. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA. 96:6879–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das, H., V. Groh, C. Kuijl, M. Sugita, C.T. Morita, T. Spies, and J.F. Bukowski. 2001. MICA engagement by human Vgamma2Vdelta2 T cells enhances their antigen-dependent effector function. Immunity. 15:83–93. [DOI] [PubMed] [Google Scholar]
- 37.Tieng, V., C. Le Bouguenec, L. du Merle, P. Bertheau, P. Desreumaux, A. Janin, D. Charron, and A. Toubert. 2002. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc. Natl. Acad. Sci. USA. 99:2977–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto, K., Y. Fujiyama, A. Andoh, T. Bamba, and H. Okabe. 2001. Oxidative stress increases MICA and MICB gene expression in the human colon carcinoma cell line (CaCo-2). Biochim. Biophys. Acta. 1526:10–12. [DOI] [PubMed] [Google Scholar]
- 39.Onda, H., S. Ohkubo, Y. Shintani, K. Ogi, K. Kikuchi, H. Tanaka, K. Yamamoto, I. Tsuji, Y. Ishibashi, T. Yamada, et al. 2001. A novel secreted tumor antigen with a glycosylphosphatidylinositol-anchored structure ubiquitously expressed in human cancers. Biochem. Biophys. Res. Commun. 285:235–243. [DOI] [PubMed] [Google Scholar]
- 40.Pende, D., P. Rivera, S. Marcenaro, C.C. Chang, R. Biassoni, R. Conte, M. Kubin, D. Cosman, S. Ferrone, L. Moretta, et al. 2002. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 62:6178–6186. [PubMed] [Google Scholar]
- 41.Girardi, M., D.E. Oppenheim, C.R. Steele, J.M. Lewis, E. Glusac, R. Filler, P. Hobby, B. Sutton, R.E. Tigelaar, and A.C. Hayday. 2001. Regulation of cutaneous malignancy by gammadelta T cells. Science. 294:605–609. [DOI] [PubMed] [Google Scholar]
- 42.Sutherland, C.L., N.J. Chalupny, and D. Cosman. 2001. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 181:185–192. [DOI] [PubMed] [Google Scholar]
- 43.Perussia, B., C. Ramoni, I. Anegon, M.C. Cuturi, J. Faust, and G. Trinchieri. 1987. Preferential proliferation of natural killer cells among peripheral blood mononuclear cells cocultured with B lymphoblastoid cell lines. Nat. Immun. Cell Growth Regul. 6:171–188. [PubMed] [Google Scholar]
- 44.Cosman, D., N. Fanger, L. Borges, M. Kubin, W. Chin, L. Peterson, and M.L. Hsu. 1997. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. 7:273–282. [DOI] [PubMed] [Google Scholar]
- 45.Huber, M.T., R. Tomazin, T. Wisner, J. Boname, and D.C. Johnson. 2002. Human cytomegalovirus US7, US8, US9, and US10 are cytoplasmic glycoproteins, not found at cell surfaces, and US9 does not mediate cell-to-cell spread. J. Virol. 76:5748–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinsella, T.M., and G.P. Nolan. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413. [DOI] [PubMed] [Google Scholar]
- 47.Burns, J.C., T. Friedmann, W. Driever, M. Burrascano, and J.K. Yee. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA. 90:8033–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mosley, B., M.P. Beckmann, C.J. March, R.L. Idzerda, S.D. Gimpel, T. VandenBos, D. Friend, A. Alpert, D. Anderson, J. Jackson, et al. 1989. The murine interleukin-4 receptor: molecular cloning and characterization of secreted and membrane bound forms. Cell. 59:335–348. [DOI] [PubMed] [Google Scholar]
- 49.Zerangue, N., M.J. Malan, S.R. Fried, P.F. Dazin, Y.N. Jan, L.Y. Jan, and B. Schwappach. 2001. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc. Natl. Acad. Sci. USA. 98:2431–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonifacino, J.S., and E.C. Dell'Angelica. 1999. Molecular bases for the recognition of tyrosine-based sorting signals. J. Cell. Biol. 145:923–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salih, H.R., H.G. Rammensee, and A. Steinle. 2002. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 169:4098–4102. [DOI] [PubMed] [Google Scholar]
- 52.Groh, V., J. Wu, C. Yee, and T. Spies. 2002. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 419:734–738. [DOI] [PubMed] [Google Scholar]
- 53.Gruhler, A., P.A. Peterson, and K. Fruh. 2000. Human cytomegalovirus immediate early glycoprotein US3 retains MHC class I molecules by transient association. Traffic. 1:318–325. [DOI] [PubMed] [Google Scholar]
- 54.Ziegler, H., R. Thale, P. Lucin, W. Muranyi, T. Flohr, H. Hengel, H. Farrell, W. Rawlinson, and U.H. Koszinowski. 1997. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 6:57–66. [DOI] [PubMed] [Google Scholar]
- 55.Ziegler, H., W. Muranyi, H.G. Burgert, E. Kremmer, and U.H. Koszinowski. 2000. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 19:870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krmpotic, A., D.H. Busch, I. Bubic, F. Gebhardt, H. Hengel, M. Hasan, A.A. Scalzo, U.H. Koszinowski, and S. Jonjic. 2002. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 3:529–535. [DOI] [PubMed] [Google Scholar]
- 57.Spriggs, M.K. 1996. One step ahead of the game: viral immunomodulatory molecules. Annu. Rev. Immunol. 14:101–130. [DOI] [PubMed] [Google Scholar]
- 58.Wagner, M., A. Gutermann, J. Podlech, M.J. Reddehase, and U.H. Koszinowski. 2002. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 196:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krmpotic, A., M. Messerle, I. Crnkovic-Mertens, B. Polic, S. Jonjic, and U.H. Koszinowski. 1999. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 190:1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]