Accumulation of Tissue Factor into Developing Thrombi In Vivo Is Dependent upon Microparticle P-Selectin Glycoprotein Ligand 1 and Platelet P-Selectin (original) (raw)
Abstract
Using a laser-induced endothelial injury model, we examined thrombus formation in the microcirculation of wild-type and genetically altered mice by real-time in vivo microscopy to analyze this complex physiologic process in a system that includes the vessel wall, the presence of flowing blood, and the absence of anticoagulants. We observe P-selectin expression, tissue factor accumulation, and fibrin generation after platelet localization in the developing thrombus in arterioles of wild-type mice. However, mice lacking P-selectin glycoprotein ligand 1 (PSGL-1) or P-selectin, or wild-type mice infused with blocking P-selectin antibodies, developed platelet thrombi containing minimal tissue factor and fibrin. To explore the delivery of tissue factor into a developing thrombus, we identified monocyte-derived microparticles in human platelet–poor plasma that express tissue factor, PSGL-1, and CD14. Fluorescently labeled mouse microparticles infused into a recipient mouse localized within the developing thrombus, indicating that one pathway for the initiation of blood coagulation in vivo involves the accumulation of tissue factor– and PSGL-1–containing microparticles in the platelet thrombus expressing P-selectin. These monocyte-derived microparticles bind to activated platelets in an interaction mediated by platelet P-selectin and microparticle PSGL-1. We propose that PSGL-1 plays a role in blood coagulation in addition to its known role in leukocyte trafficking.
Keywords: blood coagulation, fibrin, thrombosis, endothelium, intravital microscopy
Introduction
P-selectin glycoprotein ligand 1 (PSGL-1)*and P-selectin are vascular cell adhesion molecules that play a critical role in leukocyte trafficking and lymphocyte migration (1–3). P-selectin is expressed on activated platelets (4–7) and binds to PSGL-1 on circulating leukocytes (8–10). Although the contribution of these proteins to the inflammatory response has been the subject of numerous studies, the role of these proteins in hemostasis and thrombosis has not been elucidated. Using a shunt model in a baboon, we have previously demonstrated that infusion of blocking P-selectin antibodies decreases fibrin generation in the developing experimental thrombus (11). This presented the first evidence for a P-selectin–dependent pathway of blood coagulation. Although P-selectin up-regulates tissue factor expression in monocytes (12), de novo synthesis of tissue factor is required and rapid fibrin generation could not be explained by leukocyte incorporation into the developing thrombus. A role for P-selectin or PSGL-1 has remained uncertain in the mechanism of fibrin formation.
After vascular injury, extravasation of blood is minimized by the rapid formation of intravascular thrombi composed of platelets and fibrin (13). Tissue factor is the initial activator of the blood coagulation pathway that culminates in the generation of a fibrin clot (14). An integral membrane protein, it is expressed on most nonvascular cells (15, 16). The classical model has been that vascular injury leads to the exposure of flowing blood to extravascular cells expressing tissue factor, with the concomitant rapid initiation of blood coagulation. Low levels of tissue factor have been identified circulating in normal plasma (17–19) and an in vitro thrombosis model has been used to demonstrate tissue factor accumulation within the fibrin-dominated thrombus upon perfusion of fresh human blood onto collagen-coated slides or sections of pig arterial media (20). The transfer of tissue factor particles from a monocyte-like cell line to the platelet surface in vitro is dependent upon CD15 and tissue factor (21), suggesting that blood-borne tissue factor accumulates in the thrombus in a mechanism that involves CD15 and tissue factor on membranous particles bound to P-selectin on platelets. Here, we test this hypothesis in in vivo models that offer opportunities to analyze the initiation of thrombus formation in a complex physiologic system that includes the vessel wall, the presence of flowing blood, and the absence of anticoagulants.
We have developed instrumentation for real-time imaging of multiple fluorescent components during thrombus formation in the microcirculation of a living mouse using intravital high speed microscopy (22). Using this system in conjunction with a laser-induced endothelial injury model (23), we demonstrate distribution of tissue factor throughout the developing thrombus in vivo, although most tissue factor is concentrated at the thrombus vessel wall interface (22). In this study of in vivo thrombus formation in genetically altered mice, we demonstrate P-selectin– and PSGL-1–dependent tissue factor accumulation and fibrin generation during arterial thrombus formation in vivo. Monocyte-derived microparticles expressing tissue factor and PSGL-1 accumulate on activated platelets expressing P-selectin, thus concentrating tissue factor to a level that triggers the initiation of blood coagulation.
Materials and Methods
Intravital Microscopy
High speed intravital digital videomicroscopy was performed as previously described (22).
Mice
PSGL-1 null mice, previously described (24, 25), backcrossed five generations into a C57B6 background (N5), have been deposited at The Jackson Laboratory for distribution (B6.Cg-SelPltm1Fur). P-selectin−/− (C57BL/6J-Selptm1Bay) and C57BL/6J mice were obtained from The Jackson Laboratory. All procedures were approved by the Animal Care and Use Committee of Beth Israel Deaconess Medical Center.
Laser-induced Vessel Wall Injury
12–16-wk-old male mice were preanesthetized with ketamine, xylazine, and atropine sulfate, and a trachea tube was inserted to facilitate spontaneous respiration (22). Nembutal was administered through the jugular vein. The cremaster muscle, exteriorized in 4–7 min, was prepared for intravital microscopy (24, 26). The microvasculature was visualized by transmission microscopy. Antibodies in 200 μl physiologic saline were infused via a jugular vein cannulus. Approximately 5 min later, an arteriole was identified and endothelial injury was induced using a pulsed nitrogen dye laser applied through the microscope objective using the Micropoint laser system (Photonic Instruments). Multiple independent thrombi were generated, a new injury always upstream to previous injuries, in different arterioles over the course of approximately 1 h.
Antibodies and Reagents
Mice were infused with 1.9 μg/g mouse body weight of rat anti–mouse P-selectin (BD Biosciences), 8 μg/g body weight of sheep anti–rabbit tissue factor (American Diagnostica, Inc.) or affinity-purified rabbit anti–mouse tissue factor (152–166) antibody, 2 μg/g body weight of mouse anti–fibrin II β chain (Accurate Chemical), 0.1 μg/g body weight of rat anti–mouse CD41 (BD Biosciences), 1 μg/g body weight of chicken anti–rat IgG (BD Biosciences), 4 μg/g body weight of donkey anti–sheep IgG (Molecular Probes), and 1 U/g body weight of hirudin (Calbiochem), where indicated. Alexa 350, Alexa 488, and Alexa 660 were conjugated to purified antibodies according to the Alexa Fluor Protein Labeling Kit (Molecular Probes). The mouse anti–fibrin II β chain–specific antibody binds to fibrin but does not bind to fibrinogen (27).
Antibodies to mouse tissue factor were prepared by standard methods (28) based upon a previous report (29). A synthetic peptide, mTF(152–166), representing residues 152–166 of mouse tissue factor with an additional NH2-terminal cysteine for purposes of chemical linking, was synthesized. The sequence was verified by automated Edman degradation and its molecular mass was confirmed at 1,643.88 amu by mass spectroscopy. Rabbits were immunized with 500 μg of the peptide/KLH conjugate, and then 200 μg of the peptide/KLH conjugate monthly. Anti-tissue factor peptide antibodies were purified by immunoaffinity chromatography using the synthetic peptide. Antiserum was applied to a KLH-Sepharose column to remove anti-KLH antibodies. Antibodies that failed to bind were applied to a peptide-Sepharose column, and bound antibody was eluted with 100 mM glycine, pH 3.
Preparation of Microparticles
Isolation of Microparticles from Human Platelet-poor Plasma.
Venous blood was drawn from healthy volunteers into 4% sodium citrate or 50 mM EDTA (1:10 vol/vol) and a complete EDTA-free protease inhibitor cocktail (Boehringer), and then centrifuged at 25°C at 2,500 g for 25 min. Platelet-poor plasma was decanted. Flow cytometry demonstrated that >95% of all particles were smaller than 1 μm.
Mixed Leukocyte– and Platelet-derived Microparticles Generated from Mouse Whole Blood.
Blood was drawn from wild-type mice by cardiac puncture and anticoagulated with sodium citrate as described above. For mouse preparations, 2% dextran in saline (molecular weight 160,000) was mixed (1:1 vol/vol) with the cell suspension for 40 min at room temperature to sediment red cells. Dextran-rich supernatant including leukocytes as well as residual platelets were washed twice and resuspended in Hank's buffer containing 1 mM calcium chloride and the protease inhibitor cocktail (Boehringer). 1.5 μg/ml calcein AM was added to the cells and microparticles were generated using 10 μM A23187 for 20 min at room temperature. Cells were removed by centrifugation at 14,000 g in an Eppendorf microcentrifuge. Flow cytometry indicated that >98% of the particles present in the supernatant were smaller than 1 μm.
Microparticles Generated from Density Gradient–purified Human Mononuclear Cells.
Blood was obtained from human volunteers into 4% sodium citrate (1:10 vol/vol) and an EDTA-free protease inhibitor cocktail, and centrifuged at 200 g for 15 min. Blood was layered on Ficoll-Paque and centrifuged at 400 g for 30 min at 4°C. The interface cells were removed, washed with RPMI 1640 medium three times at 4°C, and resuspended in RPMI 1640. Platelet to mononuclear cell ratio, determined microscopically, was typically ∼0.5:1. The cells were then incubated for 6 h with 100 ng/ml LPS (Escherichia coli serotype 055:B5; Sigma-Aldrich) at 37°C under 5% CO2, or 5.5 h with LPS followed by 20 min with 10 μM A23187 and a protease inhibitor cocktail. Cell suspensions were centrifuged for isolation of microparticles as described above.
Microparticles Generated from WEHI Cells.
WEHI cells (American Type Culture Collection WEHI 274.1) were incubated for 40 min with calcein AM in Hank's buffer and cultured in DMEM. Microparticles were generated with the addition of 10 μM A23187 for 20 min and a protease inhibitor cocktail. Cell suspensions were centrifuged for isolation of microparticles as described above.
Mouse Microparticle Incorporation into Arterial Thrombi
Calcein-labeled microparticles were generated from density gradient–purified mononuclear cells or from WEHI cells in cell culture. Microparticles were isolated by ultracentrifugation at 150,000 g for 2 h at 10°C. The pellet was resuspended in sterile saline and evaluated for fluorescence intensity and size by flow cytometry before infusion into an anesthetized mouse.
Purification and Analysis of Tissue Factor and PSGL-1–bearing Microparticles
Tissue factor–bearing microparticles were isolated from human platelet-poor plasma or mononuclear cell supernatant using polystyrene beads coated with anti-tissue factor antibodies. Polystyrene beads (4.5-μm diameter; Polysciences) were washed three times with PBS and incubated with polyclonal rabbit anti–human tissue factor antibodies (American Diagnostics, Inc.) at 100 μg/ml at 24°C for 1 h. The beads were twice pelleted by centrifugation and washed in PBS containing 1% BSA. To generate control beads, parallel preparations were performed with a polystyrene bead coated with nonimmune IgG. Adsorption of antibodies to beads was confirmed by flow cytometry. Mononuclear cell supernatant or platelet-poor plasma was mixed with beads coated with anti-tissue factor antibodies or nonimmune IgG for 18 h at 24°C after preincubation of mononuclear cell supernatant or platelet-poor plasma with 8 μg/ml human IgG or rabbit IgG for 30 min to block Fc receptors. The beads were centrifuged, washed twice with PBS/0.1% BSA, and resuspended in the same buffer before evaluation by flow cytometry and tissue factor assay. Microparticles were analyzed with a FACSCalibur® (Becton Dickinson) flow cytometer by forward scatter and side scatter. Microparticle concentration was determined as previously described (30).
Beads were incubated with PE-conjugated anti–PSGL-1 antibody at 24°C for 40 min, and then examined for fluorescence on the FACSCalibur® flow cytometer. Background fluorescence was determined by preincubation of the beads with a 50-fold excess of the same antibody but lacking a fluorescent label before the addition of PE-conjugated anti–PSGL-1 antibody. Data were analyzed using CellQuest™ software.
Tissue factor activity was assayed with a one stage clotting time (31). The values were converted to arbitrary units of procoagulant activity by comparison with a standard curve obtained using a rabbit brain thromboplastin standard.
For analysis of PSGL-1 function on microparticles, P-selectin IgG chimera–coated beads were prepared as the antibody-coated beads, which were incubated with calcein-labeled microparticles that had been pretreated with either 2 mM EDTA, 10 μg/ml anti–PSGL-1 monoclonal antibody (BD Biosciences), or 10 μg/ml of its isotype control.
Results
We have previously described the use of real-time in vivo fluorescence and brightfield microscopy to capture digital videos of thrombotic events in arterioles in the mouse cremaster microcirculation (22). Using this system, we have explored the role of P-selectin and PSGL-1 in a pathway to fibrin formation in experimental thrombi generated in living mice.
Platelets Accumulate and Express P-Selectin at the Site of Endothelial Injury.
P-selectin appears on the surface of activated platelets that have undergone degranulation of α granules (7). Therefore, we studied P-selectin expression on platelets incorporated into a developing thrombus in vivo to determine whether platelet P-selectin is exposed to flowing blood. Alexa 350–conjugated P-selectin antibody was infused into the circulation of an anesthetized mouse. Intravital widefield microscopy was performed to visualize P-selectin fluorescence in one channel and the brightfield image in a second channel after endothelial injury. Platelets initially adhere and accumulate on the surface of the injured vessel wall (22). In the current experiments depicting the earliest events of thrombus formation, no leukocyte rolling was observed on the arterial wall or the arterial thrombus. Platelet P-selectin expression was observed within the thrombus during thrombus development in a wild-type mouse (Fig. 1 A). However, in control experiments, no P-selectin antibody was detected in the thrombus of a P-selectin null mouse (Fig. 1 B), indicating that the antibody is not trapped nonspecifically within the thrombus. The exposure of P-selectin in the developing thrombus of a wild-type mouse indicates platelet activation and degranulation on the time scale of thrombus formation under the conditions used. Although only representative images are depicted, these experiments have been performed in four mice of each genotype.
Figure 1.

Expression of P-selectin in the developing thrombus. P-selectin was localized in the developing thrombus with Alexa 350–conjugated rat anti–mouse P-selectin antibody. The Alexa 350 fluorochrome image, collected digitally and presented as a red pseudocolor, is merged with the brightfield image. The images depicted are 160 s (wild-type mouse) and 200 s (P-selectin null mouse) after laser-induced endothelial injury. The boundaries of the thrombus are indicated by the arrowheads. A, wild-type mouse; B, P-selectin null mouse.
Tissue Factor Accumulates in the Developing Thrombus in Wild-type Mice but Not PSGL-1 Null nor P-Selectin Null Mice.
We investigated tissue factor accumulation in wild-type mice and mice lacking PSGL-1 or P-selectin. Considering the critical nature of these experiments, we compared our initial results using a commercially available sheep anti–rabbit tissue factor antibody that cross reacts with mouse tissue factor (22) with rabbit antibodies against a synthetic peptide based upon residues 152–166 of mouse tissue factor, rabbit anti–mTF (152–166) antibodies, which we prepared and characterized. Rabbit anti–mTF (152–166) antibodies were immunoaffinity purified and characterized by Western blot analysis. This antibody is reactive against a 43,000 molecular weight band in mouse fibroblasts, corresponding to tissue factor (Fig. 2 A, left). A minor higher molecular weight band might be a tissue factor dimer. Furthermore, this antibody is reactive with a recombinant mouse tissue factor-Ig chimera (unpublished data). Rabbit anti–mTF (152–166) antibodies do not inhibit tissue factor activity, as measured in a linked chromagenic assay for tissue factor stimulation of Factor X activation. Anti-mTF (152–166) antibodies detected tissue factor in arterial thrombi in vivo. Nonimmune IgG was minimally reactive with thrombi under identical conditions (Fig. 2 A, middle). These experiments were conducted with four mice. Images of tissue factor in thrombi during intravital microscopy using the rabbit anti–mTF (152–166) antibodies were similar to those obtained with the sheep anti–rabbit tissue factor antibody (Fig. 2 A, right). We have used both anti-tissue factor antibodies independently in these intravital experiments.
Figure 2.
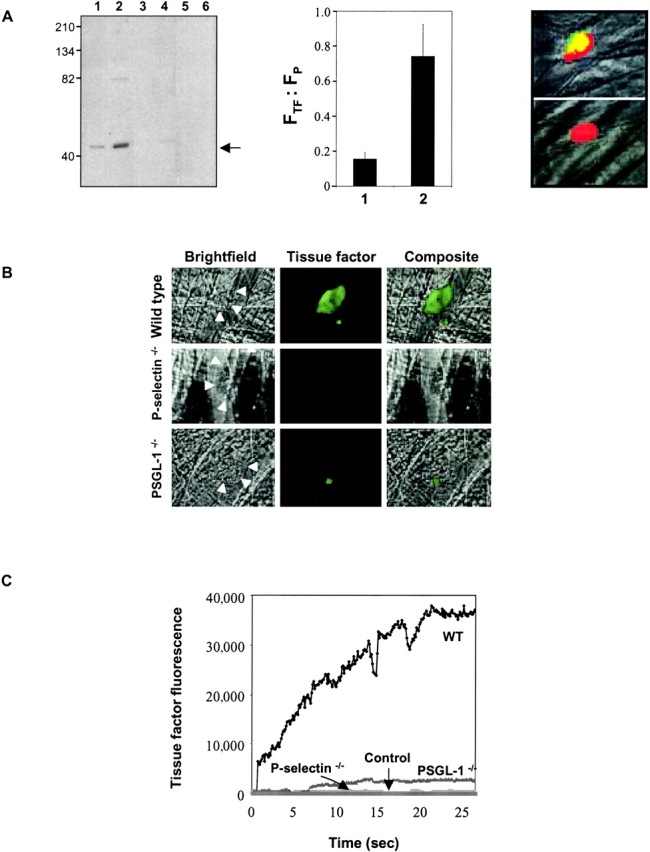
Tissue factor accumulation in the developing arterial thrombus. (A) Left: characterization of rabbit anti–mTF (152–166) antibodies. Western blot of tissue factor with affinity-purified rabbit anti–mTF (152–166) antibodies. STO cells, red cells, and platelets were lysed and the proteins in the lysate were separated on a 10% SDS-PAGE gel under reducing conditions. Lanes 1 and 5, 5 μg STO cell lysate; lanes 2 and 6, 7 μg STO cell lysate; lane 3, 5 μg murine red cell lysate; lane 4, 7 μg murine red cell lysate. Lanes 1–4 were developed with anti–mouse tissue factor antibody and lanes 5 and 6 were developed with an irrelevant antibody, anti-CD41. Arrow indicates molecular weight of tissue factor. Middle: comparison of tissue factor fluorescence detected with rabbit anti–mTF (152–166) antibodies (bar 2) or nonimmune IgG control (bar 1). The ratio of the integrated tissue factor fluorescence (FTF) to the integrated platelet fluorescence (FP) in thrombi generated in wild-type mice is shown based upon multiple independent experiments. P < 0.0042. Right: image of tissue factor in a thrombus detected using rabbit anti–mTF (152–166) antibodies. Top: Thrombus detected with anti-mTF (152–166) antibodies and anti-CD41 antibody directed against platelets. Bottom: Thrombus imaged with nonimmune IgG and anti-CD41 antibody directed against platelets. Red, platelets; green, tissue factor; yellow, platelets plus tissue factor. (B) Intravital images of arterial thrombi. Tissue factor was detected using Alexa 488–conjugated anti-tissue factor antibodies infused into the systemic circulation. The fluorescence image, recorded digitally, is presented as the green pseudocolor. Brightfield images of the thrombi are indicated by arrowheads. Images are representative of 12 thrombi formed in 2 arterioles in 2 mice of each genotype. (C) Time course of tissue factor antigen accumulation in the developing arterial thrombus in wild-type and genetically altered mice. Tissue factor was detected using sheep anti–tissue factor antibodies infused into the systemic circulation followed by Alexa 660–conjugated donkey anti–sheep IgG. Sheep IgG was substituted for anti-tissue factor antibody, control. Each curve represents the raw digital data of a single representative experiment. WT, wild-type mouse; PSGL−/−, PSGL-1 null mouse; P-selectin−/−, P-selectin null mouse.
We previously demonstrated that blocking antibodies to P-selectin infused into a baboon inhibited the formation of a fibrin clot in a shunt thrombosis model although platelet deposition was unaffected (11). Because fibrin generation is initiated by tissue factor, we examined the role of P-selectin and PSGL-1 in tissue factor accumulation in the developing thrombus. Alexa 488–conjugated sheep anti–rabbit tissue factor antibodies were infused into the circulation of a wild-type mouse. After 5 min, vascular injury was induced in a cremaster muscle arteriole. Tissue factor accumulated into the thrombus after vessel injury (Fig. 2 B), confirming our earlier results (22).
Endothelial injury led to thrombus formation in both the PSGL-1 null mouse and the P-selectin null mouse. However, minimal tissue factor was incorporated into the thrombus generated in either deficient mouse strain (Fig. 2 B). The initial time course of tissue factor accumulation in the developing thrombus of a wild-type mouse, PSGL-1 null mouse, and P-selectin null mouse indicates that minimal tissue factor is associated with thrombi formed in PSGL-1 null mice or P-selectin null mice, in contrast to that of wild-type mice (Fig. 2 C).
Because tissue factor synthesis and appearance might be altered by the level of P-selectin (32) or possibly PSGL-1 expression, we studied wild-type mice to determine whether blockade of P-selectin function just before thrombus formation would interfere with tissue factor accumulation in the thrombus. Thrombi (n = 7) were formed in wild-type mice and an additional seven thrombi were formed after infusion of anti–P-selectin antibody. Data are the mean ± SEM. P-selectin antibody infused into wild-type mice before thrombus formation inhibited tissue factor accumulation in thrombi by 80% compared with tissue factor accumulation in thrombi formed in the absence of P-selectin antibody (Fig. 3 A). These results are qualitatively similar to that observed with the P-selectin null mouse in the absence of anti–P-selectin antibodies.
Figure 3.

Anti–P-selectin antibodies inhibit tissue factor and fibrin accumulation in the developing thrombus in wild-type mice. (A) Tissue factor in the thrombus formed was quantitated using Alexa 488–conjugated sheep anti–tissue factor antibodies from images derived by high speed widefield microscopy. Thrombi were formed in the absence of anti–P-selectin antibody in a wild-type mouse (bar 1). Subsequently, anti–P-selectin antibody (0.5 μg/g body weight) was infused and thrombi were formed in the presence of antibody (bar 2) between 5 and 30 min after antibody infusion. Data are at 60 s after injury and from seven thrombi formed in four arterioles before infusion of antibody and seven thrombi formed in four arterioles after infusion of antibody. Two mice were used. Mean ± SEM. (B) Experimental protocol was the same as described above except that fibrin was detected using Alexa 660–conjugated anti-fibrin–specific antibodies.
Fibrin Accumulates in the Developing Thrombus in Wild-type Mice but Is Decreased in PSGL-1 Null and P-Selectin Null Mice.
To determine whether the tissue factor observed in the thrombus was biologically active, we studied the kinetics of fibrin accumulation in the developing thrombus using fibrin-specific antibodies (27). Alexa 660–conjugated fibrin-specific antibody was infused into the anesthetized mouse 5 min before the initiation of thrombus formation. Fibrin was detected in the developing thrombus in the wild-type mouse (Fig. 4 A, left), as previously demonstrated (22). Identical experiments performed using PSGL-1 null mice, P-selectin null mice, and wild-type mice treated with hirudin to inhibit thrombin showed minimal fibrin accumulation in the developing thrombus at 90 s (Fig. 4 A, left). We have quantitated double label experiments of platelet fluorescence and fibrin fluorescence from multiple experiments (Fig. 4 A, right). The ratio of the integrated fibrin fluorescence to the integrated platelet fluorescence based upon results from individual thrombi derived from the average of two mice of each genotype and a total of six thrombi is shown. To determine fluorescence background in the absence of fibrin, hirudin, which blocks fibrin formation, was used in wild-type mice and the integrated fibrin fluorescence to the integrated platelet fluorescence ratio was determined and subtracted from these ratios in wild-type mice, PSGL-1 null mice, and P-selectin null mice. The integrated fibrin fluorescence to the integrated platelet fluorescence ratio in thrombi from the P-selectin null mice and in thrombi from the PSGL-1 null mice was approximately 0, in contrast to this ratio determined in wild-type mice. Statistical analysis using the t test shows that this integrated fibrin fluorescence to the integrated platelet fluorescence ratio decrease is significant (P < 0.05). Error bars indicate standard error of the mean. Although the size and rate of enlargement of the platelet thrombi vary in these different experiments, only the wild-type mouse shows the development of significant amounts of fibrin colocalized with platelets (Fig. 4 B, yellow).
Figure 4.
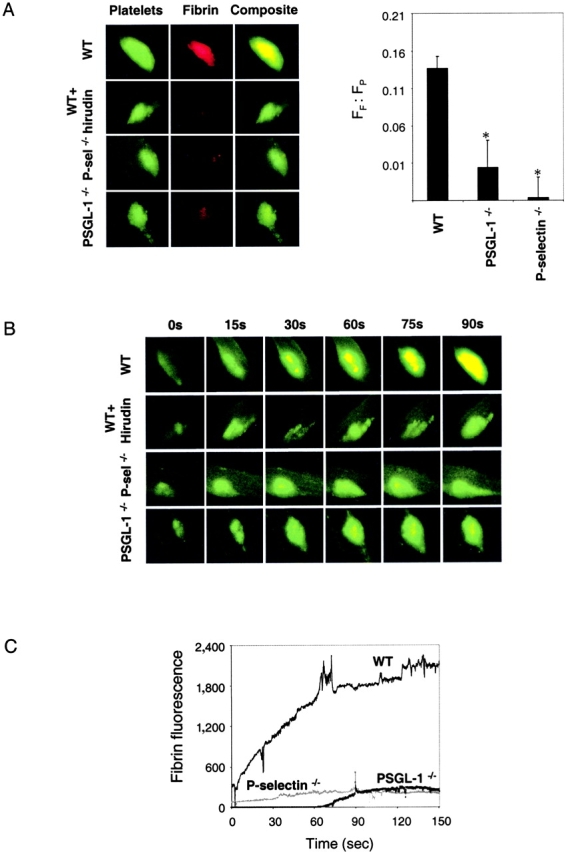
Fibrin deposition in the developing thrombus. (A) Left: Alexa 660–conjugated anti–mouse fibrin antibody and rat anti–mouse CD41 detected with Alexa 488–conjugated chicken anti–rat IgG were used to detect fibrin (red) and platelets (green). Overlap of the platelet and fibrin images is shown as a composite (yellow). When used, hirudin (1 U/g body weight) was infused immediately before initial thrombus formation. Fibrin and platelets were observed in thrombi formed 60 s after vessel injury. Images are representative of 10 thrombi formed in 6 arterioles in 3 mice of each genotype. Right: the ratio of the integrated fibrin fluorescence (FF) to the integrated platelet fluorescence (FP) in thrombi generated in wild-type mice, PSGL-1 null mice, and P-selectin null mice is shown (black) based upon multiple independent experiments. The fluorescence background in the absence of fibrin was determined in wild-type mice treated with hirudin. Error bars indicate standard error of the mean whereas * indicates statistical significance. (B) Images of the developing thrombus from 0–90 s. Platelets (green), fibrin (red), and platelet/fibrin composite (yellow). (C) Time course of fibrin formation in the developing thrombi of wild-type and genetically altered mice. Each curve represents the raw digital data of a single representative experiment. WT, wild-type mouse; PSGL−/−, PSGL-1 null mouse; P-selectin−/−, P-selectin null mouse.
We have previously demonstrated that anti–P-selectin antibodies inhibited fibrin deposition during experimental thrombosis in a baboon model (11). To revisit this experiment in a mouse model, P-selectin antibody was infused into a wild-type mouse before thrombus formation and fibrin was detected with a fibrin-specific antibody. Thrombi (n = 7) were formed in wild-type mice and an additional seven thrombi were formed after infusion of anti–P-selectin antibody. Data are the mean ± SEM. Thrombi formed in the presence of P-selectin antibody contained 60% less fibrin than thrombi formed in the absence of P-selectin antibody (Fig. 3 B). These results are quantitatively similar to those observed with the P-selectin null mouse in the absence of anti–P-selectin antibodies.
The time course of fibrin accumulation in the developing thrombus of a wild-type mouse, PSGL-1 null mouse, and P-selectin null mouse indicates that fibrin appearance correlates to the presence of tissue factor (Fig. 4 C). In the absence of tissue factor accumulation in the developing thrombus, as in the PSGL-1 null and P-selectin null mice (Fig. 2 C), there is minimal fibrin accumulation.
To compare the time course and localization of platelet deposition, tissue factor accumulation, and fibrin formation in the thrombus of a wild-type mouse or a mouse deficient in PSGL-1 or P-selectin, we monitored platelets, tissue factor, and fibrin simultaneously within the context of the brightfield image of the microcirculation. Intravital four channel video images were obtained that visualized platelets (red), tissue factor (green), and fibrin (blue) in the microcirculation (black/white). Colocalization of tissue factor and platelets (yellow), tissue factor and fibrin (turquoise), fibrin and platelets (magenta), and fibrin, tissue factor, and platelets (white) are also depicted. Images were collected continuously before, during, and after laser injury. The fluorescence intensities of tissue factor, fibrin, and platelets are not calibrated, and thus do not reflect the relative concentration of components to each other. Furthermore, the signal to noise level is too low to be certain of the timing of tissue factor detection relative to platelet and fibrin appearance. At time 0 s, the baseline fluorescence in each fluorescence channel was adjusted in the initial image at the time of injury to zero fluorescence. In this example of thrombus formation in a wild-type mouse (Fig. 5 , left) at time +20 s, platelets and tissue factor are accumulating on the vessel wall. As the platelet thrombus expands at time +75 s and time +85 s, fibrin appears within the thrombus. At time +100 s, platelet thrombus expansion continues as the quantity of fibrin and tissue factor on the proximal edge increases while fibrin and tissue factor extend distally within the platelet thrombus. Fibrin extends through much of the platelet thrombus, although the distal end of the thrombus remains exclusively composed of platelets.
Figure 5.
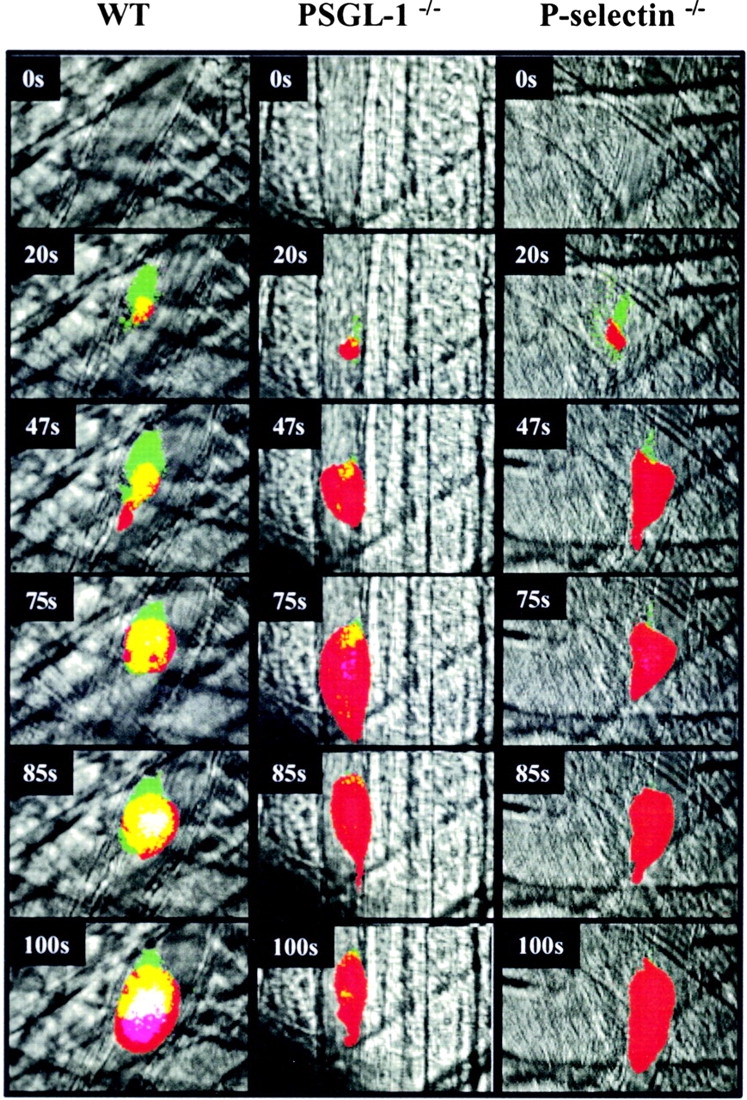
Intravital imaging of platelet, tissue factor, and fibrin deposition in the developing thrombus of a wild-type mouse, PSGL-1 null mouse, or P-selectin null mouse after endothelial injury. Alexa 660–conjugated CD41 Fab fragments, Alexa 488–conjugated sheep anti–tissue factor antibodies, and Alexa 350–conjugated mouse anti–fibrin antibodies were infused into the systemic circulation. Representative composite images of the developing thrombus are shown. Red, platelets; green, tissue factor; blue, fibrin; yellow, platelets plus tissue factor; turquoise, tissue factor plus fibrin; magenta, platelets plus fibrin; white, platelets plus fibrin plus tissue factor. Fluorescence intensity in each channel was zeroed in images obtained before thrombus formation. Videos were continuously collected in four channels before and during laser injury and during thrombus development. To simplify analysis of the composite image, the dynamic range of the intensity of each pseudocolor was minimized. Images are representative of four thrombi formed in each of the mouse strains. Left, wild-type mouse; middle, PSGL-1 null mouse; right, P-selectin null mouse.
Similar experiments were performed using either PSGL-1 null or P-selectin null mice. In the PSGL-1 null mouse, platelets accumulated at the site of injury but minimal tissue factor or fibrin is observed in the growing thrombus (Fig. 5, middle). Similar observations were made in the growing arterial thrombus of the P-selectin null mouse (Fig. 5, right). These results emphasize that the tissue factor that accumulates at the leading edge of the thrombus and along the vascular wall is dependent upon the presence of both PSGL-1 and P-selectin. The fibrin that forms correlates to the amount of tissue factor observed. These in vivo experiments demonstrate that platelets accumulate rapidly in the developing thrombi in wild-type mice, PSGL-1 null mice, and P-selectin null mice. However, only in wild-type mice is this event followed by significant fibrin deposition.
Mouse Microparticles Accumulate in the Developing Thrombus.
To determine the mechanism by which P-selectin and PSGL-1 influence tissue factor accumulation in the developing thrombus, we examined the source of tissue factor. Rauch et al. (21) have previously shown that anti-CD15 antibodies inhibit blood-borne tissue factor association with platelets in vitro, so we considered sources of tissue factor associated with CD15. CD15, a carbohydrate antigen, is broadly distributed on cell surface glycoproteins and glycolipids on myeloid cells and is a component of the counterreceptor of L-selectin, E-selectin, and P-selectin (33, 34). Because tissue factor is known to be inducible on the surface of monocytes and may also be expressed on granulocytes, we examined developing arterial thrombi for leukocytes and leukocyte-derived microparticles as a potential source of tissue factor. In our in vivo model of arterial thrombosis, no leukocytes either interacted with or were incorporated into the thrombus during the initial phase of thrombus development when tissue factor first appears in the thrombus (35). Therefore, we hypothesized that subcellular components circulating in blood that are derived from leukocytes, including microparticles, might include tissue factor physically associated with PSGL-1. Such tissue factor- and PSGL-1–bearing species would bind to P-selectin expressed on activated platelets in the arterial thrombus via PSGL-1, concentrating tissue factor in the thrombus.
Blood cells were isolated from anticoagulated mouse blood and incubated with calcein AM to introduce a fluorescent probe into the cytoplasm (Fig. 6 A, top left). After activation of these cells with the calcium ionophore A23187, a large number of fluorescent microparticles were generated (Fig. 6 A, bottom left). These microparticles were isolated by centrifugation (Fig. 6 A, bottom middle) and their size was compared with calibrated microspheres of 0.93-μm diameter (Fig. 6 A, top middle). ∼95% of the microparticles were smaller than 1 μm, as estimated by scattering measurements by flow cytometry. A histogram of the microparticles indicates their intense fluorescence (Fig. 6 A, right).
Figure 6.
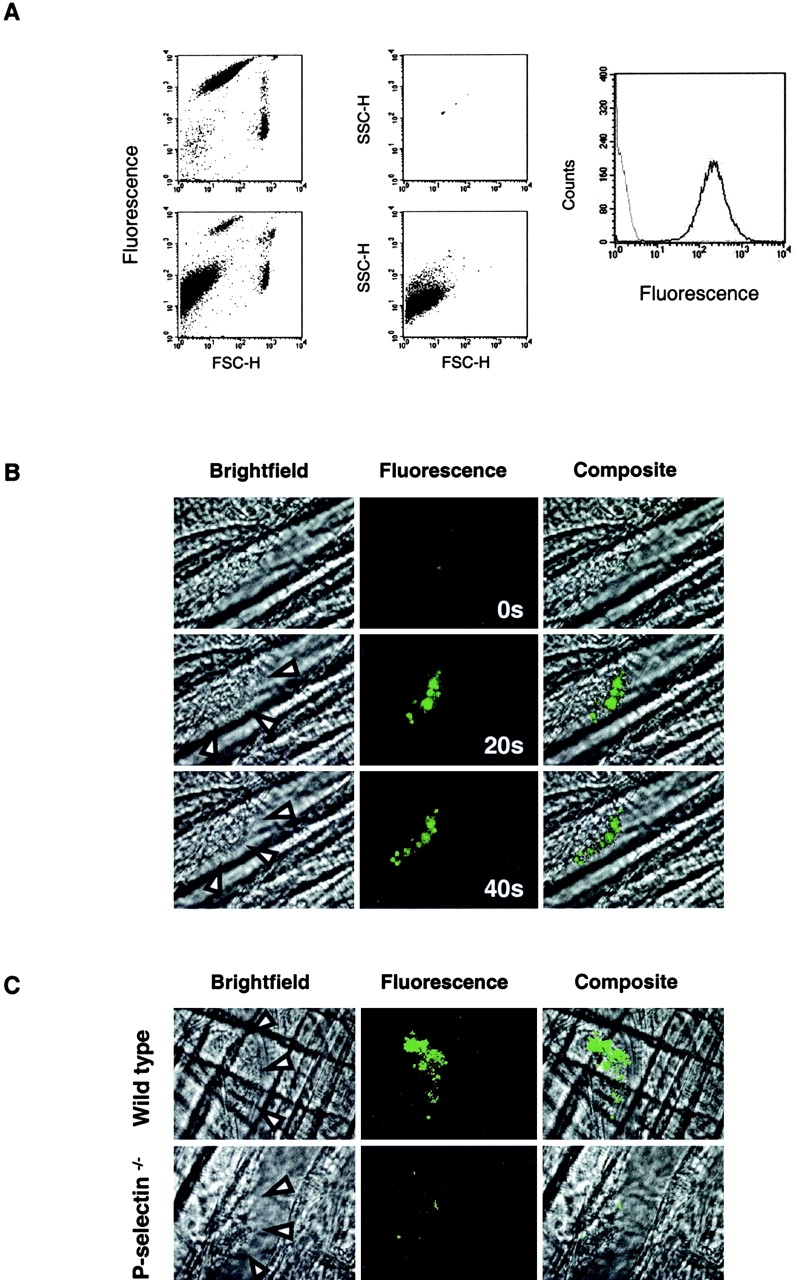
Calcein-labeled mouse microparticles accumulate into arterial thrombi. A leukocyte-rich blood cell preparation was labeled with calcein AM. (A) Flow cytometry. Two distinct populations are shown on the top left: a small, highly labeled cell population (platelets) and a larger population with lower calcein labeling (leukocytes). Upon the addition of A23187, a third population of calcein-labeled particles appeared (bottom left). 0.93 μm calibration microbeads were used to determine the limits of size of particles <1 μm (top middle). After the addition of A23187, the cells were removed by centrifugation, with >95% of the remaining particles in the supernatant <1 μm (bottom middle). These microparticles exhibited calcein fluorescence (black) compared with particles generated under the same conditions without the addition of calcein (gray; right). (B) Calcein-labeled microparticles were injected into the mouse circulation and their incorporation into arterial thrombi monitored over time by real-time widefield fluorescence videomicroscopy. Images are shown at 0, 20, and 40 s after thrombus formation. Left, brightfield image; arrow heads, the lumenal edge of the thrombus; middle, fluorescence image; right, composite brightfield and fluorescence images. Experiments are representative of three independent experiments. (C) Calcein-labeled microparticles derived from WEHI cells, a murine monocyte-like cell line, were infused into wild-type mice (top) or P-selectin null mice (bottom). The images were obtained 60 s after endothelial injury and the initiation of thrombus formation. Left, brightfield image; arrow heads, the lumenal edge of the thrombus; middle, fluorescence image; right, composite brightfield and fluorescence images.
These microparticles were infused into a wild-type mouse while the cremaster muscle vascular window was under direct observation. Although fluorescent microparticles could be seen rapidly flowing in the microcirculation, there was no interaction of these particles with the vessel wall of the arteriole (Fig. 6 B, 0s). Upon endothelial injury and thrombus formation, fluorescent microparticles became rapidly associated with the developing thrombus (Fig. 6 B).
Because microparticles derived from mononuclear cells are contaminated with microparticles derived from platelets that contaminate the cell preparation, we prepared microparticles from a mouse monocyte-like cell line, WEHI. WEHI cells express functional PSGL-1 and were the source of mouse PSGL-1 cDNA (36). WEHI cells in culture were loaded with calcein AM. After activation of these cells with the calcium ionophore A23187, a large number of fluorescent microparticles were generated and isolated by centrifugation. Light scattering measurements via flow cytometry revealed that these microparticles were <1 μm in diameter. The WEHI microparticle preparation was infused into either a wild-type mouse or a P-selectin null mouse while the cremaster muscle vascular window was under direct observation. Although fluorescent microparticles could be seen rapidly flowing in the microcirculation, there was no interaction of these particles with the vessel wall of the arteriole. Upon endothelial injury in the arteriole and thrombus formation, fluorescent microparticles became associated with the leading edge of the thrombus generated in wild-type mice (Fig. 6 C). However, microparticles did not accumulate in the P-selectin null mice although occasional transient interaction of microparticles with the lumenal edge of the thrombus was observed (Fig. 6 C). These results indicate that PSGL-1–bearing microparticles in the circulating blood localize in thrombi that express P-selectin through P-selectin–PSGL-1 interaction but do not accumulate in thrombi lacking P-selectin.
Tissue Factor and PSGL-1 on Blood Microparticles.
We examined microparticles in mouse and human plasma for the coexpression of both tissue factor antigen and PSGL-1 antigen. Anticipating low levels of such species in normal plasma, we developed sensitive methodology for detecting tissue factor and PSGL-1 by examining microparticles derived from human mononuclear cells. Supernatant from these cells were examined for tissue factor activity and for the presence of particles. As shown in Fig. 7 A, left, the supernatant of the mononuclear cells contained 1,410 particles/μl and low but measurable tissue factor activity. This activity was inhibited by anti-tissue factor antibodies. If the mononuclear cells were stimulated with LPS, the cell supernatant included 1,869 particles/μl with significantly higher tissue factor activity. This is consistent with the known induction of tissue factor biosynthesis by LPS (37). The concomitant stimulation of mononuclear cells with both LPS and the calcium ionophore A23187 led to 5,000 particles/μl and a 20-fold stimulation of tissue factor activity. Tissue factor activity could be removed from the cell supernatant by ultracentrifugation, indicating that the tissue factor measured is not a protein free in solution (Fig. 7 A, right). These results are consistent with tissue factor–bearing microparticles in the cell supernatant of stimulated and unstimulated mononuclear cells.
Figure 7.
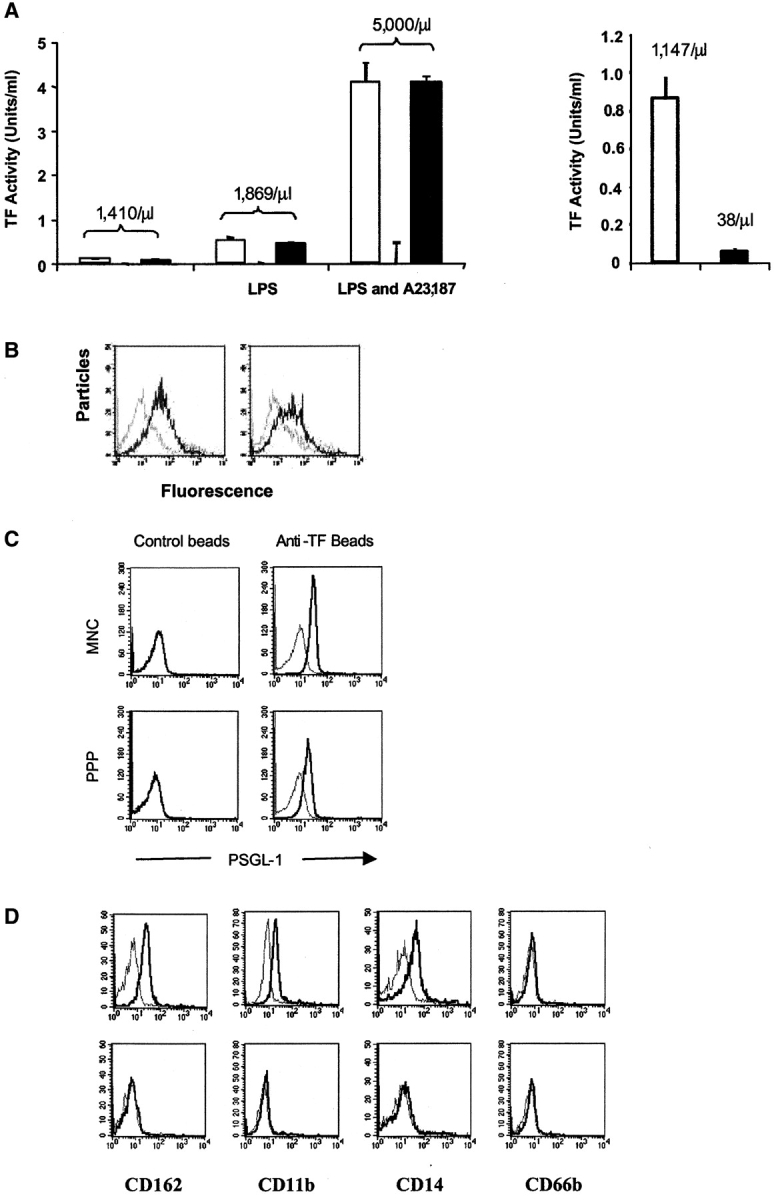
Functional tissue factor and PSGL-1 on microparticles and presence of microparticles positive for tissue factor and PSGL-1. (A) Monocytes produce tissue factor–bearing microparticles. The medium from monocytes isolated and cultured in the absence of any additions, in the presence of LPS, or in the presence of LPS and A23187 was centrifuged for 30 s at 14,000 g to remove cells. The microparticles in the supernatant were counted and assayed for the presence of tissue factor activity. Left: open bars, tissue factor activity in the supernatant; gray bars, tissue factor activity after addition of anti-tissue factor antibody; black bars, tissue factor activity after the addition of nonimmune IgG. Right: supernatant was analyzed for the presence of microparticles before and after ultracentrifugation at 150,000 g for 120 min. Open bar, before ultracentrifugation; black bar, after centrifugation. The number of particles per microliter is indicated. (B) Microparticles express functional PSGL-1. P-selectin Ig chimera adsorbed to beads was incubated with calcein-labeled microparticles in the presence of buffer (black) or EDTA (gray; left) and anti–PSGL-1 antibody (gray) or isotype-matched control antibody (black; right). (C) Flow cytometric analysis of tissue factor–positive microparticles concentrated from human platelet–poor plasma (PPP; bottom) and mononuclear cell supernatant (MNC; top). These microparticles were incubated with beads containing tissue factor antibody (right) and beads containing nonimmune IgG (left). Black, fluorescently labeled anti–PSGL-1 antibody; gray, fluorescently labeled anti–PSGL-1 antibody plus 50-fold excess of unlabeled anti–PSGL-1 antibody. (D) Flow cytometric analysis of tissue factor–positive microparticles concentrated from human platelet null plasma. CD162 (PSGL-1), CD11b, CD14, CD66b. Top, beads coated with anti-tissue factor antibody; bottom, beads coated with nonimmune IgG; black, fluorescently labeled specific antibody; gray, fluorescently labeled specific antibody plus 50 fold excess of unlabeled specific antibody.
Calcein-labeled microparticles were evaluated for the presence of functional PSGL-1. Using P-selectin IgG beads, we captured fluorescent microparticles in the presence of calcium (Fig. 7 B). In the presence of EDTA (Fig. 7, left) or a blocking antibody to PSGL-1 (Fig. 7 B, right), the interaction of microparticles with P-selectin IgG was inhibited.
Having shown the presence of functional tissue factor and functional PSGL-1 on microparticles, we examined mononuclear cell supernatant and platelet-poor plasma for the presence of PSGL-1 antigen after concentration of tissue factor–positive microparticles on beads containing anti-tissue factor. Anti-tissue factor beads were incubated with mononuclear cell supernatant or platelet-poor plasma, and the beads were examined by flow cytometry for PSGL-1 antigen. Tissue factor–positive microparticles from mononuclear cell supernatant or platelet-poor plasma both exhibited PSGL-1 antigen (Fig. 7 C). Control beads containing an isotype- matched control antibody in place of anti-tissue factor antibody did not bind to microparticles expressing PSGL-1. These results demonstrate the presence of microparticles in platelet-poor plasma or mononuclear cell supernatant that contain both tissue factor and PSGL-1.
Tissue factor–positive microparticles from platelet-poor plasma were examined to identify their cellular source (Fig. 7 D). These microparticles were positive for PSGL-1 (CD162), CD11b, and CD14, consistent with the phenotype of monocytes. They were negative for CD66b, a granulocyte marker.
Discussion
P-selectin and PSGL-1 are cell adhesion molecules that play a critical role in the initial capture and tethering events associated with leukocyte interaction with the endothelium of the vasculature. P-selectin, present in the storage granules of platelets and endothelial cells, is translocated upon cell activation to the plasma membrane where it is expressed on the membrane surface. PSGL-1, a mucinous glycoprotein, is constitutively expressed on the leukocyte plasma membrane. During the initial phases of inflammation, leukocyte rolling is mediated on the vasculature by these adhesion molecules. PSGL-1 appears to be the dominant P-selectin ligand during leukocyte rolling, although this glycoprotein also serves as an E-selectin ligand for leukocyte rolling and lymphocyte migration in vivo (24, 25, 38). In this study, we have broadened the known functions of PSGL-1 to propose a role in tissue factor accumulation and fibrin formation during the development of experimental thrombi through the concentration of PSGL-1–bearing microparticles in the developing thrombus. This provides a molecular basis for our previous observation of the ability of anti–P-selectin antibodies to inhibit fibrin formation during thrombogenesis (11).
The longstanding model of blood coagulation has been that (a) blood clotting is initiated via tissue factor, (b) tissue factor is not exposed to blood but is constitutively expressed on nonvascular cells, and (c) upon vascular injury, tissue factor on nonvascular cells comes in contact with flowing blood, initiating a series of linked, enzymatic reactions that culminate in the generation of fibrin and a fibrin clot. A number of recent observations are not consistent with this model. First, tissue factor circulates in blood at levels of ∼100–150 pg per ml (17–19). Second, tissue factor is associated with thrombi formed on pig arterial media or collagen-coated glass slides exposed to flowing human blood in vitro (20). The association of tissue factor–positive granules with platelets in this thrombus was inhibited by anti-CD15 (21), suggesting a role for this carbohydrate antigen in this interaction. Third, this model does not include a role for P-selectin. Yet, antibodies to P-selectin inhibit fibrin formation (11), P-selectin null mice have a prolonged bleeding time (39), and thrombi formed in vitro indicate architectural differences when formed from blood of wild-type and P-selectin null mice (40). Using digital videomicroscopy to image the microcirculation of a living mouse, we have analyzed the role of PSGL-1 and P-selectin in thrombosis in vivo.
In this and an earlier study, we have observed platelet deposition, P-selectin expression, tissue factor localization, and fibrin formation during thrombus formation in wild-type mice after vascular injury of the arterial vessel wall (22). We now show that tissue factor accumulation in the thrombus is minimal in mice lacking either PSGL-1 or P-selectin. During the earliest stages of thrombus development in wild-type mice, tissue factor accumulates at the leading edge of the developing thrombus, where plasma tissue factor first comes in contact with platelet P-selectin in the thrombus, and at the thrombus-vessel wall interface, where tissue factor might be derived from the vessel wall or from plasma captured by endothelial cell P-selectin. We show that tissue factor in both sites is minimal in P-selectin null mice and PSGL-1 null mice. Andre et al. (32) demonstrated that elevated soluble P-selectin levels maintained in a mouse for 22 h or P-selectin constitutively expressed on the mouse endothelium leads to increased circulating microparticles, some expressing tissue factor. We considered that the minimal tissue factor in the thrombi of mice lacking either PSGL-1 or P-selectin in our experiments might be due to decreased numbers of tissue factor–bearing microparticles. However, we show that an inhibitory P-selectin antibody infused into a wild-type mouse just before endothelial injury blocks tissue factor and fibrin accumulation in the thrombus. These results parallel the results of P-selectin antibody inhibition of fibrin clot formation in a baboon model (11). Thus, chronic deficiency of P-selectin, as in the P-selectin null mouse, is not responsible for the absence of tissue factor in the thrombus. Because both the P-selectin null mouse and PSGL-1 null mouse demonstrate decreased tissue factor accumulation in the developing thrombus, direct interaction between P-selectin and tissue factor is not responsible for tissue factor accumulation and is not the basis for tissue factor–mediated transfer of microparticles to platelets (21). Furthermore, the report of tissue factor in platelets (41) and inference that platelet tissue factor contributes to thrombus formation does not explain the role of PSGL-1 in tissue factor accumulation in thrombi in our in vivo experiments. The tissue factor accumulated in the developing thrombus is biologically active because the quantity of tissue factor antigen correlates with the fibrin level that forms in the thrombus. Based upon these in vivo studies, we support the concept of Giesen et al. (20) that one pathway for the propagation of blood coagulation after endothelial injury involves the accumulation and localization of blood-borne tissue factor on microparticles onto the platelet thrombus.
Although the absence of PSGL-1 or P-selectin greatly reduces the observed tissue factor in the developing thrombus, some tissue factor can be detected. This raises the question whether there exists an alternate mechanism that can deliver microparticles to the platelet thrombus or whether our observation is due to platelet tissue factor (41). Given the minimal amount of tissue factor and fibrin observed in the P-selectin null mice and the PSGL-1 null mice, it would not appear that this possible alternate pathway is critical for fibrin formation.
Based upon the known presence of low concentrations of tissue factor in normal blood, the presence of tissue factor in thrombi formed in vitro and inhibition of tissue factor–bearing microparticles interaction with platelets by anti-CD15 antibodies (21), we hypothesized that PSGL-1 might play a role in the concentration of tissue factor–positive leukocyte-derived microparticles into the developing thrombus. CD15, a carbohydrate antigen, is broadly expressed on glycoproteins and glycolipids on myeloid cells. CD15 antibody inhibited the binding of platelets to tissue factor–positive microparticles generated from THP-1 cells, suggesting a role for CD15 in this interaction (21). The P-selectin ligand expressed on myeloid cells, along with most other plasma membrane glycoproteins on these cells, includes CD15 as a component (33). This P-selectin ligand has been identified as a glycoprotein, PSGL-1 (9, 10). PSGL-1, an integral membrane protein including sialylated and fucosylated core 2 glycans, is found on the plasma membrane of neutrophils, monocytes, and certain lymphocyte subsets. Because monocytes express PSGL-1 and can be induced to express tissue factor, the PSGL-1–dependent delivery of tissue factor to the platelet thrombus expressing P-selectin could be mediated by blood leukocytes. However, in our in vivo imaging experiments, we observe no leukocyte incorporation into the developing thrombus during the initial minutes after arterial injury (35). Because tissue factor–positive microparticles have been shown in in vitro experiments to be associated with platelets in the thrombus (21), these particles might express PSGL-1 necessary for fibrin formation.
Blood under normal and pathophysiologic conditions contains procoagulant microparticles (42). Several lines of evidence suggest that microparticles derived from platelets (43–45), leukocytes (46–48), endothelial cells (49, 50), and smooth muscle cells (51) could play a role in blood coagulation. Furthermore, it is well established that microparticles express phosphatidylserine on the membrane surface that can provide the catalytic surface for the tenase and prothrombinase complexes, along with antigens characteristic of their cellular origin (52).
We have generated mouse microparticles that are intensely fluorescently labeled and infused them into a living mouse while monitoring the microcirculation of the cremaster muscle. In the absence of endothelial injury, there was no evidence of microparticle–vessel wall interaction in the arterial microcirculation. This parallels the observation that leukocyte rolling is not present on the arterial limb. However, upon endothelial injury and thrombus formation, we observe rapid accumulation of fluorescent microparticles into the developing thrombus. Although only a very small fraction of the blood microparticles were fluorescently labeled, real-time video imaging demonstrated their instant capture solely on the growing thrombus in a mechanism dependent upon P-selectin. No interaction was observed between microparticles and the endothelium. Microparticles were only transiently associated with developing thrombi in the P-selectin null mouse.
To explore this in a system where ample starting material was available, we first looked for tissue factor–positive, PSGL-1+ microparticles in human platelet–poor plasma. These microparticles are <1 μm in diameter. Low concentrations of both tissue factor and PSGL-1 precluded direct measurement of these antigens on the particle surface by flow cytometry. Therefore, we concentrated tissue factor–positive microparticles on polystyrene beads coated with anti-tissue factor antibody. These beads were subsequently shown to be positive for PSGL-1 antigen by flow cytometry. This link between tissue factor and PSGL-1, coupled with our data showing that there is functional PSGL-1 and active tissue factor on microparticles and that such particles are incorporated into arterial thrombi, implicate PSGL-1 in a new role as a cell adhesion molecule on microparticles involved with delivering tissue factor to the developing thrombus.
The tissue factor and Factor VIIa concentrations in normal blood are low. Although encryption of tissue factor may also contribute to the absence of spontaneous coagulation in flowing blood (53), we hypothesize that upon reaching a threshold level during concentration in the platelet thrombus, tissue factor can complex with Factor VIIa and initiate blood coagulation, culminating in the generation of fibrin. It remains to be seen whether tissue factor within the thrombus is part of a physiologically critical pathway of fibrin generation or whether it is a supplement to mechanisms involving tissue factor within the damaged vessel wall.
P-selectin and PSGL-1 are important cell adhesion molecules that mediate the initial steps in the inflammatory pathway and lymphocyte migration (24, 25, 54). Our results demonstrate that these adhesion molecules also play a role in thrombus formation in capturing tissue factor–bearing plasma microparticles into the site of vascular injury where platelet P-selectin can bind to PSGL-1 on these microparticles. Although much attention has been focused on the anti-inflammatory applications of the blockade of the selectins and their ligands, inhibition of P-selectin or PSGL-1 function might be a useful pharmacologic target for antithrombotic therapy.
Acknowledgments
We are grateful to Dr. Samuel Rapaport for helpful discussions and insight and Dr. Joshua Goldstein for performing some preliminary experiments.
This work was supported by grants from the National Institutes of Health (NIH; HL51926 and HL69435). Q. Liu was the recipient of a postdoctoral fellowship from the Heart and Stroke Foundation of Canada. P. Gross was the recipient of a postdoctoral fellowship from the Canadian Institute of Health Research. J. Chou was the recipient of a Howard Hughes Medical Student Fellowship. E. Vandendries is the recipient of a Clinical Scientist Development Award from the NIH. The microscope was obtained with partial support from the NIH (S10RR15680).
Q. Liu's present address is Department of Dermatology, Massachusetts General Hospital, 55 Fruit St., Boston, MA 02114.
K. Croce's present address is Department of Medicine, Brigham and Women's Hospital, 75 Francis St., Boston, MA 02115.
P. Gross' present address is Department of Medicine, University of Toronto, 30 Bond St., Toronto, Ontario M5B 1WB, Canada.
Footnotes
*
Abbreviation used in this paper: PSGL-1, P-selectin glycoprotein ligand 1.
References
- 1.McEver, R.P. 1994. Selectins. Curr. Opin. Immunol. 6:75–84. [DOI] [PubMed] [Google Scholar]
- 2.Kansas, G.S. 1996. Selectins and their ligands: current concepts and controversies. Blood. 88:3259–3287. [PubMed] [Google Scholar]
- 3.Yang, J., B.C. Furie, and B. Furie. 1999. The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb. Haemost. 81:1–7. [PubMed] [Google Scholar]
- 4.Hsu-Lin, S., C.L. Berman, B.C. Furie, D. August, and B. Furie. 1984. A platelet membrane protein expressed during platelet activation and secretion. Studies using a monoclonal antibody specific for thrombin-activated platelets. J. Biol. Chem. 259:9121–9126. [PubMed] [Google Scholar]
- 5.McEver, R.P., and M.N. Martin. 1984. A monoclonal antibody to a membrane glycoprotein binds only to activated platelets. J. Biol. Chem. 259:9799–9804. [PubMed] [Google Scholar]
- 6.Stenberg, P.E., R.P. McEver, M.A. Shuman, Y.V. Jacques, and D.F. Bainton. 1985. A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J. Cell Biol. 101:880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berman, C.L., E.L. Yeo, J.D. Wencel-Drake, B.C. Furie, M.H. Ginsberg, and B. Furie. 1986. A platelet alpha granule membrane protein that is associated with the plasma membrane after activation. Characterization and subcellular localization of platelet activation-dependent granule-external membrane protein. J. Clin. Invest. 78:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsen, E., A. Celi, G.E. Gilbert, B.C. Furie, J.K. Erban, R. Bonfanti, D.D. Wagner, and B. Furie. 1989. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 59:305–312. [DOI] [PubMed] [Google Scholar]
- 9.Sako, D., X.J. Chang, K.M. Barone, G. Vachino, H.M. White, G. Shaw, G.M. Veldman, K.M. Bean, T.J. Ahern, B. Furie, et al. 1993. Expression cloning of a functional glycoprotein ligand for P-selectin. Cell. 75:1179–1186. [DOI] [PubMed] [Google Scholar]
- 10.Moore, K., N. Stults, S. Diaz, D. Smith, R. Cummings, A. Varki, and R. McEver. 1992. Identification of a specific glycoprotein ligand for P-selectin (CD62) on myeloid cells. J. Cell. Biol. 118:445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palabrica, T., R. Lobb, B.C. Furie, M. Aronovitz, C. Benjamin, Y.M. Hsu, S.A. Sajer, and B. Furie. 1992. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature. 359:848–851. [DOI] [PubMed] [Google Scholar]
- 12.Celi, A., G. Pellegrini, R. Lorenzet, A. De Blasi, N. Ready, B.C. Furie, and B. Furie. 1994. P-selectin induces the expression of tissue factor on monocytes. Proc. Natl. Acad. Sci. USA. 91:8767–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furie, B., and B.C. Furie. 1988. The molecular basis of blood coagulation. Cell. 53:505–518. [DOI] [PubMed] [Google Scholar]
- 14.Morrissey, J.H. 2001. Tissue factor: an enzyme cofactor and a true receptor. Thromb. Haemost. 86:66–74. [PubMed] [Google Scholar]
- 15.Morrissey, J.H., H. Fakhrai, and T.S. Edgington. 1987. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. 50:129–135. [DOI] [PubMed] [Google Scholar]
- 16.Spicer, E.K., R. Horton, L. Bloem, R. Bach, K.R. Williams, A. Guha, J. Kraus, T.C. Lin, Y. Nemerson, and W.H. Konigsberg. 1987. Isolation of cDNA clones coding for human tissue factor: primary structure of the protein and cDNA. Proc. Natl. Acad. Sci. USA. 84:5148–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koyama, T., K. Nishida, S. Ohdama, M. Sawada, N. Murakami, S. Hirosawa, R. Kuriyama, K. Matsuzawa, R. Hasegawa, and N. Aoki. 1994. Determination of plasma tissue factor antigen and its clinical significance. Br. J. Haematol. 87:343–347. [DOI] [PubMed] [Google Scholar]
- 18.Fareed, J., D.D. Callas, D. Hoppensteads, and E.W. Bermes. 1995. Tissue factor antigen levels in various biological fluids. Blood Coagul. Fibrinolysis. 6:S32–S36. [DOI] [PubMed] [Google Scholar]
- 19.Zumbach, M., M. Hofmann, V. Borcea, T. Luther, M. Kotzsch, M. Muller, O. Hergesell, K. Andrassy, E. Ritz, R. Ziegler, et al. 1997. Tissue factor antigen is elevated in patients with microvascular complications of diabetes mellitus. Exp. Clin. Endocrinol. Diabetes. 105:206–212. [DOI] [PubMed] [Google Scholar]
- 20.Giesen, P.L., U. Rauch, B. Bohrmann, D. Kling, M. Roque, J.T. Fallon, J.J. Badimon, J. Himber, M.A. Riederer, and Y. Nemerson. 1999. Blood-borne tissue factor: another view of thrombosis. Proc. Natl. Acad. Sci. USA. 96:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rauch, U., D. Bonderman, B. Bohrmann, J.J. Badimon, J. Himber, M.A. Riederer, and Y. Nemerson. 2000. Transfer of tissue factor from leukocytes to platelets is mediated by CD15 and tissue factor. Blood. 96:170–175. [PubMed] [Google Scholar]
- 22.Falati, S., P. Gross, G. Merrill-Skoloff, B.C. Furie, and B. Furie. 2002. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat. Med. 8:1175–1181. [DOI] [PubMed] [Google Scholar]
- 23.Rosen, E.D., S. Raymond, A. Zollman, F. Noria, M. Sandoval-Cooper, A. Shulman, J.L. Merz, and F.J. Castellino. 2001. Laser-induced noninvasive vascular injury models in mice generate platelet- and coagulation-dependent thrombi. Am. J. Pathol. 158:1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang, J., T. Hirata, K. Croce, G. Merrill-Skoloff, B. Tchernychev, E. Williams, R. Flaumenhaft, B.C. Furie, and B. Furie. 1999. Targeted gene disruption demonstrates that P-selectin glycoprotein ligand 1 (PSGL-1) is required for P-selectin–mediated but not E-selectin–mediated neutrophil rolling and migration. J. Exp. Med. 190:1769–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirata, T., G. Merrill-Skoloff, M. Aab, J. Yang, B.C. Furie, and B. Furie. 2000. P-selectin glycoprotein ligand 1 (PSGL-1) is a physiological ligand for E-selectin in mediating T helper 1 lymphocyte migration. J. Exp. Med. 192:1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ley, K. 1995. Gene-targeted mice in leukocyte adhesion research. Microcirculation. 2:141–150. [DOI] [PubMed] [Google Scholar]
- 27.Kudryk, B., A. Rohoza, M. Ahadi, J. Chin, and M.E. Wiebe. 1984. Specificity of a monoclonal antibody for the NH2-terminal region of fibrin. Mol. Immunol. 21:89–94. [DOI] [PubMed] [Google Scholar]
- 28.Bristol, J.A., B.C. Furie, and B. Furie. 1993. Propeptide processing during factor IX biosynthesis. Effect of point mutations adjacent to the propeptide cleavage site. J. Biol. Chem. 268:7577–7584. [PubMed] [Google Scholar]
- 29.Dackiw, A.P., I.D. McGilvray, M. Woodside, A.B. Nathens, J.C. Marshall, and O.D. Rotstein. 1996. Prevention of endotoxin-induced mortality by antitissue factor immunization. Arch. Surg. 131:1273–1278. [DOI] [PubMed] [Google Scholar]
- 30.Liu, Q., M.M. Rooney, A. Kasirer-Friede, E. Brown, S.T. Lord, and M.M. Frojmovic. 1998. Role of the gamma chain Ala-Gly-Asp-Val and Aα chain Arg-Gly-Asp-Ser sites of fibrinogen in coaggregation of platelets and fibrinogen-coated beads. Biochim. Biophys. Acta. 1385:33–42. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzet, R., J. Niemetz, A.J. Marcus, and M.J. Broekman. 1986. Enhancement of mononuclear procoagulant activity by platelet 12-hydroxyeicosatetraenoic acid. J. Clin. Invest. 78:418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andre, P., D. Hartwell, I. Hrachovinova, S. Saffaripour, and D.D. Wagner. 2000. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc. Natl. Acad. Sci. USA. 97:13835–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larsen, E., T. Palabrica, S. Sajer, G.E. Gilbert, D.D. Wagner, B.C. Furie, and B. Furie. 1990. PADGEM-dependent adhesion of platelets to monocytes and neutrophils is mediated by a lineage-specific carbohydrate, LNF III (CD15). Cell. 63:467–474. [DOI] [PubMed] [Google Scholar]
- 34.Foxall, C., S.R. Watson, D. Dowbenko, C. Fennie, L.A. Lasky, M. Kiso, A. Hasegawa, D. Asa, and B.K. Brandley. 1992. The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. J. Cell Biol. 117:895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross, P.L., B.C. Furie, and B. Furie. 2002. Kinetics of platelet activation, P-selectin expression and leukocyte rolling during arterial thrombus formation in vivo. Blood. 100:23a. [Google Scholar]
- 36.Yang, J., J. Galipeau, C.A. Kozak, B.C. Furie, and B. Furie. 1996. Mouse P-selectin glycoprotein ligand-1: molecular cloning, chromosomal localization, and expression of a functional P-selectin receptor. Blood. 87:4176–4186. [PubMed] [Google Scholar]
- 37.Semeraro, N., A. Biondi, R. Lorenzet, D. Locati, A. Mantovani, and M.B. Donati. 1983. Direct induction of tissue factor synthesis by endotoxin in human macrophages from diverse anatomical sites. Immunology. 50:529–535. [PMC free article] [PubMed] [Google Scholar]
- 38.Xia, L., M. Sperandio, T. Yago, J.M. McDaniel, R.D. Cummings, S. Pearson-White, K. Ley, and R.P. McEver. 2002. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J. Clin. Invest. 109:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramaniam, M., P.S. Frenette, S. Saffaripour, R.C. Johnson, R.O. Hynes, and D.D. Wagner. 1996. Defects in hemostasis in P-selectin-deficient mice. Blood. 87:1238–1242. [PubMed] [Google Scholar]
- 40.Ruggeri, Z., M. Subramanian, J. Dent, D. Wagner, and E. Saldivar. 2000. P-Selectin and the three dimensional structure of platelet thrombi. Blood. 96(Suppl. 1):812a. [Google Scholar]
- 41.Muller, I., A. Zillman, M. Kotzsch, M. Spannagl, S. Zahler, T. Luter, and B. Engelmann. 2001. Collagen stimulation triggers the rapid activation of platelet-associated tissue factor. Thrombosis and Haemostasis Supplement July. 201:P2887. [Google Scholar]
- 42.Nieuwland, R., R.J. Berckmans, S. McGregor, A.N. Boing, F.P. Romijn, R.G. Westendorp, C.E. Hack, and A. Sturk. 2000. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 95:930–935. [PubMed] [Google Scholar]
- 43.Sims, P.J., T. Wiedmer, C.T. Esmon, H.J. Weiss, and S.J. Shattil. 1989. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane: studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J. Biol. Chem. 254:17049–17057. [PubMed] [Google Scholar]
- 44.Thiagarajan, P., and J.F. Tait. 1991. Collagen-induced exposure of anionic phospholipid in platelets and platelet-derived microparticles. J. Biol. Chem. 266:24302–24307. [PubMed] [Google Scholar]
- 45.Siljander, P., O. Carpen, and R. Lassila. 1996. Platelet-derived microparticles associate with fibrin during thrombosis. Blood. 87:4651–4663. [PubMed] [Google Scholar]
- 46.Satta, N., F. Toti, O. Feugeas, A. Bohbot, J. Dachary-Prigent, V. Eschwege, H. Hedman, and J.M. Freyssinet. 1994. Monocyte vesiculation is a possible mechanism for dissemination of membrane-associated procoagulant activities and adhesion molecules after stimulation by lipopolysaccharide. J. Immunol. 153:3245–3255. [PubMed] [Google Scholar]
- 47.Berckmans, R.J., R. Nieuwland, A.N. Boing, F.R.H.T.M. Romijn, C.E. Hack, and A. Sturk. 2001. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb. Haemost. 85:639–646. [PubMed] [Google Scholar]
- 48.Mesri, M., and D.C. Altieri. 1999. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J. Biol. Chem. 274:23111–23118. [DOI] [PubMed] [Google Scholar]
- 49.Combes, V., A.-C. Simon, G.-E. Grau, D. Arnoux, L. Camoin, F. Sabatier, M. Mutin, M. Sanmarco, J. Sampol, and F. Dignat-George. 1999. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J. Clin. Invest. 104:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sabatier, F., V. Roux, F. Anfosso, L. Camoin, J. Sampol, and F. Dignat-George. 2002. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood. 99:3962–3970. [DOI] [PubMed] [Google Scholar]
- 51.Schecter, A.D., B. Spirn, M. Rossikhina, P.L. Giesen, V. Bogdanov, J.T. Fallon, E.A. Fisher, L.M. Schnapp, Y. Nemerson, and M.B. Taubman. 2000. Release of active tissue factor by human arterial smooth muscle cells. Circ. Res. 87:126–132. [DOI] [PubMed] [Google Scholar]
- 52.Gilbert, G.E., P.J. Sims, T. Wiedmer, B. Furie, B.C. Furie, and S.J. Shattil. 1991. Platelet-derived microparticles express high affinity receptors for factor VIII. J. Biol. Chem. 266:17261–17268. [PubMed] [Google Scholar]
- 53.Le, D.T., S.I. Rapaport, and L.V. Rao. 1992. Relations between factor VIIa binding and expression of factor VIIa/tissue factor catalytic activity on cell surfaces. J. Biol. Chem. 267:15447–15454. [PubMed] [Google Scholar]
- 54.Mayadas, T.N., R.C. Johnson, H. Rayburn, R.O. Hynes, and D.D. Wagner. 1993. Leukocyte rolling and extravasation are severely compromised in P-selectin-deficient mice. Cell. 74:541–554. [DOI] [PubMed] [Google Scholar]