Induction of NFATc2 Expression by Interleukin 6 Promotes T Helper Type 2 Differentiation (original) (raw)
Abstract
Interleukin (IL)-6 is produced by professional antigen-presenting cells (APCs) such as B cells, macrophages, and dendritic cells. It has been previously shown that APC-derived IL-6 promotes the differentiation of naive CD4+ T cells into effector T helper type 2 (Th2) cells. Here, we have studied the molecular mechanism for IL-6–mediated Th2 differentiation. During the activation of CD4+ T cells, IL-6 induces the production of IL-4, which promotes the differentiation of these cells into effector Th2 cells. Regulation of IL-4 gene expression by IL-6 is mediated by nuclear factor of activated T cells (NFAT), as inhibition of NFAT prevents IL-6–driven IL-4 production and Th2 differentiation. IL-6 upregulates NFAT transcriptional activity by increasing the levels of NFATc2. The ability of IL-6 to promote Th2 differentiation is impaired in CD4+ T cells that lack NFATc2, demonstrating that NFATc2 is required for regulation of IL-4 gene expression by IL-6. Regulation of NFATc2 expression and NFAT transcriptional activity represents a novel pathway by which IL-6 can modulate gene expression.
Keywords: CD4+ T cells, IL-4, NFAT, cytokines, gene regulation
Introduction
Cytokines can be pleiotropic in a given cell type due, in part, to the activation of several signaling pathways. IL-6 binds to the surface IL-6 receptor (IL-6R)α, leading to the dimerization of gp130/IL-6Rβ (1). Dimerization of gp130 by IL-6 causes the activation of two signaling pathways: (a) the Janus kinase (JAK)*/signal transducers and activators of transcription (STAT) pathway and (b) the CCAAT/enhancer binding protein (C/EBP) pathway. Activation of the extracellular signal–regulated kinase (ERK) pathway by IL-6 appears to mediate phosphorylation and activation of C/EBPβ (NF-IL6; references 2–4). IL-6 also induces the expression of C/EBPδ (NF-IL6β), another member of the C/EBP family of transcription factors (5). C/EBPδ together with C/EBPβ regulate type-1 IL-6 responsive genes (6, 7). Activation of Jak1, 2, and Tyk2 by IL-6 results in the phosphorylation and activation of STAT3 and, to much lesser extent, STAT1 leading to the induction of type-2 IL-6 responsive gene expression (8–11).
IL-6 is produced by APCs such as B cells, macrophages, and dendritic cells. It can also be secreted by nonprofessional APCs such as glial, epithelial, endothelial, and some tumor cells (for a review, see reference 12). IL-6 is crucial for the acute phase response in hepatocytes, is involved in the differentiation of B cells and myeloid cells, and induces growth of osteoblasts, hematopoietic, and neural cells (12). It has been recently shown that IL-6 can confer multidrug resistance to breast cancer cells (13).
IL-6 also influences several aspects of the T cell immune response. This cytokine can act as a survival factor for resting T cells, but not for activated CD4+ T cells (14, 15). IL-6 can also protect resting CD8+ T cells from death (16). We have recently shown that IL-6 inhibits the differentiation of CD4+ T cells into effector Th1 cells. IL-6 induces the expression of suppressor of cytokine signaling (SOCS)1 which inhibits IFN-γ signaling (17). IL-6 also directs the differentiation of CD4+ T cells into IL-4–producing effector Th2 cells (18), but the molecular mechanism has not yet been defined. Whereas the inhibition of Th1 differentiation by IL-6 is IL-4 independent (17), IL-4 is required for the promotion of Th2 differentiation by IL-6 (18). Thus, it is likely that IL-6 utilizes two different mechanisms to inhibit Th1 differentiation and promote Th2 differentiation.
Here we show that IL-6 promotes Th2 differentiation by inducing IL-4 gene expression during the activation of CD4+ T cells. Several studies have shown the presence of nuclear factor of activated T cells (NFAT) binding motifs in the IL-4 promoter and the positive role of NFAT in the regulation of IL-4 gene transcription (19–21). Four members of the NFATc family have been described (NFATc1, c2, c3, and c4). IL-4 production appears to be normal in mice that lack NFATc3 (22). In contrast, reduced IL-4 production by Th2 cells has been observed in splenocytes from NFATc1–deficient chimeric mice (23, 24). The role of NFATc2 in IL-4 regulation is less clear. Although some studies have reported increased production of IL-4 in the absence of NFATc2 (25), other studies have shown an initial decrease in IL-4 synthesis in these mice (26, 27). Disruption of NFATc2 and NFATc3 results in increased IL-4 production (28), whereas disruption of both NFATc1 and NFATc2 abrogates production of a large number of cytokines including IL-4 (29).
In this study we show that IL-6 regulates IL-4 gene expression by activating NFAT. IL-6 induces the specific expression of NFATc2 mRNA and protein. Examination of NFATc2–deficient mice revealed that NFATc2 is required for the induction of IL-4 gene expression and Th2 differentiation by IL-6. Thus, the regulation of NFATc2 expression by IL-6 represents a novel mechanism by which this cytokine regulates NFAT-mediated transcription and IL-4 gene expression.
Materials and Methods
Reagents.
Anti-CD3 mAb (145–2C11), anti-CD28 (BD PharMingen), IL-6 (R&D Systems), IL-4 (R&D Systems), IL-2 (R&D Systems), IL-5 (R&D Systems), IL-7 (R&D Systems), anti–IL-6 mAb (R&D Systems), anti–IL-4 mAb (BD PharMingen), PMA (Sigma-Aldrich), and ionomycin (Sigma-Aldrich) were used for T cell activation and differentiation.
Mice.
Wild-type B10.BR mice (The Jackson Laboratory) were used as the source of CD4+ T cells except when indicated. NFAT-, NF-κB-, and activator protein 1 (AP-1)-luciferase reporter transgenic mice in the B10.BR background were used to determine transcriptional activities (30–33). The dominant negative (dn)NFAT transgenic mice (34) were backcrossed onto the B10.BR background. C57Bl/6 NFATc2−/− mice have been published previously (27). Procedures that involved mice were approved by institutional guidelines for animal care.
Cell Preparation and Activation.
Total CD4+ T cells were isolated from spleen and lymph nodes by negative selection as described (33) using a combination of anti-MHC class II (m5/115), anti-CD8 (TIB105), anti-NK1.1 (BD PharMingen), and anti-Mac1 mAb (BD PharMingen). Isolation of naive CD4+ T cells was performed by purification of CD4+ T cells by negative selection, followed by staining with a FITC-conjugated anti-CD4 mAb (BD PharMingen) and a PE-conjugated anti-CD44 (Caltag), and cell sorting. CD4+CD44low population was isolated as naive CD4+ T cells. Cells were activated with plate-bound anti-CD3 (5 μg/ml) and soluble anti-CD28 (1 μg/ml) mAbs in the presence or absence of the corresponding cytokine. For the generation of effector Th2 cells, the cells were extensively washed after four days of activation, and restimulated with plate-bound anti-CD3 mAb (5 μg/ml) alone for 24 h.
Analysis of naive and memory CD4+ T cells in spleen was performed by staining with a Red613-conjugated anti-CD4, FITC-conjugated anti-CD45RB, and PE-conjugated anti-CD44 mAbs and flow cytometry. APCs were generated by treatment of splenocytes with mitomycin C (50 μg/ml) for 50 min at 37°C followed by extensive washes.
Determination of Cytokine Production.
ELISAs were performed with purified anti–IL-4 mAb (2 μg/ml) as capture antibody, the corresponding biotinylated anti–IL-4 mAb (1 μg/ml; BD PharMingen), horseradish peroxidase-conjugated streptavidin (Sigma-Aldrich), and the TMB microwell peroxidase substrate and stop solution (Kirkegaard & Perry Labs, Inc.) according to the recommended protocol (BD PharMingen). Recombinant mouse IL-4 (R&D Systems) was used as a standard.
Ribonuclease Protection Assay.
Total RNA was extracted from cells using Ultraspec RNA Isolation Reagent (Biotecx Laboratory) as recommended by the manufacturer. Ribonuclease protection assay (RPA) was performed using the mCK-1 template kit (BD PharMingen) according to the manufacturer's protocol. Briefly, 3 μg of total RNA was hybridized overnight with [32P]UTP radiolabeled in vitro transcribed RNA probes. Overlapping single stranded (ss)RNA on hybridized double-stranded (ds)RNAs was digested with RNases A and T1 and the protected dsRNA duplexes were purified and resolved on urea-denaturing gels. Gels were dried and exposed to Eastman Kodak Co. M r film for autoradiographic analysis.
Electrophoretic Mobility Shift Assay.
Nuclear extracts were prepared from stimulated and unstimulated CD4+ T cells as described previously (35, 36). Binding reactions were performed using 2 μg of nuclear proteins and a [32P]dCTP end-labeled double-stranded oligonucleotide probe containing an NFAT binding site from the proximal IL-4 gene promoter (37), an AP-1 binding site from the collagenase gene promoter (38, 39), or a cAMP response element binding protein (CREB) site from the somatostatin gene promoter (40). 1 μl of anti-glutathione S-transferase (GST) Ab, anti-NFATc2 Ab (Upstate Biotechnology), or anti-NFATc1 mAb (Affinity BioReagents, Inc.) was present during the binding reactions for supershift analysis. Samples were electrophoresed under nondenaturing conditions and exposed to film for autoradiography.
Northern Blot Analysis.
Total RNA from CD4+ T cells was isolated as described above. 10 μg of RNA were separated and transferred to a nylon membrane. Membranes were hybridized with a [32P]-labeled mouse NFATc2 cDNA probe (41). Blots were stripped and reprobed with a γ-actin cDNA probe as a loading control.
Western Blot Analysis.
Whole cell lysates were separated by SDS-PAGE, transferred to membranes, and blotted as described previously (42) with anti-NFATc2 (Upstate Biotechnology), anti-NFATc1 (Affinity BioReagents, Inc.), anti-NFATc3 (Santa Cruz Biotechnology, Inc.), anti–GATA-3 (Santa Cruz Biotechnology, Inc.), anti-actin (Sigma-Aldrich), and anti-Flag (M2; Sigma-Aldrich) Abs.
Luciferase Assay.
Relative luciferase activity was determined as described previously (31). Relative light units (RLU) were measured with a TD 20/20 Luminometer (Turner Designs).
Confocal Fluorescence Microscopy.
CD4+ T cells were purified from lymph nodes and spleens and stimulated for 48 h with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6, cytospun onto slides, and fixed in 3.7% formaldehyde. Slides were placed in methanol for 10 min at –20°C. Cells were permeabilized in 0.1% Triton X-100 for 15 min, blocked in 1% BSA for 1 h, incubated with either anti-NFATc2 rabbit antiserum (Upstate Biotechnology), anti-NFATc1 mouse mAb (Affinity BioReagents, Inc.), or anti-NFATc3 Ab (Santa Cruz Biotechnology, Inc.) for 1 h, washed twice in PBS, then incubated with either Cy3-conjugated anti–rabbit or anti–mouse secondary mAbs for 30 min. After washing twice, nuclei were stained with YOYO-1-iodide (Molecular Probes, Inc.) in PBS with 50 μg/ml RNase for 5 min. Slides were washed twice in PBS, briefly rinsed in H2O, allowed to dry, then coverslipped using Vectashield mounting medium (Vector Laboratories). Images of NFAT expression were created using a BioRad MRC 1024 confocal workstation, including an Olympus BX50 fluorescence microscope and LaserSharp scanning software.
Results
IL-6 Upregulates IL-4 Expression During CD4+ T Cell Activation.
Previously, we have shown that IL-6 is able to differentiate naive CD4+ T cells into effector Th2 cells (18). CD4+ T cells differentiated with anti-CD3 and anti-CD28 mAbs in the presence of IL-6 for 4 d produced high levels of IL-4 after restimulation with anti-CD3 mAb (Fig. 1 A). Polarization of Th2 cells by exogenous IL-6 was as strong as that obtained with exogenous IL-4 (Fig. 1 A). IL-6–mediated Th2 differentiation, however, is dependent on endogenous IL-4, as the presence of a neutralizing anti–IL-4 mAb prevents this effect of IL-6 (18).
Figure 1.

IL-6 induces IL-4 gene expression and cytokine production in CD4+ T cells. (A) CD4+ T cells (106 cells/ml) were stimulated with immobilized anti-CD3 mAb (5 μg/ml), soluble anti-CD28 mAb (1 μg/ml) and medium alone (−), IL-4 (103 U/ml), or IL-6 (100 ng/ml). After 4 d, cells were extensively washed and restimulated for 24 h with immobilized anti-CD3 mAb (5 μg/ml). Supernatants were analyzed for IL-4 production by ELISA. (B) CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6. Supernatants were harvested and IL-4 production was determined by ELISA at different time points after activation. (C) Total RNA extracted from unstimulated CD4+ T cells and from CD4+ T cells stimulated for 3 d with anti-CD3 and anti-CD28 mAbs (anti-CD3/CD28) in the presence or absence of IL-6 was examined by RPA. mRNA levels for IL-4, IL-10, IL-13, and L32 are shown. (D) RPA analysis of total RNA from unstimulated CD4+ T cells or from CD4+ T cells stimulated for 48 h in the absence or presence of IL-6 and/or anti–IL-4 mAb (10 μg/ml). (E) CD4+ T cells were differentiated in the absence (−), or presence of IL-4, IL-6, anti–IL-4, anti–IL-4 and IL-4 (anti–IL4/IL-4), or anti–IL-4 and IL-6 (anti–IL4/IL-6). After 4 d, the cells were washed and restimulated with anti-CD3 mAb alone. IL-4 production was determined by ELISA 24 h after restimulation.
It was possible that IL-6 directly triggered initial IL-4 production by CD4+ T cells during activation and that the secreted IL-4 promoted further Th2 differentiation. To address this hypothesis we examined the effect of IL-6 on IL-4 production during the activation of CD4+ T cells. We first measured the levels of IL-4 produced by CD4+ T cells activated with anti-CD3 and anti-CD28 mAbs for different periods of time. Although it is difficult to detect IL-4 during activation due to its continuous consumption, we observed increased levels of IL-4 production by cells activated in the presence of IL-6 (Fig. 1 B). Analysis of intracellular IL-4 staining showed that the presence of IL-6 during the stimulation of CD4+ T cells also caused an increase in the number of cells producing IL-4 (data not shown). RPA was used to examine the effect of IL-6 on IL-4 gene expression during activation of CD4+ T cells. Increased levels of IL-4 mRNA were detected in CD4+ T cells stimulated in the presence of IL-6 compared with those in cells stimulated in the absence of IL-6 (Fig. 1 C).
As the differentiation of Th2 CD4+ T cells by IL-6 was dependent on the endogenous production of IL-4 by these cells, we wanted to demonstrate that the induction of early IL-4 expression by IL-6 did not require endogenous IL-4. Thus, we examined the effect of IL-6 on early IL-4 gene expression in the presence of a neutralizing anti–IL-4 mAb. The increased IL-4 mRNA levels induced by IL-6 were also observed in the presence of the anti–IL-4 mAb early during activation (Fig. 1 D). To show the neutralizing activity of the anti–IL-4 mAb, we tested its ability to prevent Th2 differentiation. CD4+ T cells were differentiated in the presence of IL-4, IL-6, or anti–IL-4 for 4 d and restimulated for 24 h. The presence of the anti–IL-4 mAb abrogated both IL-4 and IL-6–mediated Th2 differentiation (Fig. 1 E). Together, these results demonstrated that IL-6 promotes Th2 differentiation by increasing the production of IL-4 by CD4+ T cells during their activation. Endogenous IL-4 was, therefore, required for IL-6–mediated Th2 polarization, but induction of IL-4 gene and protein expression by IL-6 in CD4+ T cells early during activation did not require endogenous IL-4.
IL-6 Induces NFAT-mediated Transcription in CD4+ T Cells.
The IL-4 gene is regulated by several transcription factors including NFAT (43). Several NFAT binding sites have been identified within the proximal murine IL-4 promoter and have been shown to be important for its induction (19–21). To determine whether IL-6 could regulate NFAT-mediated transcription, we examined the effect of IL-6 on NFAT transcriptional activity using CD4+ T cells isolated from NFAT-luciferase reporter transgenic mice (30, 31). High levels of NFAT transcriptional activity was detected in cells stimulated with anti-CD3 and anti-CD28 mAbs in the presence of IL-6 compared with the activity in cells stimulated in the absence of IL-6 (Fig. 2 A). The increased NFAT activity (4–6-fold) was not due to an increased number of cells, as we observed only a slight increase (15–20%) in cell viability in the presence of IL-6 (Fig. 2 B). Unlike NFAT, IL-6 only caused a marginal increase in NF-κB– and AP-1–mediated transcription (15–20%; Fig. 2 C), showing the specific effect of IL-6 on NFAT-mediated transcription.
Figure 2.
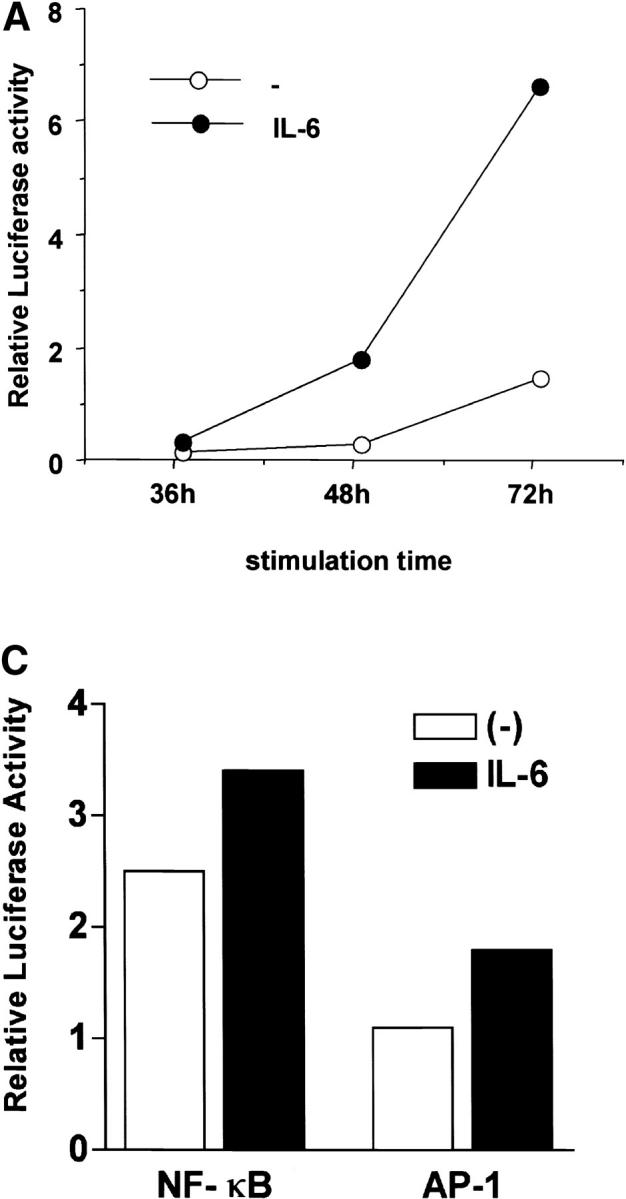
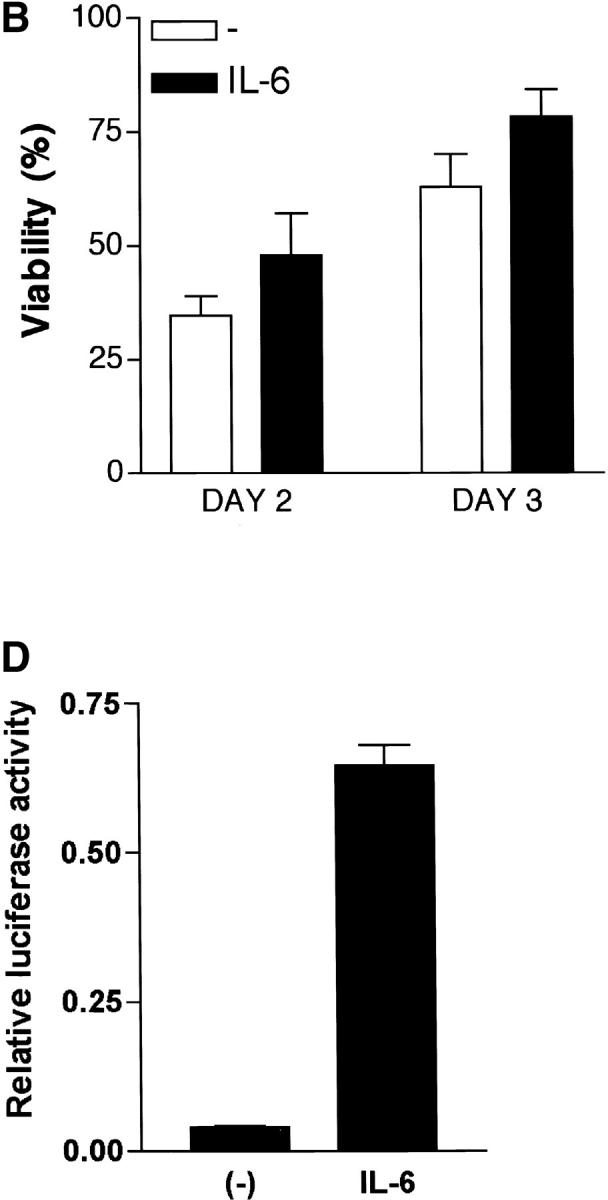
Regulation of NFAT transcriptional activity by IL-6. (A) 5 × 105 CD4+ T cells from NFAT-luciferase reporter transgenic mice were stimulated with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6. Relative luciferase activity was measured at different periods of stimulation. (B) Viability of CD4+ T cells stimulated as in panel A was determined by Trypan Blue exclusion. (C) CD4+ T cells from AP-1- or NF-κB-luciferase reporter transgenic mice were stimulated as in panel A and luciferase activity was determined after 48 h. (D) Naive CD4+CD44low T cells were isolated from NFAT-luciferase mice by cell sorting. 4 × 105 cells were stimulated with anti-CD3 and anti-CD28 mAbs for 48 h in the absence (−) or presence of IL-6 or IL-4 and relative luciferase activity was determined.
To show that the activation of NFAT by IL-6 was not due to a potential effect of this cytokine on a select subpopulation of CD4+ T cells (e.g., memory cells), we examined the NFAT activity in a purified naive population. Naive CD4+ T cells from NFAT-luciferase transgenic mice were isolated by cell sorting, stimulated in the presence or absence of IL-6 and luciferase activity was determined after 48 h. IL-6 also caused a marked induction of NFAT-mediated transcription in purified naive CD4+ T cells (Fig. 2 D). Thus, IL-6 specifically enhanced NFAT-mediated transcription during the activation of naive CD4+ T cells, suggesting that IL-6 regulates IL-4 gene expression by increasing NFAT activity.
NFAT Is Regulated by IL-6 Produced by APCs, but Not by IL-4.
We have shown that early induction of IL-4 expression by IL-6 is independent of endogenous IL-4 (Fig. 1 D). To test whether regulation of NFAT by IL-6 was also independent of IL-4, we examined the effect of a neutralizing anti–IL-4 mAb on NFAT transcriptional activity. CD4+ T cells from NFAT-luciferase reporter transgenic mice were stimulated with anti-CD3 and anti-CD28 mAbs for different periods of time in the absence or presence of IL-6 and an anti–IL-4 mAb. The presence of the anti-IL4 mAb did not prevent activation of NFAT by IL-6 (Fig. 3 A). However, the anti-IL4 mAb completely abrogated the effect of IL-6 on GATA-3 (Fig. 3 B), a Th2-specific transcription factor involved in the regulation of IL-4 expression (44). Thus, activation of NFAT by IL-6 represented an early event independent of IL-4, while upregulation of GATA-3 by IL-6 is an indirect effect mediated by endogenous IL-4. To further confirm that NFAT-driven transcription was not regulated by IL-4, CD4+ T cells from NFAT-luciferase transgenic were stimulated in the presence or absence of IL-4 for 2 or 3 d and luciferase activity was assayed. In contrast to IL-6, IL-4 did not increase NFAT transcriptional activity (Fig. 3 C). Similar results were obtained in purified naive CD4+ T cells (data not shown). We also tested the effect of other Th2 cytokines (e.g., IL-5) and T cell growth factors (IL-2 and IL-7) on NFAT transcriptional activity. None of these cytokines enhanced transcription mediated by NFAT (Fig. 3 D). Moreover, no effect on NFAT transcriptional activity by Th1-related cytokines (e.g., IL-12 and IFN-γ) was detected (data not shown). Thus, IL-6 specifically regulated NFAT activation.
Figure 3.





Regulation of NFAT by IL-6 is independent of IL-4. (A) 5 × 105 NFAT-luciferase CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAbs and medium alone (−), IL-6, anti–IL-4 mAb, or IL-6 and anti–IL-4- mAb (IL-6/anti–IL4) and luciferase activity was measured 48 h later. (B) CD4+ T cells were stimulated in the presence of medium (−), IL-4, IL-6, anti–IL-4 mAb, or IL-6 and anti–IL-4 mAb for 2 d. GATA-3 expression was determined by Western blot using whole cell extracts. Blots were stripped and reprobed for actin. (C) CD4+ T cells from NFAT-luciferase transgenic mice were stimulated with anti-CD3 and anti-CD28 mAbs in the presence of medium alone (−), IL-6, or IL-4. After 2 or 3 d, relative luciferase activity was measured. (D) NFAT- luciferase CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAbs in the presence of medium alone (−), IL-2 (25 U/ml), IL-5 (100 ng/ml), or IL-7 (100 ng/ml). Relative luciferase activity was determined after 36 or 48 h. (E) 5 × 105 CD4+ T cells from NFAT luciferase transgenic mice were stimulated with soluble anti-CD3 mAb (1 μg/ml) and wild-type splenic APCs (2 × 105 cells) in the presence or absence of a neutralizing anti–IL-6 mAb (10 μg/ml). Relative luciferase activity was then measured after 36 or 48 h.
IL-6 is produced by professional APCs such as macrophages, B cells, dendritic cells, and nonprofessional APCs such as epithelial, endothelial, and some tumor cells (12). We examined whether physiological levels of IL-6 produced by accessory cells contributed to NFAT activation. CD4+ T cells from the NFAT-luciferase transgenic mice were stimulated with a soluble anti-CD3 mAb and wild-type APCs in the presence or absence of a neutralizing anti–IL-6 mAb. The presence of the anti-IL-6 mAb caused a dramatic reduction of NFAT transcriptional activity in stimulated CD4+ T cells (Fig. 3 D). Thus, APC-derived IL-6 largely contributed to the regulation of NFAT during the activation of CD4+ T cells.
Inhibition of NFAT Prevents IL-6–induced Th2 Differentiation.
To demonstrate that NFAT activation was required for IL-6–mediated Th2 differentiation we used transgenic mice expressing a dominant negative form of NFAT (dnNFAT) that suppresses NFAT transcriptional activity, but not the activity of other transcription factors such as AP-1 or NF-κB (34). Although expression of this transgene is driven by the proximal lck promoter, two lines of dnNFAT transgenic mice (no. 4 and no. 7) also expressed dnNFAT in CD4+ T cells (Fig. 4 A). To test whether expression of dnNFAT could inhibit activation of NFAT in CD4+ T cells we examined NFAT DNA binding activity in nuclear extracts from dnNFAT transgenic and negative littermate control cells by electrophoretic mobility shift assay (EMSA). Levels of NFAT DNA binding were dramatically reduced (70–90%) in activated CD4+ T cells from dnNFAT mice compared with the levels in cells from negative littermate control mice (Fig. 4 B). DNA binding activity of CREB was also examined as a control (Fig. 4 B). We also examined the effect of dnNFAT on NFAT-mediated transcription using CD4+ T cells isolated from NFAT-luciferase transgenic and NFAT-luciferase × dnNFAT doubly transgenic mice. The presence of dnNFAT blocked NFAT transcriptional activity in CD4+ T cells from doubly transgenic mice that were stimulated with PMA and ionomycin (Fig. 4 C). Moreover, the expression of dnNFAT prevented the enhancement of NFAT transcriptional activity by IL-6 in CD4+ T cells stimulated with anti-CD3 and anti-CD28 mAbs (Fig. 4 D).
Figure 4.



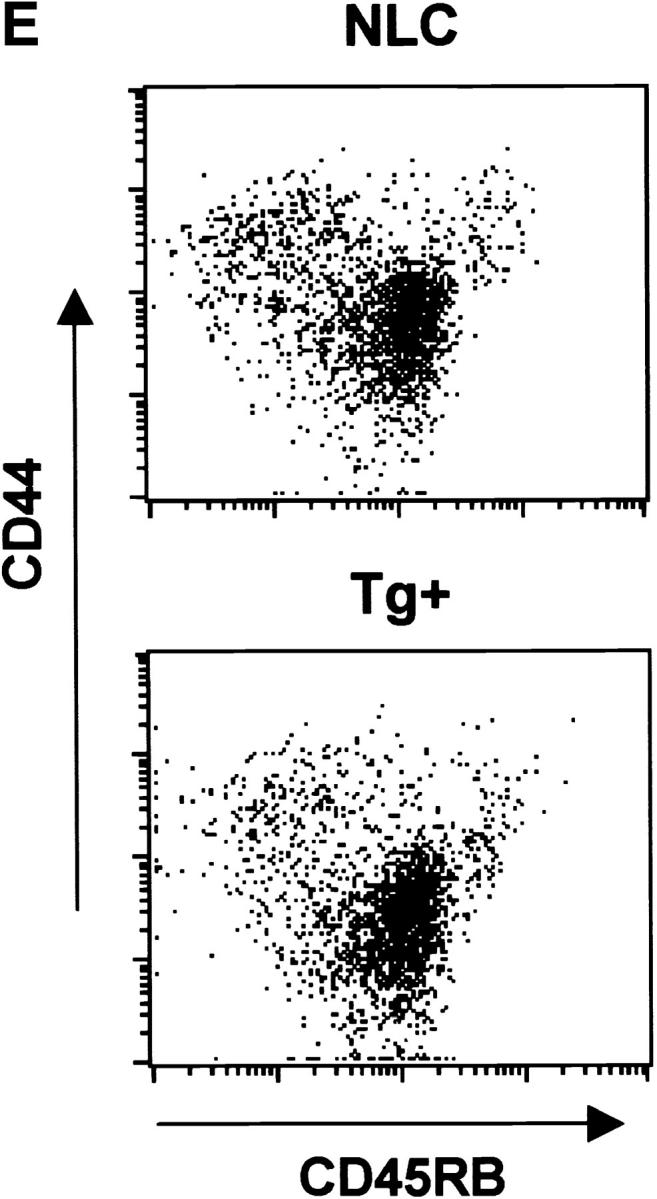
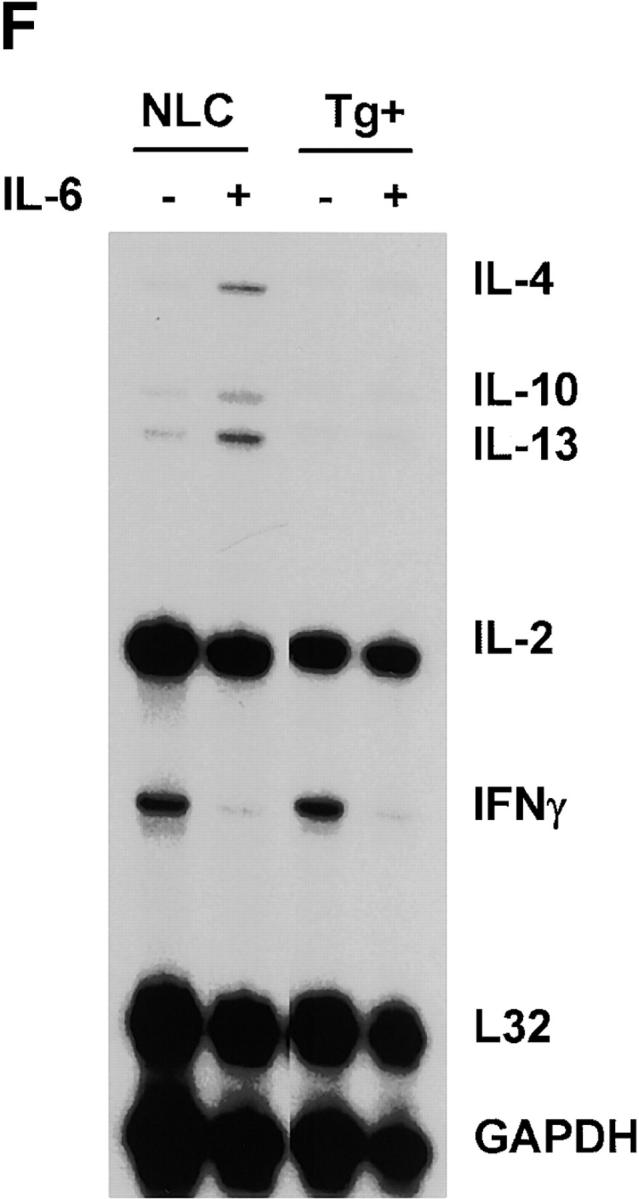
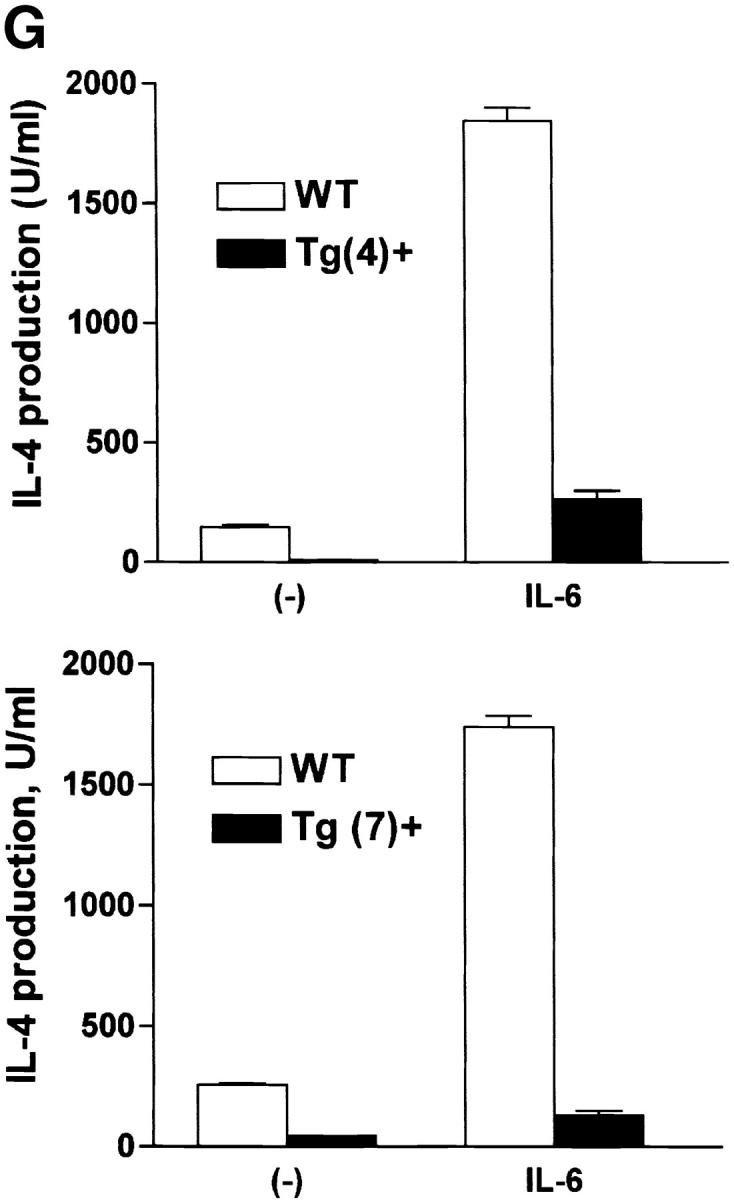
Expression of dnNFAT inhibits IL-6–driven IL-4 production and Th2 differentiation. (A) Whole extracts were isolated from CD4+ T cells from negative littermate control (NLC) and lines no. 4 and no. 7 of dnNFAT transgenic (Tg+) mice. Expression of the dnNFAT transgene was analyzed by Western blotting using an Ab directed against the Flag tag on dn-NFAT. (B) CD4+ T cells from negative littermate control (NLC) or dnNFAT transgenic mice (Tg+) were stimulated for 12 h with anti-CD3 and anti-CD28 mAbs. Nuclear extracts were prepared and analyzed for NFAT (top panel) and CREB (bottom panel) DNA binding activity by EMSA. (C) CD4+ T cells were isolated from NFAT-luciferase × dnNFAT doubly transgenic (Tg+) mice and NFAT-luciferase single transgenic (NLC) mice. 5 × 105 CD4+ T cells were stimulated with PMA (5 ng/ml) and ionomycin (250 ng/ml) and luciferase activity was measured after 24 or 36 h. (D) NFAT luciferase activity was measured in 5 × 105 CD4+ T cells from NFAT-luciferase × dnNFAT doubly transgenic (Tg+) mice or NFAT-luciferase transgenic (NLC) mice stimulated for 2 d with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6. (E) CD4+ T cells were purified from negative littermate control (NLC) or dnNFAT transgenic mice (Tg+), stained with anti-CD4, CD44, and CD45RB mAbs and analyzed by flow cytometry. Dot plots represent the CD44/CD45RB distribution in gated CD4+ T cells. (F) Cytokine mRNA levels were analyzed by RPA in CD4+ T cells from negative littermate control (NLC) and dnNFAT (Tg+) mice that were stimulated with anti-CD3 and anti-CD28 mAbs for 3 d in the presence or absence of IL-6. (G) CD4+ T cells from negative littermate control (NLC) or dnNFAT (Tg+) mice were stimulated for 4 d with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6, washed, and restimulated with anti-CD3 mAb alone for 24 h IL-4 production in supernatants was determined by ELISA. WT, wild-type.
The percentage and number of CD4+ and CD8+ T cells present in lymph nodes and spleen were similar in dnNFAT transgenic mice compared with control mice (data not shown). There was also a normal distribution of naive (CD44lowCD45RBhigh) and memory (CD44highCD45RBlow) CD4+ T cells in dnNFAT transgenic mice (Fig. 4 E). Furthermore, the expression of dnNFAT did not significantly affect the proliferation of CD4+ T cells (data not shown). We therefore addressed whether inhibition of NFAT prevented the induction of IL-4 gene expression by IL-6. CD4+ T cells from negative littermate control or dnNFAT transgenic mice were stimulated in the presence or absence of IL-6 for 2 d and total RNA was analyzed for cytokine gene expression by RPA. In contrast to wild-type CD4+ T cells, IL-6 was unable to induce IL-4 gene expression in activated CD4+ T cells from dnNFAT mice (Fig. 4 F). Upregulation of IL-10 and IL-13 gene expression by IL-6 was also inhibited in dnNFAT transgenic CD4+ T cells. Notably, the inhibition of IFN-γ gene expression by IL-6 that we have described previously (17) was not affected in dnNFAT CD4+ T cells (Fig. 4 F), suggesting that IL-6 utilizes separate pathways to inhibit Th1 and promote Th2 differentiation.
To determine whether NFAT was required for induction of Th2 polarization by IL-6, we examined the ability of IL-6 to promote Th2 differentiation in dnNFAT transgenic mice. CD4+ T cells were isolated from negative littermate control or dnNFAT transgenic mice, stimulated in the presence or absence of IL-6 for 4 d, and restimulated for 24 h with anti-CD3 mAb alone. Effector CD4+ T cells from negative littermate control mice that were differentiated with IL-6 produced large amounts of IL-4 (Fig. 4 G), while IL-6–mediated Th2 differentiation was abrogated in CD4+ T cells from two lines of dnNFAT transgenic mice (Fig. 4 G). The presence of dnNFAT, however, only caused a partial inhibition of Th2 differentiation by exogenous IL-4 (data not shown). Thus, NFAT plays a critical role for the upregulation of IL-4 gene expression by IL-6 early during activation, but other transcription factors (e.g., GATA3, c-maf) are likely to be more important for further IL-4–driven Th2 differentiation.
IL-6 Selectively Induces the Expression of NFATc2 in CD4+ T Cells.
To understand the molecular mechanism by which IL-6 regulates NFAT activity, we examined NFAT DNA binding using nuclear extracts from CD4+ T cells stimulated with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6. The levels of NFAT nuclear complexes found in CD4+ T cells activated in the presence of IL-6 were substantially higher compared with those in cells activated in the absence of IL-6 (Fig. 5 A). In contrast, the levels of CREB or AP-1 DNA binding were not significantly affected by IL-6 (Fig. 5 A). The relative abundance of NFATc2 and NFATc1 in the NFAT nuclear complexes from cells stimulated in the presence or absence of IL-6 was determined by supershift analysis using specific NFAT Abs. An anti-NFATc2 Ab prevented DNA binding of nuclear NFAT complexes from cells stimulated in the presence of IL-6 while an anti-NFATc1 Ab had a lesser effect (Fig. 5 B). These results suggested that NFATc2 was the predominant component of NFAT complexes in cells activated in the presence of IL-6.
Figure 5.




IL-6 induces NFATc2 expression. (A) Nuclear extracts were prepared from unstimulated CD4+ T cells or from CD4+ T cells that were stimulated with anti-CD3 and anti-CD28 mAbs in the presence of absence of IL-6 for 2 d. Extracts were incubated with a double-stranded oligonucleotide containing the NFAT (top panel), AP-1 (middle panel), or CREB (bottom panel) binding sequence and DNA binding complexes were resolved by EMSA. (B) CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6. After 2 d nuclear extracts were prepared and NFAT and AP-1 DNA binding activity was determined by EMSA. Binding reactions were performed in the presence of control anti-GST Ab (CT), anti-NFATc2 (c2) Ab, or anti-NFATc1 (c1) mAb. (C) NFATc2 (left panels), NFATc1 (middle panels), and NFATc3 (right panels) expression in CD4+ T cells stimulated for 2 d in the absence or presence of IL-6 or IL-4 was determined by confocal microscopy using specific anti-NFAT Abs and the DNA marker YOYO for nuclear detection. Green color represents YOYO, red color indicates NFAT staining, and yellow color indicates colocalization of YOYO and NFAT (40×). (D) CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAbs in the presence or absence of IL-6 for 2 d. Whole cell extracts were prepared and examined for NFATc2, NFATc1, and NFATc3 protein levels by Western blot. Blots were stripped and reprobed for determination of NFATc1 and NFATc3 expression. (E) Northern blot analysis of NFATc2 gene expression using total RNA from unstimulated CD4+ T cells and cells stimulated in the presence or absence of IL-6. γ-actin expression was examined as a loading control.
To confirm the selective nuclear accumulation of NFATc2 in cells activated in the presence of IL-6 we examined the expression of NFATc2 by confocal microscopy. Increased nuclear levels (yellow color) of NFATc2 were found in CD4+ T cells stimulated in the presence of IL-6. Interestingly, the cytoplasmic levels (red color) of NFATc2 in CD4+ T cells stimulated in the presence of IL-6 were also higher than those in cells activated in the absence of IL-6 (Fig. 5 C). IL-6 did not significantly affect the levels of NFATc1 or NFATc3 (Fig. 5 C). Moreover, NFATc2 levels were not affected by IL-4 (Fig. 5 C). Together, these results suggested that the selective nuclear accumulation of NFATc2 induced by IL-6 could be due to an up-regulation of NFATc2 expression.
We examined NFATc2 protein levels by Western blot analysis using whole extracts. CD4+ T cells stimulated in the presence of IL-6 contained higher levels of NFATc2 compared with those stimulated in the absence of IL-6 (Fig. 5 D). The more diffuse (faster moving) NFATc2 band in cells stimulated in the presence of IL-6 suggested that there were increased levels of dephosphorylated (i.e., nuclear) NFATc2 in these cells. In contrast, the levels of NFATc1 were not affected by IL-6 (Fig. 5 D), and the marginal increase in NFATc3 levels was not consistent among experiments (Fig. 5 D).
To address whether the elevated levels of NFATc2 were due to an increase of NFATc2 gene expression, we analyzed NFATc2 mRNA levels by Northern blot. In correlation with increased NFATc2 protein expression, high NFATc2 mRNA levels were found in CD4+ T cells activated in the presence of IL-6, while low levels of NFATc2 mRNA were detectable in CD4+ T cells stimulated in the absence of IL-6 (Fig. 5 E). Together, these results showed that IL-6 increased the levels of NFATc2 mRNA and protein in CD4+ T cells during activation, suggesting that NFATc2 could be responsible for IL-6–induced Th2 differentiation.
NFATc2 Is Required for IL-6–mediated Th2 Differentiation.
To establish whether NFATc2 was indeed required for IL-6 to induce IL-4 expression and Th2 differentiation, we analyzed CD4+ T cells lacking NFATc2 (NFATc2−/−). We first examined whether NFATc2 was required for induction of early IL-4 gene expression by IL-6. CD4+ T cells from wild-type and NFATc2−/− mice were stimulated for 2 d in the absence or presence of IL-6. IL-4 mRNA levels were strongly induced (sixfold) by IL-6 in wild-type CD4+ T cells (Fig. 6 A). However, the presence of IL-6 did not cause a substantial increase in IL-4 mRNA (twofold) in NFATc2−/− CD4+ T cells (Fig. 6 A). Thus, IL-6–induced IL-4 gene expression requires NFATc2. The elevated expression of IL-4 in NFATc2−/− CD4+ T cells compared with wild-type cells stimulated in the absence of IL-6 correlated with previous observations (25, 26). These increased levels of IL-4 in CD4+ T cells lacking NFATc2 might be due to compensation by other NFATc family members. We observed increased levels of NFATc1 and NFATc3 in NFATc2−/− CD4+ T cells (Fig. 6 B). Interestingly, IL-6 did not affect NFATc1 or NFATc3 levels in NFATc2−/− CD4+ T cells, supporting the specific effect of IL-6 on NFATc2.
Figure 6.




NFATc2 is required for IL-6–mediated Th2 differentiation. (A) Total RNA from wild-type (WT) or NFATc2-deficient (NFATc2−/−) CD4+ T cells that were stimulated for 2 d in the presence or absence of IL-6 was subjected to RPA analysis. (B) NFATc1 and NFATc3 protein levels were determined by Western blot analysis of whole cell lysates from wild-type and NFATc2−/− CD4+ T cells stimulated for 4 d in the presence or absence of IL-6. Actin is shown as a loading control. (C) CD4+ T cells from wild-type or NFATc2−/− mice were stimulated for 4 d with anti-CD3 and anti-CD28 mAbs in the absence (−) or presence of IL-6. After 4 d cells were washed, restimulated with anti-CD3, and IL-4 production was determined after 24 h. Results are representative of three experiments. (D) NFAT-mediated differentiation of Th2 cells by IL-6.
We examined whether the lack of NFATc2 interfered with the differentiation of CD4+ T cells into effector Th2 cells by IL-6. CD4+ T cells were isolated from wild-type mice and NFATc2−/− mice (27), differentiated in the absence or the presence of IL-6, and then restimulated. Whereas IL-6 promoted strong Th2 differentiation in wild-type CD4+ T cells (60-fold induction of IL-4 production), the ability of IL-6 to promote Th2 differentiation of NFATc2−/− CD4+ T cells was severely compromised (fivefold induction of IL-4 production; Fig. 6 C). Thus, we conclude that NFATc2 is required for complete Th2 differentiation by IL-6.
Discussion
IL-6 is produced by several cell types including macrophages, dendritic, B, epithelial, endothelial, and some tumor cells and is involved in a diversity of biological processes (for a review, see reference 12). Its pleiotropic character is probably due to its ability to regulate several signaling pathways and transcription factors that control the expression of a number of genes. Previously, we have shown that IL-6 can differentiate CD4+ T cells into effector Th2 cells, but this differentiation is dependent on endogenous IL-4 (18). In the current study we have investigated the molecular mechanism of Th2 differentiation by IL-6. We show that IL-6 induces the expression of IL-4 in CD4+ T cells during activation. Regulation of IL-4 expression by IL-6 is mediated by activation of NFAT since inhibition of NFAT abrogates this effect of IL-6 on IL-4.
The JAK/STAT3 and the mitogen-activated protein (MAP) kinase/C/EBP are the two major signaling pathways triggered by IL-6 (12, 45). Regulation of NFAT represents a novel signaling pathway by which IL-6 can exert some of its functions. Upregulation of NFAT activity by IL-6 appears to be due to the induction of NFATc2 expression since the levels of NFATc1 and NFATc3 were not affected by this cytokine. NFATc2 was originally identified as a preexisting (NFATp) cytoplasmic transcription factor that is dephosphorylated and translocated into the nucleus of T cells in response to increased levels of intracellular Ca2+ (41). Whereas expression of NFATc1 is inducible in splenocytes and thymocytes after short treatment with PMA and ionomycin (46), no changes in NFATc2 levels have been previously reported. We now show that expression of NFATc2 increases during the activation of CD4+ T cells (Fig. 5 D) and IL-6 highly upregulates the levels of both mRNA and protein (Fig. 5, D and E). Unlike IL-6, other cytokines such as IL-4, IL-5, IL-2, IL-7, IL-12, and IFN-γ do not affect NFAT levels, indicating that NFATc2 expression is induced by specific signals triggered by IL-6. The regulation of NFATc2 gene expression by IL-6 could be mediated by C/EBP since potential C/EBP binding sites are present in the promoter of the human NFATc2 gene (unpublished data). Thus, regulation of IL-4 gene expression by IL-6 might be indirectly mediated by specific C/EBP family members. Induction of IL-4 gene expression by exogenous IL-4 also appears to be mediated by an indirect mechanism. IL-4 activates STAT6 which, in turn, induces the expression of GATA-3 (47).
IL-6 also induces expression of GATA-3. However, while the effect of IL-6 on NFATc2 expression is independent of endogenous IL-4, induction of GATA-3 expression by IL-6 is requires IL-4. We propose that during the early stages of the immune response, IL-6 derived from professional or nonprofessional APCs enhances NFAT activity in naive CD4+ T cells leading to an upregulation of endogenous IL-4 gene expression. IL-4 then increases the expression of GATA-3 for complete Th2 differentiation (Fig. 6 D).
The role of NFATc2 in the regulation of IL-4 gene transcription is somewhat controversial. Several studies have shown that NFATc2 can bind to NFAT motifs within the proximal IL-4 promoter and control its activity in T cells (19–21). In addition, NFATc2 binds to several regulatory elements within a specific 3′ IL-4 enhancer (48) and DNase I hypersensitivity sites located within the spacer region between the IL-4 and IL-13 genes (49). Due to the aberrant IL-4 expression in lymphoid cells from mice lacking NFATc2, it has been proposed that NFATc2 might be a negative regulator of IL-4 expression and Th2 effector function (25, 28). However, other studies show that in the initial phase of activation, synthesis of IL-4 mRNA is reduced in lymphoid cells from NFATc2-deficient mice (26, 27). We have observed that effector CD4+ T cells that lack NFATc2 contain higher levels of NFATc1 and NFATc3 compared with wild-type cells (Fig. 6 B), probably to compensate for the lack of NFATc2. Thus, it is likely that the increased levels of these two NFAT members may be responsible for the increased expression and production of IL-4 in NFATc2−/− mice. In correlation, IL-4 production is completely abolished in CD4+ T cells lacking NFATc1 and NFATc2 (29). Here we show that increased levels of NFATc2 induced by IL-6 early during activation correlate with increased NFAT transcriptional activity and IL-4 production in CD4+ T cells. These results support a positive regulatory role for NFATc2 in IL-4 gene expression. More importantly, the ability of IL-6 to promote Th2 differentiation was largely impaired in the absence of NFATc2, indicating that NFATc2 is indeed required for regulation of IL-4 by IL-6.
In addition to its role in promoting Th2 differentiation, we have recently shown that IL-6 inhibits Th1 differentiation (17). Whereas differentiation of Th2 by IL-6 is dependent on endogenous production of IL-4, inhibition of Th1 differentiation by IL-6 is not. IL-6 inhibits Th1 differentiation by interfering with IFN-γ signaling through up-regulation of SOCS1. In the absence of SOCS1, IL-6 fails to inhibit Th1 differentiation, but it retains its ability to drive Th2 differentiation (17). In this study we show that IL-6 promotes Th2 differentiation by activating NFAT which induces endogenous IL-4 gene expression. Activation of NFAT, however, is not required for inhibition of IFN-γ gene expression and Th1 differentiation by IL-6 (Fig. 4 G). IL-6 therefore is a key element in the regulation of the Th1/Th2 balance, as it is able to promote Th2 differentiation and inhibit Th1 differentiation by two independent molecular mechanisms involving different signaling pathways.
Acknowledgments
We thank C. Charland for expert flow cytometry analysis and D. Taatjes for helpful discussion on the confocal analysis.
This work was supported by a National Institute of Health Program Project grant (M. Rincón PO1 AI45666), and the COBRE Program of the National Center for Research Resources (M. Rincón P20 RR15557). R.J. Davis is an Investigator of the Howard Hughes Medical Institute. S. Diehl is supported by an Environmental Pathology Training Grant from the National Institute of Environmental Health Science (T32ES07122).
C.-W. Chow's present address is Department of Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY 10461.
Footnotes
*
Abbreviations used in this paper: AP-1, activator protein 1; C/EBP, CCAAT/enhancer binding protein; CREB, cAMP element binding protein; EMSA, electrophoretic mobility shift assay; JAK, Janus kinase; NFAT, nuclear factor of activated T cells; RPA, ribonuclease protection assay; STAT, signal transducer and activator of transcription.
References
- 1.Taga, T., M. Hibi, Y. Hirata, K. Yamasaki, K. Yasukawa, T. Matsuda, T. Hirano, and T. Kishimoto. 1989. Interleukin-6-triggers the association of its receptor with a possible signal transducer, gp 130. Cell. 58:573–581. [DOI] [PubMed] [Google Scholar]
- 2.Daeipour, M., G. Kumar, M.C. Amaral, and A.E. Nel. 1993. Recombinant IL-6 activates p42 and p44 mitogen-activated protein kinases in the IL-6 responsive B cell line, AF-10. J. Immunol. 150:4743–4753. [PubMed] [Google Scholar]
- 3.Akira, S., H. Issihiki, T. Sugita, O. Tanabe, S. Kinoshita, Y. Nishio, T. Nakajima, T. Hirano, and T. Kishimoto. 1990. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 9:1897–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poli, V., F.P. Mancini, and R. Cortese. 1990. IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell. 63:643–653. [DOI] [PubMed] [Google Scholar]
- 5.Ramji, D.P., A. Vitelli, F. Tronche, R. Cortese, and G. Ciliberto. 1993. The two C/EBP isoforms, IL-6DBP/NF-IL6 and C/EBP delta/NF-IL6 beta, are induced by IL-6 to promote acute phase gene transcription via different mechanisms. Nucleic Acids Res. 21:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinoshita, S., S. Akira, and T. Kishimoto. 1992. A member of the C/EBP family, NF-IL6 beta, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc. Natl. Acad. Sci. USA. 89:1473–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poli, V. 1998. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 273:29279–29282. [DOI] [PubMed] [Google Scholar]
- 8.Lutticken, C., U.M. Wegenka, J. Yuan, J. Buschmann, C. Schindler, A. Ziemiecki, A.G. Harpur, A.F. Wilks, K. Yasukawa, T. Taga, et al. 1994. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 263:89–92. [DOI] [PubMed] [Google Scholar]
- 9.Stahl, N., T.G. Boulton, T. Farruggella, N.Y. Ip, S. Davis, B.A. Witthuhn, F.W. Quelle, O. Silvennoinen, G. Barbieri, S. Pellegrini, et al. 1994. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science. 263:92–95. [DOI] [PubMed] [Google Scholar]
- 10.Akira, S., Y. Nishio, M. Inoue, X.J. Wang, S. Wei, T. Matsusaka, K. Yoshida, T. Sudo, M. Naruto, and T. Kishimoto. 1994. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 77:63–71. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima, K., T. Matsuda, Y. Fujitani, H. Kojima, Y. Yamanaka, K. Nakae, T. Takeda, and T. Hirano. 1995. Signal transduction through IL-6 receptor: involvement of multiple protein kinases, stat factors, and a novel H7-sensitive pathway. Ann. NY Acad. Sci. 762:55–70. [DOI] [PubMed] [Google Scholar]
- 12.Hirano, T. 1998. Interleukin 6 and its receptor: ten years later. Int. Rev. Immunol. 16:249–284. [DOI] [PubMed] [Google Scholar]
- 13.Conze, D., L. Weiss, P.S. Regen, A. Bhushan, D. Weaver, P. Johnson, and M. Rincón. 2001. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 61:8851–8858. [PubMed] [Google Scholar]
- 14.Teague, T.K., P. Marrack, J.W. Kappler, and A.T. Vella. 1997. IL-6 rescues resting mouse T cells from apoptosis. J. Immunol. 158:5791–5796. [PubMed] [Google Scholar]
- 15.Teague, T.K., B.C. Schaefer, D. Hildeman, J. Bender, T. Mitchell, J.W. Kappler, and P. Marrack. 2000. Activation-induced inhibition of interleukin 6-mediated T cell survival and signal transducer and activator of transcription 1 signaling. J. Exp. Med. 191:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conze, D., J. Lumsden, H. Enslen, R.J. Davis, G. Le Gros, and M. Rincon. 2000. Activation of p38 MAP kinase in T cells facilitates the immune response to the influenza virus. Mol. Immunol. 37:503–513. [DOI] [PubMed] [Google Scholar]
- 17.Diehl, S., J. Anguita, A. Hoffmeyer, T. Zapton, J.N. Ihle, E. Fikrig, and M. Rincón. 2000. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity. 13:805–815. [DOI] [PubMed] [Google Scholar]
- 18.Rincón, M., J. Anguita, T. Nakamura, E. Fikrig, and R.A. Flavell. 1997. IL-6 directs the differentiation of IL-4-producing CD4+ T cells. J. Exp. Med. 185:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chuvpilo, S., C. Schomberg, R. Gerwing, A. Heinfling, R. Reeves, F. Grummt, and E. Serfling. 1993. Multiple closely-linked NFAT/octamer and HMG I(Y) binding sites are part of interleukin-4 promoter. Nucleic Acids Res. 21:5694–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szabo, S.J., J.S. Gold, T.L. Murphy, and K.M. Murphy. 1993. Identification of cis-acting regulatory elements controlling interleukin-4 gene expression in T cells: roles for NF-Y and NF-ATc. Mol. Cell. Biol. 13:4793–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rooney, J.W., T. Hoey, and L.H. Glimcher. 1995. Coordinate and cooperative roles for NFAT and AP-1 in the regulation of murine IL-4 gene. Immunity. 2:473–483. [DOI] [PubMed] [Google Scholar]
- 22.Oukka, M., I.-C. Ho, F.C. de la Brousse, T. Hoey, M.J. Grusby, and L.H. Glimcher. 1998. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity. 9:295–304. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida, H., H. Nishina, H. Takimoto, L.E.M. Marengère, A.C. Wakeham, D. Bouchard, Y.-Y. Kong, T. Ohteki, A. Shahinian, M. Bachmann, et al. 1998. The transcription factor NF-ATc1 regulates lymphocytes proliferation and Th2 cytokine production. Immunity. 8:115–124. [DOI] [PubMed] [Google Scholar]
- 24.Ranger, A.M., M.R. Hodge, E.M. Gravallese, M. Oukka, L. Davidson, F.W. Alt, F.C. de la Brousse, T. Hoey, M. Grusby, and L.H. Glimcher. 1998. Delayed lymphoid repopulation with defects in IL-4-driven responses produced by inactivation of NF-ATc. Immunity. 8:125–134. [DOI] [PubMed] [Google Scholar]
- 25.Kiani, A., J.P.B. Viola, A.H. Lichtman, and A. Rao. 1997. Down-regulation of IL-4 gene transcription and control of Th2 cell differentiation by a mechanism involving NFAT1. Immunity. 7:849–860. [DOI] [PubMed] [Google Scholar]
- 26.Hodge, M.R., A.M. Ranger, F. Charles de la Brousse, T. Hoey, J. Grusby, and L.H. Glimcher. 1996. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity. 4:397–405. [DOI] [PubMed] [Google Scholar]
- 27.Schuh, K., B. Kneitz, J. Heyer, F. Sieblet, C. Fischer, E. Jankevies, E. Rúde, E. Schmitt, A. Schimpl, and E. Serfling. 1997. NF-ATp plays a prominent role in the transcriptional induction of Th2-type lymphokines. Immunol. Lett. 57:171–175. [DOI] [PubMed] [Google Scholar]
- 28.Ranger, A.M., M. Oukka, J. Rengarajan, and L.H. Glimcher. 1998. Inhibitory function of two NFAT family members in lymphoid homeostasis and Th2 development. Immunity. 9:627–635. [DOI] [PubMed] [Google Scholar]
- 29.Peng, S.L., A.J. Gerth, A.M. Ranger, and L.H. Glimcher. 2001. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity. 14:13–20. [DOI] [PubMed] [Google Scholar]
- 30.Rincón, M., and R.A. Flavell. 1996. Regulation of AP-1 and NFAT transcription factors during thymic selection of T cells. Mol. Cell. Biol. 16:1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rincón, M., and R.A. Flavell. 1997. Transcription mediated by NFAT is highly inducible in effector CD4+ Th2 cells but not in Th1 cells. Mol. Cell. Biol. 17:1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millet, I., R. Phillips, R.S. Sherwin, S. Ghosh, R.E. Voll, R.A. Flavell, A. Vignery, and M. Rincón. 2000. Inhibition of NF-kB activity and enhancement of apoptosis by the neuropeptide calcitonin gene-related peptide. J. Biol. Chem. 275:15114–15121. [DOI] [PubMed] [Google Scholar]
- 33.Rincón, M., and R.A. Flavell. 1994. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 13:4370–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow, C.-W., M. Rincón, and R.J. Davis. 1999. Requirement for transcription factor NFAT in interleukin-2 expression. Mol. Cell. Biol. 19:2300–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schreiber, E., P. Matthias, M.M. Müller, and W. Shaffner. 1989. Rapid detection of octamer binding proteins with ‘mini’-extracts prepared from a small number of cells. Nucleic Acids Res. 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tugores, A., M.A. Alonso, F. Sánchez-Madrid, and M.O. de Landázuri. 1992. Human T cell activation through the activation-inducer molecule/CD69 enhances the activity of transcription factor AP-1. J. Immunol. 148:2300–2306. [PubMed] [Google Scholar]
- 37.Rooney, J.W., M.R. Hodge, P.G. McCaffrey, A. Rao, and L.H. Glimcher. 1994. A common factor regulates both Th1- and Th2-specific cytokine gene expression. EMBO J. 13:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angel, P., M. Imagawa, R. Chiu, B. Stein, R.J. Imbra, H.J. Rahmsdorf, C. Jonat, P. Herrlich, and M. Karin. 1987. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 49:729–739. [DOI] [PubMed] [Google Scholar]
- 39.Lee, W., P. Mitchell, and R. Tjian. 1987. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 49:741–752. [DOI] [PubMed] [Google Scholar]
- 40.Montminy, M.R., and L.M. Bilezikijan. 1987. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 328:175–178. [DOI] [PubMed] [Google Scholar]
- 41.McCaffrey, P.G., C. Luo, T.K. Kerpolla, J. Jain, T.M. Badalian, A.M. Ho, E. Burgeon, W.S. Lane, J.N. Lambert, T. Curran, et al. 1993. Isolation of cyclosporin-sensitive T cell transcription factor NFATp. Science. 262:750–754. [DOI] [PubMed] [Google Scholar]
- 42.Chow, C.W., C. Dong, R.A. Flavell, and R.J. Davis. 2000. c-Jun NH(2)-terminal kinase inhibits targeting of the protein phosphatase calcineurin to NFATc1. Mol. Cell. Biol. 20:5227–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy, K.M., W. Ouyang, J.D. Farrar, J. Yang, S. Ranganath, H. Asnagli, M. Afkarian, and T.L. Murphy. 2000. Signaling and transcription in T helper development. Annu. Rev. Immunol. 18:451–594. [DOI] [PubMed] [Google Scholar]
- 44.Zheng, W.-P., and R.A. Flavell. 1997. The transcription factor GATA-3 is necessary and sufficient for Th2 gene expression in CD4+ T cells. Cell. 89:587–596. [DOI] [PubMed] [Google Scholar]
- 45.Heinrich, P.C., I. Behrmann, G. Muller-Newen, F. Schaper, and L. Graeve. 1998. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 334:297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Northrop, J.P., S.N. Ho, L. Chen, D.J. Thomas, G.P. Nolan, A. Admon, and G.R. Crabtree. 1994. NF-AT components define a family of transcription factors targeted in T-cell activation. Nature. 369:497–502. [DOI] [PubMed] [Google Scholar]
- 47.Ouyang, W., S.H. Ranganath, K. Weindel, D. Bhattacharya, T.L. Murphy, W.C. Sha, and K.M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 9:745–755. [DOI] [PubMed] [Google Scholar]
- 48.Agarwal, S., O. Avni, and A. Rao. 2000. Cell-type-restricted binding of the transcription factor NFAT to a distal IL-4 enhancer in vivo. Immunity. 12:643–652. [DOI] [PubMed] [Google Scholar]
- 49.Takemoto, N., Y. Kamogawa, H. Jun Lee, H. Kurata, K. Arai, A. O'Garra, N. Arai, and S. Miyatake. 2000. Cutting edge: chromatin remodeling at the IL-4/IL-13 intergenic regulatory region for Th2-specific cytokine gene cluster. J. Immunol. 165:6687–6691. [DOI] [PubMed] [Google Scholar]