Physiological β Cell Death Triggers Priming of Self-reactive T Cells by Dendritic Cells in a Type-1 Diabetes Model (original) (raw)
Abstract
The prelude to type-1 diabetes is leukocyte infiltration into the pancreatic islets, or insulitis. This process begins in pancreatic lymph nodes when T lymphocytes reactive to islet β cells encounter antigen-presenting cells (APCs) displaying peptides derived from β cell proteins. We show here that a ripple of physiological β cell death, which occurs at 2 wk of age in all mouse strains, precipitates the arrival of such APCs, and that the relevant APC is a dendritic cell of CD11c+CD11b+CD8α− phenotype. These findings have significant implications concerning the nature of the diabetes-provoking deficits in NOD mice, the identity of the primordial diabetogenic antigens, and our understanding of the balance between immunity and tolerance in a pathological context.
Keywords: autoimmune response, pancreas, PLN, antigen transport, apoptosis
Introduction
In type-I diabetes, the insulin-producing β cells of the pancreatic islets of Langherhans are destroyed by T lymphocyte–provoked mechanisms (1). During the first stage of this biphasic autoimmune disease, termed insulitis, the islets are invaded by a mix of leukocytes, including DCs, macrophages, B cells, and CD4+ and CD8+ T cells. After a relatively benign interlude, the islet infiltrate converts to a pathological lesion, resulting in the eventual destruction of a majority of the β cells. The ensuing loss of glucose homeostasis leads to a series of debilitating disorders collectively known as diabetes.
In immune responses in general, naive T cells are activated by DCs in peripheral lymphoid organs (2). Because circulating naive T cells cannot usually access peripheral nonlymphoid tissues, the tissue-derived antigens they recognize must be transported to T cell zones of nearby lymphoid organs and processed into MHC–peptide complexes. Upon antigen recognition and activation, the T cells proliferate and differentiate into effector cells, which circulate through nonlymphoid tissues and are retained where they rerecognize cognate antigen.
The autoimmune response that results in β cell destruction follows the same basic scheme, with β cell–specific T cells being primed in pancreatic LNs (PLNs) and activated T cells invading the islets shortly thereafter (3, 4). In the BDC2.5 TCR transgenic (tg) mouse model of autoimmune diabetes (derived from a diabetogenic, β cell–reactive, CD4+ T cell clone; references 5, 6), the priming of potentially diabetogenic CD4+ T cells is subject to temporal regulation, commencing with striking synchronicity from 15 to 18 d of age (3). Analogous observations have been made in at least three other TCR tg models in which CD4+ or CD8+ T cells, specific for β cell epitopes presented by H-2b, H-2d, or H-2g7 molecules, are first sensitized in PLNs at ∼15 d of age (3, 7–9). Together, these observations argue that the temporal control of islet-specific T cell priming is not restricted to a particular TCR tg system, applies to both MHC class I– and class II–restricted β cell–specific T cells, and is not a unique property of the NOD genetic background.
The lack of autoreactive T cell priming in the PLNs of day 10 mice suggests that one of the following players in the autoimmune response is either absent or functionally impaired at this time: the antigen, the T cell, or the APC. A deficiency in antigen expression cannot account for this phenomenon because relevant β cell antigens are expressed in neonates (7, 8, 10), and their expression levels do not greatly increase between 10 and 20 d of age (reference 7; unpublished data). A functional incompetence of β cell–reactive T cells in 10-d-old mice has also been ruled out by transfer experiments (3). This suggests that changes in APCs underlie the initiation of autoantigen exposure because of altered ability to pick up, process, transport, or present pancreatic antigens, or because the relevant β cell antigens are not made available to them for some reason. Here, we explore alterations in APC function and β cell death as possible triggers for activation of diabetogenic T cells in the PLN.
Materials and Methods
Mice.
The generation and maintenance of BDC2.5/NOD and BDC2.5/B6.H2g7/g7 TCR tg mice were described previously (5, 11). The BDC2.5 TCR recognizes a β cell peptide associated with I-Ag7. For typing of BDC2.5 mice, monoclonal antibodies directed against CD4 and Vβ4 were used. Caspase-12 knockout mice were obtained from J. Yuan (Harvard Medical School, Boston, MA) on the C57/Bl6 background and were crossed to congenic C57Bl/6.H2g7/g7 or NOD backgrounds. All mice were maintained under barrier conditions in the Joslin Diabetes Center Animal Facility.
Flow Cytometric Analysis of DCs.
LN and pancreatic cell suspensions were prepared as described previously (12). Organs were teased and digested in Ca2+-free buffer containing Collagenase P or D (Roche Molecular Biochemicals) at 37°C for 20 min. Digested material was pipetted vigorously and filtered in cold wash buffer containing 5–10 mM EDTA. Cells were washed, and erythrocytes were lysed in pancreatic suspensions using NH4Cl. After blocking with purified α-FcγR (2.4G2), bulk organ suspensions were stained for multicolor flow cytometry in PBS/2% FCS using cocktails of monoclonal antibodies. Antibodies specific for MHC class II (10-3.6), CD40 (3/23), CD54 (3E2), CD80 (16-10A1), CD11c (HL3), and CD4 (L3T4) were purchased from BD Biosciences. Antibodies specific for B220 (CD45R), CD11b (M1/70), and CD8α (CT-CD8α) were purchased from Caltag. Directly conjugated isotype controls included mouse IgG2a, rat IgG2a, and Armenian hamster IgG (BD Biosciences). For dead cell exclusion, suspensions were incubated with Hoescht 33342 (HO; Calbiochem) for 5 min on ice before analysis, and dead cells, which stain brightly with HO, were detected with the UV laser. Cell suspensions were prepared rapidly and analyzed immediately to avoid maturation and/or fixation artifacts. Dead cells (HOhi) and B220hi cells were excluded during analysis. For DC analysis, samples were acquired with a MoFlo (DakoCytomation) and analyzed using the Summit software.
Analysis of Antigen Transport.
Mice were anesthetized and their abdomens were opened to access the pancreas for injection. Pancreata were injected with fluorescein-conjugated ovalbumin (Fluo.Ova) (500 μg/adult, 250 μg/juvenile; Molecular Probes), 0.5 μm YG carboxylate polystyrene microspheres (0.25% solid; Polysciences, Inc.), or CFSE-labeled adult NOD hepatocytes (106/adult, 0.5 × 106/juvenile). Hepatocytes were prepared by digesting teased livers in collagenase-containing buffer at 37°C for 10 min. Liver suspensions were depleted of erythrocytes using NH4Cl and washed hepatocytes were resuspended in warm PBS/0.1% BSA. CFSE-labeling was performed as described previously (3). Cells were washed and resuspended in PBS. Before injection, >90% hepatocytes stained positively with trypan blue.
Endocytosis Assay.
PLN cell suspensions were prepared from NOD mice using collagenase to extract DCs from tissues (as described in the Flow Cytomeric Analysis of DCs section). Cells were incubated at 37 or 4°C in RPMI 1640 complete medium (5% FCS) without antigen or with graded doses of Fluo.Ova for 30 min. Cells were washed three times in cold medium and stained with antibodies specific for CD11c and MHC class II on ice for cytofluorimetry. The fraction of Fluo.Ova+ cells and the mean fluorescence intensity of Fluo.Ova were determined.
Assessment of T Cell Activation.
T cell activation was assessed using an adoptive transfer system with CFSE-labeled lymphocytes as described previously (3). Draining (PLNs) and nondraining LNs (inguinal lymph nodes [ILNs]) were harvested ∼66 h after transfer and single cell suspensions were prepared using glass slide disruption. Bulk LN cells were stained with α-CD4 or α-Vβ4 and α-CD44 for flow cytometric measurement of T cell activation. The extent of T cell proliferation was determined simultaneously by CFSE dilution analysis. For T cell analysis, samples were acquired using Coulter instrumentation and analyzed with Expo software. T cells were transferred 24–48 h after administration of drugs or antigen. We observed high levels of CD44 expression on all BDC2.5 T cells (gated on transferred cells as follows: CD4+ CFSE+ or Vβ4+CFSE+) diluting CFSE in PLNs. For dying islet cell injections, pancreatic islets of Langerhans were isolated from 4–6-wk-old NOD mice at the Joslin Diabetes Center Islet Core, resuspended in PBS, and subjected to four cycles of freezing and thawing to induce 100% necrosis in samples. Each recipient mouse received the equivalent of 100 islets intrapancreatically.
Drugs.
Streptozotocin (STZ; Sigma-Aldrich) was resuspended in sodium citrate buffer immediately before intraperitoneal injection and administered 1× at 80 mg/kg. Benzyloxylcarbonyl-V-A-d-_O_-methyl fluoromethyl ketone (ZVAD; Calbiochem) was dissolved in DMSO and injected intraperitoneally. Each mouse received 100 μl of a 10 μM ZVAD solution.
Immunizations.
NOD mice were anesthetized, and a single hind footpad was injected with 10 μg of the mimic peptide, 1040-63 (Peptron; reference 13), 24 h after ZVAD administration.
Results
DCs in PLNs from 10- versus 20-d-old Mice.
Given that DCs transport tissue antigens to LNs and activate cognate T cells encountered there, it is reasonable to speculate that the absence of T cell priming in PLNs of 10-d-old mice and its presence in PLNs of 20-d-old mice may reflect alterations in local DC representation or function. To investigate this possibility, we compared the composition of DC subsets in days 10 and 20 PLNs from NOD mice using multicolor cytofluorimetry. Single cell suspensions were analyzed without preenrichment to prevent any loss of potentially important cells; instead, they were stained with appropriate reagents for excluding dead cells and lymphocytes. Using a combination of classical DC markers, we identified four subsets of myeloid cells in day 20 PLNs, distinguished by surface expression of CD11c and/or CD11b (Fig. 1 , top left) as follows: subset 1, CD11b+CD11c−; subset 2, CD11b2+CD11c+; subset 3, CD11b+CD11c+++; and subset 4, CD11b−CD11c2+. Subset 1 is partially comprised of F4/80+ macrophages (unpublished data). To characterize DC populations of the PLN-expressing lymphoid markers, we stained suspensions with Abs specific for CD8α and CD4 in combination with the DC markers CD11c and CD11b. Distinct CD8α+ CD11c+ DCs (Fig. 1, subset 5, bottom left) lacking CD11b expression (not depicted) were identified. PLN DCs lacking CD8α were subdivided into CD11cloCD8α− (subset 6) and CD11chiCD8α− (subset 7) subsets (Fig. 1, bottom left). A population of CD4+ DCs (CD4+ CD11c+++CD11b+) was also detected within subset 3 of day 20 PLNs (unpublished data).
Figure 1.
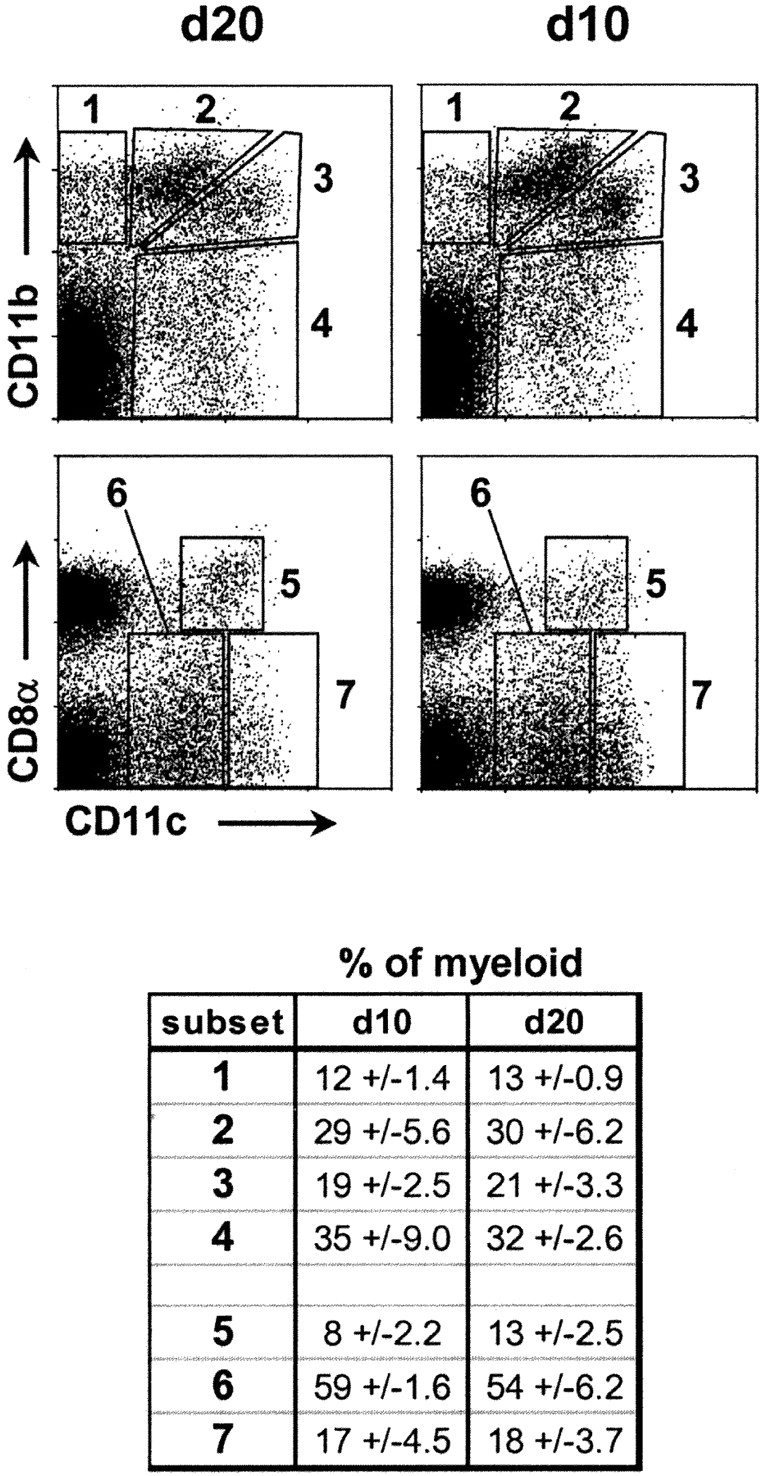
Composition of PLN DCs at days 10 (d10) and 20 (d20). DC subsets in PLNs of 10- and 20-d-old NOD mice were stained with CD11c (x axis) and CD11b or CD8α (y axis). Dotplots (gated on B220−HO−) are representative of results from >15 experiments. Values in table indicate the proportion of each subset expressed as the percentage of total myeloid (CD11b+CD11c+) cells (average ± SD of three independent experiments).
We characterized the makeup of day 10 PLNs, and found DC subsets 1–7 to be readily detectable (Fig. 1, right). In addition, no significant differences were measured in the relative abundances (expressed as percentage of total myeloid cells) of subsets 1–7 between days 10 and 20 PLNs (Fig. 1, table). PLNs were also sampled before day 10 (as early as day 4) and between days 11 and 19; in all cases, the relative levels of subsets 1–7 were essentially identical to those found in 10- and 20-d old (unpublished data). Therefore, these results indicate that the major PLN DC subsets are all present from shortly after birth and do not substantially change in relative abundance between days 4 and 20. The absolute cellularity of all DC subsets increased approximately threefold between days 10 and 20, whereas the absolute cellularity of the total PLN augmented approximately fivefold during this period (d10 PLN ≈ 3 × 105 cells; day 20 ≈ 1.5 × 106 cells). Thus, DCs comprise an increasingly smaller proportion of the total PLN as this organ develops.
To investigate the possibility that islet-specific T cell priming does not occur in PLNs of 10-d-old animals because of ineffective DC maturation, we examined the surface expression of classical DC maturation markers by each of the subsets described in Fig. 1. As shown for subsets 1–4 in Fig. 2 , the expression profiles of CD40, CD54, CD80, CD86, and MHC class II molecules on subsets 1, 3, and 4 were not markedly different between days 10 and 20. Data for subsets 5–7 prompted a similar conclusion (unpublished data). However, MHC class II and CD86 molecules were consistently up-regulated between days 10 and 20 by DCs belonging to subset 2 (CD11b2+CD11c+).
Figure 2.
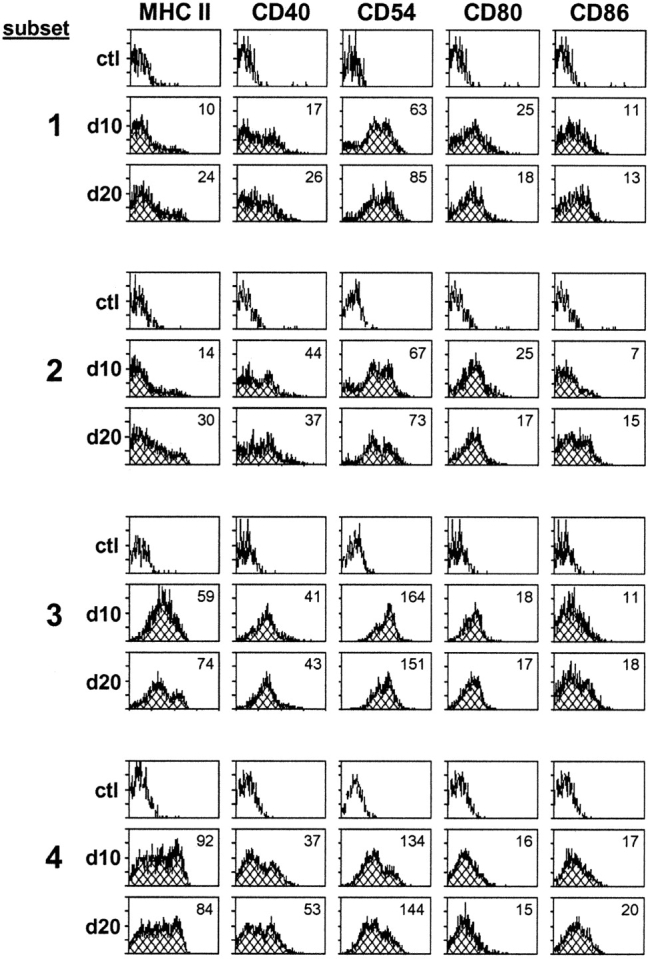
Expression of maturation markers by PLN DCs at days 10 (d10) and 20 (d20). Surface expression of MHC class II, CD40, CD54, CD80, and CD86 molecules compared on subsets 1–4 described in Fig. 1 from PLNs of 10- and 20-d-old NOD mice. Isotype controls are shown for each subset. Value in top right corner of each histogram indicates mean fluorescence intensity of specified marker. Histograms represent results of 4–10 experiments.
Transport of Pancreatic Antigens to PLNs in 10- and 20-d-old NOD Mice.
Pancreatic antigens must be shuttled to the PLNs and presented to circulating naive T cells to initiate insulitis. Because little is known of antigen transit from the pancreas to the draining LN, we sought to examine this pathway in some detail. First, we established a method for monitoring specific antigens as they accumulated in the PLNs after drainage from the pancreas; cytofluorimetry was used to quantitatively assess transport of various forms of antigens to the PLNs after direct injection into the adult pancreas.
Injection of Fluo.Ova into the pancreas resulted in its detection in a clear number of PLN CD11c+ DCs (Fig. 3) . The extent was maximal ∼20 h after administration (unpublished data), a time course reminiscent of similar studies of antigen transport after airway exposure (14). This encouraged the use of a similar approach to examine transport of antigens more relevant to diabetes, specifically two particulate antigens that may mimic fragments of dying β cells as follows: (a) fluorescently labeled hepatocytes (Fluo.Cells) and (b) fluorescently labeled microspheres (Fluo.Beads). We used primary hepatocytes as surrogate β cells because the latter are difficult to obtain in sufficient numbers for detection in a cytofluorimetry-based migration assay. Trypan-positive hepatocytes, generated by collagenase treatment of liver, were labeled with CFSE and injected directly into the pancreas. The transport kinetics of Fluo.Cells and Fluo.Beads, monitored by cytofluorimetry, differed from those of soluble antigen, taking ∼40 h for optimal detection in the PLNs. The majority of APCs bearing Fluo.Ova and Fluo.Cells were CD11c+, whereas the APCs containing Fluo.Beads were both CD11c+ and CD11c− (Fig. 3 a). We also examined the relative abundances and absolute numbers of DCs in the PLNs after intrapancreatic injection of antigen (Fig. 3 b). The numbers of PLN DCs do not significantly change after administration of Fluo.Ova, Fluo.Cells, or Fluo.Beads.
Figure 3.
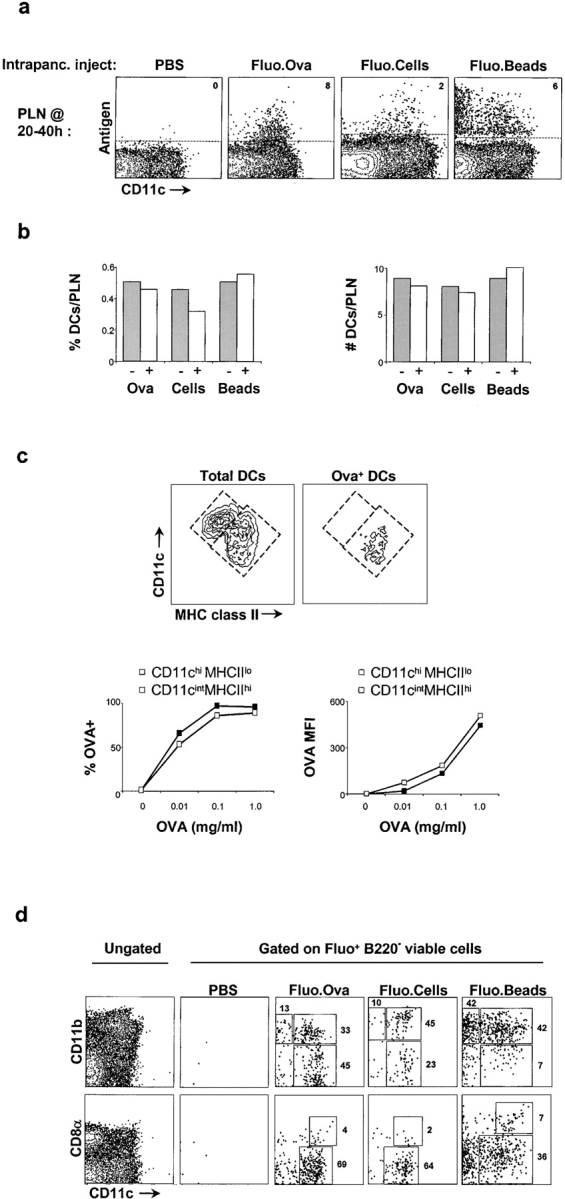
Transport of pancreatic antigens to draining LNs. (a) Flow cytometric assessment of antigen transport to PLN after intrapancreatic injection of PBS, Fluo.Ova, Fluo.Cells, or Fluo.Beads into ∼20-d-old NOD mice. Antigen-bearing cells (Fluo+) are plotted versus CD11c+ cells. PLNs were harvested ∼20 h after Fluo.Ova administration and ∼40 h after administration of Fluo.Cells or Fluo.Beads. Value shown in top right corner of the dotplots is the percentage of antigen-bearing cells (Fluo+) among CD11c+ cells. (b) Quantification of DCs in 4-wk-old PLNs before and after administration of Fluo.Ova, Fluo.Cells, and Fluo.Beads. The relative abundance (left) and absolute number (right) of DCs (CD11c+ cells) per PLN are shown. (c) Characterization of antigen capture by distinct DC subsets in vivo and in vitro. Cell suspensions from PLNs were stained with CD11c (y axis) and MHC class II (x axis). Left contour plot depicts total DCs in the PLNs, whereas right contour plot depicts Fluo.Ova+ DCs in PLN after intrapancreatic injection. Fluo.Ova was associated with CD11cintMHCIIhi DCs but not CD11chiMHCIIlo DCs in vivo. The line graphs depict the in vitro endocytic capacity of CD11chiMHCIIlo (open squares) and CD11cintMHCIIhi (closed squares) DC populations. The fraction of Fluo.Ova+ cells within and the amount of antigen captured by each DC population were similar. (d) Phenotypic characterization of antigen-bearing (Fluo+) cells in 4-wk-old NOD PLNs. The leftmost panel represents total PLN cells (no Fluo or FL1 gate) after a mock intrapancreatic injection. The other dotplots display antigen-bearing (Fluo+) cells in PLNs after intrapancreatic injection of specified antigen. Values in dotplots depict the percentage of antigen-bearing cells (Fluo+) within the individual gates. Data are representative of four to six experiments.
To discern whether soluble antigen (Fluo.Ova) was acquired by DCs in the PLNs or at the site of antigen application, we followed an approach described in a study of airway antigen transport (14). As in the lung study, we found that Fluo.Ova could be detected in CD11cint MHCIIhi DCs ∼20 h after intrapancreatic injection of the soluble antigen but not in CD11chiMHCIIlo DCs (Fig. 3 c, top). We tested the ability of these two populations to capture Fluo.Ova ex vivo and found that their endocytic capacities were nearly identical (Fig. 3 c, bottom). If Fluo.Ova was getting to the PLNs by passive leakage, we would have expected to see this antigen associated with both of the PLN DC populations because they both avidly endocytose Fluo.Ova upon deliberate exposure. Because this was clearly not the case, we concluded that Fluo.Ova+ PLN DCs captured their antigen in the pancreas and migrated to the draining LNs rather than capturing their antigen within the LNs.
Next, we pinpointed the specific DC subsets mediating β cell–derived antigen transport in 20-d-old mice. Fluo.Ova and Fluo.Cells were detected largely in CD11c+ DCs, whereas Fluo.Beads were seen in both CD11b+ macrophages and CD11b+ DCs (Fig. 3 d, top row). Soluble protein (Fluo.Ova) and particulate antigens (Fluo.Cells, Fluo.Beads) were associated with both CD8α+ and CD8α− DCs (Fig. 3 d, bottom row); however, the CD8α+ DCs played a more minor role than CD8α− DCs. All three of the antigens tested were absent from the CD4+CD11b+ DC population corresponding to subset 3 delineated in the first paragraph of Results (unpublished data).
Having established that fragments of dying cells are transported from the pancreas to the PLNs by DCs in day 20 mice, we asked whether the initiation of insulitis might be controlled by a developmental change in the ability of DCs to capture and transport dying cells. To test this, we injected Fluo.Cells into pancreata of 8- and 20-d-old recipients and monitored Fluo.Cell-trafficking to PLNs ∼42 h later. As in 20-d-old, CD11c+ DCs, primarily CD8α−, were largely responsible for ferrying Fluo.Cells to the PLNs in 10-d-old (Fig. 4 , left). The frequency of CD11b+ DCs transporting Fluo.Cells increased from 21 to 40% between days 10 and 20. These data demonstrate that pancreatic DCs are indeed equipped to capture and transport dying cells to PLNs by 10 d of age.
Figure 4.

Antigen transport by day 10 (d10) pancreatic DCs. Flow cytometric analysis of antigen-bearing cells (Fluo+) in ∼10- and 20-d-old NOD mice 40 h after intrapancreatic injection of Fluo.Cells into NOD mice. Total PLN cells are shown in the left column and antigen-bearing (Fluo+) cells in the right column for both ages. The values in top right corner of dot plot indicate the percentage of Fluo+ DCs expressing CD11b (top plots) or CD8α (bottom plots) in gate above dotted line. Data are representative of two to four experiments.
A Role for β Cell Death in Triggering Diabetogenic T Cells.
A potential trigger for initiation of the antiislet response that culminates in diabetes development might be β cell death (for review see reference 15). In addition to the steady-state β cell turnover known to occur throughout life (16), it has been reported that rodents (and humans) exhibit a transient ripple of β cell death shortly after birth, thought to be a normal physiological process reflecting tissue remodeling or metabolic perturbations (17–21). In mice, this occurs at ∼12 d of age, only a brief time before the window of initial priming of naive, potentially diabetogenic T cells in the PLNs. Thus, it is tempting to speculate that the increase in β cell death at 12 d of age augments capture, transport, and presentation of β cell antigens by DCs, leading to antiislet T cell activation from 15 to 18 d of age.
We tested this notion with different strategies using the BDC2.5 TCR tg system. The still-unidentified antigen recognized by BDC2.5 T cells appears to be associated with particulate fractions of β cells, possibly on the cytoplasmic face of secretory granules (22). Although the BDC2.5 antigen is known to be synthesized shortly after birth (10), its subcellular localization suggests that it might be more likely encountered when the integrity of the β cell membrane is disrupted, for example, during programmed cell death or cell injury. To assess this possibility, we transferred CFSE-labeled naive BDC2.5 T cells into 6-d-old NOD mice that were unmanipulated or had been intrapancreatically implanted with dead islets. The activation of these transferred cells and their proliferation in draining and distal LNs was ascertained by flow cytometry 66 h after transfer, as a dilution of the CFSE label with each cell division. In the latter case, a much higher fraction (47 vs. 3%) of the transferred T cells was activated and proliferated in the PLNs, a clear response to dying β cells (Fig. 5 a). The intrapancreatic injection of buffer alone resulted in a small increase in BDC2.5 T cell activation (3 vs. 13%) in PLNs, suggesting that the surgical procedure may have caused some β cell death (Fig. 5 a). Transferred BDC2.5 T cells remained naive in ILNs in all recipients (Fig. 5 a). We also transferred CFSE-labeled naive BDC2.5 T cells into 6-d-old recipients that had or had not been treated with STZ, a cytotoxic drug to which β cells are particularly sensitive. A single injection of STZ resulted in a much more robust PLN activation (64 vs. 2%) of the transferred T cells (Fig. 5 b). These results indicate that β cell death within the pancreas is capable of provoking priming of islet-reactive CD4+ T cells circulating through PLNs before 10 d of age.
Figure 5.

The presence of dead β cells provokes activation of naive BDC2.5 T cells in neonatal PLNs. (a) Activation of transferred naive BDC2.5 T cells (gated on CD4+Vβ4+ cells) in PLNs and ILNs after intrapancreatic implantation of dead islets or buffer into 6-d-old NOD mice. Proliferation was assessed in all experiments ∼66 h after transfer by CFSE dilution in CD4+ T cells. (b) Activation of transferred naive BDC2.5 T cells (gated on CD4+Vβ4+ cells) in PLNs after intraperitoneal injection of STZ into 6-d-old NOD mice.
If experimental induction of β cell death could promote activation of β cell–reactive T cells circulating through the PLNs of perinatal mice, might physiological β cell death be a trigger for the antiislet reactivity that initiates insulitis and eventually culminates in diabetes? To address this question, we treated mice with the irreversible pancaspase inhibitor, ZVAD, to block caspase-dependent β cell death in situ, and examined priming of transferred BDC2.5 T cells in the PLNs. NOD mice received a single injection of ZVAD at a dose shown previously to inhibit liver abscess formation in a mouse model of parasite infection (23). To ascertain that this systemic ZVAD treatment was effectively blocking caspase activity, we examined the effects in the thymus, because apoptosis associated with thymocyte selection is readily detectable in this tissue. Using cytofluorimetry, we found that a single dose of ZVAD inhibited death of double-negative thymocytes in vivo by 50% (unpublished data), suggesting that the drug was having an effect, though one that was probably only partial. To minimize any potential effects of ZVAD on donor T cells, we administered the inhibitor 24–48 h before transfer of T cells. A single injection of ZVAD into 21–42-d-old recipients before T cell transfer impaired activation of naive BDC2.5 T cells by 40% on average (P < 0.0001; Fig. 6 a, left histograms). Treatment of 12-d-old mice with ZVAD impaired the activation of naive BDC2.5 T cells by 50% (Fig. 6 a, bar graph). Systemic ZVAD treatment did not alter resident APCs or transferred T cell function because no significant difference was observed in T cell activation in popliteal LNs of ZVAD-treated and untreated animals that had been immunized with a mimotope recognized by BDC2.5 T cells (Fig. 6 a, right histograms; reference 13). Therefore, spontaneous priming of BDC2.5 T cells in the PLNs of young mice is controlled, at least in part, by caspase-mediated apoptosis of β cells.
Figure 6.

β cell death is required for optimum BDC2.5 T cell priming. (a) Effect of systemic ZVAD treatment on naive BDC2.5 T cell activation in PLNs of 14–42-d-old NOD mice. The left histograms depict BDC2.5 T cell proliferation, assessed by CFSE dilution in CD4+Vβ4+ cells, in the PLNs of 21–42-d-old mice after ZVAD treatment (administered at day 2). BDC2.5 T cell activation was impaired by 40% on average (P < 0.0001). The histograms in right column display T cell proliferation in popliteal LNs of ZVAD-treated NOD mice after footpad immunization with BDC2.5 peptide mimic. BDC2.5 T cells do not proliferate in popliteal LNs of unimmunized recipients (shaded histogram). Bar graph depicts BDC2.5 T cell proliferation in the PLNs or ILNs of 14-d-old after ZVAD treatment at day 2. (b) BDC2.5 T cell activation in PLNs of caspase-12–deficient B6.H2g7/g7 mice (KO) and wild-type littermate controls (WT). Proliferation of transferred naive BDC2.5 T cells, assessed by CFSE dilution in CD4+Vβ4+ cells, was reduced by 56% on average in KO recipients (P = 0.006). Graph on right shows the proliferation indices for naive wild-type BDC2.5 T cells after transfer into 17–25-d-old caspase-12 KO mice or wild-type littermates on the B6.H2g7/g7 background. Each symbol represents an individual mouse.
Because pancreatic β cells seem particularly prone to death in response to protein overexpression (24), we speculated that the physiological β cell death in young mice might be mediated by caspase-12, a death-promoting enzyme specifically activated by ER stress (25). Therefore, we backcrossed the caspase-12 knockout mutation (25) onto the NOD and B6.H2g7 backgrounds, intercrossed, and transferred naive, CFSE-labeled, BDC2.5 T cells into caspase-12–deficient mice and control littermates. Lack of caspase-12–impaired T cell proliferation in 17–25-d-old mice by 56% on average (P = 0.006; Fig. 6 b). These results suggest that the caspase-12–dependent ER stress pathway of β cell death may control, at least partially, the availability of β cell–derived antigens in the PLNs.
NOD and Other Strains.
On the one hand, it has been reported that NOD mice show defective clearance of apoptotic cells by macrophages (26). On the other hand, it was shown that the time of insulitis onset was the same for BDC2.5 TCR tgs on the NOD and B6.H-2g7 genetic backgrounds (6). Therefore, we wondered how the release and transport of β cell antigens to the PLNs and the activation of islet-reactive T cells compared in these two strains. We found that the ability to activate transferred naive BDC2.5 T cells occurred with essentially superimposable kinetics in the PLNs of NOD or B6.H2g7 recipients, with a transition past 15 d of age in both strains (Fig. 7 a). Therefore, developmental regulation of anti–β cell T cell priming is not a unique fate of autoimmuneprone NOD mice. As with NOD mice again, we found that inhibition of apoptosis in young B6.H2g7 mice lead to a marked reduction (50%) in the activation of transferred naive BDC2.5 T cells in the PLN (Fig. 7 b). Thus, priming of potentially diabetogenic T cells is governed by a physiological process common to multiple mouse strains, and does not reflect NOD-specific aberrations in APCs or β cells.
Figure 7.

Regulation of autoreactive T cell priming not determined by NOD background. (a) Naive BDC2.5 T cell activation was assessed in PLNs of NOD and B6.H2g7/g7 mice at different ages ranging from 7 to 28 d of age. CFSE dilution in BDC2.5 T cells (gated on CD4+Vβ4+ cells) was measured in all recipients ∼66 h after transfer. (b) Effect of systemic ZVAD treatment on naive BDC2.5 T cell activation (measured by CFSE dilution in CD4+Vβ4+ cells) in PLNs of 21-d-old B6.H2g7/g7 recipients. Systemic ZVAD treatment caused a 50% decrease in proliferation of BDC2.5 T cells in PLNs of B6.H2g7/g7 mice.
Discussion
Two New Features.
Results from several previous studies have converged to suggest the following scenario of insulitis initiation in the NOD mouse model of type-1 diabetes (and its TCR tg derivatives). Like all antigen-ignorant lymphocytes, naive β cell–reactive T cells circulate through the blood and lymphoid tissues of juvenile mice. Through some process beginning at ∼2 wk of age, antigens derived from β cells are taken up by APCs in the pancreatic islets, inducing their migration to the immediately draining LNs, the PLNs, and their maturation en route. In the PLNs, the APCs present β cell–derived antigens to naive β cell–reactive T cells in circulation and activate them. Upon activation, the T cells acquire the ability to migrate through the tissues; in the islets, they reencounter cognate antigen, become reactivated, and are retained, thereby initiating insulitis (for review see reference 15). Findings from the present set of experiments elucidate this scenario by revealing two important new features.
First, the stimulus initiating insulitis appears to be a ripple of β cell death that occurs physiologically in juvenile mice of all strains. The abrupt onset of islet infiltration in BDC2.5 TCR tg animals from 15 to 18 d of age reflects the acquisition of a new competence by APCs in the PLNs to present β cell–derived antigens to circulating β cell–reactive T cells (3). Incompetence before this time could reflect a deficiency in the APCs and/or the antigens. We show here that PLN APCs from juvenile mice are of standard composition (Figs. 1 and 2) and that, within days after birth, APCs are fully capable of taking up antigen in the pancreas (Figs. 3–5) and offering it to CD4+ T cells in the PLN (Fig. 5). This finding is consistent with previous works indicating that the machinery for presentation of MHC class I–restricted peptides to CD8+ T cells in the PLNs (7, 9) and spleen (27) of neonatal mice is intact. Clearly, the relevant APCs are there and are functional in juvenile mice. Likewise, we have shown previously that the β cell–derived antigen recognized by BDC2.5 T cells is present within days after birth (reference 10; unpublished data). However, it remained possible that, although synthesized, the antigen is not made available for presentation until the abrupt occurrence of some event at ∼2 wk of age.
An intriguing possibility was that an augmentation of β cell death that occurs in the islets around this time is the precipitating event, a normal physiological process termed “tissue remodeling.” Several groups have reported such an increase in rodents, peaking ∼12 d after birth (17–20). Significant β cell death at this age was first surmised from mathematical modeling studies (17); confirmation came from quantifying dying cells on pancreas sections (18, 19). The frequency of defunct cells was low even at peak times (4–13%), but, given that they can be very difficult to detect (owing to rapid engulfment and disposal), it is possible that a significant fraction (estimated at one third) of the β cells in juvenile rodents is turning over at this time. Interestingly, an analogous perinatal wave of β cell death, peaking at birth, has been demonstrated in the islets of humans, leading to the suggestion that extension of this wave might be the cause of hyperinsulinemia in infancy (21). Because dead cells can be a potent activation signal for APCs, in particular DCs, this ripple of β cell death could provide a trigger for mobilization of potentially diabetogenic T cells.
Our studies provide both “gain of function” and “loss of function” arguments in favor of this notion. On the one hand, extraneously introducing dead β cells into the pancreas (Fig. 5 a) or provoking death specifically of β cells in vivo via injection of STZ (Fig. 5 b) prematurely precipitated the appearance of the BDC2.5 antigen on the surface of PLN APCs as early as 6 d of age. These results are in line with previous conclusions that experimental induction of β cell death via CTLs (7) or STZ (9) promoted activation of naive CD8+ β cell–specific T cells in the PLNs of neonatal mice, and that STZ treatment exacerbated insulitis in adult mice by enhancing the priming of T cells responsive to β cell antigens (28). These results argue that induction of β cell death can lead to the priming of naive diabetogenic T cells, but stops short of demonstrating that it actually does so during spontaneous disease development. On the other hand, inhibiting caspase-dependent cell death either by treatment of mice with ZVAD (Fig. 6 a) or by using a caspase-12–deficient mouse line (Fig. 6 b) delayed the arrival of BDC2.5–antigen-loaded APCs in the PLNs of juvenile mice. However, neither pharmacological nor genetic inhibition of caspase activity was able to halt progression to insulitis or diabetes; indeed, in both cases, inhibition of the PLN activation of transferred BDC2.5 T cells was only partial. The incomplete effects of ZVAD in halting BDC2.5 T cell priming were not very surprising given that systemic administration of this drug did not completely block cell death (reference 23; unpublished data), even when the drug was continuously infused into the target tissue (29). The incomplete effects of the caspase-12 deficiency also have ample precedent; drug-induced, ER stress-mediated apoptosis of diverse cell-types was far from totally inhibited in the knock-out mice (25). The partiality of both types of caspase inhibition may indicate that additional caspases or other death signaling molecules are involved in these processes, or at least can be mobilized in a state of deficiency. It is known, for example, that the death of some cell types can change from an apoptotic to a necrotic mechanism by depleting ATP or blocking the activity of caspases. Most relevant here is that, although transiently inhibiting cell death in several contexts, ZVAD did not promote a long-term increase in cell viability (30–33).
The second important feature revealed by the present studies is that the APC that ferries β cell debris from the pancreas to the PLNs is a DC of CD11b+CD8α− phenotype. Although it has been known for some time that DCs express a wide array of receptors for internalizing diverse ligands (34), the uptake of dying cells by DCs originally came as somewhat of a surprise because macrophages were thought to be the specialists in clearing cellular debris at that time (35). Several analyses subsequently demonstrated that cell-associated antigens could be captured, processed, and presented by splenic DCs (36, 37). Tissue-restricted antigens have been detected in DCs within regional lymphatics and tissue-draining LNs (38, 39); however, the specific mechanisms by which DCs encounter and transport parenchymal cell antigens has remained unclear.
We examined the transport of dying parenchymal cells from the pancreas to the PLNs, and found that CD11c+ DCs were pivotal in this process (Fig. 3 a), both CD11b+ and CD11b− DCs being involved (Fig. 3 b). Interestingly, the relative abundance of CD11b+ DCs carrying dying cell fragments to the PLNs augmented between days 10 and 20, and these cells up-regulated expression of MHC class II molecules and CD86 during the same time window. Similarly, we observed a mobilization of CD11b+ DCs from the pancreas into the PLNs in response to STZ-induced β cell death (unpublished data). These results are of interest in light of a recent paper that CD11b+ DCs were more efficient than CD11b− DCs at activating an islet-specific T cell hybridoma after induction of a limited amount of β cell death in vivo (28). Fragments of endogenous dead cells have been detected in both CD8α+ and CD8α− DCs, although engulfment of intravenously administered cells was attributed to the CD8α+ subset of spleen DCs (37, 40). In contrast, we found that CD8α+ DCs contributed only minimally to the transport of dying cells from the pancreas to the PLNs (Fig. 4). This is supported by our observation that DCs residing in the pancreas, where antigen is initially encountered, were largely CD11b+CD8α− as follows: 60% of pancreatic DCs expressed CD11b; and 96% lacked CD8α expression (unpublished data). Likewise, stomach DCs that captured parietal cell antigens expressed CD11b but not CD8α (39). These discrepancies might be due to tissue-specific differences in DC subsets or to differences in the nature of the engulfed dying cell. However, in general, it seems that our findings on the pancreas–PLN axis are very reminiscent of what occurs with stomach antigens.
Implications.
This new information may have some profound implications for how insulitis is initiated in prelude to type-1 diabetes. One of the more important implications relates to the factors responsible for the predisposition of NOD mice to spontaneously develop diabetes. A ripple of physiological β cell death occurs at ∼2 wk of age in the islets of all mouse strains examined thus far, including B6 (20). This may be somewhat more pronounced in NOD mice (20), which might reflect a reported deficit in the phagocytosis of apoptotic cells by macrophages derived from these animals (26). However, any such differences seem not to read out further downstream. Results presented in Fig. 7 and as described previously (6) argue that caspase-dependent release of β cell–derived antigens to the PLNs and spontaneous initiation of islet invasion by a β cell–reactive T cell clone, respectively, are indistinguishable on the B6.H-2g7 and NOD genetic backgrounds. It would appear most likely that NOD mice are diabetes-prone not because of an aberrant self stimulation but rather of an abnormal self response; i.e., they have an autoreactive T lymphocyte repertoire insufficiently purged of cells capable of responding to β cell antigens. This interpretation is supported by recent claims of defects in the negative selection of NOD thymocytes (41, 42).
Another important implication of our findings concerns the antigens responsible for triggering type-1 diabetes. There has been much debate on this topic, with a primary role being assigned to insulin, glutamic acid decarboxylase, heat shock protein, or a variety of other proteins (43, 44). The indiscriminate release of β cell antigens implied in a scenario rooted in physiological cell death could explain why it has been so difficult to convincingly identify the primordial diabetogen. If the ripple of β cell death that occurs in the islets of juvenile mice is indeed translated into an influx into the PLNs of DCs displaying β cell–derived antigens, and assuming that a multitude of proteins is released from dying cells, then multiple β cell antigens should be simultaneously presented after this event (45). The coordinated priming of T cells with reactivities to diverse β cell proteins (BDC2.5, OT-I, clone-4, 8.3) at ∼15 d of age suggests that this is in fact the case (3, 7–9). This conclusion may seem at odds with older works about a temporal sequence to the priming of β cell–reactive T cells of different specificities (46, 47), but this hierarchy could conceivably reflect differing sensitivities of the assays used to read out the responses. Release of antigens to the PLNs by a normal physiological process also eliminates the need to invoke molecular mimicry by microbial T cell epitopes to initiate the autoimmune process, as done, for example, by Mintern et al. (7). This notion has attracted much interest but has only limited direct experimental support thus far (48), although microbes might still precipitate insulitis prematurely or cause it to convert more rapidly to diabetes.
Finally, it is important to explain why immunity, rather than tolerance, issues from this priming process. A popular paradigm has been that necrotic cells are immunogenic and apoptotic cells tolerogenic. However, apoptotic cells can also provoke an immune response under the appropriate conditions, for example, when present in high enough numbers, physiologically stressed, or exposed to inflammatory cytokines such as TNF-α and IL-1β (45). In addition, several studies have linked the presentation of autoantigens and production of autoantibodies to apoptotic cells, notably in systemic lupus erythematosus (49). An additional consideration is that the response elicited from T cells by the presentation of antigens released from dying β cells in the PLNs may not represent immunity in the strict sense. In both NOD mice and TCR tg derivatives, in particular BDC2.5, there can be a long lag between the initiation of insulitis and the development of diabetes because the T cell infiltrate remains “innocuous” or benign (50, 51). The explanation for the T cells' good behavior remains controversial, being variously attributed to a TH2 phenotype (51), IL-10 production (52), or other factors (53). Perhaps this behavior represents a state of immune deviation, something between true immunity and true tolerance, the result of being primed by DCs loaded in the pancreas with apoptotic β cell fragments. Another possibility is that regulatory T cells also emanate from the priming process, akin to the scenario invoked by Hugues et al. (28).
Curing type-I diabetes is an important objective; preventing it is a goal of even greater import. Elucidating the events that precipitate T cell invasion of the islets will provide the necessary framework for achieving this goal. Here, we have identified one such critical event, an event that probably takes place in all mouse strains, but is provocative in NOD mice because of their autoreactive repertoire.
Acknowledgments
We thank G. Losyev for assistance with flow cytometry, E. Hyatt for assistance with animal husbandry, K. Elder and J. White for administrative support, and T. Lund, A. Goldrath, and A. Herman for critical discussions about this work.
S. Turley is a recipient of a Cancer Research Institute postdoctoral fellowship. This work was supported by funds from the National Institutes of Health (RO1DK59658-02) and by the Joslin Diabetes and Endocrinology Research Center core facilities (P30DK36836-16).
Abbreviations used in this paper: Fluo.Beads, fluorescently labeled microspheres; Fluo.Cells, fluorescently labeled hepatocytes; Fluo.Ova, fluorescein-conjugated ovalbumin; HO, Hoescht 33342; ILN, inguinal LN; PLN, pancreatic LN, STZ, streptozotocin; tg, transgenic; ZVAD, benzyloxylcarbonyl-V-A-d-_O_-methyl fluoromethyl ketone.
References
- 1.Tisch, R., and H. McDevitt. 1996. Insulin-dependent diabetes mellitus. Cell. 85:291–297. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins, M.K., A. Khoruts, E. Ingulli, D.L. Mueller, S.J. McSorley, R.L. Reinhardt, A. Itano, and K.A. Pape. 2001. In vivo activation of antigen-specific CD4 T cells. Annu. Rev. Immunol. 19:23–45. [DOI] [PubMed] [Google Scholar]
- 3.Hoglund, P., J. Mintern, C. Waltzinger, W. Heath, C. Benoist, and D. Mathis. 1999. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J. Exp. Med. 189:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gagnerault, M.C., J.J. Luan, C. Lotton, and F. Lepault. 2002. Pancreatic lymph nodes are required for priming of β cell reactive T cells in NOD mice. J. Exp. Med. 196:369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katz, J.D., B. Wang, K. Haskins, C. Benoist, and D. Mathis. 1993. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 74:1089–1100. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez, A., J.D. Katz, M.G. Mattei, H. Kikutani, C. Benoist, and D. Mathis. 1997. Genetic control of diabetes progression. Immunity. 7:873–883. [DOI] [PubMed] [Google Scholar]
- 7.Mintern, J.D., R.M. Sutherland, A.M. Lew, K. Shortman, F.R. Carbone, and W.R. Heath. 2002. Constitutive, but not inflammatory, cross-presentation is disabled in the pancreas of young mice. Eur. J. Immunol. 32:1044–1051. [DOI] [PubMed] [Google Scholar]
- 8.Morgan, D.J., C. Kurts, H.T.C. Kreuwel, K.L. Holst, W.R. Heath, and L.A. Sherman. 1999. Ontogeny of T cell tolerance to peripherally expressed antigens. Proc. Natl. Acad. Sci. USA. 96:3854–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang, Y., B. O'Brien, J. Trudeau, R. Tan, P. Santamaria, and J.P. Dutz. 2000. In situ beta cell death promotes priming of diabetogenic CD8 T lymphocytes. J. Immunol. 168:1466–1472. [DOI] [PubMed] [Google Scholar]
- 10.Katz, J.D., C. Benoist, and D. Mathis. 1995. T helper cell subsets in insulin-dependent diabetes. Science. 268:1185–1188. [DOI] [PubMed] [Google Scholar]
- 11.Luhder, F., J. Katz, C. Benoist, and D. Mathis. 1998. MHC class II molecules can protect from diabetes by positively selecting T cells with additional specificities. J. Exp. Med. 187:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vremec, D., and K. Shortman. 1997. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159:565–573. [PubMed] [Google Scholar]
- 13.Judkowski, V., C. Pinilla, K. Schroder, L. Tucker, N. Sarvetnick, and D.B. Wilson. 2001. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 166:908–917. [DOI] [PubMed] [Google Scholar]
- 14.Vermaelen, K.Y., I. Carro-Muino, B.N. Lambrecht, and R.A. Pauwels. 2001. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 193:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathis, D., L. Vence, and C. Benoist. 2001. Beta-cell death during progression to diabetes. Nature. 414:792–798. [DOI] [PubMed] [Google Scholar]
- 16.Slack, J.M. 1995. Developmental biology of the pancreas. Development. 121:1569–1580. [DOI] [PubMed] [Google Scholar]
- 17.Finegood, D.T., L. Scaglia, and S. Bonner-Weir. 1995. Dynamics of β-cell mass in the growing rat pancreas. Diabetes. 44:249–256. [DOI] [PubMed] [Google Scholar]
- 18.Scaglia, L., C.J. Cahill, D.T. Finegood, and S. Bonner-Weir. 1997. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology. 138:1736–1741. [DOI] [PubMed] [Google Scholar]
- 19.Petrik, J., E. Arany, T.J. McDonald, and D.J. Hill. 1998. Apoptosis in the pancreatic islet cells of the neonatal rat is associated with a reduced expression of insulin-like growth factor II that may act as a survival factor. Endocrinology. 139:2994–3004. [DOI] [PubMed] [Google Scholar]
- 20.Trudeau, J.D., J.P. Dutz, E. Arany, D.J. Hill, W.E. Fieldus, and D.T. Finegood. 2000. Neonatal β-cell apoptosis: a trigger for autoimmune diabetes? Diabetes. 49:1–7. [DOI] [PubMed] [Google Scholar]
- 21.Kassem, S.A., I. Ariel, P.S. Thornton, I. Scheimberg, and B. Glaser. 2000. β-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. 49:1325–1333. [DOI] [PubMed] [Google Scholar]
- 22.Bergman, B., and K. Haskins. 1994. Islet-specific T-cell clones from the NOD mouse respond to beta-granule antigen. Diabetes. 43:197–203. [DOI] [PubMed] [Google Scholar]
- 23.Yan, L., and S.L. Stanley, Jr. 2001. Blockade of caspases inhibits amebic liver abscess formation in a mouse model of disease. Infect. Immun. 69:7911–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parham, P. 1988. Intolerable secretion in tolerant transgenic mice. Nature. 333:500–503. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa, T., H. Zhu, N. Morishima, E. Li, J. Xu, B.A. Yankner, and J. Yuan. 2000. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-b. Nature. 403:98–103. [DOI] [PubMed] [Google Scholar]
- 26.O'Brien, B.A., Y. Huang, X. Geng, J.P. Dutz, and D.T. Finegood. 2002. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes. 51:2481–2488. [DOI] [PubMed] [Google Scholar]
- 27.Dadaglio, G., C.M. Sun, R. Lo-Man, C.A. Siegrist, and C. Leclerc. 2002. Efficient in vivo priming of specific cytotoxic T cell responses by neonatal dendritic cells. J. Immunol. 168:2219–2224. [DOI] [PubMed] [Google Scholar]
- 28.Hugues, S., E. Mougneau, W. Ferlin, D. Jeske, P. Hofman, D. Homann, L. Beaun, C. Schrike, M. Von Herrath, A. Lehuen, and N. Glaichenhaus. 2002. Tolerance to islet antigens and prevention from diabetes induced by limited apoptosis of pancreatic beta cells. Immunity. 16:169–181. [DOI] [PubMed] [Google Scholar]
- 29.Li, M., V.O. Ona, C. Guegan, M. Chen, V. Jackson-Lewis, L.J. Andrews, A.J. Olszewski, P.E. Stieg, J.P. Lee, S. Przedborski, and R.M. Friedlander. 2000. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 288:335–339. [DOI] [PubMed] [Google Scholar]
- 30.Leist, M., B. Single, A.F. Castoldi, S. Kuhnle, and P. Nicotera. 1997. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185:1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melino, G., F. Bernassola, R.A. Knight, M.T. Corasaniti, G. Nistico, and A. Finazzi-Agro. 1997. S-nitrosylation regulates apoptosis. Nature. 388:432–433. [DOI] [PubMed] [Google Scholar]
- 32.Lemaire, C., K. Andreau, V. Souvannavong, and A. Adam. 1998. Inhibition of caspase activity induces a switch from apoptosis to necrosis. FEBS Lett. 425:266–270. [DOI] [PubMed] [Google Scholar]
- 33.Cauwels, A., B. Janssen, A. Waeytens, C. Cuvelier, and P. Brouckaert. 2003. Caspase inhibition causes hyperacute tumor necrosis factor-induced shock via oxidative stress and phospholipase A2. Nat. Immunol. 4:387–393. [DOI] [PubMed] [Google Scholar]
- 34.Mellman, I., and R.M. Steinman. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell. 106:255–258. [DOI] [PubMed] [Google Scholar]
- 35.Aderem, A., and D.M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. [DOI] [PubMed] [Google Scholar]
- 36.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iyoda, T., S. Shimoyama, K. Liu, Y. Omatsu, Y. Akiyama, Y. Maeda, K. Takahara, R.M. Steinman, and K. Inaba. 2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang, F.-P., N. Platt, M. Wykes, J.R. Major, T.J. Powell, C.D. Jenkins, and G.G. MacPherson. 2000. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 191:435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scheinecker, C., R. McHugh, E.M. Shevach, and R.N. Germain. 2002. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J. Exp. Med. 196:1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inaba, K., S. Turley, F. Yamaide, T. Iyoda, K. Mahnke, M. Inaba, M. Pack, M. Subklewe, B. Sauter, D. Sheff, et al. 1998. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 188:2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kishimoto, H., and J. Sprent. 2001. A defect in central tolerance in NOD mice. Nat. Immunol. 2:1025–1031. [DOI] [PubMed] [Google Scholar]
- 42.Lesage, S., S.B. Hartley, S. Akkaraju, J. Wilson, M. Townsend, and C.C. Goodnow. 2002. Failure to censor forbidden clones of CD4 T cells in autoimmune diabetes. J. Exp. Med. 196:1175–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wegmann, D.R. 1996. The immune response to islets in experimental diabetes and insulin-dependent diabetes mellitus. Curr. Opin. Immunol. 8:860–864. [DOI] [PubMed] [Google Scholar]
- 44.Winer, S., H. Tsui, A. Lau, A. Song, X. Li, R.K. Cheung, A. Sampson, F. Afifiyan, A. Elford, G. Jackowski, et al. 2003. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat. Med. 9:198–205. [DOI] [PubMed] [Google Scholar]
- 45.Bellone, M., G. Iezzi, P. Rovere, G. Galati, A. Ronchetti, M.P. Protti, J. Davoust, C. Rugarli, and A.A. Manfredi. 1997. Processing of engulfed apoptotic bodies yields T cell epitopes. J. Immunol. 159:5391–5399. [PubMed] [Google Scholar]
- 46.Kaufman, D.L., M. Clare-Salzler, J. Tian, T. Forsthuber, G.S.P. Ting, P. Robinson, M.A. Atkinson, E.E. Sercarz, A.J. Tobin, and P.V. Lehmann. 1993. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 366:69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tisch, R., X.-D. Yang, S.M. Singer, R.S. Liblau, L. Fugger, and H.O. McDevitt. 1993. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 366:72–75. [DOI] [PubMed] [Google Scholar]
- 48.Benoist, C., and D. Mathis. 2001. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat. Imm. 2:797–801. [DOI] [PubMed] [Google Scholar]
- 49.Pittoni, V., and D. Isenberg. 1998. Apoptosis and antiphospholipid antibodies. Semin. Arthritis Rheum. 28:163–178. [DOI] [PubMed] [Google Scholar]
- 50.André, I., A. Gonzalez, B. Wang, J. Katz, C. Benoist, and D. Mathis. 1996. Checkpoints in the progression of autoimmune disease: Lessons from diabetes models. Proc. Natl. Acad. Sci. USA. 93:2260–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dilts, S.M., and K.J. Lafferty. 1999. Autoimmune diabetes: the involvement of benign and malignant autoimmunity. J. Autoimmun. 12:229–232. [DOI] [PubMed] [Google Scholar]
- 52.Phillips, J.M., N.M. Parish, M. Drage, and A. Cooke. 2001. Cutting edge: interactions through the IL-10 receptor regulate autoimmune diabetes. J. Immunol. 167:6087–6091. [DOI] [PubMed] [Google Scholar]
- 53.André-Schmutz, I., C. Hindelang, C. Benoist, and D. Mathis. 1999. Cellular and molecular changes accompanying the progression from insulitis to diabetes. Eur. J. Immunol. 29:245–255. [DOI] [PubMed] [Google Scholar]