Tuberculosis Toxin Blocking Phagosome Maturation Inhibits a Novel Ca2+/Calmodulin-PI3K hVPS34 Cascade (original) (raw)
Abstract
The capacity of Mycobacterium tuberculosis to infect latently over one billion people and cause two million fatalities annually rests with its ability to block phagosomal maturation into the phagolysosome in infected macrophages. Here we describe how M. tuberculosis toxin lipoarabinomannan (LAM) causes phagosome maturation arrest, interfering with a new pathway connecting intracellular signaling and membrane trafficking. LAM from virulent M. tuberculosis, but not from avirulent mycobacteria, blocked cytosolic Ca2+ increase. Ca2+ and calmodulin were required for a newly uncovered Ca2+/calmodulin phosphatidylinositol (PI)3 kinase hVPS34 cascade, essential for production of PI 3 phosphate (PI3P) on liposomes in vitro and on phagosomes in vivo. The interference of the trafficking toxin LAM with the calmodulin-dependent production of PI3P described here ensures long-term M. tuberculosis residence in vacuoles sequestered away from the bactericidal and antigen-processing organelles in infected macrophages.
Keywords: phosphatidylinositol 3-kinase, calmodulin, calcium, Mycobacterium tuberculosis, EEA1
Introduction
Phagolysosome biogenesis depends on interactions of the phagosome with the intracellular sorting pathways delivering late endosomal and lysosomal constituents to the maturing phagosome (1, 2). It has been shown that two specific rab5 effectors play an essential role in phagosomal maturation (2, 3): the phosphatidylinositol (PI)3 kinase hVPS34 (4), and the endosomal tethering molecule EEA1, which associates with endosomal membranes via its PI3 phosphate (PI3P) binding FYVE domain (5). It has been reported that by a hitherto unknown mechanism_, Mycobacterium tuberculosis_ inhibits EEA1 recruitment to phagosomes in infected macrophages, thus precluding phagolysosome formation (3). The inhibition by M. tuberculosis of EEA1 recruitment to the phagosome obstructs a pathway, dependent on EEA1 and Syntaxin 6, of delivery to phagosomes of lysosomal hydrolases and V0-ATPase proteolipid (6). The resulting block in phagosomal maturation contributes to the long-term survival and persistence of the tubercle bacillus in host macrophages (7).
It has been established that Ca2+ affects phagosomal maturation (8, 9). However, the mechanism of how Ca2+ affects phagolysosome formation remains to be delineated. The initial clues come from the studies showing that inhibition of cytosolic Ca2+ rise blocks phagosomal acquisition of late endosomal and lysosomal markers and lumenal acidification of the phagosome (8, 10). Significantly, infection of macrophages with M. tuberculosis prevents Ca2+ fluxes and inhibits activation of the downstream Ca2+/calmodulin effectors such as Ca2+/calmodulin kinase II (CaMKII; 8, 9). Here, we investigated potential connections between M. tuberculosis effects on Ca2+ fluxes and our previous reports that the M. tuberculosis glycosylated PI lipoarabinomannan (LAM) prevents EEA1 recruitment to phagosomes, which in turn leads to a block in phagosome maturation (3, 6). We examined whether the M. tuberculosis product LAM affected Ca2+ fluxes and whether and how Ca2+ signaling influenced EEA1 recruitment to phagosomes.
Materials and Methods
Ratiometric [Ca2+]c Imaging.
J774 cells were loaded for 30 min with 5 μM Fura-2 acetoxy-methyl ester. The cells were illuminated at 340 nm for 250 msec and 380 nm for 100 msec in 5-s intervals using a TILL Polychrome monochromator, and 340 380 fluorescence ratios were calculated using TILL software. For FcR clustering-induced [Ca2+]c rise, Fura-2–preloaded J774 cells were incubated at 4°C with 55 μg/ml mouse IgG for 30 min in HBSS, 1% BSA. After the addition of 20 μg/ml LAM or 10 μM _N,N_-dimethylsphingosine (DMS), FcR clustering was initiated by adding 30 μg/ml Texas Red anti–mouse F(ab′)2 to the cells at 37°C. The cells were illuminated at 340 nm for 150 msec and 380 nm for 50 msec with 1-s intervals between time points. FcR aggregation was examined by fluorescence microscopy at the end of experiment.
Live Cell Imaging, In Vivo PI3P Localization by Confocal Microscopy, and Immunofluorescence Microscopy.
Intracellular PI3P was imaged using p40PX-EGFP (11). 24 h after transfection, RAW macrophages transfected with the p40PX-EGFP fusion (11) were imaged live during and after phagocytosis of 1 μm Texas Red–labeled latex beads in an UltraView LCI Confocal System. Immunofluorescence microscopy was performed as previously described (3).
EEA1 Binding to Liposomes.
Liposomes were prepared by mixing phosphatidylserine (PS), PI, and PI3P, as indicated. 1 mg/ml dried lipid mixtures, resuspended in 50 mM Hepes, pH 7.2, 100 mM NaCl, and 0.5 mM EDTA, were sonicated for 5 min and liposomes were collected by centrifugation at 12,000 rpm for 30 min, and resuspended at 2 mg/ml (total lipid) in 200 mM sucrose, 25 mM Hepes, pH 7.4, 125 mM K+ acetate, 2.5 mM magnesium acetate, 0.5 mM CaCl2, 1 mM DTT, 1 mM sodium vanadate, 20 mM NaF, protease inhibitor cocktail, and an ATP regenerating system. Liposomes were incubated with 2 mg/ml cytosol at room temperature for 15 min and centrifuged. Resuspended pellets were analyzed by immunoblotting.
Protein Binding to Calmodulin Agarose.
0.5 mg/ml J774 cytosol was incubated for 3 h at 4°C with 50 μl calmodulin agarose beads in the presence of 0.5 mM CaCl2 or 2 mM EGTA, and 100 μM W7, W5, or 0.4% DMSO (control). Samples washed in binding buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, protease inhibitor cocktail, 1 mM sodium orthovanadate, 0.5 mM CaCl2, or 2 mM EGTA), were analyzed by immunoblotting.
Phagosome Purification, Immunoblotting, and Antibody Sources.
Latex bead phagosomes (LBC) were isolated and characterized as previously described (12, 13). Immunoblotting was performed as previously described (3). Antibodies were: EEA1 (provided by S. Corvera, University of Massachusetts, Amherst, MA), Syntaxin 8 (provided by W. Hong, Institute of Molecular and Cell Biology Singapore, Singapore), Syntaxin 3 (provided by P. Tuma, Johns Hopkins, Baltimore, MD), and hVPS34 and p150 (provided by J. Backer, Albert Einstein College of Medicine, Bronx, NY).
Online Supplemental Material.
Video shows effects of calmodulin inhibition on PI3P levels on phagosomes. RAW 264.7 cells were transfected with P40PX-EGFP (green) and allowed to phagocytose Texas Red–labeled latex beads (red). Frames were taken 48 s apart. The movie is played at 3 fps. This movie corresponds to Fig. 4, G–L. Cells were treated with W7 at indicated time point. Video 1 is available at http://www.jem.org/cgi/content/full/jem.20030527/DC1.
Figure 4.
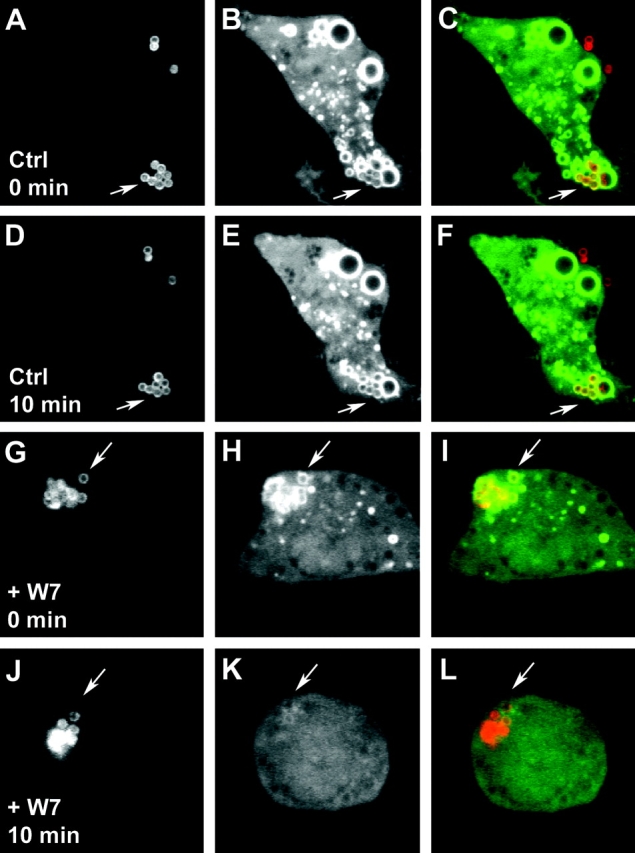
Calmodulin positively regulates PI3P on phagosomes. RAW 264.7 macrophages, transfected with the PI3P probe p40PX-EGFP, were imaged live while phagocytosing latex beads using an UltraView confocal microscope. After latex bead internalization and determination of PI3P positivity of the LBC, solvent without (Ctrl) or with 25 μM W7 (+W7) were added to the chamber. Panels: a, d, g, and j, Texas Red–labeled latex beads; b, e, h, and k, GFP probe for PI3P; c, f, i, and l, merged red and green channel images. Note localization of PI3P GFP probe on LBC after bead internalization (a–c and g–i), continued presence of the PI3P GFP probe on phagosomes 10 min after the addition of solvent alone (d–f), and the disappearance of the PI3P GFP probe when W7 was added (j–l). Arrows depict representative LBC in each experimental set. Examples of three independent experiments with comparable results are shown. Also, see Video 1 (available at http://www.jem.org/cgi/content/full/jem.20030527/DC1), which corresponds to panels g–l.
Results
M. tuberculosis Glycosylated Phosphatidylinositol LAM Inhibits Cytosolic Ca2+ Rise in Macrophages.
We considered two separate lines of investigation demonstrating: (a) that M. tuberculosis LAM blocks phagosome maturation (3) by inhibiting EEA1-dependent trafficking pathway from TGN to phagosomes (6), and (b) that _M. tuberculosis_–engendered inhibition of Ca2+ fluxes in macrophages leads to phagosome maturation arrest (8, 9). This led us to hypothesize that the phagosome maturation block might be mediated by LAM action on cytosolic Ca2+. We tested this possibility by measuring intracellular Ca2+ concentrations ([Ca2+]c) in J774 murine macrophage cell line after the addition of the Ca2+ ionophore A23187. Cells were preincubated for 30 min in Ca2+ containing HBSS with or without 20 μg/ml M. tuberculosis LAM. In a separate set of samples, the LAM equivalent from a nonpathogenic mycobacterial species, Mycobacterium smegmatis, was used instead of M. tuberculosis LAM. Fig. 1, A–D , shows that LAM from M. tuberculosis inhibits both the rate and the maximum [Ca2+]c rise. This effect is specific for M. tuberculosis LAM, as the LAM equivalent from M. smegmatis did not block [Ca2+]c increase (Fig. 1, A–D). These observations demonstrate that LAM, a product of M. tuberculosis, prevents increase of [Ca2+]c. This finding, combined with the previous demonstrations that LAM inhibits phagosomal maturation (3, 6) and that inhibition of Ca2+ fluxes is responsible for M. tuberculosis phagosome maturation block (8, 9), indicates that mycobacterial phagosome maturation arrest is caused by LAM-mediated inhibition of [Ca2+]c increases.
Figure 1.

M. tuberculosis LAM inhibits [Ca2**+**]c rise. J774 cells were preincubated 30 min without or with 20 μg/ml M. tuberculosis LAM (Mt LAM) or M. smegmatis LAM (Ms LAM). (A) Ratio (340/380 nm) images at different time points after the addition of 1 μM A23187 untreated (control), Mt LAM–treated, or Ms LAM–treated J774 cells. (B) [Ca2+]c kinetics (mean ± SEM, n = 30 cells). Lines: black, untreated cells; red, Mt LAM-treated cells; green, Ms LAM–treated cells. (C) Rate of [Ca2+]c increase ± SEM (n = 3 independent experiments, mean of 30 cells per experiment). (D) Maximum initial [Ca2+]c increase ± SEM (n = 3 independent experiments, mean of 30 cells per experiment). (E) [Ca2+]c kinetics after FcR cross-linking (refer to Materials and Methods) on J774 cells treated with 10 μM DMS (blue line), 20 μg/ml Mt LAM (red line), or control (black line). Mean of 65 cells (same field). FcR clustering revealed by Texas Red anti–mouse F(ab')2 fragment in presence (E”) or absence (E') of Mt LAM. (F) [Ca2+]c increase upon FcR cross-linking in the absence or presence of 10 μM DMS or 20 μg/ml Mt. LAM. Four independent experiments, mean of 65 cells per experiment, are shown. P-values, paired t test (www.graphpad.com).
To complement ionophore studies, we tested effects of LAM on [Ca2+]c rise using FcR clustering as a physiological stimulus (14). J774 cells were split and left to adhere to coverslips for 2 h. Cytosolic Ca2+ rise, elicited by FcR clustering (14), showed strong sphingosine kinase (SK) dependence as DMS, an SK inhibitor, reduced by 60% the increase of [Ca2+]c (Fig. 1, E and F). Next, we tested the effects of M. tuberculosis LAM by adding it 5 min before inducing FcR clustering. Fig. 1, E and F, show that LAM reduced the increase of [Ca2+]c without affecting receptor clustering (Fig. 1 E, E' and E”). These results show that M. tuberculosis LAM inhibits Ca2+ signaling induced by physiological stimuli. Furthermore, a significant portion of LAM-induced inhibition overlapped with the SK-dependent [Ca2+]c rise. In this context, it is noteworthy that a recent report suggests that M. tuberculosis affects Ca2+ increases by interfering with SK signaling (15).
Calmodulin and CaMKII Are Required for EEA1 Recruitment to Phagosomal Membrane.
Next, we investigated whether the LAM-mediated block of Ca2+ increase inhibited EEA1 recruitment to phagosomes. Previous studies have implicated calmodulin and its downstream effector CaMKII as the Ca2+ responsive elements affected by M. tuberculosis (9). Following this lead, we investigated whether calmodulin played a role in EEA1 recruitment to phagosomes. Macrophages were allowed to phagocytose complement opsonized latex beads for different periods of time in the presence or absence of W7, a specific inhibitor of Ca2+/calmodulin interactions with its binding partners. After phagocytosis, LBC were purified, using established protocols of flotation in sucrose gradients (3, 12, 13), and probed for EEA1 by immunoblotting (Fig. 2 A). W7 treatment decreased the amount of EEA1 associated with phagosomes (Fig. 2 A). The role of calmodulin in EEA1 recruitment to phagosomes was confirmed by immunofluorescence microscopy (Fig. 2, B–E). Quantitation of EEA1 association with phagosomes (Fig. 2 F) showed a decrease in the numbers of LBC positive for EEA1 in W7-treated macrophages. As a control, LBC were also stained for Syntaxin 8, an endosomal SNARE (16) previously used as an EEA1-independent control for phagosomes (3, 17). W7 did not affect Syntaxin 8 association with LBC, indicating that decrease in EEA1, observed in the presence of the calmodulin inhibitor, was not a result of phagocytosis inhibition but was due to diminished EEA1 recruitment. These experiments demonstrate that calmodulin promotes association of EEA1 with phagosomes in vivo.
Figure 2.

Ca2+/calmodulin and CaMKII positively regulate EEA1 recruitment to phagosomes. (A) Immunoblotting analysis and quantitation (mean ± SE) of EEA1 on purified LBC isolated from 25 μM W7-treated or -untreated macrophages. (B–E) Immunofluorescence images of EEA1 recruited to phagosomes (B and D) and corresponding phase contrast images (C and E) in W7-treated (D and E) or -untreated (B and C) macrophages. Triangles indicate EEA1 colocalization with phagosomes. (F) Phagosome colocalization quantitation of EEA1 and Syntaxin 8 by immunofluorescence (mean ± SE; n = 1,145 phagosomes, 35 fields). (G) Immunoblotting analysis of EEA1 and Syntaxin 3 on purified phagosomes from 2 μM KN62-treated and -untreated cells.
One of the major effectors of Ca2+/calmodulin is the multifunctional Ser/Thr kinase, CaMKII. An increase of [Ca2+]c induces the binding of Ca2+/calmodulin to CaMKII, which in turn relieves CaMKII from autoinhibition, resulting in autophosphorylation of CaMKII and activation of the kinase (18). The role of CaMKII in EEA1 recruitment to phagosomes was investigated using LBC isolated from macrophages treated with the CaMKII-specific inhibitor KN62. KN62 interferes with the binding of Ca2+/calmodulin specifically to CaMKII, thus preventing activation of CaMKII. Fig. 2 G shows that treatment of macrophages with KN62 for 1 h decreased the amount of EEA1 on phagosomes. Syntaxin 3 was used as a loading control. This result indicates that CaMKII is necessary for EEA1 recruitment to phagosomes.
Ca2+/Calmodulin Affects Generation of PI3P In Vitro.
How does calmodulin affect EEA1 recruitment to phagosomes? Ca2+/calmodulin binds directly to EEA1 via its IQ domain, but this association has been reported to repel EEA1 from membranes (19). Interestingly, Ca2+/calmodulin has been shown to positively regulate the levels of PI3P in CHO cells (20). Next, we examined whether calmodulin acted directly on EEA1, or affected EEA1 binding to membranes indirectly, via a PI3 kinase. Two types of liposomes, PS/PI and PS/PI3P were prepared and then incubated with cytosol. After 30 min of incubation, liposomes were pelleted and probed for EEA1 by immunoblotting. Wortmannin, a specific inhibitor of PI3 kinase, inhibited EEA1 binding to PS/PI liposomes (Fig. 3 A), indicating the importance of PI3 kinase activity in this assay. Next, the liposomes were incubated with J774 cytosol in the presence or absence of W7. After 15 min of incubation, liposomes were pelleted and probed for EEA1 by immunoblotting (Fig. 3 A). The association of EEA1 with PI containing liposomes was reduced in the presence of W7, whereas its association with liposomes containing preformed PI3P liposomes was not affected (Fig. 3 A). These results show that calmodulin enhancement of EEA1 recruitment to membranes occurs at a PI3 kinase step, as W7 inhibited the binding of EEA1 to liposomes containing the PI3 kinase substrate PI, but did not affect its binding to liposomes with preexisting PI3P.
Figure 3.

Ca2+/calmodulin and PI3 kinase–dependent association of EEA1 with liposomes and Ca2+-dependent binding of calmodulin to a protein complex containing PI3 kinase hVPS34. (A) Quantitation (mean ± SE) of EEA1 on phosphatidylserine (PS) and phosphatidylinositol (PI) or PS and PI3P liposomes after incubation with J774 cytosol in the presence or absence of 100 nM PI3 kinase inhibitor wortmannin or 100 μM calmodulin inhibitor W7. (B) Immunoblotting analysis of hVPS34 and p150 on calmodulin agarose beads after incubation with J774 cytosol in the presence of EGTA (lane 1), Ca2+ (lanes 2, 3, and 4), W7 (lane 3), and W5 (lane 4).
Ca2+-dependent Association of Calmodulin with a hVPS34- and p150-containing Complex.
The only type III PI3 kinase acting on endomembranes, hVPS34 (21), has been shown to play a role in phagosome maturation and EEA1 recruitment (3, 22). Although calmodulin has previously been shown to bind the type I PI3 kinase p85/p110 (20), its association with hVPS34 has not been investigated. We used calmodulin agarose beads to examine whether calmodulin binds the PI3 kinase hVPS34. Beads were incubated with J774 cytosol in the presence or absence of Ca2+, or with and without W7 in the presence of Ca2+. The bound material was probed for hVPS34 by immunoblotting (Fig. 3 B). The PI3 kinase hVPS34 bound to calmodulin in a Ca2+-dependent manner (Fig. 3 B, lanes 1 and 2). The binding was sensitive to W7 (Fig. 3 B, lanes 2 and 3), and to a much lower extent to W5 (Fig. 3 B, lane 4), a less active structural analogue of W7. These data indicate that hVPS34 interacts directly or indirectly with calmodulin. The PI3 kinase hVPS34 is also known to associate with a Ser/Thr kinase, p150, which modulates hVPS34 function (23). Next, we tested whether this subunit of the PI3 kinase was also present in the complexes bound to calmodulin beads (Fig. 3 B). Indeed, binding of p150 was observed, and its retention on calmodulin beads was also sensitive to W7 (Fig. 3 B). The association of p150 and hVPS34 with calmodulin was Ca2+ dependent (Fig. 3 B).
Calmodulin Affects PI3P Dynamics on Phagosomal Membranes.
Next, the role of calmodulin in the in vivo distribution and levels of PI3P was probed by live microscopy using green fluorescent protein (GFP) fused to a PI3P binding domain. Macrophages were transfected with a GFP fusion with the p40phox PX domain (PX-GFP), which binds exclusively to PI3P on endomembranes (11). Fig. 4 and Video 1 (available at http://www.jem.org/cgi/content/full/jem.20030527/DC1) show that upon the addition of W7 to the cells, the PX-GFP probe dissociated from the phagosomes. The effect was not limited to phagosomal organelles, as other endomembranes were also affected. These observations show that calmodulin plays a role in PI3P generation and maintenance in vivo.
Discussion
Here, we have reported a new signaling cascade, connecting intracellular Ca2+, calmodulin, and CaMKII with the recruitment of hVPS34 and production of PI3P on phagosomes. This signaling pathway is important for phagosome maturation and is a target for inhibition by M. tuberculosis. Based on our results, we conclude that the inhibitory effects of LAM on cytosolic Ca2+ increases can account for the known Ca2+ block associated with phagocytosis of M. tuberculosis (8). This action of LAM precludes interaction of PI3 kinase hVPS34 with calmodulin necessary for the downstream recruitment of EEA1. Because EEA1, in combination with Syntaxin 6, is necessary for the delivery of lysosomal components from the TGN to the phagosome (6), mycobacterial phagosome maturation arrest is caused by LAM-mediated disruption of Ca2+/calmodulin-dependent regulation of PI3P and EEA1 on phagosomes.
ManLAM has been reported to prevent Ca2+ rise in B10R macrophages (24). The inhibition of Ca2+ fluxes shown here is restricted to LAM isolated from virulent M. tuberculosis. There are several structural differences between LAM from M. tuberculosis and nonpathogenic mycobacteria such as M. smegmatis. These include acyl chains and the polysaccharides modifying the inositol ring (25). For instance, the polysaccharide portion of M. tuberculosis LAM has mannose termini whereas M. smegmatis LAM lacks mannose caps but has PI residues (25).
It is worth noting that an early study suggested that LAMP-1 acquisition by phagosome is independent of calcium in macrophages (26). However, more recent reports have demonstrated that cytosolic Ca2+ is important for phagosome maturation (27), including acidification (8, 10) and phagosome acquisition of lysosomal markers such as CD63 and Cathepsin D (8). The role of LAM in inhibiting these processes might be attributed to its partitioning into the host cell endomembranes (7), with its strongest action at the point of origination, the mycobacterial phagosome. Because dead mycobacteria do not block [Ca2+]c rise (8), it is likely that LAM has to be actively shed by live organisms to maintain appropriate concentration and localization. Alternatively, LAM might be extracted from or trapped in a denatured or cross-linked cell envelope during bacterial killing.
PI3P is crucial for phagosome maturation (3, 22). Inhibition of PI3P production, using wortmannin or blocking antibodies against hVPS34, prevents EEA1 recruitment, blocking phagosome maturation (2, 3, 22). This involves inhibition of delivery of hydrolases and H+-ATPase proteolipid subunit from the TGN (3, 6), albeit a subset of wortmannin-insensitive markers (such as LAMP, Syntaxin 8, and Syntaxin 13) is not affected (3, 6). The effects on PI3 kinase may also affect the recruitment to phagosomes of other PI3P binding proteins such as p40phox subunit of NADPH oxidase (28), which positively regulates superoxide production (29). Thus, Ca2+-dependent regulation of PI3K may not only promote phagolysosome biogenesis but may also regulate assembly of NADPH oxidase and oxidative burst. Interference with Ca2+ signaling by M. tuberculosis LAM could thus have a dual effect by diminishing the oxidative burst during mycobacterial uptake by macrophages (30) and blocking mycobacterial phagosome maturation (7).
In summary, the data presented here, combined with a number of recent reports (3, 6, 8–10, 15) are consistent with a model in which M. tuberculosis LAM inhibits a cascade consisting of cytosolic Ca2+ transients, calmodulin, PI3 kinase hVPS34, and EEA1. This pathway is necessary for conversion of phagosomes into phagolysosomes, as EEA1 and Syntaxin 6 cooperate in the delivery of lysosomal components to the phagosome (3, 6). Thus, M. tuberculosis LAM acts as a trafficking toxin causing mycobacterial phagosome arrest by interfering with the Ca2+/calmodulin PI3 kinase cascade. This is the first molecular definition of the mode of action of an M. tuberculosis toxin. Understanding the mechanism of mycobacterial phagosome maturation block will help in designing new antituberculosis therapies.
Acknowledgments
We thank J. Backer, J. Murray, M. Yaffe, S. Corvera, W. Hong, P. Tuma, and J. Belisle for reagents. LAM was prepared at Colorado State University with the support of National Institute of Allergy and Infectious Diseases contract NO1 AI-75320, “Tuberculosis Research Materials and Vaccine Testing.”
This work was supported by grant AI45148 from National Institutes of Health.
The online version of this article contains supplemental material.
References
- 1.Meresse, S., O. Steele-Mortimer, E. Moreno, M. Desjardins, B. Finlay, and J.P. Gorvel. 1999. Controlling the maturation of pathogen-containing vacuoles: a matter of life and death. Nat. Cell Biol. 1:E183–E188. [DOI] [PubMed] [Google Scholar]
- 2.Vieira, O.V., R.J. Botelho, and S. Grinstein. 2002. Phagosome maturation: aging gracefully. Biochem. J. 366:689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fratti, R.A., J.M. Backer, J. Gruenberg, S. Corvera, and V. Deretic. 2001. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J. Cell Biol. 154:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volinia, S., R. Dhand, B. Vanhaesebroeck, L.K. MacDougall, R. Stein, M.J. Zvelebil, J. Domin, C. Panaretou, and M.D. Waterfield. 1995. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 14:3339–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christoforidis, S., H.M. McBride, R.D. Burgoyne, and M. Zerial. 1999. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 397:621–625. [DOI] [PubMed] [Google Scholar]
- 6.Fratti, R.A., J. Chua, I. Vergne, and V. Deretic. 2003. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. USA. 100:5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell, D.G., H.C. Mwandumba, and E.E. Rhoades. 2002. Mycobacterium and the coat of many lipids. J. Cell Biol. 158:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malik, Z.A., G.M. Denning, and D.J. Kusner. 2000. Inhibition of Ca(2+) signaling by Mycobacterium tuberculosis is associated with reduced phagosome–lysosome fusion and increased survival within human macrophages. J. Exp. Med. 191:287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malik, Z.A., S.S. Iyer, and D.J. Kusner. 2001. Mycobacterium tuberculosis phagosomes exhibit altered calmodulin-dependent signal transduction: contribution to inhibition of phagosome-lysosome fusion and intracellular survival in human macrophages. J. Immunol. 166:3392–3401. [DOI] [PubMed] [Google Scholar]
- 10.Downey, G.P., R.J. Botelho, J.R. Butler, Y. Moltyaner, P. Chien, A.D. Schreiber, and S. Grinstein. 1999. Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcgammaRIIA receptors. J. Biol. Chem. 274:28436–28444. [DOI] [PubMed] [Google Scholar]
- 11.Kanai, F., H. Liu, S.J. Field, H. Akbary, T. Matsuo, G.E. Brown, L.C. Cantley, and M.B. Yaffe. 2001. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat. Cell Biol. 3:675–678. [DOI] [PubMed] [Google Scholar]
- 12.Via, L.E., D. Deretic, R.J. Ulmer, N.S. Hibler, L.A. Huber, and V. Deretic. 1997. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J. Biol. Chem. 272:13326–13331. [DOI] [PubMed] [Google Scholar]
- 13.Desjardins, M., L.A. Huber, R.G. Parton, and G. Griffiths. 1994. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124:677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melendez, A., R.A. Floto, D.J. Gillooly, M.M. Harnett, and J.M. Allen. 1998. FcgammaRI coupling to phospholipase D initiates sphingosine kinase-mediated calcium mobilization and vesicular trafficking. J. Biol. Chem. 273:9393–9402. [DOI] [PubMed] [Google Scholar]
- 15.Malik, Z.A., C.R. Thompson, S. Hashimi, B. Porter, S.S. Iyer, and D.J. Kusner. 2003. Cutting edge: Mycobacterium tuberculosis blocks Ca(2+) signaling and phagosome maturation in human macrophages via specific inhibition of sphingosine kinase. J. Immunol. 170:2811–2815. [DOI] [PubMed] [Google Scholar]
- 16.Subramaniam, V.N., E. Loh, H. Horstmann, A. Habermann, Y. Xu, J. Coe, G. Griffiths, and W. Hong. 2000. Preferential association of syntaxin 8 with the early endosome. J. Cell Sci. 113:997–1008. [DOI] [PubMed] [Google Scholar]
- 17.Fratti, R.A., J. Chua, and V. Deretic. 2002. Cellubrevin alterations and Mycobacterium tuberculosis phagosome maturation arrest. J. Biol. Chem. 277:17320–17326. [DOI] [PubMed] [Google Scholar]
- 18.Braun, A.P., and H. Schulman. 1995. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu. Rev. Physiol. 57:417–445. [DOI] [PubMed] [Google Scholar]
- 19.Mills, I.G., S. Urbe, and M.J. Clague. 2001. Relationships between EEA1 binding partners and their role in endosome fusion. J. Cell Sci. 114:1959–1965. [DOI] [PubMed] [Google Scholar]
- 20.Joyal, J.L., D.J. Burks, S. Pons, W.F. Matter, C.J. Vlahos, M.F. White, and D.B. Sacks. 1997. Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 272:28183–28186. [DOI] [PubMed] [Google Scholar]
- 21.Vanhaesebroeck, B., S.J. Leevers, K. Ahmadi, J. Timms, R. Katso, P.C. Driscoll, R. Woscholski, P.J. Parker, and M.D. Waterfield. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70:535–602. [DOI] [PubMed] [Google Scholar]
- 22.Vieira, O.V., R.J. Botelho, L. Rameh, S.M. Brachmann, T. Matsuo, H.W. Davidson, A. Schreiber, J.M. Backer, L.C. Cantley, and S. Grinstein. 2001. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 155:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panaretou, C., J. Domin, S. Cockcroft, and M.D. Waterfield. 1997. Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3-kinase. Substrate presentation by phosphatidylinositol transfer protein to the p150.Ptdins 3-kinase complex. J. Biol. Chem. 272:2477–2485. [DOI] [PubMed] [Google Scholar]
- 24.Rojas, M., L.F. Garcia, J. Nigou, G. Puzo, and M. Olivier. 2000. Mannosylated lipoarabinomannan antagonizes Mycobacterium tuberculosis-induced macrophage apoptosis by altering Ca2+-dependent cell signaling. J. Infect. Dis. 182:240–251. [DOI] [PubMed] [Google Scholar]
- 25.Vercellone, A., J. Nigou, and G. Puzo. 1998. Relationships between the structure and the roles of lipoarabinomannans and related glycoconjugates in tuberculosis pathogenesis. Front. Biosci. 3:e149–e163. [DOI] [PubMed] [Google Scholar]
- 26.Zimmerli, S., M. Majeed, M. Gustavsson, O. Stendahl, D.A. Sanan, and J.D. Ernst. 1996. Phagosome–lysosome fusion is a calcium-independent event in macrophages. J. Cell Biol. 132:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worth, R.G., M.K. Kim, A.L. Kindzelskii, H.R. Petty, and A.D. Schreiber. 2003. Signal sequence within Fcgamma RIIA controls calcium wave propagation patterns: apparent role in phagolysosome fusion. Proc. Natl. Acad. Sci. USA. 100:4533–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wishart, M.J., G.S. Taylor, and J.E. Dixon. 2001. Phoxy lipids: revealing PX domains as phosphoinositide binding modules. Cell. 105:817–820. [DOI] [PubMed] [Google Scholar]
- 29.Kuribayashi, F., H. Nunoi, K. Wakamatsu, S. Tsunawaki, K. Sato, T. Ito, and H. Sumimoto. 2002. The adaptor protein p40(phox) as a positive regulator of the superoxide-producing phagocyte oxidase. EMBO J. 21:6312–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon, A.H., and P.D. Hart. 1994. Stimulation or inhibition of the respiratory burst in cultured macrophages in a mycobacterium model: initial stimulation is followed by inhibition after phagocytosis. Infect. Immun. 62:4650–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]