Retinoids Regulate Survival and Antigen Presentation by Immature Dendritic Cells (original) (raw)
Abstract
Maturation of dendritic cells (DCs) is a critical step for the induction of an immune response. We have examined the role of retinoid nuclear receptor pathways in this process. Retinoids induce DC apoptosis, in the absence of inflammatory signals, through retinoic acid receptor (RAR)α/retinoic X receptor (RXR) heterodimers. In contrast, via a cross talk with inflammatory cytokines, retinoids increase DNA binding activity of nuclear factor κB in DCs, trigger membrane major histocompatibility complex class II and costimulatory molecule expression, induce the differentiation of immature DCs into mature DCs, and enhance antigen-specific T cell response. This maturation of DCs is mediated via a RXR-dependent/RAR-independent pathway and via an RARα/RXR pathway distinct from the one responsible for apoptosis. Apoptosis and activation, mediated through distinct nuclear retinoid receptor pathways, can be dissociated from each other with selective synthetic retinoids. We identify a novel cellular function for retinoids and suggest that selective retinoids might be of interest for controlling antigen presentation.
Keywords: human, cellular activation, antigen presentation, immunomodulators, nuclear receptors
Introduction
The generation of adaptive immune response against foreign antigen requires the participation of both antigen-specific T cells and antigen-presenting cells. DCs have the capacity to sample antigen at the boundary with the “external milieu” and migrate toward T cell areas of the draining lymph node. There, they can provide processed antigen together with the costimulatory signals required for the proliferation of antigen-specific T cells (1). The maturation process of DCs is thus believed to be critical for the induction of an immune response. Retinoids, through their cognate nuclear retinoid receptors, transactivate numerous genes and exert potent effects in development and hemopoiesis (2). Retinol (Rol; vitamin A) is metabolized intracellularly via two distinct pathways forming its active derivatives: (a) retinoic acids (RAs), all-trans RA (tRA) and 9-cis RA (9cRA), whose effects are transduced by nuclear retinoid receptors (RA receptors [RARs] and retinoic X receptors [RXRs]; 2), and (b) retro-retinoids, which do not bind to known receptors (3). Rol was shown to enhance T cell–mediated host versus graft response in mice, augment specific antitumor immunity, and impair acquired immunological tolerance of foreign cells (4–7). Conversely, several studies have shown that vitamin A–deficient rodents fail to elicit full immune response to viral and bacterial pathogens (8), and field studies have convincingly shown that Rol supplementation reduces mortality from infectious diseases among children in areas where vitamin A deficiency is endemic (9–12). The cellular and molecular mechanisms that underlie the effect of retinoids on the immune system remains elusive. Vitamin A was shown to enhance B and T cell survival and proliferation in vitro (3, 13, 14). These effects are triggered by retro-retinoids, and B and T cells do not synthesize RA in appreciable amounts (3, 13, 14). 9cRA did not affect thymocyte proliferation, but was shown to be a potent negative regulator of activation-induced T cell apoptosis (15), likely through RAR/RXR heterodimers (16). The effects of retinoids on immune responses might also be mediated by DCs. Mice fed with a diet containing vitamin A acetate or 13-cis-RA presented with an increase in the number of accessory cells in the paracortical region of the lymph nodes and in splenic marginal zone (17). The antigen-presenting cell function of these cells was increased as evidenced by both alloproliferative and allocytotoxic responses in vitro (17). Activation of Langerhans cells (LCs) after topical administration of RA have been reported (18). Opposite effects of retinoids has also been reported, which include decreased antigen-presenting cell function in vitro, a toxic effect on epidermal LCs, and decreased response to vaccinal antigen in vivo (19–23).
This study aimed to investigate the various potential effects of retinoids on DCs and dissect the nuclear pathways that underlie these effects. Here we report that retinoids, through their cognate nuclear receptors, act in vitro on immature DCs to induce either their activation or their apoptosis, in the presence or absence of inflammatory signals, respectively. Using receptor-selective retinoid ligands (see Table I), we show that the effects of retinoids on immature DCs are mediated through distinct retinoid receptor pathways that can be dissociated from each other. Our data identify new pathways and potential tools for the regulation of antigen presentation by DCs.
Table I.
Functional Characteristics of Retinoids
| Retinoid nuclear receptors | |||||||
|---|---|---|---|---|---|---|---|
| Ligands | RARαAF2 | RARβAF2 | RARγAF2 | RXRAF2 | (RXR)2DR1 | RARα-RXRDR5 | References |
| Natural ligands | |||||||
| tRA | + | + | + | 0 | 0 | + | 2 |
| 9cRA | + | + | + | + | + | + | 2 |
| Synthetic ligands | |||||||
| BMS753 | + | 0 | 0 | 0 | 0 | + | 30 |
| BMS614 | −a | 0 | 0 | 0 | 0 | − | 28 |
| BMS641 | 0 | + | 0 | 0 | ND | ND | 27 |
| BMS453 | − | + | − | 0 | ND | ND | 27 |
| BMS961 | 0 | (+) | + | 0 | 0 | 0 | 30 |
| BMS493 | − | − | − | 0 | ND | ND | 26 |
| SR11237 | 0 | 0 | 0 | + | + | 0 | 28–30 |
| BMS749 | − | − | − | + | + | 0 | 40 |
Materials and Methods
Culture of DCs.
Immature DCs with the LC phenotype (CD1a+, E-cadherin+, CLA+, CMHIIlo, CD86−, TNFRIlo) among which 20–40% express Lag and Langerin (see Fig. 4), with high pinocytic and weak antigen presentation activities, were obtained as previously described (24, 25) from purified blood CD14+ human monocytes cultured for 5–7 d in RPMI 1640 supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% heat-inactivated FCS Myoclone (all from GIBCO BRL), 100 ng/ml GM-CSF (Sandoz AG), 10 ng/ml IL-4 and 10 ng /ml TGFβ 1 (R&D Systems), and referred to as LC-type DC. These immature DCs mature after CD40L triggering (as a surrogate cognate signal; 25).
Figure 4.

Retinoid receptor pathways transduce LC-type DC activation in a cross talk with TNFα. (a) Retinoids cooperate with TNFα to up-regulate MHC class II and CD86 on immature LC-type DCs. Day 6 immature LC-type DCs were cultured for 40 h with TNFα and/or retinoids or vehicle as indicated. Cells were washed and stained with HLA-DR-FITC and CD86-PE or isotype controls for 15 min at 4°C. 104 events were then analyzed with a FACSCalibur™ (Becton Dickinson) using CELLQuest™ software (Becton Dickinson). Percents are given for DRhi/CD86hi cells gated as indicated. Data are representative of 10 experiments on different donors. (b) Synergy between retinoids and TNFα is dose dependent. In the presence of TNFα, Rol (•) tRA (▪), 9cRA (□), the RARα agonist BMS753 (○), the RARβ agonist BMS641 (▵), and the RXR agonist SR11237 (▴) up-regulate MHC class II and costimulatory CD86 molecules on immature LC-type DCs. The percentages of DRhi/CD86hi cells were plotted against increasing concentrations (nM) of each retinoid. Results correspond to one representative experiment. (c–e) Two distinct retinoid receptor pathways transduce LC-type DC activation in synergy with TNFα. Cells were cultured for 40 h in the presence of 10 ng/ml TNFα with 100 nM tRA, BMS753, or 9cRA, or with 1,000 nM SR11237, with or without 1,000 nM of the pan-RAR antagonist BMS493 or 1,000 nM of the RARα ligand BMS614. (c and d) Percentages of DRhi/CD86hi cells are represented and results are expressed as the mean ± SD of at least three experiments on different donors. (e) Cells were cultured in the presence of 10 ng/ml TNFα with 1,000 nM SR11237 and increasing doses (nM) of BMS614 (•) or BMS 493 (○), or, as controls, with 100 nM BMS753 and increasing doses (nM) of BMS493 (□), or with vehicle (ethanol) and increasing doses (nM) of BMS614 (▪). The percentages of DRhi/CD86hi cells are presented. Results are from one representative experiment.
Retinoids and Cytokines.
tRA was from Sigma-Aldrich and Rol and 9cRA were from Calbiochem. RARα (BMS614), RARβ (BMS 641 and BMS453), RARγ (BMS961) agonists, RARα (BMS 614) and pan-RAR (BMS493) antagonists, and the pan-RXR agonist (SR11237) were provided by Bristol-Myers Squibb (see Table 1; references 26–30). All retinoids were stored dissolved in ethanol at 10−3 M. Human recombinant TNFα and IL-1β were from R&D Systems. Human holoRBP, fully saturated by Rol, was purified from plasma as previously described (31) and stored in Tris-HCl 20 mM buffer, pH 7.4. Murine fibroblast cell lines transfected with human CD40L were provided by F. Brière (Schering-Plough, Dardilly, France).
Flow Cytometry Analysis.
105 cells were incubated in 96-well plates (Becton Dickinson) for 15 min at 4°C in PBS, 2% human AB serum, and 0.01 M NaN3, with FITC-conjugated anti-DR (Immunotech) and PE-conjugated CD86 (BD Biosciences) at the appropriate concentration, or with control isotype-matched irrelevant mAbs at the same concentration. After washing, 104 events were analyzed with a FACSCalibur™ (Becton Dickinson) using CELLQuest™ software (Becton Dickinson). For apoptosis studies, 4 × 105 cells were washed and either nuclei were stained with propidium iodide (PI; 50 μg/ml in PBS 0.1 NaCi, 0.1% Triton X-100 for 20 min at 37°C) or, alternatively, cells were incubated for 15 min at 4°C in PBS, 2% human AB serum, and 0.01 M NaN3, with annexin V and 2 μg/ml PI. 2 × 104 events were then analyzed with a FACSCalibur™ (Becton Dickinson) using CELLQuest™ software (Becton Dickinson).
Confocal Microscopy.
Cells were washed in Ca2+/Mg++-free PBS and were allowed to adhere to glass slides coated with 50 μg/ml poly-l-lysine (Sigma-Aldrich) for 30 min. Slides were washed and fixed with PBS 4% paraformaldehyde for 30 min, incubated with PBS 0.1 M glycine for 10 min, and kept at 4°C in PBS 0.01 M sodium azide. For staining, slides were incubated for 30 min at room temperature with mouse anti–human Langerin (provided by S. Saeland, Schering-Plough Laboratory for Immunological Research, Dardilly, France; 32, 33) followed by goat anti–mouse Cy3. Slides were washed and incubated with 10% mouse serum for 20 min and then incubated with FITC-conjugated mAb to HLA-DR (Immunotech). Slides were mounted with Fluoprep (Biomerieux SA) and analyzed with a confocal laser microscope system (LSM510; Carl Zeiss MicroImaging, Inc.) attached to a microscope (Axiovert 200M; Carl Zeiss MicroImaging, Inc.).
Antigen Presentation Assays.
DCs were collected, washed three times, pulsed for 8 h with tetanus toxin or medium alone, then for 40 h with or without retinoids and 10 ng/ml TNFα. Cells were washed two times in RPMI with 10% human AB serum, resuspended in RPMI with 10% human AB serum, and added in triplicate at various concentrations to 105 autologous (autologous antigen presentation assay) or allogenous (MLR) T cells/well in 96-well tissue culture plates (Falcon). T cells were isolated by the standard Ficoll-Paque method followed by magnetic depletion of non–T cells (MACS; Miltenyi Biotec). [3H]thymidine (Amersham Biosciences) incorporation was measured in newly synthesized DNA over 18 h, using pulses initiated at days 4 or 5 of the culture with 1 mCi/well of [3H]thymidine. Cells were then harvested with a 96-well Harvester (Amersham Biosciences), collected on glass fiber filter (Amersham Biosciences), and the incorporation of thymidine was measured with a Beta-plate microscintillation counter (LKB; Amersham Biosciences).
Electrophoretic Mobility Shift Assay.
4 × 106 cells were washed once in cold PBS and allowed to swell on ice for 10 min in buffer A (10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and 2 mM PMSF) containing the following protease inhibitors: leupeptin, aprotinin, pepstatin, and antipain, each at a concentration of 4 μg/ml. Samples were then centrifuged and the pellet was suspended in 20 μl buffer C (20 mM Hepes, pH 7.8, 420 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, and 2 mM PMSF) containing the protease inhibitors. The mixture was left for 20 min on ice. Cell debris were removed by centrifugation and the resulting nuclear extracts were stored at −70°C. Protein concentration was measured with the Bio-Rad protein assay (Bio-Rad Laboratories). Nuclear extracts (5–10 μg protein) were assayed for DNA binding activity in a total volume of 20 μl binding buffer (20 mM Tris-HCl, pH 8, 60 mM KCl, 2 mM MgCl2, 0.3 mM DTT, 12% glycerol, and 3 μg poly dI-dC). Nuclear extracts were incubated separately for 15 min at 30°C with the double stranded labeled probes for nuclear factor (NF)-κB (5′-GATCCCAAGAGGGATTTCACCTAAATCC-3′). The samples were then loaded onto a nondenaturing 5% polyacrylamide gel and subjected to electrophoresis at 14 V/cm in a low ionic strength buffer (0.5X TBE). Gels were dried and examined with a PhosphorImager (Molecular Dynamics).
Quantitation of IL-12 Production by ELISA.
Supernatants were stored at −70°C until cytokine measurements. Production of bioactive IL-12 p70 were measured in duplicate using ELISA Quantikine Kits (R&D Systems) according to the manufacturer's instructions. Sensitivity of IL-12 detection was 0.5 pg/ml.
Statistics.
Student's t test was used to interpret the significance of differences between experimental groups (presented as mean ± SD). P value was two tailed and considered significant when <0.05.
Results
Apoptosis of Immature DCs Is Mediated through RARα-RXR Nuclear RAR Heterodimers.
Immature DCs with the LC (LC-type DCs) phenotype (refer to Materials and Methods) cultured with retinoids rapidly died. 1 μM Rol significantly induced the death of immature LC-type DCs with a 20 and 40% reduction of viable cell numbers after 2 and 3 d, respectively (Fig. 1 a). The active derivatives of Rol, tRAs and 9cRAs, induced a similar dose- and time-dependent cell death as determined by the reduction of viable cell number and the percentage of apoptotic nuclei (Table II and unpublished data). This effect was restricted to DCs at their immature stage of differentiation because neither mature DCs (CD40L-treated) nor monocytes (from days 0 to 4 of culture with cytokines) died after exposure to retinoids (unpublished data). Dying cells expressed annexin V (Fig. 1 b) and death was blocked by the caspase inhibitor Z-Vad-fmk (Table II), but not by anti-Fas blocking antibody ZB4, even though LCs express Fas (unpublished data), indicating a FasL-independent apoptosis. Moreover a pan-RAR (α, β, and γ) antagonist (BMS493; reference 26), inhibited Rol-induced and tRA-induced apoptosis (Fig. 1 and Table II), demonstrating that apoptosis was mediated through RAs and their receptors.
Figure 1.

Retinoids induce dose- and time-dependent apoptosis of LC-type DCs. (a) Day 6 immature LC-type DCs were cultured in the absence (•) or presence of increasing amounts (▵, 10 nM; ▴, 100 nM; ○, 1,000 nM) of Rol and viable cells were counted each day with trypan blue exclusion. (b) After day 2 of culture in the presence of the indicated amounts of tRA, cells were washed and incubated with anti–annexin V antibody and 2 μg/ml PI. 104 total events (without gating) were then analyzed with a FACSCalibur™ (Becton Dickinson) using CELLQuest™ software (Becton Dickinson). Data are representative of three experiments on different donors.
Table II.
Inhibition of Retinoid-induced Apoptosis by RAR Antagonists, Caspase Inhibitor, TNFα, and CD40L
| 0 | Rol1 μM | tRA1 μM | BMS7531 μM | BMS753 100 nM + SR11237 100 nM | |
|---|---|---|---|---|---|
| Medium | ≤5 | 17 ± 8 | 38 ± 10 | 38 ± 8 | 38 ± 5 |
| BMS614 (1 μM) | ≤5 | — | 15 ± 8 | 9 ± 5 | 7 ± 3 |
| BMS493 (1 μM) | ≤5 | 4 ± 1 | 10 ± 6 | 5 ± 3 | 4 ± 2 |
| Z-Vad (50 nM) | ≤5 | — | 9 ± 4 | 9 ± 7 | 11 ± 4 |
| TNFα (10 ng/ml) | ≤5 | 4.5 ± 1 | 8 ± 7 | 11 ± 1 | 14 ± 2 |
| CD40La | ≤5 | — | ≤5 | ≤5 | ≤5 |
Retinoid nuclear receptor signal transduction is most frequently transduced by heterodimers composed of RARs and RXRs (2). RT-PCR analysis revealed that LC-type cells expressed transcripts for all three RARs (α, β, and γ) and all three RXRs (α, β, and γ; unpublished data). A panel of synthetic retinoid ligands (Table I) was then used to further investigate which receptors (among RARs and RXRs) were involved in the effect of retinoids. The results are summarized in Tables III and IV. Apoptosis of immature LC-type DCs was induced by a selective RARα agonist (BMS753; reference 30), but not by RARβ agonists (BMS 641, BMS453; reference 27), an RARγ BMS961 agonist (30), or a pan-RXR agonist (SR11237; references 28–30; Table III). An RARα-selective antagonist (BMS 614, slt; reference 28) inhibited tRA and BMS753-induced apoptosis (Table II). These results indicate that the apoptotic effect of RA is transduced by RARα. The pan-RXR agonist (SR11237), which had no effects on its own, had a synergistic effect on LC-type DC apoptosis induced by the selective RARα agonist BMS753 (Table IV). The RARβ agonists BMS641 and BMS453, but not the selective RARγ agonist (BMS961), also synergize with the pan-RXR agonist to induce LC-type DC apoptosis (Table IV), albeit with a lower efficiency. Thus, the retinoid-triggered LC-type DC apoptosis is transduced by RARα/RXR heterodimers in which the transcriptional activity of RXR is subordinated to ligand binding to its RAR partner (27, 28, 34). Furthermore, RARβ-RXR heterodimers can also transduce the apoptotic effect of retinoids, albeit with a lower efficiency.
Table III.
Rol-induced Apoptosis of LC-type DCs Is Mediated via RARα-RXR Heterodimers
| BMS ligandsa | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EtOH | Rol | tRA | 9cRA | 753 | 453 | 641 | 961 | SR11237 | 614 | 493 | |
| 1 nM | — | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | — |
| 10 nM | — | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 |
| 50 nM | — | — | — | — | 18 ± 3 | — | — | — | — | — | — |
| 100 nM | — | ≤5 | 14 ± 6 | 8 ± 3 | 27 ± 9 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 |
| 1 μM | ≤5 | 17 ± 8 | 36 ± 10 | 30 ± 9 | 37 ± 7 | 10 ± 6 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 |
Table IV.
Rol-induced Apoptosis of LC-type DCs: Synergistic Effect between a pan-RXR Agonist (SR11237) and Selective RARα and RARβ Agonists
| SR11237 100 nM + BMS ligandsa | ||||||
|---|---|---|---|---|---|---|
| 753 | 453 | 641 | 614 | 493 | 961 | |
| 1 nM | 6 ± 3 | ≤5 | ≤5 | ≤5 | ≤5 | ≤5 |
| 10 nM | 25 ± 10 | 5 | ≤5 | ≤5 | ≤5 | ≤5 |
| 100 nM | 32 ± 7 | 20 ± 6 | 16 ± 3 | ≤5 | ≤5 | ≤5 |
| 1 μM | 47 ± 10 | 19 ± 7 | 30 ± 2 | ≤5 | ≤5 | ≤5 |
Retinoid-induced Apoptosis Requires Supraphysiological Levels of Retinoids and Is Inhibited by Inflammatory Cytokines.
The above data may account, at least in part, for the reported inhibitory effect of retinoids on the in vitro allogenous MLR (19, 23). However, they cannot account for stimulations of the immune response that have been observed upon retinoid administration in vivo (4, 5, 17). Major differences between in vitro and in vivo conditions might be the availability of retinoids and the environmental inflammatory signals encountered by DCs. Inflammatory cytokines, such as TNFα and IL-1β, are known to enhance migration of immature LCs from the skin to the lymph nodes (1, 35) and were shown to contribute to immature DC activation (1, 36). Therefore, we investigated the effect of retinoids on immature LC-type DCs in the presence of such cytokines. We found that TNFα inhibited in a dose-dependent manner (>50% inhibition at 1 ng/ml TNFα) the proapoptotic effects of Rol and tRA, as well as of 9cRA and of the RARα selective agonist BMS753 (Fig. 2 , a and b, and Table II). Other stimuli known to contribute to LC activation such as IL-1β (1–10 ng/ml; not depicted) and the cognate signal CD40L also inhibited the apoptotic effect of retinoids (Table II). Therefore, inflammatory or cognate signals antagonize the effects of retinoids on immature LC survival. We also investigated whether concentrations of Rol required to induce apoptosis are present in physiological conditions. Rol is distributed to peripheral tissues in vivo as a complex with the Rol binding protein (holoRBP). To approximate the availability of retinoids in vivo, we thus investigated the effects of purified holoRBP in this model. We did not observe apoptosis of immature LC-like cells at concentrations of holoRBP similar to those observed in steady-state serum in normal individuals (1–2 μM). Therefore, the concentration required to induce apoptosis might be above physiological levels (Fig. 3) .
Figure 2.
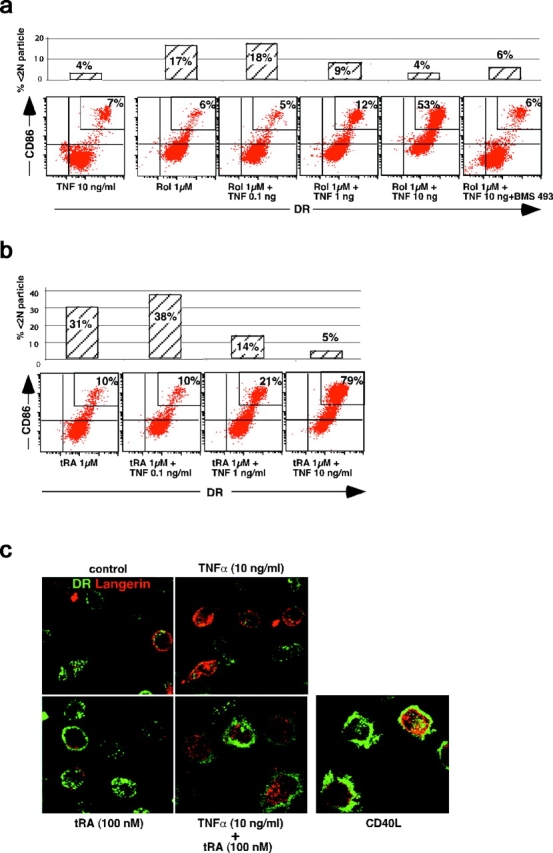
Rol and tRA cooperate with TNFα to induce a dose-dependent increase in the percentage of DRhi-CD86hi LC-type DCs, which parallels inhibition of apoptosis. Cells were cultured in the presence of 1 μM Rol (a) or tRA (b) and increasing concentration of TNFα. Expression of HLA-DR, CD86 (see Fig. 1), and apoptosis (percent <2 N particle; see Table II) were determined at the same time point on separate aliquots of the same culture. Data from a representative experiment out of three on different donors. (c) Day 6 immature LC-type DCs were cultured for 40 h with TNFα and/or tRA, or CD40L transfected fibroblasts. Cells were then analyzed by confocal microscopy for MHC class II (green) and Langerin (red) expression. Although tRA or TNFα on their own do not induce significant morphological changes, the combination of them strikingly induces the formation of many thin dendrites, MHC class II+, at the cell surface. These morphological changes resemble those that accompany the maturation induced after exposure to CD40L. Note the membrane staining for Langerin on immature cells.
Figure 3.
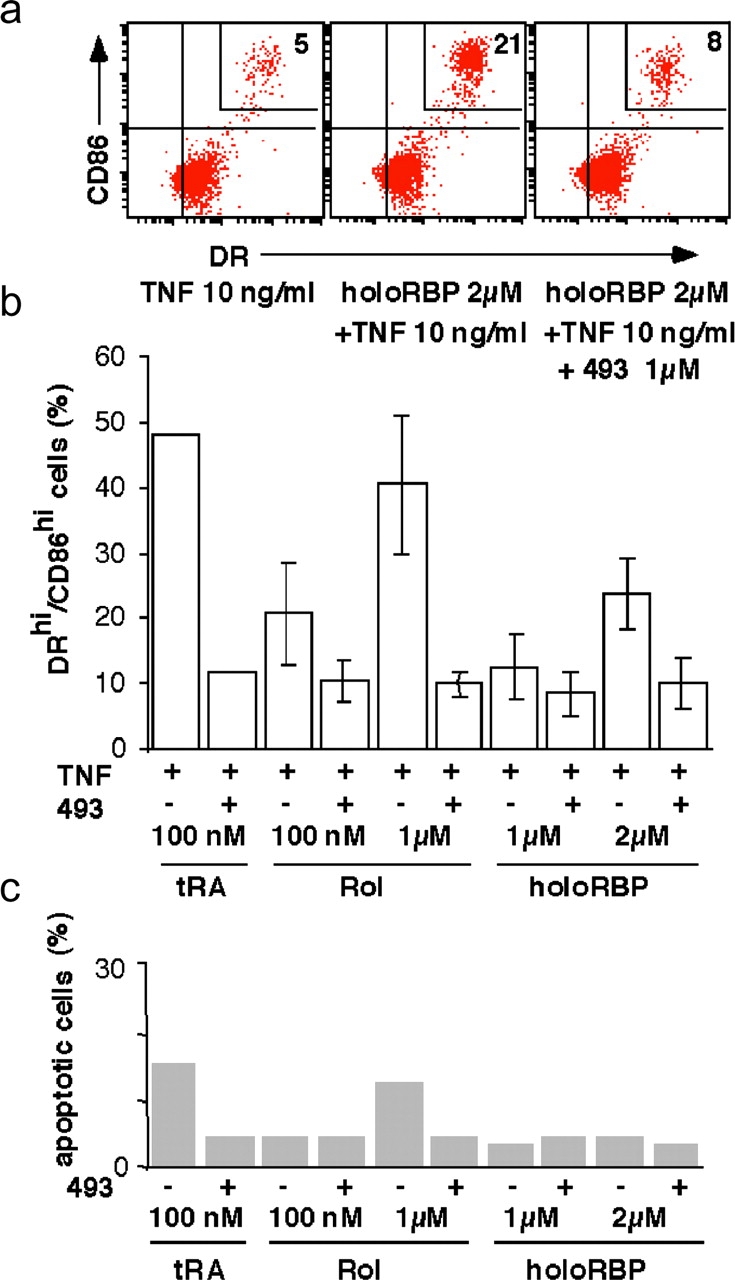
HoloRBP cooperates with TNFα to up-regulate MHC class II and CD86 on immature LCs. Day 6 immature LCs were cultured in the presence of 2 μM holoRBP purified from human plasma (a) or 1 μM Rol, 1 μM holoRBP, or 2 μM holoRBP (b) in the presence of 10 ng/ml TNFα without (−) or with (+) 1 μM BMS493, and analyzed as indicated in a. Data from one representative experiment is shown. (b) Value are mean and SD of two separate experiments on different donors. (c) Day 6 immature LC-type DCs were cultured for 2 d with the indicated retinoids and then washed and incubated with anti–annexin V antibody and 2 μg/ml PI. 104 total events (without gating) were then analyzed with a FACSCalibur™ (Becton Dickinson) using CELLQuest™ software (Becton Dickinson) as described in Fig. 1.
Synergistic Activation of Immature DCs by Retinoids and Inflammatory Cytokines.
Next, we investigated the effects of retinoids on immature LC activation. On its own, TNFα, as well as LPS and IL-1β, is not sufficient to induce significant activation (class II and costimulatory molecule expression and enhancement of antigen presentation) of immature LC-type DCs in vitro (Fig. 2, a–c; reference 25) because TGFβ1 inhibits the activation of these cells in response to inflammatory stimuli (25, 37, 38). In the absence or at a low concentration of TNFα (≤1 ng/ml), Rol and tRA (Fig. 2, a and b), as well as 9cRA, RAR, and RXR agonists (Fig. 4 a), did not significantly induce class II or CD86 up-regulation on immature LC-type DCs.
Strikingly, Rol and tRA, in the presence of TNFα (≥1 ng/ml), induced a dose-dependent increase in the membrane expression of class II and CD86 by LC-type DCs, which paralleled inhibition of apoptosis (Fig. 2, a and b), suggesting immunological maturation of LCs. Confocal microscopy examination (Fig. 2 c) confirmed that exposure to tRA and TNFα induced class II antigen translocation from cytoplasmic compartments to the cell surface, and showed that this was accompanied by other typical changes observed in maturing DCs such as an increase in cell size and the formation of numerous thin dendrites (1). These effects are similar to what is observed after DC maturation in response to CD40L (Fig. 2 c). 9cRA and the selective RARα agonist BMS753 had similar effects as Rol and tRA on immature LC activation in the presence of TNFα (Fig. 4, a and b). The activation induced by Rol, RAs, and BMS753 was inhibited by the pan-RAR antagonist BMS493 (Figs. 2 a and 4 c). Together, the fact that these chemically distinct compounds exert the same effects and that this effect is inhibited by the same pan-RAR antagonist (BMS493) demonstrates that this effect was mediated through RAs and their nuclear receptors.
To investigate whether these effects of retinoids on DC activation might be of physiological relevance we then investigated the effects of purified holoRBP in this model. Interestingly, the addition of holoRBP (at a concentration corresponding to its steady-state serum level in normal individuals) in the presence of TNFα also induced a dose-dependent activation of immature DCs (Fig. 3). The activation induced by holoRBP as well as by free Rol was inhibited by the pan-RAR antagonist BMS493, demonstrating that their effect was mediated through RAs and their receptors (Fig. 3).
Interestingly, the selective RXR agonist SR11237 also induced LC-type DC activation in the presence of TNFα (at 1–10 ng/ml; Fig. 4, a and b). In contrast to what is observed with RAR agonists, the effect of SR11237 was not inhibited by the pan-RAR antagonist BMS493 or by the RARα antagonist BMS614. Rather, BMS614 appeared to synergize with SR11237 to activate LCs (Fig. 4, c–e), whereas BMS614 had no effect on its own (Fig. 4 a).
We have previously shown that TNFα receptor I (p55, CD120a) expression on LC-type DCs was very low, due to the effects of TGFβ1 (25) exposure to retinoids thus might have accounted for the increased response to TNFα by increasing TNFα receptor expression. However, flow cytometry analysis did not reveal any change in CD120a and CD120b expression after exposure to retinoids (unpublished data).
Retinoids Increase DNA Binding Activity of NF-κB Complexes in Response to TNFα.
As nuclear translocation and binding of NF-κB complexes to their DNA response element was shown to be involved in DC activation (1, 39), the effect of retinoids might be to increase the DNA binding capacity of NF-κB. Interestingly, the addition of TNFα after a short (6-h) exposure to tRA, BMS753 or SR11237 induced a strong increase in the binding of nuclear NF-κB complexes to a cognate DNA probe when compared with nuclear extracts from retinoid-untreated cells, or from cells treated with the RARα antagonist BMS614 (Fig. 5) . Retinoids on their own had no detectable effect on NF-κB complex binding activity. The addition of 10 nM N-tosyl-L-phenylalanine chloromethyl ketone, which has been reported to inhibit translocation of NF-κB and activation of DCs (39), inhibited by 50% the increase of class II and CD86 on LC-type DCs treated with tRA and TNFα without any evidence of toxicity (unpublished data).
Figure 5.

tRA, the RARα agonist BMS753, the RXR agonist SR11237, but not vehicle (EtOH) or the RARα antagonist BMS614, synergize with TNFα to induce nuclear translocation of NF-κB. Day 6 immature LC-type DCs were washed and incubated for 6 h in fresh complete medium alone or with vehicle (ethanol) or retinoids at a 1-μM concentration. Cells were washed, incubated for 30 min with 50 ng/ml TNFα or medium alone as control, and then processed for electrophoretic mobility shift assay as described in Materials and Methods.
Retinoids Enhance Antigen Presentation to T Cells in the Presence of TNFα.
LC-type DCs treated with retinoids and TNFα significantly increased allogeneic T lymphocyte proliferation (MLR; Fig. 6 , a and b). To investigate the effect of retinoids on an antigen-specific immune response, we studied the presentation of tetanus toxoid (TT) by LC-type DCs to fresh autologous lymphocytes of immune individuals. TT-pulsed LC-type DCs exposed to TNFα and tRA, or TNFα and SR11237 (Fig. 6 c), induced an increase in the proliferative response of autologous T lymphocytes as compared with pulsed untreated LC-type DCs or LC-type DCs incubated with either TNFα alone or retinoids alone. Note that even though LC-type DCs were washed several times before coculture, a direct effect of retinoids on T cells cannot be easily ruled out because retinoids are highly lipophilic. However, such an effect appears unlikely, as retinoids and/or TNFα-treated LC-type DCs did not stimulate autologous T lymphocytes in the absence of antigen (Fig. 6 c). Furthermore, T cells do not synthesize RAs in appreciable amounts (14). Therefore, we conclude from the above results that retinoids cooperate with inflammatory signals to induce activation of immature LC-type DCs and enhance their ability for antigen presentation.
Figure 6.

TNFα cooperates with retinoids to increase antigen presentation by DCs. (a) Rol- and TNFα-treated LC-type DCs increase alloreactive proliferative response. Immature LC-type DCs were cultured with TNFα and/or Rol or vehicle for 40 h. Cells were washed four times in RPMI with 10% human AB serum and added in triplicate at stimulator/effector ratio of 1 and 4% to 105 purified T cells/well from the same donor or from a second donor, in 96-well tissue culture plates. SD are indicated. *, P < 0.05. (b) TNFα cooperate with tRA and RXR agonist to increase alloreactive proliferative response. Day 6 immature LC-type DCs were cultured with TNFα and/or retinoids or vehicle (ethanol) for 40 h. Cells were then washed four times in medium containing 10% human AB serum and added in triplicate at stimulator/effector ratio of 1 and 4% to 105 purified T cells/well from the same donor or from a second donor, in 96-well tissue culture plates. Background thymidine incorporation was always <10% of alloreactive response. SD were <15%. *, P < 0.01. Results are from one representative experiment out of nine on different donors. (c) TNFα cooperates with retinoids to increase antigen presentation by immature LC-type DCs. Day 6 immature LC-type DCs were pulsed for 8 h with TT or medium alone, and then cultured with TNFα and/or retinoids for 40 h. Cells were then washed four times in medium containing10% human AB serum and added at stimulator/effector ratio of 4 and 16% to 105 purified T cells/well from the same donor, in triplicate in 96-well tissue culture plates. T cell proliferation was measured as indicated in Materials and Methods. Background thymidine incorporation, in the absence of pulse with TT, is indicated (no TT) and was always <10% of the antigen-specific response. SD were <15%. *, P < 0.01. Results are from one representative experiment out of three. (d) Production of IL-12 upon CD40 triggering. Day 6 immature DCs were incubated for 40 h with 100 nM tRA, 1,000 nM of the RXR agonist SR11237, and/or 10 ng/ml TNFα and either CD40 liter-transfected fibroblasts or CD32-transfected fibroblasts as control. Supernatants were then collected and analyzed for bioactive p70 IL-12 production using ELISA. Results are mean and SD of five experiments on different donors.
IL-12 Production by LC-type DCs Is Not Influenced by Exposure to Retinoids.
DCs activate helper T cells through antigen-restricted class II/TCR and costimulatory molecule interaction (1). It has been proposed that activated helper T cells then in turn activate DCs/LCs through CD40L–CD40 interaction (40), resulting in the production of IL-12 by DCs/LCs (1, 40). We investigated whether retinoids may directly influence IL-12 production by LC type-DCs in the absence of T cells. Neither TNFα or retinoids alone, nor the combination of the two, at doses that induced apoptosis or activation, resulted in a significant production of IL-12 in culture supernatants in the absence of CD40 triggering (Fig. 6 d). Conversely, CD40 triggering increased the production of IL-12 by LC-type DCs, but this increase was not modified by the addition of retinoids and/or TNFα (Fig. 6 d and unpublished data). Thus retinoids, which enhance the early phase of LC–T cell antigen-restricted interaction mediated through class II/TCR and costimulatory molecules, may not replace, nor influence directly, the outcome of the LC–T cell interaction mediated through CD40L–CD40 interaction.
Retinoid Receptor Pathways That Can Be Dissociated from the Apoptotic One Mediate the Effects of Retinoids on DC Activation.
The pan-RXR agonist (SR11237) cooperated with TNFα to induce expression of CD86 and class II Ag (Fig. 3, a and b), increase DNA binding activity of NF-κB complexes (Fig. 5), and enhance presentation to antigen-specific T cells (Fig. 6, b and c). The pan-RAR antagonist BMS493 had only a weak effect on this TNFα-SR11237 synergism for LC-type DC activation (Fig. 4, c–e). In contrast, BMS493 strongly inhibited tRA- and BMS753-induced apoptosis (Table II), as well as RAR-dependent activation of LC-type DCs (see below). Moreover, another synthetic retinoid (BMS749), which acts as both a pan-RXR agonist and a pan-RAR antagonist (41), also cooperated with TNFα to activate LC-type DCs (unpublished data). Note that neither SR11237 (Table II) nor BMS749 (not depicted) exhibited an apoptotic effect on their own. Thus, activation of immature LC-type DCs in synergy with TNFα can be achieved via an RXR pathway that is RAR independent and not involved in apoptosis.
On the other hand, the synergistic effects between either Rol, tRA, or the RARα agonist BMS753 and TNFα on LC-type DC activation, were inhibited by the pan-RAR antagonist BMS493 (Fig. 3 and unpublished data), demonstrating that an RARα-dependent pathway could also induce activation. Interestingly, the RARα antagonist BMS614 synergized with the RXR agonist SR11237 for TNFα-induced LC-type DC activation (Fig. 4, c–e) and antigen presentation (Fig. 6, b and c). This synergistic effect was inhibited by BMS493 (Fig. 4 d), therefore indicating the involvement of RXR/RARα heterodimers. Note that BMS614 did not induce, but inhibited retinoid-induced apoptosis (Table II). Natural retinoids and selective ligands are chemically distinct compounds (2). The fact that they exert the same effects on cell activation in the presence of TNFα and that these effects inhibited the pan-RAR antagonist (BMS493) is strong evidence that the target of retinoids are nuclear retinoid receptors.
Together these results demonstrate that the effects of retinoids on LC-type DC activation are efficiently transduced through both (a) an RXR pathway that is RAR independent and (b) an RARα/RXR heterodimer-dependent pathway, the latter responding to bona fide RARα agonists (tRA and BMS753) as well as to the RARα antagonist BMS614 in the presence of the pan-RXR agonist SR11237.
The RARβ agonists (BMS641 and BMS453) had a weak effect on the activation of immature LC-type DCs (Fig. 4 a and unpublished data) and the selective RARγ agonist (BMS961) had no effect whatsoever (unpublished data). Interestingly, the pan-RAR antagonist BMS493 only partially blocked 9cRA-induced activation (Fig. 4 c) to a level similar to that obtained with the RXR agonist SR11237. This, considered together with the fact that 9cRA binds to both RAR and RXR, suggests that 9cRA is able to trigger both the RAR-dependent and the RAR-independent pathways of LC activation.
Discussion
Our present data demonstrate that retinoids act on immature LC-type DCs generated in vitro to regulate either apoptosis or antigen presentation in the absence or presence of inflammatory cytokines, respectively. These effects are mediated by the active derivatives of vitamin A and their cognate nuclear receptors. This identifies immature DCs, such as LCs, as a key cellular target of retinoids and possibly of vitamin A in the immune system.
DCs are essential antigen-presenting cells that initiate immune responses. Immature LCs internalize antigens and, after being activated by inflammatory stimuli and other indicators of cell damage, migrate to T cell areas of secondary lymphoid organs where they can initiate the immune response (1). Epithelial cells produce active TGFβ (42), which is required for the differentiation of malpighian epithelium–associated immature LCs (43), but TGFβ1 inhibits the inflammatory stimuli–mediated activation of immature LCs (25, 37, 38). Our results suggest a potential role for retinoids in the activation of immature DCs, such as LCs, in malpighian epithelia (where most pathogens are encountered), as upon the addition of retinoids immature LC-type DCs are synergistically activated in the presence of a moderate amount of inflammatory mediators (1–10 ng/ml TNFα or IL-1β), likely to be produced at the site of local inflammation. Upon the addition of Rol as a complex with its carrier protein RBP at a concentration close to that observed in the serum of normal individuals (2 μM), a proportion of immature LCs are also synergistically activated in the presence of inflammatory mediators. The mechanism we describe here may therefore be of physiological relevance and may allow cell damage to be taken into account at the earliest phase of the cognate immune response by efficiently triggering an antigen-specific T lymphocyte–mediated immune response.
In contrast, in the absence of inflammatory stimuli, retinoids on their own do not activate immature LC-type DCs, rather, increasing concentration to nonphysiological levels leads to apoptosis in a dose-dependent manner. We have obtained similar effects of retinoids (apoptosis in the absence of inflammatory stimuli and enhancement of TNFα-induced activation; unpublished data) in another model of DCs obtained from human monocytes treated by GM-CSF and IL-4 (36, 44). Therefore, the opposite effects of retinoids on the immune response that have been described, both in vitro and in vivo (4–12, 19–23), could be related both to the presence and absence of inflammatory signals and to the doses of retinoids. The distinct effects of retinoids on immature DCs might have been related to a signaling imbalance, such as the delivering of an activation signal by retinoids in the absence of an appropriate antiapoptotic stimuli. However, the results of this study do not support this hypothesis because the nuclear retinoid receptor pathways that transduce these effects are distinct.
We demonstrate that the retinoid signal triggering caspase-dependent immature LC-type DC apoptosis in the absence of inflammatory signals is transduced by RARα/RXR heterodimers, in which the transcriptional activity of RXR upon binding of an agonistic ligand is subordinated (27, 28, 34) to the binding of an agonist ligand to its RARα partner (Table II and Fig. 7) . As expected, apoptosis mediated through RARα/RXR heterodimers is inhibited by RAR antagonists BMS493 and BMS614, and an RXR agonist (SR11237) does not induce apoptosis on its own. On the other hand, the retinoid signals that cooperate with TNFα to induce LC-type DC activation (as evidenced by increased DNA binding activity of NF-κB complexes, an increase of cell surface expression of class II and costimulatory molecules, and enhancement of antigen presentation) are mediated by two distinct pathways that can be dissociated from the apoptotic one by using synthetic ligands (Fig. 7). A first pathway is RXR dependent and RAR independent, as it is induced by RXR-selective agonists (SR11237 and BMS749) and not inhibited by RAR antagonists (BMS493 and BMS614). The retinoid signal may be transduced by RXR homodimers acting on DR1 response elements, and/or possibly through heterodimers between RXR and other nuclear receptors, such as PPARs or several orphan receptors (2, 41, 45, 46). Note that on its own, RXR agonist does not induce apoptosis. A second pathway involves RARα/RXR heterodimers as evidenced by the induction of LC-type DC activation by the RARα selective agonist BMS753, and the synergistic effect of the RARα-selective ligand BMS614 and the RXR-selective agonist SR11237 (Fig. 4).
Figure 7.

Effect of retinoids on immature DC survival and activation. Schematic representation of retinoid receptor pathways that transduce apoptosis or activation of immature DCs (LC).
This RARα/RXR pathway is, however, different from the RARα/RXR pathway responsible for apoptosis of immature LC-type DCs because apoptosis is induced by RARα agonists through RAR-RXR heterodimer and is inhibited by BMS614, whereas in a sharp contrast activation is synergistically triggered by BMS614 in combination with an RXR agonist (SR11237).
In this respect, note that BMS614, which inhibits transactivation by RARα from DR5 response elements in transfection experiments, was already shown in another instance (28) to synergize with the RXR agonist SR11237 via RARα/RXR heterodimers to induce the differentiation of myeloid cells. Thus, the ligand binding requirements of RARα/RXR heterodimers to induce apoptosis and activation of immature LC-type DCs are different (Fig. 6), indicating that the responsive genes and/or coactivators proteins involved in these two processes are most probably different.
Activation of immature DCs in the present model results from the cooperation of signals transduced by retinoid receptors and by cytokine receptors. The molecular basis of this effect is not known. Of note, the present results show that retinoids and TNFα synergistically induce NF-κB binding to its DNA response element, which has been shown to be linked to DC activation (39). Future studies will be needed to explore the mechanisms of this synergistic effect. One possible mechanism would have been the up-regulation of TNFα receptor p55 by retinoids, however neither p55 nor p75 receptor expression at the cell membrane as measured by flow cytometry were influenced by retinoid treatment (unpublished data).
The availability of selective retinoid nuclear receptor ligands, which dissociate apoptosis and activation of immature LC-type DCs, may open new avenues for the therapeutic control of immune response, e.g., to improve efficiency of vaccination against certain pathogens or tumor antigen. For example, an RXR-selective agonist (SR11237) has no effect on its own on LC-type DC apoptosis, whereas it can induce LC-type DC activation, and the RARα ligand BMS614 in combination with the RXR agonist SR11237 stimulates antigen presentation in the presence of inflammatory cytokines (Fig. 5 c), whereas it inhibits the RARα/RXR-mediated apoptotic effects of natural and synthetic retinoids (Table II). The physiological role of vitamin A and retinoids on DCs remains unknown. However, it is of note that Vitamin A deficiency impairs resistance to infection and increases the risk of death, particularly to pathogens encountered at epithelial barriers, and induces an immune defect in response to infection, involving T, B, and NK cells (8–12). Thus, future studies should explore whether a retinoid-related defect in immature DC activation might contribute to the immune defect and the increased mortality due to infection in vitamin A deficiency.
Acknowledgments
We thank Drs. G. Richards, C. Benoist, P. Kastner, and S. Chan for a critical reading of this paper. We are grateful to Bristol-Myers Squibb for gifts of synthetic retinoids and support to IGBMC.
The work at Necker was supported by the CNRS, the INSERM, the Université Paris-V, the Comité de Paris de la Ligue Nationale contre le Cancer, and the Histiocytosis Association of America. F. Geissmann was supported by a fellowship from INSERM. P. Revy is a scientist from CNRS. The work at IGBMC was supported by the CNRS, the INSERM, the Hôpital Universitaire de Strasbourg, the Association pour la Recherche sur le Cancer, the Collège de France, and the Fondation pour la Recherche Médicale.
Abbreviations used in this paper: 9cRA, 9-cis retinoic acid; LC, Langerhans cell; NF, nuclear factor; PI, propidium iodide; RA, retinoic acid; RAR, RA receptor; Rol, retinol; RXR, retinoic X receptor; tRA, all-trans RA; TT, tetanus toxoid.
References
- 1.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- 2.Chambon, P. 1996. A decade of molecular biology of retinoic acid receptors. FASEB J. 10:940–954. [PubMed] [Google Scholar]
- 3.Buck, J., F. Derguini, E. Levi, K. Nakanishi, and U. Hammerling. 1991. Intracellular signaling by 14-hydroxy-4,14-retro-retinol. Science. 254:1654–1656. [DOI] [PubMed] [Google Scholar]
- 4.Malkovsky, M., A.J. Edwards, R. Hunt, L. Palmer, and P.B. Medawar. 1983. T-cell-mediated enhancement of host-versus-graft reactivity in mice fed a diet enriched in vitamin A acetate. Nature. 302:338–340. [DOI] [PubMed] [Google Scholar]
- 5.Malkovsky, M., C. Dore, R. Hunt, L. Palmer, P. Chandler, and P.B. Medawar. 1983. Enhancement of specific antitumor immunity in mice fed a diet enriched in vitamin A acetate. Proc. Natl. Acad. Sci. USA. 80:6322–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malkovsky, M., R. Hunt, L. Palmer, C. Dore, and P.B. Medawar. 1984. Retinyl acetate-mediated augmentation of resistance to a transplantable 3-methylcholanthrene-induced fibrosarcoma. The dose response and time course. Transplantation. 38:158–161. [DOI] [PubMed] [Google Scholar]
- 7.Malkovsky, M., P.B. Medawar, D.R. Thatcher, J. Toy, R. Hunt, L.S. Rayfield, and C. Dore. 1985. Acquired immunological tolerance of foreign cells is impaired by recombinant interleukin 2 or vitamin A acetate. Proc. Natl. Acad. Sci. USA. 82:536–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross, A.C., and C.B. Stephensen. 1996. Vitamin A and retinoids in antiviral responses. FASEB J. 10:979–985. [PubMed] [Google Scholar]
- 9.Rahmathullah, L., B.A. Underwood, R.D. Thulasiraj, R.C. Milton, K. Ramaswamy, R. Rahmathullah, and G. Babu. 1990. Reduced mortality among children in southern India receiving a small weekly dose of vitamin A. N. Engl. J. Med. 323:929–935. [DOI] [PubMed] [Google Scholar]
- 10.Semba, R.D. 1994. Vitamin A, immunity, and infection. Clin. Infect. Dis. 19:489–499. [DOI] [PubMed] [Google Scholar]
- 11.Sommer, A. 1992. Vitamin A deficiency and childhood mortality. Lancet. 340:488–489. [DOI] [PubMed] [Google Scholar]
- 12.Underwood, B.A., and P. Arthur. 1996. The contribution of vitamin A to public health. FASEB J. 10:1040–1048. [PubMed] [Google Scholar]
- 13.Buck, J., A. Myc, A. Garbe, and G. Cathomas. 1991. Differences in the action and metabolism between retinol and retinoic acid in B lymphocytes. J. Cell Biol. 115:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garbe, A., J. Buck, and U. Hammerling. 1992. Retinoids are important cofactors in T cell activation. J. Exp. Med. 176:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang, Y., M.S. Vacchio, and J.D. Ashwell. 1993. 9-cis-retinoic acid inhibits activation-driven T-cell apoptosis: implications for retinoid X receptor involvement in thymocyte development. Proc. Natl. Acad. Sci. USA. 90:6170–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bissonnette, R.P., T. Brunner, S.B. Lazarchik, N.J. Yoo, M.F. Boehm, D.R. Green, and R.A. Heyman. 1995. 9-cis retinoic acid inhibition of activation-induced apoptosis is mediated via regulation of fas ligand and requires retinoic acid receptor and retinoid X receptor activation. Mol. Cell. Biol. 15:5576–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katz, D.R., M. Drzymala, J.A. Turton, R.M. Hicks, R. Hunt, L. Palmer, and M. Malkovsky. 1987. Regulation of accessory cell function by retinoids in murine immune responses. Br. J. Exp. Pathol. 68:343–350. [PMC free article] [PubMed] [Google Scholar]
- 18.Meunier, L., K. Bohjanen, J.J. Voorhees, and K.D. Cooper. 1994. Retinoic acid upregulates human Langerhans cell antigen presentation and surface expression of HLA-DR and CD11c, a beta 2 integrin critically involved in T-cell activation. J. Invest. Dermatol. 103:775–779. [DOI] [PubMed] [Google Scholar]
- 19.Bedford, P.A., and S.C. Knight. 1989. The effect of retinoids on dendritic cell function. Clin. Exp. Immunol. 75:481–486. [PMC free article] [PubMed] [Google Scholar]
- 20.Hachisuka, H., and H. Uno. 1987. Effects of retinoic acid on the epidermal Langerhans cells and beta-glucuronidase activity in macaque skin. Am. J. Dermatopathol. 9:316–323. [DOI] [PubMed] [Google Scholar]
- 21.Semba, R.D., Z. Munasir, J. Beeler, A. Akib, Muhilal, S. Audet, and A. Sommer. 1995. Reduced seroconversion to measles in infants given vitamin A with measles vaccination. Lancet. 345:1330–1332. [DOI] [PubMed] [Google Scholar]
- 22.Walsh, L.J., G.J. Seymour, and R.N. Powell. 1985. The in vitro effect of retinol on human gingival epithelium. II. Modulation of Langerhans cell markers and interleukin-1 production. J. Invest. Dermatol. 85:501–506. [DOI] [PubMed] [Google Scholar]
- 23.Williams, N.A., and T.J. Hill. 1991. Effects on murine epidermal Langerhans cells of drugs known to cause recrudescent herpes simplex virus infection in a mouse model. J. Invest. Dermatol. 97:933–937. [DOI] [PubMed] [Google Scholar]
- 24.Geissmann, F., C. Prost, J.P. Monnet, M. Dy, N. Brousse, and O. Hermine. 1998. Transforming growth factor β 1, in the presence of granulocyte/macrophage colony-stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J. Exp. Med. 187:961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geissmann, F., P. Revy, A. Regnault, Y. Lepelletier, M. Dy, N. Brousse, S. Amigorena, O. Hermine, and A. Durandy. 1999. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J. Immunol. 162:4567–4575. [PubMed] [Google Scholar]
- 26.Chazaud, C., P. Chambon, and P. Dolle. 1999. Retinoic acid is required in the mouse embryo for left-right asymmetry determination and heart morphogenesis. Development. 126:2589–2596. [DOI] [PubMed] [Google Scholar]
- 27.Chen, J.Y., S. Penco, J. Ostrowski, P. Balaguer, M. Pons, J.E. Starrett, P. Reczek, P. Chambon, and H. Gronemeyer. 1995. RAR-specific agonist/antagonists which dissociate transactivation and AP1 transrepression inhibit anchorage-independent cell proliferation. EMBO J. 14:1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen, J.Y., J. Clifford, C. Zusi, J. Starrett, D. Tortolani, J. Ostrowski, P.R. Reczek, P. Chambon, and H. Gronemeyer. 1996. Two distinct actions of retinoid-receptor ligands. Nature. 382:819–822. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann, J.M., L. Jong, A. Fanjul, J.F. Cameron, X.P. Lu, P. Haefner, M.I. Dawson, and M. Pfahl. 1992. Retinoids selective for retinoid X receptor response pathways. Science. 258:1944–1946. [DOI] [PubMed] [Google Scholar]
- 30.Taneja, R., B. Roy, J.L. Plassat, C.F. Zusi, J. Ostrowski, P.R. Reczek, and P. Chambon. 1996. Cell-type and promoter-context dependent retinoic acid receptor (RAR) redundancies for RAR beta 2 and Hoxa-1 activation in F9 and P19 cells can be artefactually generated by gene knockouts. Proc. Natl. Acad. Sci. USA. 93:6197–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malpeli, G., C. Folli, and R. Berni. 1996. Retinoid binding to retinol-binding protein and the interference with the interaction with transthyretin. Biochim. Biophys. Acta. 1294:48–54. [DOI] [PubMed] [Google Scholar]
- 32.Valladeau, J., V. Duvert-Frances, J.J. Pin, C. Dezutter-Dambuyant, C. Vincent, C. Massacrier, J. Vincent, K. Yoneda, J. Banchereau, C. Caux, et al. 1999. The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface. Eur. J. Immunol. 29:2695–2704. [DOI] [PubMed] [Google Scholar]
- 33.Valladeau, J., O. Ravel, C. Dezutter-Dambuyant, K. Moore, M. Kleijmeer, Y. Liu, V. Duvert-Frances, C. Vincent, D. Schmitt, J. Davoust, et al. 2000. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 12:71–81. [DOI] [PubMed] [Google Scholar]
- 34.Vivat, V., C. Zechel, J.M. Wurtz, W. Bourguet, H. Kagechika, H. Umemiya, K. Shudo, D. Moras, H. Gronemeyer, and P. Chambon. 1997. A mutation mimicking ligand-induced conformational change yields a constitutive RXR that senses allosteric effects in heterodimers. EMBO J. 16:5697–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roake, J.A., A.S. Rao, P.J. Morris, C.P. Larsen, D.F. Hankins, and J.M. Austyn. 1995. Dendritic cell loss from nonlymphoid tissues after systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J. Exp. Med. 181:2237–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sallusto, F., M. Cella, C. Danieli, and A. Lanzavecchia. 1995. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J. Exp. Med. 182:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Epstein, S.P., R.L. Baer, G.J. Thorbecke, and D.V. Belsito. 1991. Immunosuppressive effects of transforming growth factor beta: inhibition of the induction of Ia antigen on Langerhans cells by cytokines and of the contact hypersensitivity response. J. Invest. Dermatol. 96:832–837. [DOI] [PubMed] [Google Scholar]
- 38.Geissmann, F., M.C. Dieu-Nosjean, C. Dezutter, J. Valladeau, S. Kayal, M. Leborgne, N. Brousse, S. Saeland, and J. Davoust. 2002. Accumulation of immature Langerhans cells in human lymph nodes draining chronically inflamed skin. J. Exp. Med. 196:417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rescigno, M., M. Martino, C.L. Sutherland, M.R. Gold, and P. Ricciardi-Castagnoli. 1998. Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 188:2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ridge, J.P., F. Di Rosa, and P. Matzinger. 1998. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 393:474–478. [DOI] [PubMed] [Google Scholar]
- 41.Benoit, G., L. Altucci, M. Flexor, S. Ruchaud, J. Lillehaug, W. Raffelsberger, H. Gronemeyer, and M. Lanotte. 1999. RAR-independent RXR signaling induces t(15;17) leukemia cell maturation. EMBO J. 18:7011–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munger, J.S., X. Huang, H. Kawakatsu, M.J. Griffiths, S.L. Dalton, J. Wu, J.F. Pittet, N. Kaminski, C. Garat, M.A. Matthay, et al. 1999. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 96:319–328. [DOI] [PubMed] [Google Scholar]
- 43.Borkowski, T.A., J.J. Letterio, A.G. Farr, and M.C. Udey. 1996. A role for endogenous transforming growth factor beta 1 in Langerhans cell biology: the skin of transforming growth factor beta 1 null mice is devoid of epidermal Langerhans cells. J. Exp. Med. 184:2417–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sallusto, F., and A. Lanzavecchia. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor α. J. Exp. Med. 179:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forman, B.M., P. Tontonoz, J. Chen, R.P. Brun, B.M. Spiegelman, and R.M. Evans. 1995. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 83:803–812. [DOI] [PubMed] [Google Scholar]
- 46.Mangelsdorf, D.J., and R.M. Evans. 1995. The RXR heterodimers and orphan receptors. Cell. 83:841–850. [DOI] [PubMed] [Google Scholar]