Arf6 and Phosphoinositol-4-Phosphate-5-Kinase Activities Permit Bypass of the Rac1 Requirement for β1 Integrin–mediated Bacterial Uptake (original) (raw)
Abstract
Efficient entry of the bacterium Yersinia pseudotuberculosis into mammalian cells requires the binding of the bacterial invasin protein to β1 integrin receptors and the activation of the small GTPase Rac1. We report here that this Rac1-dependent pathway involves recruitment of phosphoinositol-4-phosphate-5-kinase (PIP5K) to form phosphoinositol-4,5-bisphosphate (PIP2) at the phagocytic cup. Reducing the concentration of PIP2 in the target cell by using a membrane-targeted PIP2-specific phosphatase lowered bacterial uptake proportionately. PIP2 formation is regulated by Arf6. An Arf6 derivative defective for nucleotide binding (Arf6N122I) interfered with uptake and decreased the level of PIP2 around extracellular bacteria bound to host cells. This reduction in PIP2 occurred in spite of fact that PIP5K appeared to be recruited efficiently to the site of bacterial binding, indicating a role for Arf6 in activation of the kinase. The elimination of the Rac1-GTP–bound form from the cell by the introduction of the Y. pseudotuberculosis YopE RhoGAP protein could be bypassed by the overproduction of either PIP5K or Arf6, although the degree of bypass was greater for Arf6 transfectants. These results indicate that both Arf6 and PIP5K are involved in integrin-dependent uptake, and that Arf6 participates in both activation of PIP5K as well as in other events associated with bacterial uptake.
Keywords: integrin, Rac1, Arf6, PIP5K, Yersinia uptake
Introduction
A number of enteric pathogens, including the Gram-negative bacterium Yersinia pseudotuberculosis, enter host cells as a central step in the disease process. Enteropathogenic Yersinia species translocate across the intestinal epithelium in mammalian hosts, allowing replication in local lymph nodes, as well as spread into deep organ sites, resulting in systemic diseases (1–4). Efficient entry into intestinal lymph nodes requires the bacterial outer membrane protein invasin (4). In culture, engagement by invasin of a subset of heterodimeric integrin receptors, each having the identical β1 chain, results in phagocytic uptake (5). Binding to invasin appears to occur at a site on the integrin that is recognized by natural ligands such as fibronectin and laminin, although the binding affinity for invasin is significantly higher than that observed for natural ligands (6, 7).
After integrin receptor engagement, intracellular signaling events are required for uptake to proceed. Multimerization of invasin stimulates bacterial uptake, implying that clustering of receptor is involved in promoting a signal (8). Consistent with a signal being sent directly from the receptor, mutations in the cytoplasmic domain of the integrin β1 chain alter the rate of uptake (9, 10). Tyrosine kinase activity is also required (11–15). Finally, Rho family GTPases regulate uptake, as Yersinia that encode the translocated YopE RhoGAP protein are strongly blocked from internalization (15, 16). Evidence has been provided that inactivation of Rac1 is the primary block in uptake caused by the translocation of YopE into target cells (15, 17).
Rho family GTPases regulate a wide variety of actin-dependent events, including phagocytosis (18). In the case of one of these family members, Cdc42, there is a clear model for how GTP binding leads to induction of actin polymerization. Phosphoinositol-4,5-bisphosphate (PIP2), in collaboration with activated Cdc42 or the SH3-SH2 adaptor protein Nck, can relieve autoinhibition from WASP family members, resulting in activation of the Arp 2/3 complex (19–23). Although Arp 2/3 is clearly recruited to the site of bacterial binding, invasin-mediated uptake can occur in the presence of dominant inhibitory forms of Cdc42 and in a cell line lacking WASP and N-WASP (17). Therefore, an alternate route must exist for how engagement of integrin receptors leads to stimulation of actin polymerization.
Rac1 is recruited and activated after Y. pseudotuberculosis contacts its integrin receptor (17), but the details of how Rac1 controls cytoskeletal events associated with invasin-mediated uptake might be more complicated than the Cdc42 model. For instance, cofilin is inactivated via activation of the Rac1 effector PAK, inhibiting actin depolymerization (24). Furthermore, Rac1 may activate Arp 2/3 via a proposed Scar2–IRSp53 complex similar in nature to the Cdc42 pathway (25). Finally, Rac1 activation appears to stimulate the formation of PIP2 in some manner (26, 27). This can lead to inhibition of actin-severing activities, uncapping of barbed actin ends (28), activation of cytoskeleton-associated proteins (29), and cross talk with the Cdc42-dependent pathway for filament assembly (19).
The synthesis of PIP2 in the cell occurs primarily at the plasma membrane through the action of type I phosphatidylinositol 4-phosphate 5-kinases (PIP5Ks), of which there are three isoforms (α, β, and γ; 30). Regulation of the kinase seems to occur at two levels. First, it has been shown that Rac1-stimulated actin polymerization in platelet extracts is mediated through the action of PIP5K (27). Second, the small GTPase Arf6 stimulates PIP5K activity, inducing cytoskeletal rearrangements thought to result from the PIP2 product (31). Stimulation of PIP5K activity appears sufficient to initiate actin polymerization, as overproduction of PIP5K in the cell results in the Arp 2/3–dependent formation of actin comet tails initiating from intracellular vesicles (32). Interestingly, a constitutively active form of Arf6 is able to induce comet tail formation (33), but this occurs in the presence of antibodies directed against PIP2. Therefore, although Arf6 and PIP5K activities are linked to actin assembly, Arf6 presumably stimulates actin dynamics in both PIP2-dependent and -independent fashions.
Arf6 and PIP2 have been implicated in controlling phagocytic events. Constitutively active and GTP-binding defective forms of Arf6 inhibit Fcγ receptor–promoted uptake in macrophages (34, 35). Similarly, PIP2 and PIP5Kα have been found localized around IgG-coated erythrocytes during phagocytosis, which is mildly inhibited by a phosphatase specific for PIP2 (36), and a catalytically inactive form of the PIP5Kα isoform has been demonstrated to inhibit Fcγ phagocytosis (37). In this study, we demonstrate that Rac1-controlled uptake of a bacterial pathogen can be regulated by levels of PIP5K and Arf6 in the cell. Efficient uptake requires that these proteins and their overproduction bypasses the requirement for Rho family GTPases in integrin-promoted uptake.
Materials and Methods
Cell Culture, DNA Constructs, and Transfections.
Culture and transfection of COS1 cells were performed as previously described (17). The HA-tagged mammalian expression plasmids pcDNA PIP5Kα WT and pcDNA PIP5Kα D227A, encoding derivatives of the mouse PIP5Kα cDNA, were obtained from C. Carpenter (Beth Israel-Deaconess Hospital, Boston, MA). Plasmids containing derivatives of PIP5Kγ were obtained from R. Anderson (University of Wisconsin Medical School, Madison, WI). HA-tagged pcDNA-Arf6 was obtained from C. D'souza-Schorey (University of Notre Dame, Notre Dame, IN). The plasmids pEGFP-Akt-PH, pEGFP-PLCδPH, pEGFP-PLCδPH K32E, pEGFP-Lyn, and pEGFP-Lyn-phosphatase were provided by T. Meyer (Stanford University, Stanford, CA). pEGFP-Lyn encodes the 10–amino acid myristoylation/palmitoylation sequence from Lyn fused to EGFP. pEGFP-Lyn-phosphatase encodes a PIP2-specific 5′-phosphatase from yeast Inp54p linked to myristoylation/palmitoylation sequence from Lyn. A CFP version of Lyn-phosphatase was obtained from A. Jeromin (Mount Sinai Hospital, Toronto, Canada). pCGT-Rac1, pCGT-Rac1 V12, and pCGT-Rac1 T17 were obtained from J. Bliska (The State University of New York, Stony Brook, NY). The bacterial expression vector pYopE, which encodes YopE under ptac promoter, was obtained from J. Mecsas (Tufts University, Boston, MA). HA-tagged pcDNA-Arf6 N122I, described in Honda et al. (31), equivalent to the nucleotide-free N121I mutation in Ras, was generated using the Stratagene Quikchange site-directed mutagenesis kit, using pcDNA-Arf6 as the template. Construction of other plasmids are detailed in the Supplemental Materials and Methods, available at http://www.jem.org/cgi/content/full/jem.20021363/DC1.
For most assays, virulence plasmid-cured Y. pseudotuberculosis YPIII(P−) (38) was cultured as previously described (17). Derivatives of plasmid-harboring Y. pseudotuberculosis YP17 (YPIII(pYV+) yopT − yopE::kan yopH::cam) were grown on Luria Bertani plates at 26°C for 2 d. For YP17/pYopE, media were supplemented with 100 μg/ml ampicillin. 24 h before infection, a single colony was inoculated into Luria Bertani broth containing 100 μg/ml ampicillin and allowed to grow with shaking at 26°C overnight. Bacteria were then subcultured in media supplemented with 2.5 mM CaCl2 until OD600 = 0.1 and then shaken at 37°C for an additional 2 h to induce the Y. pseudotuberculosis type III secretion system (15). No isopropyl-β-d-thiogalactopyranoside was added to the bacteria to induce YopE expression because leaky expression of YopE from pYopE was sufficient to inhibit bacterial uptake.
Immunofluorescence Protection Assay of Bacterial Uptake.
Immunofluorescence-based bacterial uptake assays were performed as previously described (17). For 20-min infections, transfected COS1 cells plated on coverslips were incubated with bacteria at a multiplicity of infection (MOI) of 50:1 at 37°C, and the incubation was allowed to proceed for 20 min before three washes with PBS and fixation, as previously described (17). For 80-min infections, the procedure was identical, except that the MOI was reduced to 10:1. The MOI was calculated based on the number of COS1 cells plated before transfection, and assuming the bacterial titer = 5 × 108 bacteria/ml for a culture grown to A600 = 0.7. Because there was some loss of the cells during the transfection process, the MOI was effectively somewhat higher than that stated. After fixation, the coverslips were blocked (17) and uptake was assayed as described in Supplemental Materials and Methods, available at http://www.jem.org/cgi/content/full/jem.20021363/DC1. When untransfected cells were being assayed for uptake, they represented the cells on the same coverslip as the transfected cells. Data are expressed as the mean percentage obtained from three different coverslips. 80 transfected cells were examined on each coverslips. Significance of results was determined by unpaired Student's t test.
Assay for Colocalization of Mammalian Proteins with Cell-associated Bacteria.
Infected cells on coverslips were processed as previously described (17). To determine the localization of PIP5Kα on phagosomes, 50 partially internalized and/or 50 completely engulfed bacteria were scored for colocalization of PIP5Kα-green fluorescent protein (GFP). Data are the mean of determinations from three coverslips for each sample. Partially internalized bacteria were identified as bacteria that had circumferential staining with anti–Y. pseudotuberculosis after permeabilization (complete cascade blue staining), but only had a portion of the bacterial cell stained with anti–Y. pseudotuberculosis when probed before permeabilization (partial tetramethylrhodamine-5-isothiocyanate [TRITC] staining). Completely engulfed bacteria showed no detectable staining with anti–Y. pseudotuberculosis added before permeabilization. For fully internalized bacteria, a phagosome was determined to show colocalization with PIP5Kα-GFP if there were GFP staining encompassing the entire bacterium. For partially internalized bacteria, colocalization of PIP5Kα-GFP was scored as positive if the region of the bacterium that resisted probing with anti–Y. pseudotuberculosis in the absence of permeabilization showed GFP fluorescence. For assays in which only extracellular (surface bound) bacteria are analyzed, the bacteria are deemed to be extracellular only if they show circumferential staining with anti–Y. pseudotuberculosis when probed before permeabilization (complete TRITC staining with no protection of staining by the mammalian plasma membrane). In assays focusing on such extracellular bacteria, PIP5Kα-GFP exhibited colocalization if there was GFP staining around any portion of a bacterium. Complete absence of GFP staining around the entire bacterium was scored as negative. Data are represented as mean percentage ± SE from three coverslips.
Online Supplemental Material.
Fig. S1 shows efficient recruitment of PIP5Kα on phagosomal membrane during yersinia uptake. Consturuction of pEYFP-Akt-PH and pEYFP-PLCδ-PH are detailed in the Supplemental Materials and Methods. Supplemental Materials and Methods and Fig. S1 are available at http://www.jem.org/cgi/content/full/jem.20021363/DC1.
Results
Uptake of Y. pseudotuberculosis Is Associated with Efficient Recruitment of PIP5Kα onto Nascent Phagosomes.
To determine the subcellular localization of PIP5K during invasin-mediated uptake, COS1 cells transfected with a plasmid expressing an epitope-tagged version of PIP5Kα were incubated with a Y. pseudotuberculosis strain that requires invasin for cell association (refer to Materials and Methods). To distinguish between bacteria actively undergoing internalization and bacteria that have been completely internalized into an intracellular compartment, an assay requiring protection of the bacteria from antibody probing in the absence of membrane permeabilization was used (refer to Materials and Methods). Bacteria that are partially protected from antibody probing (Fig. 1 A and B, Yersinia that have red-colored poles) are defined as being partially internalized. We observed that PIP5Kα was recruited around partially formed phagosomes with almost 100% efficiency (Fig. 1 E). The enzyme persisted on the phagosome after bacterial internalization, although completely internalized Y. pseudotuberculosis showed reduced colocalization with PIP5Kα (Fig. 1 E).
Figure 1.
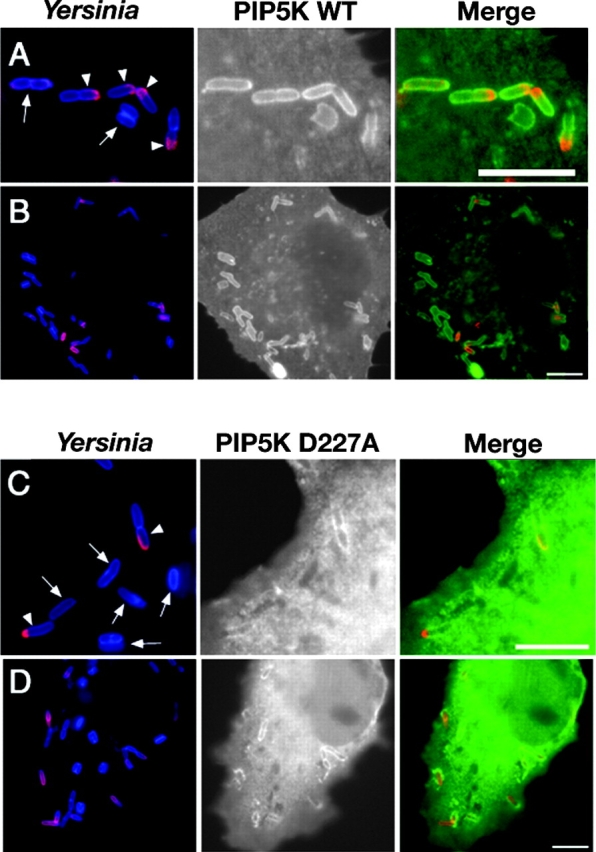

Efficient recruitment of PIP5Kα onto phagosomes containing Y. pseudotuberculosis. COS1 transfected with HA-tagged WT PIP5Kα (A and B) or the kinase-dead PIP5Kα D227A (C and D) were incubated with Y. pseudotuberculosis for 80 min. Immunofluorescence microscopy was performed to distinguish the extracellular portion (red) from the internalized portion (blue) of a single bacterium (refer to Materials and Methods). Arrows, fully internalized bacteria; arrowheads, partially internalized bacteria. (A–D) Left panels (Yersinia) contain merged images showing extracellular (red) portions of individual bacteria accessible to anti-Yersinia antibody before cell permeabilization. Note that completely engulfed bacteria show no red staining. Center panels show the localization of PIP5Kα derivatives. Right panels (Merge) contain merged images combining extracellular part of bacteria (red) and localization of PIP5Kα (green). Both PIP5Kα WT (A) and PIP5KαD227A (C) colocalize with nascent phagosomes containing partially internalized bacteria (arrowheads). Bars, 10 μm. (E) Comparison of the percentage of internalized or partially internalized bacteria (defined in Materials and Methods) staining positively for PIP5Kα WT or PIP5KαD227A. Data are mean ± SE from triplicate coverslips.
It appeared that the kinase activity of PIP5Kα enhanced the affinity of the protein for the Y. pseudotuberculosis phagosomes (Fig. 1 E), reminiscent of previous results (39). When COS1 cells harboring a kinase-dead variant of PIP5Kα (PIP5KαD227A) were challenged with Y. pseudotuberculosis, the protein associated as efficiently as the WT protein on a per phagosome basis (Fig. 1 E), but PIP5KαD227A was lost from a significant fraction of the internalized phagosomes (Fig. 1 E, 25% of phagosomes associated with PIP5KαD227A). Consistent with recent evidence that the γ isoform of PIP5K is associated with integrin-dependent signaling processes (41, 42), we found that transfected PIP5Kγ also colocalized about nascent phagosomes (see Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20021363/DC1). The intensity of colocalization of the γ isoform was significantly higher than for a fusion between the Lyn myristoylation domain and GFP, a marker of enhanced membrane thickness (see Supplemental Materials and Methods, available at http://www.jem.org/cgi/content/full/jem.20021363/DC1; P < 1.14E-07), arguing against fortuitous concentration of the kinase.
Overexpression of the kinase-dead PIP5Kα resulted in only a small reduction in uptake efficiency using the simple assay described above (unpublished data; P = 0.05), and showed much less dramatic effects than had been previously reported in regards to Fcγ receptor–mediated phagocytosis (37). As it has been reported that the related kinase-dead PIP5Kβ isoform has interfering effects on endocytosis (40) and that expression of derivatives of the γ isoform interfered with cell spreading (41, 42), we also analyzed kinase-dead versions of these isoforms. They showed no detectable interference in our assay system (unpublished data).
Localized and Transient Production of PIP2 on Nascent Phagosomes.
To determine the localization of PIP2 during bacterial uptake, Y. pseudotuberculosis was incubated with COS1 cells transfected with a construct expressing the phospholipase C δ pleckstrin homology domain (PLCδ-PH) tagged with GFP (43, 44). As was observed with PIP5Kα, PIP2 was concentrated on partially formed phagosomes (Fig. 2 A). To verify the specificity of the PIP2 visualization by GFP-PLCδ-PH, a GFP-PLCδ-PH K32E mutant that is defective in PIP2 binding was used. GFP-PLCδ-PH K32E did not colocalize with internalized bacteria at all, and only weakly with partially internalized bacteria (compare Fig. 2, B and C). This detection of PIP2 at bacterial adhesion sites was due to increased plasma membrane density of the lipid, rather than to simple accumulation of bulk membrane material around the bacterium. When a control marker for plasma membrane, Lyn-GFP, was compared with GFP-PLCδ-PH there was a clear enhancement of the signal for PIP2 relative to the plasma membrane marker. This was demonstrated by determining the ratio of average GFP pixel density at bacterial adhesion sites relative to sites on the membrane in which there were no adherent bacteria. The increased accumulation of PIP2 (2.3 ± 0.32 SEM, n = 22) was significantly higher than that for plasma membrane marker Lyn-GFP (2.0 ± 0.3 SEM, n = 21, P = 0.003). This result is very similar to what had been observed for accumulation of PIP2 around phagocytic cups during Fcγ receptor–mediated internalization of erythrocytes (36).
Figure 2.
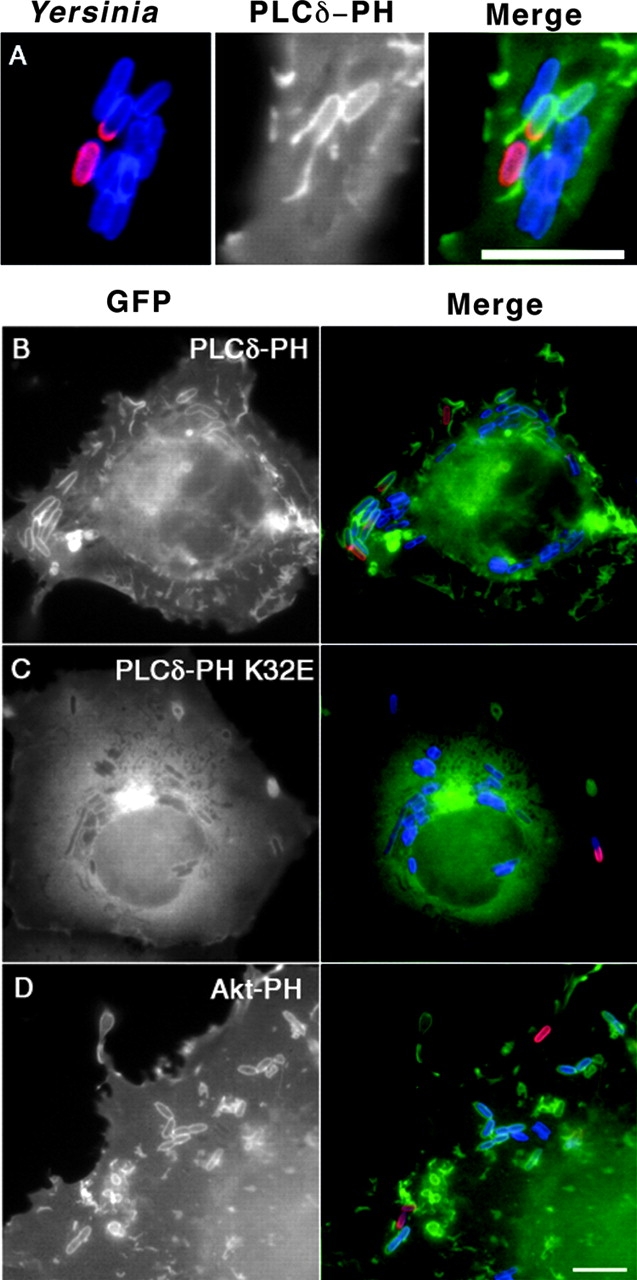

Transient localization of PIP2 on phagosomal membrane during Yersinia uptake. (A–D) COS1 cells transfected with GFP-PLCδ-PH (A and B), GFP-PLCδ-PH K32E (C), or GFP-Akt-PH (D) were incubated with Y. pseudotuberculosis (YPIII(P−)) for 20 min. Fluorescence channels corresponding to the extracellular part (red) and internalized part of bacteria (blue; refer to Materials and Methods) are merged on the left in A. The right panels (Merge; A–E) show merged images corresponding to exposed portions of bacteria (red), internalized portions of bacteria (blue), and GFP fusion protein (green). Bars, 10 μm. (E) Quantification of the percentage of phagosomes that colocalized with GFP-Akt-PH (□) or GFP-PLCδ-PH (▪). COS1 transfected with GFP-PLCδ-PH or GFP-Akt-PH were incubated with Y. pseudotuberculosis (YPIII(P−)) for 20 min, washed, and then incubated with bacteria-free medium for the indicated periods of time before fixation. Internalized bacteria, as determined by resistance to staining by anti–Y. pseudotuberculosis added before permeabilization, were then scored for GFP staining. Data are mean ± SE from triplicate coverslips.
PIP2 was rapidly lost from the fully internalized phagosomes, as had been observed previously for Fcγ receptor–mediated phagocytosis (Fig. 2 E; reference 36). To examine the turnover of PIP2 during bacterial uptake, we performed a pulse-chase experiment involving a 20-min infection (pulse) followed by a postwash incubation (chase) for up to 60 min. At all time points, half-internalized bacteria had intense levels of PIP2 (unpublished data). After 20 min of uptake and 0 min of chase, less than half of the enclosed phagosomes were PIP2 +, whereas after 60 min of chase, only 10% of the phagosomes were PIP2 + (Fig. 2 E).
To determine if the rapid loss of PIP2 from _Yersinia_-containing phagosome was specific to PIP2 or due to disruption of the integrity of phagosomal membrane, we repeated the pulse-chase infection protocol by using GFP-tagged PH domain from Akt (GFP-Akt-PH) to visualize the location of phosphoinositol-3,4,5-phosphate (PI(3,4,5)P)3 and PI(3,4)P2 in cells (Fig. 2 D). GFP-Akt-PH staining was observed on up to 75% of internalized Yersinia after 60 min of chase, indicating that the integrity of phagosomal membrane remained intact during the chase period (Fig. 2 E).
Uptake Efficiency of Y. pseudotuberculosis Is a Function of PIP2 Concentration in the Cell.
To determine if PIP2 plays a functional role during invasin-promoted uptake, the effect of lowering the cellular concentration of PIP2 on bacterial uptake was examined. To this end, COS1 cells were transfected with a plasma membrane–targeted PIP2-specific phosphatase, Lyn-CFP-Inp54p, which has been successfully used to reduce PIP2 levels (refer to Materials and Methods; 44). Expression of Lyn-CFP-phosphatase resulted in a 40% reduction of uptake efficiency compared with the control cells transfected with Lyn-CFP (Fig. 3 A; P = 0.001). In contrast, there was no significant effect of the phosphatase on the ability of bacteria to bind to cells (Fig. 3 A).
Figure 3.

PIP2-specific phosphatase partially inhibits Y. pseudotuberculosis uptake. (A) Blockage of uptake by degradation of PIP2. COS1 cells transfected with clones expressing either dominant interfering Rac1T17N, a membrane-targeted Lyn-CFP, or Lyn-CFP-Inp54p were incubated with Y. pseudotuberculosis (YPIII(P−)) for 20 min. The immunofluorescence protection assay was then performed (refer to Materials and Methods) to determine the percentage of cell-associated bacteria that remained extracellular relative to the total number of cell-associated bacteria (refer to Materials and Methods). Untransfected cells were used as control. Data represent mean ± SE from triplicate coverslips. (B) Lyn-CFP-Inp54p is specific and results in partial loss of PIP2 from Y. pseudotuberculosis phagosomes. COS1 cells cotransfected with YFP-PLCδ-PH or YFP-Akt-PH, and either Lyn-CFP or Lyn-CFP-Inp54p (phosphatase) were incubated with Y. pseudotuberculosis (YPIII(P−)) for 20 min. Extracellular bacteria were detected by rabbit polyclonal anti–Y. pseudotuberculosis antibody, followed by anti–rabbit conjugated with Texas Red antibody in the absence of permeabilization (refer to Materials and Methods). The presence of extracellular bacteria with membrane-associated PIP2 (YFP-PLCδ-PH) or PIP3 (YFP-Akt-PH), as visualized in a YFP filter, was scored in cells transfected with either Lyn-CFP or Lyn-CFP-Inp54p (phosphatase). Data are mean ± SE from triplicate coverslips.
The above result indicates the involvement of PIP2 in Yersinia uptake, but the reduction in uptake was not as severe as observed for cells transfected with the dominant interfering Rac1N17 derivative, which caused a 74% reduction relative to controls (Fig. 3 A; reference 17). The substantial level of uptake in the presence of the phosphatase might be due to a PIP2-independent uptake pathway, or the phosphatase might be inefficient at lowering PIP2 levels in the cell. To distinguish between these possibilities, the ability of PIP2 to be formed at the bacterial adhesion site before uptake was analyzed in cells transfected with the phosphatase (Fig. 3 B). This was assayed by introducing Y. pseudotuberculosis onto COS1 cells transfected with YFP-PLCδ-PH in the presence of either Lyn-CFP-phosphatase or a Lyn-CFP control, and determining the efficiency at which YFP-PLCδ-PH colocalized with bound bacteria. Cells transfected with the PIP2-phosphatase showed a 33% reduction relative to the control in colocalization of YFP-PLCδ-PH with bound bacteria (Fig. 3 B, %PLC-PH positive; P = 0.003). The effect of the phosphatase was specific, as it had no effect on the level of colocalization of 3-PI lipids (Fig. 3 B, %Akt-PH positive). Thus, the residual uptake seen in the PIP2-phosphatase–transfected cells can be explained by the fact that the levels of PIP2 remaining in such cells might be sufficient to support uptake.
Arf6 Localizes to the Nascent Phagosomes and Regulates Integrin-mediated Uptake.
The activity of PIP5K is stimulated in extracts by the GTP-bound form of Arf6 (31). Therefore, the effect of overproduction of Arf6 derivatives was tested next. Uptake was greatly enhanced by transfection of cells with a plasmid harboring Arf6, as the uptake efficiency in Arf6 WT-transfected cells (81 ± 2%, n = 3) was increased by 47% when compared with untransfected cells (55 ± 4%, n = 3; Fig. 4 A; P = 0.0002). The number of cell-associated WT Yersinia bound per COS1 cell, on the other hand, was identical in Arf6 WT-transfected cells and in untransfected cells in this experiment (Fig. 4 A). The activated form of Arf6, Arf6Q67L, had similar stimulatory effects on uptake (unpublished data). To determine if this stimulation were the result of WT Arf6-inducing nonspecific bacterial uptake, the Arf6 WT-transfected cells were challenged with Y. pseudotuberculosis invD911A, which allows low affinity binding of the bacteria to host cells without subsequent uptake. The transfectants were unable to internalize the bound bacteria, arguing against Arf6 causing nonspecific stimulation of uptake (see Fig. 6 J).
Figure 4.
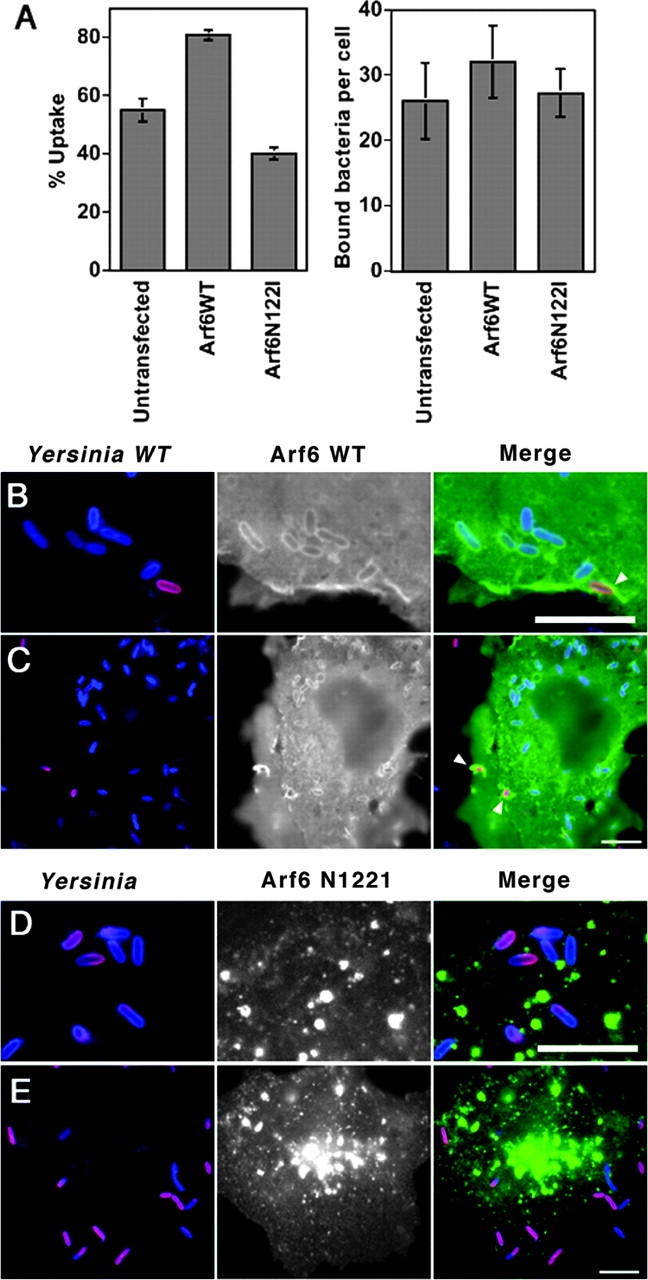
Involvement of Arf6 in invasin-mediated uptake of Yersinia. COS1 cells transfected with plasmids encoding HA-tagged WT Arf6 (B and C) or nucleotide-free Arf6 N122I (D and E) were challenged with Y. pseudotuberculosis (YPIII(P−)) for 20 min, fixed, and processed for the immunofluorescence protection assay using rabbit polyclonal anti-Yersinia and mouse monoclonal anti-HA antibodies (refer to Materials and Methods). (A) Effect of overexpressing WT or a nucleotide-free Arf6 on Yersinia uptake. Bacterial uptake and cell association were determined by immunofluorescence protection assay (refer to Materials and Methods). Data are mean ± SE from triplicate coverslips. (B and C) WT Arf6 localizes to phagosomes bearing Y. pseudotuberculosis. Localization of Arf6 around extracellular bacteria (arrowheads) is evident. (D and E). The nucleotide-free Arf6 N122I does not colocalize with phagosomes containing Y. pseudotuberculosis. Arf6 N122I shows a punctate pattern revealing intracellular aggregates. Bars, 10 μm.
Figure 6.
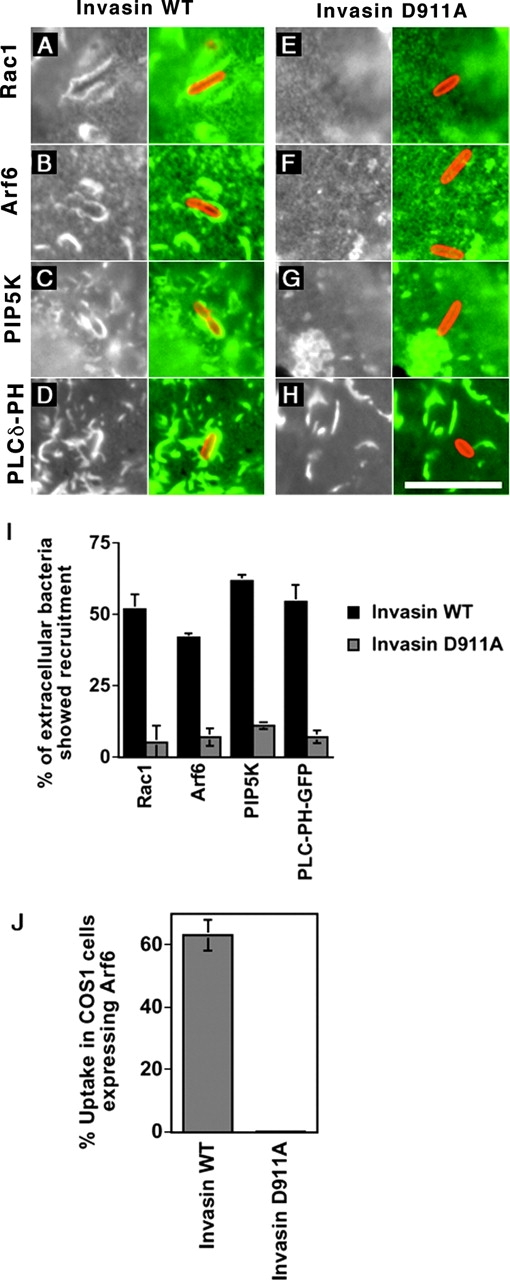
Recruitment of signaling molecules at the site of bacterial adhesion requires high affinity binding of bacterial invasin to β1 integrin receptors. (A–H) COS1 cells transfected with Rac1 (A and E), Arf6-HA (B and F), HA- PIP5Kα (C and G), or GFP-PLCδ-PH (D and H) were incubated for 20 min at 37°C with Y. pseudotuberculosis (YPIII(P−); A–D) or the invD911A mutant (E–H). Extracellular bacteria were detected by rabbit polyclonal anti–Y. pseudotuberculosis antibody, Rac1 by mouse monoclonal anti–Rac1 antibody, HA-PIP5Kα, or HA-Arf6 by mouse monoclonal anti–HA antibody. Bar, 10 μm. (I) Quantification of the percentage of extracellular bacteria expressing WT invasin (solid bars) or mutant D911A (shaded bars) staining positively for Rac1, Arf6, PIP5Kα, or GFP-PLCδ-PH. Data are mean ± SE from triplicate coverslips. (J). Effect of overexpressing Arf6 on the uptake of Y. pseudotuberculosis invD911A. COS1 cells transfected with an Arf6-expressing plasmid were incubated with YPIII(P−)invD911A for 30 min at 37°C. The cells were fixed and subjected to the immunofluorescence protection assay to determine the efficiency of bacterial internalization (refer to Materials and Methods).
To further examine the role of Arf6 in invasin-mediated uptake, the effect of the dominant interfering Arf6N122I mutant that assumes a guanine nucleotide-free state was investigated. Transfection of Arf6N122I caused a 27% decrease in uptake efficiency (40 ± 2%, n = 3) relative to untransfected cells (55 ± 4%, n = 3; Fig. 4 A; P = 0.002). The uptake efficiency in the presence of this derivative, therefore, was <50% of that observed in transfectants harboring the WT form of Arf6 (Fig. 4 A). Under these conditions, there were approximately six times as many extracellular bacteria in the Arf6N122I transfectants as were seen with the Arf6 WT transfectants (unpublished data). COS1 cells overproducing Arf6 N122I had about the same number of cell-associated bacteria as untransfected COS1 cells, arguing that altered bacterial association with the target cells could not explain the defective uptake exhibited by cells expressing this mutant (Fig. 4 A).
Consistent with Arf6 playing a functional role in invasin-mediated uptake, we observed that Arf6 WT (Fig. 4, B and C) efficiently colocalized with Y. pseudotuberculosis phagosomes. The dominant interfering Arf6N122I, however, localized to intracellular vesicles and was not associated with phagosomes (Fig. 4, D and E).
The Lowering of PIP2 Formation on Phagosomes by Dominant Interfering Forms of Arf6 and Rac1 Occurs via Different Mechanisms.
As both Arf6 and Rac1 are implicated in regulating the activity of PIP5K, the role of these proteins in controlling PIP2 production on nascent phagosomes was analyzed. COS1 cells transfected with GFP-PLCδ-PH together with Rac1 WT, Rac1 N17, Arf6 WT, or Arf6 N122I were challenged with Y. pseudotuberculosis for 20 min and localization of PIP2 around extracellularly bound bacteria was analyzed. Significantly fewer extracellular bacteria had PIP2 staining in cells transfected with Rac1N17 (41 ± 7%, n = 3) than cells transfected with Rac1 WT (63 ± 6%, n = 3; Fig. 5, A, B, and I ; P = 0.015). Similarly for Arf6, fewer bacterial adhesion sites stained positively for PIP2 in Arf6 N122I-transfected cells (53 ± 3%, n = 3) than Arf6 WT-transfected cells (71 ± 1%, n = 3; Fig. 5, C, D, and I; P = 0.0006). These results suggest that Rac1 T17N and Arf6 N122I interfere with the endogenous production of PIP2 at the phagosomal membrane.
Figure 5.
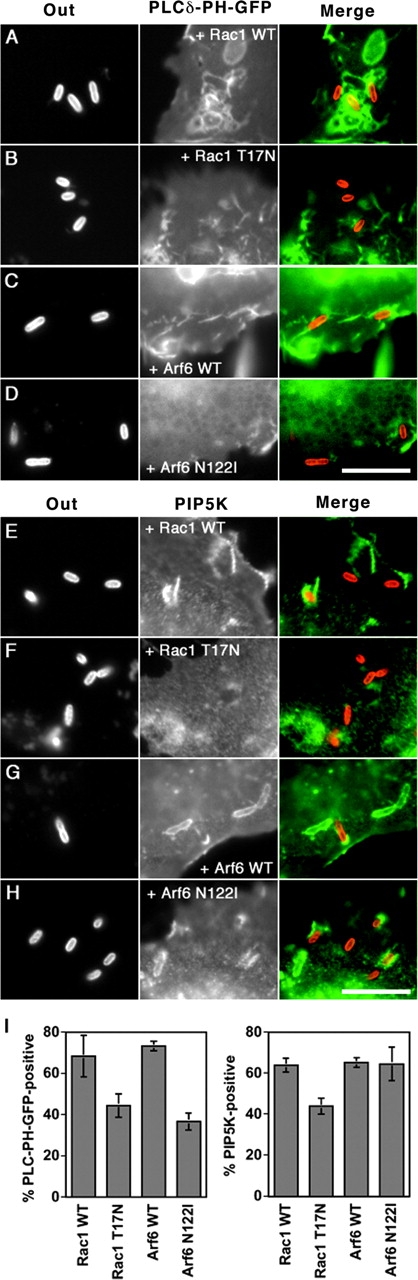
Rac1 and Arf6 control localized PIP2 production at nascent phagosome through distinct mechanisms. COS1 cells were cotransfected with GFP-tagged PLCδ-PH (A–D) or HA-tagged PIP5Kα (E–H) along with WT Rac1 (A and E), Rac1T17N (B and F), Arf6-HA (C and G), or Arf6N122I-HA (D and H). The cultures were then incubated at 37°C with Y. pseudotuberculosis (YPIII(P−)) for 20 min. Left panels of A–H (Out) display extracellular portion of bacteria, middle panels show the localization of GFP-PLCδ-PH (A–D) or PIP5Kα (E–H), and right panels of A–H (PLCδ-PH-GFP or PIP5Kα) are merged images of the left and middle panels. (A and B) A dominant interfering Rac1 causes reduced PIP2 colocalization at sites of bacterial adhesion. (C and D) Nucleotide-free Arf6 reduces PIP2 colocalization at sites of bacterial adhesion. (E and F) A dominant interfering Rac1 derivative inhibits the recruitment of PIP5Kα to sites of bacterial adhesion. (G and H) Nucleotide-free Arf6 derivative has no affect on PIP5Kα recruitment to bacterial adhesion sites. Bars, 10 μm. (I) Comparison of the percentage of extracellular bacteria that colocalized with GFP-PLCδ-PH or PIP5Kα in cells in the presence of WT or dominant interfering Rac1 or Arf6 forms. Data are mean ± SE from triplicate coverslips.
To understand how the dominant inhibitory forms interfered with PIP2 production, the recruitment of PIP5Kα onto phagosomal membranes surrounding extracellular bacteria was analyzed. Double transfectants of COS1 cells, in which cells harbored clones of PIP5Kα WT plus either Rac1 WT, Rac1T17N, Arf6 WT, or Arf6 N122I, were infected with Y. pseudotuberculosis for 20 min. Extracellular bacteria were then scored for the presence of PIP5Kα. Rac1T17N-transfected cells showed a reduction of PIP5Kα recruitment (44 ± 4%, n = 3) relative to Rac1WT-transfected cells (64 ± 4%, n = 3; Fig. 5, E, F, and I; P = 0.003). In contrast, the levels of PIP5Kα recruitment in Arf6 N122I-transfected cells (65 ± 8%, n = 3) and in Arf6 WT-transfected cells (65 ± 2%, n = 3) were identical (Fig. 5, G–I). These results indicate that Rac1 regulates the recruitment of PIP5Kα to the phagosome, whereas Arf6 controls the level of PIP5Kα activity of membrane-recruited kinase.
Recruitment of Rac1, PIP5Kα, Arf6, and PIP2 onto Nascent Phagosomes Requires High Affinity Binding to β1 Integrins.
As described above, the Y. pseudotuberculosis invD911A mutant that is defective for integrin binding adheres to host cells, but does not promote uptake (Fig. 6 J). To examine whether low affinity binding to integrins is sufficient to recruit factors involved in uptake, COS1 cells harboring clones expressing GFP-PLCδ-PH, PIP5Kα WT, Rac1 WT, or Arf6 WT were challenged for 20 min with Y. pseudotuberculosis inv + or invD911A. To compensate for the fact that bacteria expressing the invasinD911A remain mostly extracellular, potentially reducing the availability of factors normally associated with nascent phagosomes, we used an MOI of 10:1 for the mutant in this experiment, instead of 50:1 used for the WT. Even allowing for such a possibility, it was clear that although the WT invasin protein stimulated recruitment of Rac1 (Fig. 6, A and E), Arf6 (Fig. 6, B and F), PIP5Kα (Fig. 6, C and G), and PIP2 (Fig. 6, D and H) around extracellular bacteria, the invasin D911A mutant failed to do so (Fig. 6 I). Experiments in which both WT and mutant were infected at identical MOI gave results identical to those displayed in Fig. 6 I (unpublished data). These data indicate that high affinity binding of invasin to integrin receptors is necessary for the recruitment of Arf6, PIP5Kα, and PIP2 to the site of bacterial adhesion.
Bypass of Rac1 Function During Y. pseudotuberculosis by Arf6 and PIP5K.
Unlike the Y. pseudotuberculosis strain used in this study, the WT strain delivers the YopE Rho GTPase activating protein (GAP) into mammalian cells upon host cell contact (15, 16). YopE inactivates Rho, Rac1, and Cdc42 by stimulating their GTP hydrolysis activity and inhibiting bacterial uptake, probably by blocking Rac1 function (15). As both Arf6 and PIP5K are able to stimulate novel cytoskeletal events (32, 33, 45), these two proteins were overproduced to determine if they could bypass the loss of Rac1 activity that results from YopE deposition. As a positive control for bypass of YopE, cells were transfected with the constitutively active RacV12, which is insensitive to YopE, to determine the maximum level of uptake in the presence of inactivation of endogenous Rac1 (15).
A Y. pseudotuberculosis strain (YP17 p_yopE_) was grown under conditions that maximize expression of YopE (15) and used to challenge COS1 transfectants. Cells overexpressing WT Arf6 under these conditions (Fig. 7 ; 14.8 ± 2.4%, n = 3) supported bacterial uptake almost as efficiently as Rac1V12-transfected cells did (Fig. 7 A; 17.6 ± 3.2%, n = 3), whereas only minimal uptake was detected in untransfected cells (Fig. 7 A; 0.6 ± 0.3%, n = 3). As noted above for a different bacterial strain background, Arf6 also stimulated the uptake efficiency of bacteria not producing YopE and the magnitude of this effect was similar to what was observed for the constitutively active Rac1V12 (Fig. 7 B; reference 17). It should be noted that even in the case of Rac1V12, bypass of the uptake inhibition caused by YopE is far from complete (compare uptake levels in Fig. 7, A and B). Nevertheless, Arf6 overproduction is clearly as efficient as overproducing a target protein that is insensitive to YopE.
Figure 7.
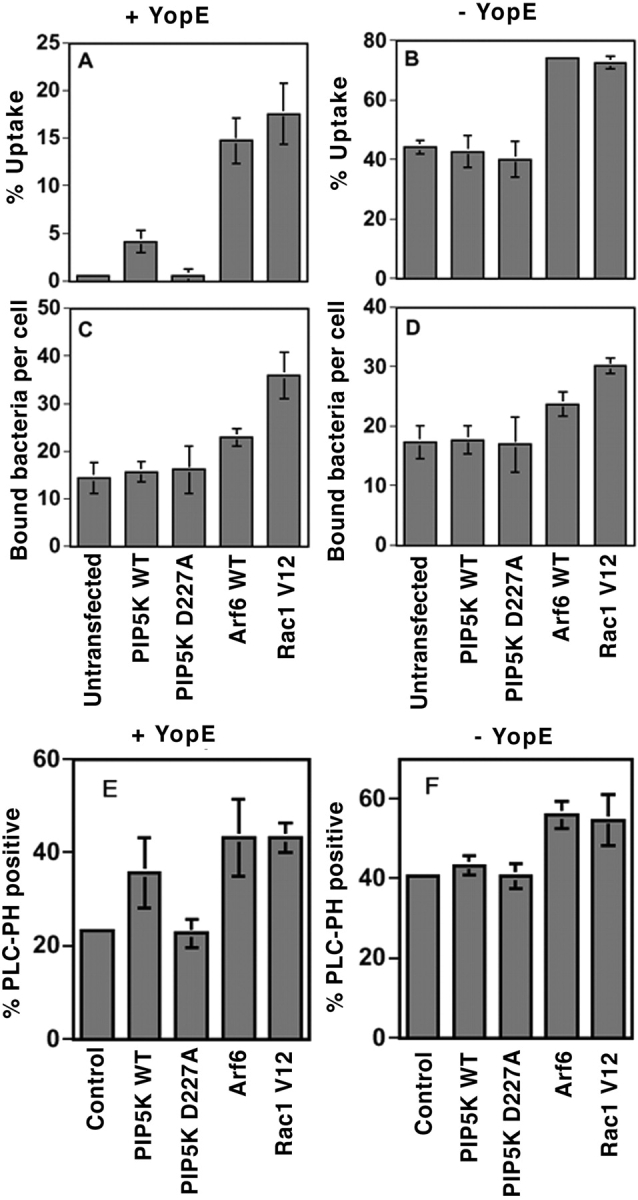
Bypass of Rac1 function by Arf6 and PIP5Kα. COS1 cells individually transfected with plasmids expressing HA-PIP5Kα WT, HA-PIP5Kα D227A, Arf6-HA, or Rac1V12 (A–D), or cotransfected with a plasmid encoding YFP-PLCδ-PH (E and F) were challenged at 37°C with YP17(p_tac-yopE_ +; A, C, and E) or YP17(yopH −, yopT −, yopE −; B, D, and F) for 30 min (A–D) or for 5 min (E and F). Untransfected cells were used as additional controls. The percentage of cell-associated bacteria that were internalized by cells (A and B) and the number of cell-associated bacteria per cell (C and D), and the percentage of extracellular bacteria that colocalized with YFP-PLCδ-PH were determined as described in Materials and Methods. Note that % Uptake as displayed in A and B are different scales. Data are mean ± SE from triplicate coverslips.
Transfectants harboring clones expressing PIP5Kα showed a behavior that was similar to that observed for the Arf6 transfectant, but the magnitude of the bypass was less dramatic. When challenged with Y. pseudotuberculosis yopE +, cells overexpressing WT PIP5Kα internalized approximately seven times as many bacteria as the untransfected control (Fig. 7 A; 4.2 ± 1.2% vs. 0.6 ± 0.3%). There was no increase in the number of bound bacteria (Fig. 7, C and D) and no such stimulation of uptake efficiency was observed in cells overexpressing the kinase-dead PIP5KαD227A mutant (Fig. 7 A; 0.6 ± 0.7%, n = 3), indicating that the kinase activity was responsible for the elevated uptake in PIP5Kα-expressing cells. The PIP5Kα-mediated stimulation of uptake, however, was only observed when Rac1 function was eliminated. Untransfected cells, as well as cells overproducing either WT or kinase-dead PIP5Kα had similar uptake levels when challenged with the isogenic Y. pseudotuberculosis strain that lacks YopE expression (Fig. 7 B; strain YP17). This indicates that PIP5K activity in the cell is limiting for uptake only when Rac1 function is eliminated.
As both overproduction of PIP5Kα and Arf6 resulted in enhanced uptake in the presence of YopE, we determined whether enhanced expression of these proteins also allowed corresponding increases in the formation of PIP2 after challenge with Y. pseudotuberculosis YopE+. To this end, Y. pseudotuberculosis strains were incubated for 5 min with COS1 cell monolayers that had been cotransfected with the plasmid encoding GFP-PLCδ-PH and each of the four plasmids used to demonstrate bypass of Rac1 function. Shorter incubation times were used than in previous experiments because cells challenged for 20 min with bacteria encoding YopE had rounded morphologies, making it difficult to score cells for PIP2 localization. There was a significant increase in the number of extracellular bacteria that stained positively for PIP2 in PIP5Kα-transfected cells (PIP5K, 36 ± 8%, n = 3; Fig. 7 E) when compared with either PIP5KαD227A-transfected cells (23 ± 3%, n = 3, P = 0.026; Fig. 7 E) or cells transfected with GFP-PLCδ-PH alone (control, 23 ± 1%, n = 3, P = 0.025; Fig. 7 E). A larger stimulation of PIP2 colocalization was observed in cells transfected with plasmids encoding Arf6 or Rac1V12 after challenge with Y. pseudotuberculosis YopE+, consistent with the more potent bypass that resulted from overproduction of these proteins compared with PIP5Kα (P = 0.007 relative to control; Fig. 7 E). In the absence of YopE, stimulation of PIP2 formation by PIP5Kα was not observed (Fig. 7 F), consistent with the lack of stimulation of bacterial uptake by PIP5Kα (Fig. 7 B). Therefore, in the presence of Rac1 inactivation, the increased uptake of bacteria that resulted from PIP5Kα overproduction was accompanied by enhanced PIP2 formation at the site of bacterial binding. In addition, conditions that resulted in Arf6 stimulation of bacterial uptake (Fig. 7, A and B) were also accompanied by increased PIP2 formation (Fig. 7, E and F).
Discussion
In this report, integrin-promoted bacterial uptake is used as a model for analyzing the internalization of a bacterial pathogen by mammalian cells. Evidence is provided that a PIP2-dependent pathway regulated by Arf6 is associated with internalization initiated by engagement of integrin receptors.
Recently, it has been hypothesized that PIP2 cannot provide the initial signal for actin polymerization because the concentration of this lipid in the plasma membrane is too high for it to play this role (46). Rather, it is proposed that PIP2 provides the information necessary to control the direction of actin polymerization relative to the plasma membrane (46). The results presented here do not argue against this model in any way, but they do point out that local concentrations of PIP2 in the plasma membrane may play an important regulatory role. PIP5Kα was rapidly recruited to the site of bacterial binding, presumably for the purpose of increasing the concentration of PIP2. In addition, partial depletion of PIP2 resulted in a proportional loss in uptake efficiency (Fig. 3). Although these results do not mean that production of PIP2 is the initiating signal for uptake, they do indicate that uptake may require rather high local concentrations of the lipid.
There are two possible reasons for PIP2 being concentrated at the site of bacterial uptake. First, this would allow the proper orientation, activation, and coalescence of a variety of PIP2-binding proteins involved in controlling actin dynamics (20, 27, 29, 47). The activity of PIP5K at the site of bacterial binding could facilitate interactions between PIP2-binding proteins and allow the concentrations of these factors to rise above a critical threshold level necessary for successful bacterial internalization. The second possibility is that PIP2 is a precursor of another signaling molecule that has to be concentrated at the site of bacterial binding to promote initiation of actin polymerization. Similar to the results in this report, Botelho et al. (36) have shown that in macrophages, PIP2 accumulates transiently around IgG-coated red blood cells before their phagocytosis. The authors associated the rapid loss of PIP2 during phagocytosis with an increase in diacylglyerol production due to phospholipase Cγ activity, and argue that products of phospholipase Cγ provide signals required for uptake. We similarly observed a rapid loss of PIP2 from the phagosomal membrane (Fig. 2), but it is possible that this may have resulted from the production of yet a third lipid signal, PI(3,4,5)P, which uses PIP2 as a precursor (Fig. 2 E). Among the PI3P products, PI(3,4,5)P may provide the unique signal necessary for integrin-mediated uptake.
We demonstrated that WT (Fig. 4) or activated Arf6 (unpublished data) stimulated integrin-mediated uptake, and that a nucleotide-free form of Arf6 (Arf6N122I) interfered with uptake. The stimulation observed is quite unusual and has not been observed in other phagocytic events. Phagocytic uptake by Fcγ receptor is regulated by Arf6 activity as well (34, 35), but it is much more sensitive to alterations in Arf6 activity than observed here. The GTP- and GDP-bound forms of Arf6 interfered with Fcγ-mediated phagocytosis in one report (35), and overexpression of the WT protein interfered in another study (34). One potential explanation for the contrast with invasin is that the work reported here involves a surface area that is considerably less than that analyzed in the previous work. In the studies on Fcγ receptor, phagocytosis of either erythrocytes or 3-μm beads was analyzed (surface areas of 100 and 28 μm2, respectively), whereas Y. pseudotuberculosis is a 1.5-μm rod (surface area = 3.4 μm2). As the phagocytosis of large particles is known to require acquisition of membrane from some intracellular sites (48, 49), it is conceivable that activated Arf6 could interfere with this event (50).
There is accumulating evidence that Arf6 regulates PIP2 concentrations in the cell (31, 50). A previous report indicated that the Arf6N122I mutant, predicted to be defective for nucleotide binding, blocked the translocation of PIP5K to membrane ruffles (31). We observed no such defect in recruitment of the kinase to surface-bound bacteria in the presence of this mutant (Fig. 5). Instead, this mutant caused a reduction in the number of bound bacteria having associated PIP2, implying that it interfered with the activation of the membrane-associated kinase and not its localization. A factor more likely to play a role in recruitment of PIP5K to the site of bacterial binding is Rac1, as the interfering Rac1N17 mutant reduced the efficiency of PIP5K recruitment to bound bacteria, with a resulting lowered efficiency of PIP2 formation at these sites. This is consistent with results regarding thrombin-induced membrane translocation of PIP5Kα in COS7 cells transfected with thrombin receptor (51). Taken together with our results on Arf6, we suggest a two-step model for PIP2 formation at sites of actin polymerization on nascent phagosomes. PIP5K is initially recruited by Rac1-GTP onto the plasma membrane, followed by subsequent activation of membrane-bound PIP5K by Arf6-GTP. A major caveat regarding this interpretation is that in these experiments, we are expressing an inhibitory protein that depresses both phagosome formation and recruitment of another molecule. As a result, we cannot easily determine whether lack of recruitment is due to lowered phagosome formation or due to the direct action of an inhibitor preventing a molecule from being recruited to the site of phagocytosis. It should be pointed out, however, that we can generate conditions in which PIP5K can be recruited to some sites of bacterial binding without colocalization of GFP-PLCδ-PH, the probe for PIP2 formation. This argues either that: (a) inhibition of phagosome formation does not necessarily result in lack of recruitment of molecules involved in uptake, or else (b) the formation of membrane invaginations that might be precursors to phagosome formation does not necessarily lead to formation of PIP2. In this particular case, therefore, some evidence exists for separation of effects.
One of the most striking results reported here is the ability of either overproduced Arf6 or PIP5Kα to overcome the uptake deficit caused by YopE deactivation of Rho family members (Fig. 7). The basis for this bypass is that both Arf6 and PIP5K appear to be capable of initiating actin polymerization events that, at least in the case of Arf6, may not require Rac1 and Cdc42 (33). Overproduction of either PIP5Kα or the activated Arf6Q67L form have been demonstrated to stimulate intracellular vesicle motility and associated actin comet formation (32, 33), although it is unclear whether this transport is part of some natural cellular process. Furthermore, bypass of Rac1 function by overproduction of Arf6 has been observed previously, as cytoskeletal rearrangements induced by constitutively active Arf6Q67L can occur in the presence of the interfering Rac1N17 protein (45).
There are two possible explanations for why overproduction of Arf6 leads to more efficient bypass of Rho family function than does overproduction of PIP5Kα: either activation of PIP5K is not the only role played by Arf6 in uptake, or overproduced PIP5Kα is not sufficiently active under conditions of YopE deposition to allow high level uptake to occur. There are several arguments in favor of the explanation that Arf6 plays roles other than simply activating PIP5K. Particle movement induced by Arf6Q67L occurs in the presence of antibodies to PIP2, indicating that cytoskeletal events promoted by Arf6 do not necessarily require PIP5K activity (33). Furthermore, Arf6Q67L is known to bind to proteins other than PIP5K, such as POR1/arfaptin 2, a protein that appears to connect the Arf6 and Rac1 signaling pathways (45). There might be alternative effectors, as well, that directly translate Arf6 signals to the cytoskeleton. Finally, overproduction of PIP5K is fully capable of inducing organelle movement without any additional activators being added to the cell, although it is not clear if organelle movements in this case can take place under conditions of Rho family depletion (32).
The PIP2 bypass of a blockage of Rac1 function has been observed in vitro. The addition of PIP2 to platelet extracts can overcome a block in actin filament formation resulting from the presence of GDPβS in Rac1-containing extracts (27), but such a bypass had not been observed previously in intact cells. As the entire process of bacterial internalization is presumably more complicated than the elongation of actin filaments mediated by PIP2, there must be some factor that is able to coordinate the movement of the plasma membrane around the bacterium and the polymerization of actin in the absence of Rac1 function. Perhaps that factor is Arf6. This protein may act as both an upstream activator of PIP5K as well as a downstream regulator, necessary to coordinate membrane traffic and cytoskeletal events involved in closure of the phagosome. This is consistent with the model that Arf6 exchange factors bind PIP2 (52), allowing activation of Arf6 at sites of high PIP2 concentrations. Any potential roles for Arf6 activity located downstream of PIP2 formation will clearly require further investigation, and are of great interest in regards to fully elucidating integrin-mediated bacterial uptake.
Acknowledgments
We thank Drs. Christopher Carpenter, Tobias Meyer, Richard Anderson, and Joan Mecsas for plasmids used in this work (see Materials and Methods). We thank Drs. Isabelle Derre, Zhao-Qing Luo, Penelope Barnes, Susan VanRheenen, and Marion Shonn for review of the manuscript.
This work was supported by the Howard Hughes Medical Institute (HHMI), MERIT award R37 AI23538 from the National Institute of Allergy and Infectious Diseases, and Program Project Award grant P30DK34928 from the National Institute of Diabetes & Kidney Diseases. R.R. Isberg is an Investigator of HHMI.
Abbreviations used in this paper: GFP, green fluorescent protein; MOI, multiplicity of infection; PI(3,4,5)P, phosphoinositol-3,4,5-phosphate; PIP2, phosphoinositol-4,5-bisphosphate; PIP5K, type I phosphatidylinositol 4-phosphate 5-kinase; PLCδ-PH, phospholipase C δ pleckstrin homology domain.
The online version of this article contains supplemental material.
References
- 1.Clark, M.A., B.H. Hirst, and M.A. Jepson. 1998. M-cell surface β1 integrin expression and invasin-mediated targeting of Yersinia pseudotuberculosis to mouse Peyer's patch M cells. Infect. Immun. 66:1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heesemann, J., K. Gaede, and I.B. Autenrieth. 1993. Experimental Yersinia enterocolitica infection in rodents: a model for human yersiniosis. APMIS. 101:417–429. [PubMed] [Google Scholar]
- 3.Marra, A., and R.R. Isberg. 1997. Invasin-dependent and invasin-independent pathways for translocation of Yersinia pseudotuberculosis across the Peyer's patch intestinal epithelium. Infect. Immun. 65:3412–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pepe, J.C., and V.L. Miller. 1993. Yersinia enterocolitica invasin: a primary role in the initiation of infection. Proc. Natl. Acad. Sci. USA. 90:6473–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isberg, R.R., and J.M. Leong. 1990. Multiple β1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell. 60:861–871. [DOI] [PubMed] [Google Scholar]
- 6.Eble, J.A., K.W. Wucherpfennig, L. Gauthier, P. Dersch, E. Krukonis, R.R. Isberg, and M.E. Hemler. 1998. Recombinant soluble human α3β1 integrin: purification, processing, regulation, and specific binding to laminin-5 and invasin in a mutually exclusive manner. Biochemistry. 37:10945–10955. [DOI] [PubMed] [Google Scholar]
- 7.Tran Van Nhieu, G., and R.R. Isberg. 1991. The Yersinia pseudotuberculosis invasin protein and human fibronectin bind to mutually exclusive sites on the α5 β1 integrin receptor. J. Biol. Chem. 266:24367–24375. [PubMed] [Google Scholar]
- 8.Dersch, P., and R.R. Isberg. 1999. A region of the Yersinia pseudotuberculosis invasin protein enhances integrin-mediated uptake into mammalian cells and promotes self-association. EMBO J. 18:1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran Van Nhieu, G., E.S. Krukonis, A.A. Reszka, A.F. Horwitz, and R.R. Isberg. 1996. Mutations in the cytoplasmic domain of the integrin β1 chain indicate a role for endocytosis factors in bacterial internalization. J. Biol. Chem. 271:7665–7672. [DOI] [PubMed] [Google Scholar]
- 10.Gustavsson, A., A. Armulik, C. Brakebusch, R. Fassler, S. Johansson, and M. Fallman. 2002. Role of the β1-integrin cytoplasmic tail in mediating invasin-promoted internalization of Yersinia. J. Cell Sci. 115:2669–2678. [DOI] [PubMed] [Google Scholar]
- 11.Rosenshine, I., V. Duronio, and B.B. Finlay. 1992. Tyrosine protein kinase inhibitors block invasin-promoted bacterial uptake by epithelial cells. Infect. Immun. 60:2211–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Persson, C., N. Carballeira, H. Wolf-Watz, and M. Fallman. 1997. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J. 16:2307–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alrutz, M.A., and R.R. Isberg. 1998. Involvement of focal adhesion kinase in invasin-mediated uptake. Proc. Natl. Acad. Sci. USA. 95:13658–13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruce-Staskal, P.J., C.L. Weidow, J.J. Gibson, and A.H. Bouton. 2002. Cas, Fak and Pyk2 function in diverse signaling cascades to promote Yersinia uptake. J. Cell Sci. 115:2689–2700. [DOI] [PubMed] [Google Scholar]
- 15.Black, D.S., and J.B. Bliska. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 37:515–527. [DOI] [PubMed] [Google Scholar]
- 16.Von Pawel-Rammingen, U., M.V. Telepnev, G. Schmidt, K. Aktories, H. Wolf-Watz, and R. Rosqvist. 2000. GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: a mechanism for disruption of actin microfilament structure. Mol. Microbiol. 36:737–748. [DOI] [PubMed] [Google Scholar]
- 17.Alrutz, M.A., A. Srivastava, K.W. Wong, C. D'Souza-Schorey, M. Tang, L.E. Ch'Ng, S.B. Snapper, and R.R. Isberg. 2001. Efficient uptake of Yersinia pseudotuberculosis via integrin receptors involves a Rac1-Arp 2/3 pathway that bypasses N-WASP function. Mol. Microbiol. 42:689–703. [DOI] [PubMed] [Google Scholar]
- 18.Hall, A., and C.D. Nobes. 2000. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355:965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohatgi, R., P. Nollau, H.Y. Ho, M.W. Kirschner, and B.J. Mayer. 2001. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J. Biol. Chem. 276:26448–26452. [DOI] [PubMed] [Google Scholar]
- 20.Rohatgi, R., H.Y. Ho, and M.W. Kirschner. 2000. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 150:1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higgs, H.N., and T.D. Pollard. 2000. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J. Cell Biol. 150:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yarar, D., W. To, A. Abo, and M.D. Welch. 1999. The Wiskott-Aldrich syndrome protein directs actin-based motility by stimulating actin nucleation with the Arp2/3 complex. Curr. Biol. 9:555–558. [DOI] [PubMed] [Google Scholar]
- 23.Higgs, H.N., and T.D. Pollard. 2001. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70:649–676. [DOI] [PubMed] [Google Scholar]
- 24.Edwards, D.C., L.C. Sanders, G.M. Bokoch, and G.N. Gill. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253–259. [DOI] [PubMed] [Google Scholar]
- 25.Miki, H., H. Yamaguchi, S. Suetsugu, and T. Takenawa. 2000. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 408:732–735. [DOI] [PubMed] [Google Scholar]
- 26.Tolias, K.F., J.H. Hartwig, H. Ishihara, Y. Shibasaki, L.C. Cantley, and C.L. Carpenter. 2000. Type Iα phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr. Biol. 10:153–156. [DOI] [PubMed] [Google Scholar]
- 27.Hartwig, J.H., G.M. Bokoch, C.L. Carpenter, P.A. Janmey, L.A. Taylor, A. Toker, and T.P. Stossel. 1995. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 82:643–653. [DOI] [PubMed] [Google Scholar]
- 28.Lin, K.M., E. Wenegieme, P.J. Lu, C.S. Chen, and H.L. Yin. 1997. Gelsolin binding to phosphatidylinositol 4,5-bisphosphate is modulated by calcium and pH. J. Biol. Chem. 272:20443–20450. [DOI] [PubMed] [Google Scholar]
- 29.Gilmore, A.P., and K. Burridge. 1996. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature. 381:531–535. [DOI] [PubMed] [Google Scholar]
- 30.Ishihara, H., Y. Shibasaki, N. Kizuki, T. Wada, Y. Yazaki, T. Asano, and Y. Oka. 1998. Type I phosphatidylinositol-4-phosphate 5-kinases. Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J. Biol. Chem. 273:8741–8748. [DOI] [PubMed] [Google Scholar]
- 31.Honda, A., M. Nogami, T. Yokozeki, M. Yamazaki, H. Nakamura, H. Watanabe, K. Kawamoto, K. Nakayama, A.J. Morris, M.A. Frohman, et al. 1999. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 99:521–532. [DOI] [PubMed] [Google Scholar]
- 32.Rozelle, A.L., L.M. Machesky, M. Yamamoto, M.H. Driessens, R.H. Insall, M.G. Roth, K. Luby-Phelps, G. Marriott, A. Hall, and H.L. Yin. 2000. Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr. Biol. 10:311–320. [DOI] [PubMed] [Google Scholar]
- 33.Schafer, D.A., C. D'Souza-Schorey, and J.A. Cooper. 2000. Actin assembly at membranes controlled by ARF6. Traffic. 1:892–903. [DOI] [PubMed] [Google Scholar]
- 34.Uchida, H., A. Kondo, Y. Yoshimura, Y. Mazaki, and H. Sabe. 2001. PAG3/Papalpha/KIAA0400, a GTPase-activating protein for ADP-ribosylation factor (ARF), regulates ARF6 in Fcγ receptor–mediated phagocytosis of macrophages. J. Exp. Med. 193:955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, Q., D. Cox, C.C. Tseng, J.G. Donaldson, and S. Greenberg. 1998. A requirement for ARF6 in Fcγ receptor-mediated phagocytosis in macrophages. J. Biol. Chem. 273:19977–19981. [DOI] [PubMed] [Google Scholar]
- 36.Botelho, R.J., M. Teruel, R. Dierckman, R. Anderson, A. Wells, J.D. York, T. Meyer, and S. Grinstein. 2000. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 151:1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coppolino, M.G., R. Dierckman, J. Loijens, R.F. Collins, M. Pouladi, J. Jongstra-Bilen, A.D. Schreiber, W.S. Trimble, R. Anderson, and S. Grinstein. 2002. Inhibition of phosphatidylinositol-4-phosphate 5-kinase Ialpha impairs localized actin remodeling and suppresses phagocytosis. J. Biol. Chem. 277:43849–43857. [DOI] [PubMed] [Google Scholar]
- 38.Bolin, I., L. Norlander, and H. Wolf-Watz. 1982. Temperature-inducible outer membrane protein of Yersinia pseudotuberculosis and Yersinia enterocolitica is associated with the virulence plasmid. Infect. Immun. 37:506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shyng, S.L., A. Barbieri, A. Gumusboga, C. Cukras, L. Pike, J.N. Davis, P.D. Stahl, and C.G. Nichols. 2000. Modulation of nucleotide sensitivity of ATP-sensitive potassium channels by phosphatidylinositol-4-phosphate 5-kinase. Proc. Natl. Acad. Sci. USA. 97:937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbieri, M.A., C.M. Heath, E.M. Peters, A. Wells, J.N. Davis, and P.D. Stahl. 2001. Phosphatidylinositol-4-phosphate 5-kinase-1β is essential for epidermal growth factor receptor-mediated endocytosis. J. Biol. Chem. 276:47212–47216. [DOI] [PubMed] [Google Scholar]
- 41.Di Paolo, G., L. Pellegrini, K. Letinic, G. Cestra, R. Zoncu, S. Voronov, S. Chang, J. Guo, M.R. Wenk, and P. De Camilli. 2002. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 420:85–89. [DOI] [PubMed] [Google Scholar]
- 42.Ling, K., R.L. Doughman, A.J. Firestone, M.W. Bunce, and R.A. Anderson. 2002. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 420:89–93. [DOI] [PubMed] [Google Scholar]
- 43.Stauffer, T.P., S. Ahn, and T. Meyer. 1998. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 8:343–346. [DOI] [PubMed] [Google Scholar]
- 44.Raucher, D., T. Stauffer, W. Chen, K. Shen, S. Guo, J.D. York, M.P. Sheetz, and T. Meyer. 2000. Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell. 100:221–228. [DOI] [PubMed] [Google Scholar]
- 45.D'Souza-Schorey, C., R.L. Boshans, M. McDonough, P.D. Stahl, and L. Van Aelst. 1997. A role for POR1, a Rac1-interacting protein, in ARF6-mediated cytoskeletal rearrangements. EMBO J. 16:5445–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Insall, R.H., and O.D. Weiner. 2001. PIP3, PIP2, and cell movement–similar messages, different meanings? Dev. Cell. 1:743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glogauer, M., J. Hartwig, and T. Stossel. 2000. Two pathways through Cdc42 couple the N-formyl receptor to actin nucleation in permeabilized human neutrophils. J. Cell Biol. 150:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cox, D., D.J. Lee, B.M. Dale, J. Calafat, and S. Greenberg. 2000. A Rab11-containing rapidly recycling compartment in macrophages that promotes phagocytosis. Proc. Natl. Acad. Sci. USA. 97:680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hackam, D.J., O.D. Rotstein, C. Sjolin, A.D. Schreiber, W.S. Trimble, and S. Grinstein. 1998. v-SNARE-dependent secretion is required for phagocytosis. Proc. Natl. Acad. Sci. USA. 95:11691–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown, F.D., A.L. Rozelle, H.L. Yin, T. Balla, and J.G. Donaldson. 2001. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol. 154:1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chatah, N.E., and C.S. Abrams. 2001. G-protein-coupled receptor activation induces the membrane translocation and activation of phosphatidylinositol-4-phosphate 5-kinase I α by a Rac- and Rho-dependent pathway. J. Biol. Chem. 276:34059–34065. [DOI] [PubMed] [Google Scholar]
- 52.Derrien, V., C. Couillault, M. Franco, S. Martineau, P. Montcourrier, R. Houlgatte, and P. Chavrier. 2002. A conserved C-terminal domain of EFA6-family ARF6-guanine nucleotide exchange factors induces lengthening of microvilli-like membrane protrusions. J. Cell Sci. 115:2867–2879. [DOI] [PubMed] [Google Scholar]