DNA-dependent Protein Kinase Activity Is Not Required for Immunoglobulin Class Switching (original) (raw)
Abstract
Class switch recombination (CSR), similar to V(D)J recombination, is thought to involve DNA double strand breaks and repair by the nonhomologous end–joining pathway. A key component of this pathway is DNA-dependent protein kinase (DNA-PK), consisting of a catalytic subunit (DNA-PKcs) and a DNA-binding heterodimer (Ku70/80). To test whether DNA-PKcs activity is essential for CSR, we examined whether IgM+ B cells from scid mice with site-directed H and L chain transgenes were able to undergo CSR. Although B cells from these mice were shown to lack DNA-PKcs activity, they were able to switch from IgM to IgG or IgA with close to the same efficiency as B cells from control transgenic and nontransgenic scid/+ mice, heterozygous for the scid mutation. We conclude that CSR, unlike V(D)J recombination, can readily occur in the absence of DNA-PKcs activity. We suggest nonhomologous end joining may not be the (primary or only) mechanism used to repair DNA breaks during CSR.
Keywords: Ig transgenes, scid B cells, nonhomologous end joining, B cell anergy
Introduction
Immunoglobulin H chain genes undergo two kinds of programmed genomic rearrangement. The first is referred to as V(D)J recombination (1). In this process, separate and distinct gene elements (V, D, and J) are assembled into coding segments (VDJ) for the variable regions of H chains. The second kind of rearrangement is referred to as class switch recombination (CSR)* (2). By means of this process, a VDJ gene segment that is initially expressed with the most upstream H chain constant (C) region gene (Cμ) is recombined with a downstream CH gene (e.g., Cγ, Cɛ, or Cα), resulting in the production of different Ig classes (e.g., IgG, IgE, or IgA). In this way, the effector function of a given antibody molecule is changed without affecting its antigen specificity.
CSR appears to result in DNA double strand breaks at switch region sites targeted for recombination (3, 4). The ends of broken DNA molecules are thought to recombine by a nonhomologous end joining (NHEJ) mechanism (for reviews see references 5, 6), similar to that used in the repair of DNA double strand breaks resulting from the initiation of V(D)J recombination or induced by ionizing radiation or chemical agents (for reviews see references 7, 8). Several proteins are known to play a critical role in NHEJ, including DNA-PK, a multiprotein complex consisting of a DNA-binding heterodimer, Ku70/80, and a large catalytic subunit, DNA-PKcs (9–12). Results from a number of studies suggest that DNA-PK is essential for CSR. Two groups have shown that insertion of VDJH and VJL coding sequences into the H and L chain loci of mice lacking Ku70 or Ku80 (Ku70−/− or Ku80−/− mice) allows B cells to develop, but these B cells are unable to undergo CSR when stimulated in vitro with reagents that induce this process (13, 14). Evidence for defective CSR has also been reported for scid pre-B cells (15) that lack DNA-PKcs activity (16, 17) and in B cells of DNA-PKcs−/− mice with a rearranged VDJH and VJλ transgene (tg; reference 18). All of these results have been cited as evidence that the mechanism of CSR is dependent on DNA-PK and involves NHEJ. However, this inference is not without some caveats. For example, B cells of Ku-deficient H/L transgenic mice show impaired proliferation (13) and this may account, to a large extent, for the inability of these cells to class switch. Also, B cells lacking DNA-PKcs were found to class switch to IgG1 normally but appeared unable to class switch to IgG2a, IgG2b, or IgG3 (18). The implication is that IgM–IgG1 switching is mechanistically distinct from that of IgM–IgG2 or IgM–IgG3. This is difficult to reconcile with the known structural similarity of different γ switch regions (19–22) and a presumed common CSR mechanism for all isotypes.
Contrary to current thinking, we report here evidence that DNA-PK activity is not required for CSR. We have found that DNA-PKcs–deficient scid B cells are not only able to class switch from IgM to IgG1, but also to IgG2a, IgG2b, IgG3, IgA, and IgE. We used scid mice with the VDJH and VJκ coding sequence of the 3H9 (23) and Vκ8 (24) tgs, each appropriately site-directed (sd) into an H and L(κ) chain allele, respectively (25, 26). Although 3H9sdVκ8sd scid mice were found to generate above normal numbers of IgM+ B cells, there was no evidence of significant class switching as they lacked IgG, IgA, and IgE. However, reconstitution of 3H9sdVκ8sd scid mice with T cells resulted in normal levels of serum IgG and IgA and significant levels of IgE, indicating that the B cells in these mice could be activated to undergo class switching in the presence of T cells and naturally occurring antigens. To assess the effect of the scid mutation on the efficiency of class switching, splenic B cells from 3H9sdVκ8sd scid mice and from control transgenic and nontransgenic scid/+ mice were stimulated in vitro to undergo class switching. Cell cultures were tested for all of the following: (1) transcripts resulting from recombination between the μ and γ1, γ2b, γ3, and α switch regions; (2) frequency of IgG1+, IgG2b+, IgG3+, and IgA+ cells; and (3) for levels of the corresponding isotypes, including IgE, in the culture medium. Our overall results indicate that the efficiency of class switching in scid transgenic B cells is close to that of control scid/+ B cells.
Materials and Methods
Mice.
C.B-17 scid mice (scid mice) with the VDJH and VJκ coding sequence of the 3H9 (23) and Vκ8 (24) tgs appropriately inserted into an H and L(κ) allele, respectively, were obtained by selective crossing of these tgs into the scid genetic background using previously established lines of 3H9sd and Vκ8sd BALB/c mice (25, 26). Mice heterozygous for the scid mutation (scid/+) and bearing the 3H9sd tg and the nonsd Vκ8 tg (24) were used as transgenic controls. We also used nontransgenic scid/+ mice as an additional control. All H/L chain transgenic scid and scid/+ mice used in this work were hemizygous for their tgs and can be formally designated as 3H9sd/+, Vκ8sd/+, scid/scid mice and 3H9sd/+, Vκ8/+, scid/+ mice. We will simply refer to these mice as 3H9sdVκ8sd scid and 3H9sdVκ8 scid/+ mice. In some experiments, 3H9sdVκ8sd scid mice were intravenously injected with a suspension of bone marrow cells and thymocytes (or thymocytes alone) from mice with deleted JH loci (JH−/−; reference 27). The strain background of the JH−/− mice used was BALB/c. These mice, here simply referred to as JH−/− mice, were obtained from Taconic. The absence of detectable B cells and serum Ig in JH−/− mice was confirmed by flow cytometry and ELISA, respectively. Mice with disrupted DNA-PKcs alleles (DNA-PK−/− mice; reference 28) were provided by G. Taccioli, Boston University School of Medicine, Boston, MA. All mice used in this work were between 9 and 17 wk of age and were maintained as specific pathogen-free mice in the Laboratory Animal Facility of the Fox Chase Cancer Center.
Cells.
Spleen cell suspensions were treated with 0.165 M NH4Cl-Tris to eliminate erythrocytes, washed, passed through a Nytex sterile nylon screen (Tetko), and resuspended in culture medium (RPMI-1640 containing 10% FCS, 5 × 10−5 M 2ME, 2 mM glutamine, and 10 mM Hepes and gentamycin at 50 μg/ml). For isotype switch analysis, spleen cells were cultured for 2 or 4 d in culture medium containing 50 μg/ml Salmonella typhimurium LPS (Sigma-Aldrich) with or without IL4 (50 or 100 ng/ml) or 1 ng/ml TGFβ (R & D Systems). Cells were deposited in 6-well plates at cell concentrations of 0.25 × 106 or 0.75 × 106 cells/ml. Bone marrow and thymus cell suspensions from JH−/− mice were prepared in the manner described previously (29).
Flow Cytometry.
Cells were stained for the B cell marker, CD45(B220), using anti-B220 (RA3–6B2) conjugated with allophycocyanin (30) and for surface IgM, IgG1, IgG2b, IgG3, or IgA using FITC-conjugated anti-IgM (331.1), anti-IgG1 (A85.1), anti-IgG2b (R12–3), anti-IgG3 (R40–82), or anti-IgA (C10–3) (BD PharMingen). The staining of cells was performed as described by Hardy (31). In some experiments, cells were stained for B220 and CD3, using anti-CD3 (500A2) conjugated with phycoerythrin (BD PharMingen). B220+IgG+, B220+IgA+, and B220−CD3+ cells were enumerated by multiparameter flow cytometry using a FACS® VantageSE flow cytometer (Becton Dickinson). To enrich for splenic B cells, we stained cells with allophycocyanin-conjugated anti-B220 and then sorted for B220+ cells using the FACS® VantageSE. Forward and light-angle scatter gates were set to exclude nonlymphoid cells. Dead cells were identified by propidium iodide staining and excluded from analysis. All analyses of flow cytometric data were performed using the FlowJo software package (TreeStar).
Reverse Transcriptase–PCR.
Total RNA and cDNA were prepared from 2-d spleen cell cultures by applying the methodology described by Kinoshita et al. (32). Briefly, total RNA was extracted using TRIzol (Invitrogen) according to manufacturer's instructions, and cDNA was synthesized with Superscript II (Invitrogen) using 2 μg of total RNA and 1 μg of Oligo(dT)12–18 Primer (Invitrogen) in a 20-μl reaction volume (one twentieth of which served as a template for subsequent RT-PCR amplification in a 25-μl reaction volume). Post-switch transcripts of different intronic (I) exons spliced to the 5′ Cμ exon, representing the excised circular DNA of different switching events, were PCR-amplified with the oligonucleotide primers described by Kinoshita et al. (32) using the following modified conditions: 33 cycles of PCR were used to amplify Iγ1-Cμ transcripts; Iγ2b-Cμ and Iγ3-Cμ transcripts were amplified after an initial denaturing step of 95°C for 6 min followed by 35 cycles of PCR (94°C for 30 s, 58°C for 1 min) using Faststart Taq DNA polymerase (Roche) with 5× G-C rich solution in the presence of 2.5 mM Mg2+. I promoter–generated transcripts representing nonexcised rearranged DNA of a class switch event (Iμ-CH transcripts) were amplified using the oligonucleotide primer pairs used by Muramatsu et al. (33) with the following PCR conditions: Iμ-Cγ3 transcripts were amplified after an initial denaturing step of 95°C for 9 min followed by 30 cycles of PCR (94°C for 30 s, 58°C for 1 min) by using AmpliTaq Gold (Perkin-Elmer) in the presence of 2.5 mM Mg2+; and for Iμ-Cα, Iμ-Cγ1, and Iμ-Cγ2b transcripts, an initial denaturing step of 95°C for 5 min followed by 30 cycles of PCR (95°C for 30 s, 58°C for 30 s, 72°C for 40 s) using Faststart Taq DNA polymerase (Roche) in the presence of 2.5 mM Mg2+. β2-microglobulin (β2M) primers (MB522, 5′-GAATGGGAAGCCGAACATACTGAACTG-3′; MB523, 5′-TGCTGATCACATGTCTCGATCC-3′) were used to amplify β2M transcripts (255 bp) to serve as an internal control for input cDNA. Conditions for β2M transcript amplification were as follows: an initial denaturing step of 95°C for 9 min followed by 20 cycles of PCR (94°C for 30 s, 58°C for 1 min) using AmpliTaq Gold (Perkin-Elmer) in the presence of 2.5 mM Mg2+. All PCR reactions were run on a programmable thermal control machine (model PTC-100; MJ Research, Inc.).
Southern Blot Analysis
RT-PCR–amplified products were separated by electrophoresis in agarose gels (1.6%), transferred to Nytran Supercharge nylon transfer membrane (Schleicher & Schuell) and detected by γ-32P-labeled oligonucleotide probes. (In some experiments [see Fig. 4], the β2M transcripts were visualized by ethidium bromide staining of the gel.) The oligonucleotide probes for circle transcripts, CμP (32), Iμ-CH transcripts, (MB554, 5′-AATGTATGGTTGTGGCTTCTGCCACCCAT CC-3′, nucleotides 4067–4098 of the Iμ region; GenBank accession no., J00440), and β2M transcripts (MB522) were labeled using a T4 Polynucleotide Kinase Kit (Invitrogen). All blots were prehybridized in a solution of 6×SSC, 0.1% SDS, 5× Denhardt's solution, and 100 μg/ml salmon sperm DNA at 60°C (58°C for β2M) for 5 h; hybridized overnight (12–15 h) at 60°C (58°C for β2M) in a solution of 6×SSC, 0.75% SDS, 5× Denhardt's solution, and 100 μg/ml salmon sperm DNA; and washed in a solution of 5× SSC and 0.1% SDS three times for 5 min at room temperature and one time for 5 min at 60°C (58°C for β2M). The membranes were exposed to a PhosphorImaging plate for quantification of bound γ-32P-labeled probes using a FujiX-BioImaging Analyzer.
Figure 4.

Detection of I promoter–generated post-switch (circle) transcripts in stimulated 3H9sdVk8sd scid spleen cells. PCR-amplified Iγ3-Cμ, Iγ1-Cμ, Iγ2b-Cμ, and Iα-Cμ transcripts in cDNA from 2-d cell cultures stimulated with LPS, LPS/IL4, and LPS/TGFβ, respectively, were readily detectable by Southern blot analysis of gel-separated PCR products. The transcripts are seen in the plus (+) lane for each genotype of mouse tested: nontransgenic scid/+ (s/+), 3H9sdVκ8 scid/+ (tg s/+), and 3H9sdVκ8sd scid (tg s/s). Post-switch circle transcripts were not detectable in nonstimulated cells taken directly from the animal (minus [−] lanes; except for the barely detectable Iγ3-Cμ transcripts in the s/+ controls). β2M, visualized by ethidium bromide staining of the gel, served as an internal control for cDNA input.
DNA-PKcs Activity.
Early B lineage cells from fetal liver and/or adult bone marrow of scid, scid/+, and DNA-PKcs−/− mice were transformed by the Abelson murine leukemia virus (A-MuLV), as described previously (34). Nuclear extracts from A-MuLV–transformed cells (20 × 106 cells), LPS/IL4-stimulated splenic B cells (20 × 106 cells), and HeLa cells (5 × 106 cells) were prepared using the Dignam method as modified by Andrews and Faller (35). Extracts were diluted 1:1 in buffer and incubated with preswollen DNA–cellulose beads (Sigma-Aldrich) to bind Ku and DNA-PKcs, as described by Woo et al. (36). To test for DNA-PKcs activity, we used the SignaTECT DNA-PK assay kit (Promega) in accordance with the manufacturer's instructions. Briefly, after the beads were incubated with nuclear extract, they were washed and resuspended in dilution buffer (12 μl) along with enzyme substrate (biotinylated p53 peptide) and γ-[32P]ATP. After a 5-min incubation, the kinase reaction was terminated and reaction mixtures were spotted onto prenumbered squares of a streptavidin-coated membrane. The membrane was washed and exposed to a PhosphorImaging plate for quantitation of bound γ-32P-labeled peptide using a FujiX-Bio Imaging Analyzer.
Serological Assays.
Mouse sera and supernatants of LPS/IL4-stimulated cell cultures were assayed for secreted IgE by ELISA using an OptEIA mouse IgE kit as directed by the manufacturer (BD PharMingen). ELISA was also used to measure the concentrations of IgM, IgG1, IgG2a, IgG2b, IgG3, and IgA in mouse sera or tissue culture media. For this, we used the same buffers and solutions as described in the above mouse IgE kit and purified mAbs specific for IgM, IgG1, IgG2a, IgG2b, IgG3, or IgA (BD PharMingen). The ELISA protocol in all experiments was as follows: the wells of 96-well plates (Nunc-Immuno™ plates) were coated with a given mAb (capture antibody), washed, and blocked with PBS containing 10% FCS. Serial dilutions of mouse sera or tissue culture media were added to the wells, and after a 2-h incubation and subsequent washing of the wells, a biotinylated version of the capture antibody (referred to as the detection antibody) was added together with avidin–horseradish peroxidase conjugate. After a 30-min incubation, the wells were washed and substrate (tetramethylbenzidine and hydrogen peroxide) was added. 30 min later, the optical density readings were taken at 450 nm using a Multiskan Ascent reader (Thermo Labsystems). Readings were compared with a standard curve of optical densities obtained by using serial dilutions of the appropriate affinity-purified mouse Ig (IgM, IgG1, IgG2a, IgG2b, IgG3, IgA, or IgE).
Results
Effect of scid Mutation on Ig Class Switching In Vivo.
Given the earlier findings of Xu et al. (37), indicating the presence of partially activated B cells in T cell–deficient 3H9sdVκ8 RAG−/− mice, it seemed likely that a similar B cell population might be present in T cell–deficient 3H9sdVκ8sd scid mice, and that this B cell population might be stimulated to differentiate and undergo class switching in the presence of T cells. Therefore, 3H9sdVκ8sd scid mice were injected intravenously with a mixture of bone marrow (3.0 × 106 cells) and thymocytes (2.0 × 106 cells) from JH−/− mice to provide a source of T cells in the absence of contaminating donor B cells. Results for one of three such experiments are illustrated in Figs. 1 B and 2. At 5 wk after injection, 3H9sdVκ8sd scid recipients were found to contain splenic T cells (Fig. 1 B), and normal to above normal concentrations of serum IgG and IgA and significant concentrations of IgE (Fig. 2) . Note that IgE concentrations in T cell–reconstituted 3H9sdVκ8sd scid mice were highly variable and ranged from 43 to 2,741 ng/ml; IgE concentrations in four age-matched scid/+ controls were somewhat higher and less variable. Before injection, 3H9sdVκ8sd scid mice contained <10 μg/ml of IgG and IgA and <20 ng/ml of IgE (Fig. 2), despite having above normal numbers of B220+IgM+ splenic B cells (Fig. 1 A and Table I). Most 3H9sdVκ8sd scid mice also lacked IgM (<10 μg/ml; unpublished data). As discussed later, the lack of serum Ig in 3H9sdVκ8sd scid mice is consistent with previous reports indicating that B cells expressing the 3H9Vκ8 antibody are inactive when these cells arise in nonautoimmune strains of mice, such as BALB/c or C.B-17 (23, 37, 38).
Figure 1.

Flow cytometric analysis of 3H9sdVk8sd scid spleen for B and T cells. (A). Contour plots show B220 versus IgM staining on spleen cells from two 3H9sdVκ8sd scid mice and from a nontransgenic scid/+ and scid mouse. The numbers below the small boxes correspond to the percentage of B220+IgM+ cells. (B). Contour plots show B220 versus CD3 staining of spleen cells from two 3H9sdVκ8sd scid mice and from two 3H9sdVκ8sd scid mice that were injected 5 wk earlier with a mixture of 3.0 × 106 bone marrow cells and 2.0 × 106 thymocytes from JH−/− mice (3H9sdVκ8sd scid + T). The numbers above the small boxes correspond to the percentage of B220−CD3+ cells.
Figure 2.

Induction of IgG, IgA, and IgE production in 3H9sdVk8sd scid mice reconstituted with T cells. 3H9sdVκ8sd scid mice were injected intravenously with a mixture of 3 × 106 bone marrow cells and 2 × 106 thymocytes from JH−/− donor mice. The concentration of total IgG (μg/ml) in each of eight individual 3H9sdVκ8sd scid recipients is indicated at day 1, and week 2, 4, and 5 after cell injection. IgA concentrations are shown for week 4 and 5; no IgA (<10 μg/ml) was detectable at day 1 and 14 after cell injection. The small inserted graph shows the serum IgE concentrations (ng/ml) in the eight 3H9sdVκ8sd scid mice before and after being injected with 3 × 106 bone marrow cells and 2 × 106 thymocytes from JH−/− donor mice. Normal concentrations of IgG, IgA and IgE in individual C.B-17 scid/+ mice are indicated with closed triangles. Flow cytometry was used to confirm that 3H9sdVκ8sd scid recipients contained T cells (Fig. 1 B) and that the JH−/− donor mice lacked B cells (not depicted). Similar results to those shown were obtained when 3H9sdVκ8sd scid mice were injected with 5 × 106 JH−/− thymocytes alone.
Table I.
Spleen Cellularity and Percentage of Splenic B Cells inscid (s/s), scid/+ (s/+), 3H9sdV_κ_8sd s/s, and 3H9sdV_κ_8 s/+ Micea
| Genotype | No. ofmice | No. of spleencells × 10−6 | Percent B220+IgM+ spleen cells |
|---|---|---|---|
| s/s | 6 | 3.5 ± 3.3 | <0.5 |
| 3H9sdVκ8sd s/s | 12 | 89 ± 14 | 83 ± 11 |
| 3H9sdVκ8 s/+ | 6 | 39 ± 8 | 34 ± 3 |
| s/+ | 9 | 107 ± 30 | 49 ± 10 |
As indicated in Table II, half of the 3H9sdVκ8sd scid recipients depicted in Fig. 2 contained above normal concentrations of all four IgG isotypes (IgG1, IgG2a, IgG2b, and IgG3); the remaining half showed marked imbalances in isotype representation, possibly reflecting a low multiplicity of IgG-producing clones. All of the recipients contained IgG of the tg allotype (IgGa), and none were found to contain IgG of the endogenous (C.B-17) allotype (IgGb; unpublished data), which is consistent with the known ability of the tgs to inhibit endogenous H (and L) chain gene rearrangement (23, 30) and the severe impairment of VH-DJH rearrangement in scid mice (39). We interpret the results of Table II and Figs. 1 B and 2 to indicate that IgM+ transgenic B cells in 3H9sdVκ8sd scid mice can be readily activated in the presence of T cells and naturally occurring antigen to differentiate and class switch to different Ig isotypes. Thus, we see no evidence for a severe impairment of Ig class switching in scid B cells using a biologically relevant system.
Table II.
Concentration of IgG1, IgG2a, IgG2b, IgG3 and Total IgG in Serum of T Cell-reconstituted 3H9sdV_κ_8sd scid Micea
| Mouse | IgG1 | IgG2a | IgG2b | IgG3 | Total IgG |
|---|---|---|---|---|---|
| μg/ml | μg/ml | μg/ml | μg/ml | μg/ml | |
| 26 | 489 | 474 | 915 | 617 | 2,495 |
| 30 | 670 | 133 | 317 | 664 | 1,784 |
| 29 | 531 | 107 | 234 | 839 | 1,711 |
| 27 | 634 | 128 | 146 | 424 | 1,332 |
| 31 | 585 | 42 | 34 | 695 | 1,356 |
| 28 | 314 | 439 | 29 | 106 | 888 |
| 33 | 350 | 16 | 306 | 42 | 714 |
| 32 | 360 | 62 | <2 | 124 | 547 |
Effect of scid Mutation on Ig Class Switching In Vitro.
The relative efficiency of class switching in 3H9sdVκ8sd scid mice could not be inferred from the preceding results because there is no normal control in these experiments. Moreover, even in a biologically relevant system such as 3H9sdVκ8sd scid mice, strong antigenic selection of transgenic B cells (or other complex variables) could potentially mask a significant impairment of class switching. Therefore, we compared the ability of 3H9sdVκ8sd scid B cells to undergo class switching in vitro relative to B cells of control transgenic and nontransgenic scid/+ mice.
Molecular Analysis.
To assess the relative efficiency with which scid B cells initiate and complete CSR, splenic B cells of 3H9sdVκ8sd scid, 3H9sdVκ8 scid/+, and nontransgenic scid/+ mice were stimulated with B cell mitogen (LPS) and cytokine (IL4 or TGFβ) or with LPS alone. Cell cultures were then analyzed 2 d later for the presence of I promoter–generated transcripts (40) resulting from recombination between the μ and γ3, γ1, γ2b, or α switch regions. The transcripts were detected by RT-PCR using the protocol described previously (32, 33). Post-switch transcripts from both nonexcised and excised rearranged DNA were amplified (Fig. 3) ; i.e., those generated from the Iμ promoter spliced to the 5′ exon of a rearranged CH gene (e.g., Iμ-Cγ3 transcripts) and those generated from the I promoter of a rearranged CH gene spliced to the 5′ exon of the Cμ gene (e.g., Iγ3-Cμ transcripts). The latter transcripts are referred to as circle transcripts because they derive from excised circular DNA (32).
Figure 3.

Illustration of intronic (I) promoter–generated transcripts associated with Ig class switching. Cartoon depicts targeting of μ and γ3 switch (S) regions for recombination, intermediate products (within brackets) and recombined products with the two types of I-generated post-switch transcripts: Iμ-Cγ3 and Iγ3-Cμ (circle) transcripts (see Results for further details).
As shown in Fig. 4, I γ3-Cμ, Iγ1-Cμ, Iγ2b-Cμ, and Iα-Cμ circle transcripts were detected in LPS-stimulated 3H9sdVκ8sd scid and scid/+ control spleen cells, cultured with the appropriate cytokine. Importantly, circle transcripts were not detectable in ex vivo (nonstimulated) 3H9sdVκ8sd scid spleen cells. Thus, the observed circle transcripts reflect recombination events initiated in vitro. In Fig. 5 , we show titration results for PCR amplification of post-switch transcripts in serially diluted cDNA from 3H9sdVκ8sd scid and 3H9sdVκ8 scid/+ cells stimulated with LPS, LPS/IL4, or LPS/TGFβ. The intensity of hybridizing signal associated with post-switch transcripts from both excised and nonexcised rearranged DNA correlated with the amount of input cDNA in the internal control (β2M). Quantitation of the amount of γ-32P-labeled probe hybridizing to a given post-switch transcript, normalized against the internal control, showed the abundance of post-switch transcripts in 3H9sdVκ8sd scid to be comparable to (or approximately two- to threefold less than) the 3H9sdVκ8 scid/+ cell controls, depending on the particular transcripts being assayed (Fig. 5 legend). Allowing for a twofold greater frequency of B cells in 3H9sdVκ8sd scid than in 3H9sdVκ8 scid/+ at the start of culture (Table I), we conclude from these results that the efficiency of class switching in scid transgenic B cells is within two to fivefold of that in scid/+ transgenic B cells.
Figure 5.

Relative abundance of I promoter–generated post-switch transcripts in stimulated 3H9sdVκ8sd scid and 3H9sdVκ**8 scid/**+ spleen cells. Post-switch transcripts of excised (e.g., Iγ3-Cμ) and nonexcised (e.g., Iμ-Cγ3) rearranged DNA were PCR amplified from threefold serial dilutions of cDNA obtained from 3H9sdVκ8sd scid (tg s/s) and 3H9sdVκ8 scid/+ (tg s/+) control cells stimulated for 2 d with LPS, LPS/IL4, or LPS/TGFβ. Post-switch transcripts and those of the internal control (β2M) were detected by Southern blot analysis of gel-separated PCR products. PhosphorImaging of the transfer membrane (Materials and Methods) showed the relative amount of a given post-switch transcript at each serial dilution to be proportional to the amount of β2M transcript, the control for amount of input cDNA. The ratio of the amount of probe hybridizing to a given post-switch transcript/β2M transcript in 3H9sdVκ8sd scid cells divided by that of the 3H9sdVκ8 scid/+ control gave the following mean values: 2.2, 0.47, and 2.1 for Iγ3-Cμ, Iγ1-Cμ, and Iγ2b-Cμ circle transcripts (Iα-Cμ circle transcripts were not titrated); and 0.56, 0.89, 0.36, and 0.31 for Iμ-Cγ3, Iμ-Cγ1, Iμ-Cγ2b, and Iμ-Cα transcripts, respectively.
Cellular Analysis.
To evaluate the effect of the scid mutation on the generation of IgG- and IgA-expressing B cells in vitro, spleen cells from individual 3H9sdVκ8sd scid, 3H9sdVκ8 scid/+, and nontransgenic scid/+ mice were separately stimulated in parallel with LPS and LPS/IL4 or with LPS and LPS/TGFβ. To compensate for the high percentage of B cells in 3H9sdVκ8sd scid spleen (Table I), 3H9sdVκ8 scid/+ and nontransgenic scid/+ control cell cultures were initiated with FACS®-sorted B220+ splenic cells. Alternatively, 3H9sdVκ8sd scid spleen cells were mixed with an equal number of spleen cells from JH−/− mice so that scid cell cultures were initiated with a similar number of splenic B cells as control scid/+ cell cultures. Cell cultures were analyzed at day 4 by flow cytometry for Ig surface–positive (Ig+) B cells of different isotypes. Gates were set to include only brightly stained B220+Ig+ cells.
As illustrated in Fig. 6 A, 4-d cultures of 3H9sdVκ8sd scid spleen cells stimulated with LPS generally showed only a two- to threefold lower percentage (∼0.65%) of B220+IgG3+ cells than control 3H9sdVκ8 scid/+ (1.2%) and nontransgenic scid/+ (2%) cell cultures initiated with sorted B220+ splenic cells. When 3H9sdVκ8sd scid spleen cells and 3H9sdVκ8 scid/+ sorted B220+ splenic cells were stimulated with LPS/IL4, we found 4-d cultures to contain comparable percentages of B220+IgG1+ cells (8–10%; Fig. 6 A) as opposed to <0.12% of IgG1-expressing cells in cultures stimulated with LPS alone (unpublished data). Nontransgenic scid/+ controls stimulated with LPS/IL4 generally contained 12–18% B220+IgG1+ cells. In cultures of 3H9sdVκ8sd scid spleen cells stimulated with LPS/TGFβ, we detected IgG2b+ and IgA+ cells. The frequency of IgG2b+ scid cells (∼1.5%) was within twofold that of the 3H9sdVκ8 scid/+ control (2.3%), as illustrated in Fig. 6 B. The nontransgenic scid/+ control in this experiment showed an unusually low percentage of IgG2b+ cells. LPS/TGFβ-stimulated cultures of 3H9sdVκ8sd scid spleen cells also contained B220+IgA+ cells at a frequency within threefold of that in the 3H9sdVκ8 scid/+ controls (Fig. 6 B, bottom row). Although the frequency of B220+IgA+ cells was extremely low (∼0.2–0.4%), no B220+IgA+ cells (<0.02%) were detected in scid or scid/+ cell cultures stimulated with LPS alone (unpublished data).
Figure 6.
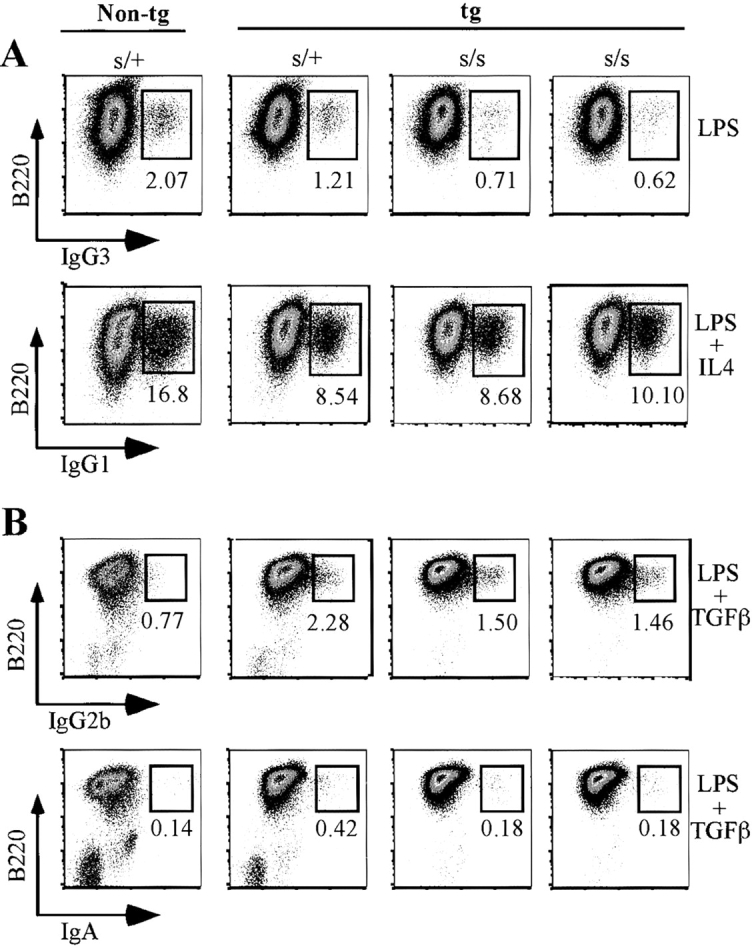
Generation of IgG- and IgA-expressing B cells in cultures of 3H9sdVκ8sd scid spleen cells. (A) Spleen cells (0.5 × 106 cells) from two individual 3H9sdVκ8sd scid (tg s/s) mice and sorted B220+ splenic cells from a 3H9sdVκ8 scid/+ (tg s/+) and nontg scid/+ (s/+) control mouse were each plated separately at 0.25 × 106 cells/ml in 6-well plates in the presence of LPS or LPS/IL4. After 4 d, each cell culture was analyzed by flow cytometry for the presence of B220+IgG3+ and B220+IgG1+ cells, respectively. The numbers at the bottom of the small boxes correspond to the percentage of IgG3+ or IgG1+ cells. (B) Spleen cells from two individual 3H9sdVκ8sd scid (tg s/s) mice were each mixed with an equal number of JH−/− spleen cells and plated separately at a total cell concentration of 0.75 × 106 cells/ml to give the same approximate number of B cells as plated in the tg s/+ and s/+ control cell cultures, containing 0.75 × 106 spleen cells/ml (based on results of Table I). Cells were cultured in the presence of LPS/TGFβ for 4 d, and then each cell culture was analyzed by flow cytometry for the presence of B220+IgG2b+ and B220+IgA+ cells. The numbers at the bottom of the small boxes correspond to the percentage of IgG2b+ or IgA+ cells. The results shown in A and B are representative of four independent experiments. Data are displayed as pseudo-color plots printed in black and white.
To compare the extent of cell proliferation in 3H9sdVκ8sd scid and control cell cultures, cell counts were made at day 1, 2, 3 and 4 of culture. Viability remained high (>90%) for all cell cultures at day 1, 2, and 3 (unpublished data) and then dropped below 80% at day 4 with scid cultures often showing somewhat lower viability than the controls. As illustrated in Table III for LPS/IL4-stimulated B cells, cell viability was generally 65–75% at day 4 with most cultures showing a four- to eightfold increase in cell concentration. Note that the scid cell cultures in experiment A showed a lower percentage of viable cells than the scid/+ controls but still contained as many viable cells as the controls. Thus, in this experiment, the scid cells appear to have proliferated to a greater extent than the scid/+ controls. Note also that in each experiment, the level of secreted IgG1 in the culture medium correlated with the yield of IgG1+ cells. We also tested for the presence of secreted IgE because the concentration of IL4 used in these experiments was sufficiently high (100 ng/ml) to expect some class switching to IgE (41, 42). As shown in Table III, low levels of IgE were detectable in three of four scid/+ cell cultures but not in any of the scid cell cultures.
Table III.
Yield of Viable Cells, IgG+ Cells, and Secreted IgG1 (and IgE) in 4-d Cultures of LPS/IL4-stimulated B Cellsa
| Exp | Genotype | Percentviability | Cells/ml× 10−6 | PercentIgG+ cells | [IgG1] | [IgE] |
|---|---|---|---|---|---|---|
| ng/ml | ng/ml | |||||
| A | s/+ | 73 | 2.2 | 18.0 | 242 | 8 |
| tg s/+ | 76 | 2.2 | 10.1 | 85 | <1 | |
| tg s/s | 58 | 2.1 | 9.2 | 97 | <1 | |
| tg s/s | 52 | 2.5 | 10.0 | 108 | <1 | |
| B | s/+ | 77 | 1.8 | 12.6 | 723 | 48 |
| tg s/+ | 68 | 1.7 | 10.2 | 587 | 20 | |
| tg s/s | 78 | 1.1 | 7.7 | 312 | <1 | |
| tg s/s | 62 | 1.8 | 8.9 | 305 | <1 |
Serological Analysis.
To test for serologic evidence of impaired Ig class switching, we compared the kinetics of Ig production (secretion) in spleen cell cultures of appropriately stimulated 3H9sdVκ8sd scid and 3H9sdVκ8 scid/+ cells. As illustrated in Fig. 7 , the concentration of IgG1 in culture supernatants of LPS/IL4-stimulated scid cells increased between day 3 and 5 from ∼20 to a range of 70–215 ng/ml. In the scid/+ control cell cultures, there was a greater increase of IgG1, from ∼20 to a range of 400–500 ng/ml. Supernatants from the same cell cultures were also tested for IgE. As indicated, IgE was not detectable in the scid cell cultures, but low amounts of IgE (15–25 ng/ml) were detectable in the scid/+ control cultures. In both LPS-stimulated scid and scid/+ control cell cultures, IgG2a concentrations increased from <5 ng/ml at day 3 to a range of 65–72 ng/ml at day 4 and appeared to plateau in the range of 72–111 ng/ml at day 5 (Fig. 7). Secreted IgG3 also increased dramatically between day 3 and 5 (from <5 to ≥200 ng/ml) with similar kinetics in scid and scid/+ cell cultures. In cultures of LPS/TGFβ-stimulated cells, IgG2b concentrations rose rapidly between day 3 and 5 (from <10 to ≥300 ng/ml), and again, with similar kinetics in both scid and scid/+ control cultures (Fig. 7). Secreted IgA was not detectable (<5 ng/ml) in LPS/TGFβ-stimulated cell cultures (unpublished data), which is consistent with a very low frequency of IgA+ cells (Fig. 6 B).
Figure 7.

Concentration of secreted IgG1 (and IgE), IgG3, IgG2a, and IgG2b in 3-, 4- and 5-d cultures of stimulated 3H9sdVκ8sd scid and 3H9sdVκ**8 scid/**+ spleen cells. Spleen cells from individual 3H9sdVκ8sd scid and 3H9sdVκ8 scid/+ control mice were stimulated separately with LPS/IL4, LPS/TGFβ, and LPS alone, and then cell culture supernatants were tested for concentrations of secreted IgG1/IgE, IgG2b/IgA, and IgG2a/IgG3, respectively. The points (diamonds for 3H9sdVκ8sd scid and squares for 3H9sdVκ8 scid/+ controls) represent the concentrations of a given isotype in individual cell cultures (each culture corresponding to an individual mouse). Points on the abscissa correspond to ≤1 ng/ml. Culture conditions and starting cell concentrations were as described in Fig. 6 B.
The results of Fig. 7 are consistent with those of Figs. 5 and 6, and indicate that the scid mutation has little or no discernible effect on IgM–IgG class switching. Whether switching to IgE is also relatively unimpaired in scid B cells is unclear from the above results, because IgE was not detected in supernatants of the LPS/IL4-stimulated scid cell cultures and was just barely detectable in the scid/+ controls (≤25 ng/ml).
Absence of DNA-PKcs Activity in scid B Lineage Cells.
Previous reports have indicated that most scid cell lines contain little or no detectable DNA-PKcs protein (43–45). However, some scid cell lines, such as bcl-2 scid pre-B cell lines, have been reported to contain near normal amounts of DNA-PKcs protein, although no DNA-PKcs activity was detectable in whole cell extracts from these lines (16, 17). To confirm that DNA-PKcs activity is indeed absent in the nucleus of scid B lineage cells, we first tested for the presence of such activity in nuclear extracts of A-MuLV–transformed scid pre-B cells. As shown in Table IV A, the amount of DNA-PKcs activity in pre-B cell lines derived from scid adult bone marrow and scid fetal liver was equivalent to that in pre-B cell lines from the bone marrow of DNA-PKcs−/− mice. Nuclear extracts of A-MuLV–transformed pre-B cells from wt mice and the human HeLa cell line served as positive controls. Note that the DNA-PKcs activity in the HeLa cell line was 15–17-fold greater than that in the pre-B cell lines from wt mice. This magnitude of difference in DNA-PKcs activity between mouse and human cell lines is consistent with previous reports (45, 46). We also tested nuclear extracts of LPS/IL4-stimulated 3H9sdVκ8sd scid B cells for DNA-PKcs activity. A suspension of 107 cells from two 3H9sdVκ8sd scid mice, consisting of 77% B220+IgM+ B cells, was aliquoted into 6-well plates at 0.25 × 106 cells/ml. The cells were cultured for 3 d in the presence of LPS and IL4. An equivalent number of sorted B220+ splenic cells from 3H9sdVκ8 scid/+ mice (controls) was cultured in parallel under the same conditions. At day 3, cells from the scid and scid/+ cultures were separately pooled and analyzed for DNA-PKcs activity. As indicated in Table IV B, there was no apparent DNA-PKcs activity in the cultured scid cells relative to that of the negative controls (DNA-PK−/− cells), though such activity was clearly detectable in the 3H9sdVκ8 scid/+ transgenic controls. To confirm that the LPS/IL4 stimulation resulted in activated B cells, small aliquots of the same cell cultures analyzed at day 3 were held for 4 d and the cells were pooled to test for the presence of B220+IgG1+ cells. We found the 3H9sdVκ8sd scid and control cell cultures to contain 10.4 and 15.7% B220+IgG1+ cells, respectively (unpublished data).
Table IV.
Lack of DNA-PKcs Activity in scid Pre-B and B Cells
| DNA-PKcsactivity | |||||
|---|---|---|---|---|---|
| Cellline | Origin | DNA-PKcsgenotype | Exp 1 | Exp 2 | |
| psl | psl | ||||
| Aa | 1 | BM | −/− | 199; 277 | 312; 388 |
| 2 | BM | −/− | 353; 229 | 423; 342 | |
| 3 | BM | s/s | 239; 344 | ||
| 4 | BM | s/s | 360; 467 | ||
| 5 | BM | +/+ | 2,613; 2,598 | ||
| 6 | FL | s/s | 371; 456 | ||
| 7 | FL | s/s | 441; 319 | ||
| 8 | FL | +/+ | 2,295; 2,012 | ||
| 9 | HeLa | +/+ | 38,163 | 36,149 | |
| Bb | |||||
| 1 | BM | −/− | 1,184; 1,103 | 234; 308 | |
| SPL | s/s | 1,393; 1,553 | 369; 337 | ||
| SPL | s/+ | 6,826; 6,088 | 2,489; 3,066 | ||
| 5 | BM | +/+ | 7,099; 7,213 | 3,520; 3,806 |
Discussion
Much of the impetus for the present work came from results of an earlier paper on leaky scid mice (29). Scid mice are leaky in that productive rearrangements at two critical loci (e.g., H and L(κ) or TCRβ and TCRα) occasionally occur within a given developing scid lymphocyte (for review see reference 47). This leakiness becomes apparent as scid mice age and results in the generation of pauciclonal B and T cells and the production of serum Ig. Of particular interest here is our earlier analysis of sera from 48 leaky scid mice showed that each mouse expressed two or more IgG isotypes (IgG1, IgG2a, IgG2b, or IgG3) in addition to IgM. None of the sera were found to contain IgM only (29) as might have been expected if IgM–IgG class switching were severely impaired. Thus, B cells in leaky scid mice showed no evidence of a severe defect in CSR.
B cells of 3H9sdVκ8sd scid mice also showed no evidence of a severe defect in CSR. We found that 3H9sdVκ8sd scid mice are able to generate normal levels of serum IgG and IgA and significant levels of serum IgE upon reconstitution with T cells. In the absence of T cells, 3H9sdVκ8sd scid mice are severely deficient in serum IgG, IgA, and IgE, even though they contain above normal numbers of splenic B cells. When 3H9sdVk8sd scid B cells were stimulated to class switch in vitro, we found that they could do so with close to the same efficiency as control 3H9sdVκ8 scid/+ cells. The implications of our findings are discussed below.
CSR and V(D)J Recombination Show Differing Dependency on DNA-PKcs.
Although the results of the present paper suggest that CSR is no more than two- to threefold less efficient in scid than wt cells, V(D)J recombination has been shown to be two to three orders of magnitude less efficient in scid than wt cells (48, 49). How can this difference be explained assuming that CSR and V(D)J recombination both utilize an NHEJ mechanism for joining DNA ends? One possible explanation is that different kinds of DNA ends are produced during CSR and V(D)J recombination. In V(D)J recombination, two kinds of DNA ends are produced: hairpin coding ends and blunt signal ends (39, 50–52). Scid cells cannot efficiently resolve hairpin coding ends (50). Thus, in V(D)J recombination, resolution of hairpin ends before their joining is one step where DNA-PKcs plays a critical role. DNA-PKcs also plays an important role in the processing and joining of open coding ends (53). In contrast to V(D)J recombination, CSR is RAG independent (15, 54) and probably does not involve the generation of hairpin ends, which is consistent with the detection of broken DNA with blunt ends following the initiation of Sμ-Sγ3 recombination (3). Blunt DNA ends, such as signal ends, can be joined in both scid (48) and DNA-PKcs–null cells (28, 55, 56) much more efficiently than coding ends. Thus, if initiation of CSR often results in blunt DNA ends, this could explain why CSR is much less dependent on DNA-PKcs activity than V(D)J recombination. However, the above reasoning assumes that an NHEJ mechanism is used exclusively to join DNA ends during CSR. But whether this is true is unclear as discussed below.
CSR Can Occur in the Absence of DNA-PKcs Activity.
DNA-PKcs activity was not detectable in 3H9sdVκ8sd scid B cells (Table IV). Therefore our finding that class switching to different IgG isotypes (and IgA) is not severely impaired in 3H9sdVκ8sd scid B cells suggests that DNA-PKcs activity is not essential for CSR. However, results from previous studies imply that DNA-PK plays a critical role in CSR. These results include the demonstration of defective CSR in B cells of H/L chain transgenic mice lacking known components of the NHEJ machinery such as Ku70, Ku80, and DNA-PKcs (13, 14, 18). There is also evidence that class switching to IgE is severely impaired in scid pre-B cell lines (15).
Although we cannot easily reconcile the apparent discrepancy between our results and those of others, we offer some possible explanations by noting some of the caveats in the different experimental systems, including our own. First, as pointed out by others (18), B cells of H/L chain transgenic mice lacking Ku70 or Ku80 show impaired proliferation and this may account, to a great extent, for the inability of these cells to class switch. Second, the reported evidence (18) for severe impairment of CSR in H/L chain transgenic B cells of mice lacking DNA-PKcs would seem to apply to all Ig isotypes except IgG1. Class switching to IgG1 was not impaired in B cells of these mice, whereas switching to IgG2a, IgG2b, and IgG3 appeared to be severely impaired; the implication being that switching between μ and γ1 switch regions is mechanistically different than switching between μ and γ2 or γ3 switch regions. This is difficult to understand from a substrate point of view, given that all γ switch regions share 49–52mer repeats (19–22). Possibly, the absence of detectable switching to isotypes other than IgG1 in the H/L chain transgenic B cells of the DNA-PKcs–null mice reflected a relatively poor response of these B cells to class switch–inducing stimuli. For example, the levels of secreted IgG1 observed in 5-d cultures of appropriately stimulated B cells from DNA-PKcs–null mice were ≤100 ng/ml and those of other isotypes were ≤10 ng/ml (18). In our work, the levels of IgG1 in 4- to 5-d cultures of LPS/Il4-stimulated B cells from 3H9sdVκ8sd scid mice were in the range of 100–300 ng/ml and those of other IgG isotypes were close to or greater than 100 ng/ml (Table III and Fig. 7).
Third, early evidence that the scid mutation severely impairs CSR (15) comes from a comparison of class switching to IgE in scid and wt pre-B cells rather than in B cells, the cell stage at which CSR normally occurs. In addition, class switching to IgE may involve more than one recombination event. Indeed, there is evidence that switching to IgG1 is often followed by a successive switch to IgE (57, 58). As switching to IgE via IgG1 would represent two successive recombination events, the overall extent of impairment in class switching to IgE could be much greater than switching to an upstream IgG constant region gene, which presumably would involve only one recombination event. On the basis of our in vivo results, it appears that class switching to IgE is not severely impaired in scid B cells. Significant levels of serum IgE were produced in T cell–reconstituted 3H9sdVκ8sd scid mice (Fig. 2). However, in attempting to assess the relative efficiency with which scid B cells switch to IgE in vitro, we found that supernatants of LPS/IL4-stimulated scid cell cultures lacked detectable IgE and that the scid/+ controls contained only very low levels of IgE (<50 ng/ml). Thus, our results are uninformative as to the extent to which the scid mutation impairs switching to IgE.
Finally, scid is not a null mutation (45, 59) and results in a truncated DNA-PKcs protein lacking DNA-PKcs activity (16, 17). Thus, it is possible that the defective scid DNA-PKcs protein, which may be present at normal levels in scid B lineage cells (16, 17), is recruited by DNA-bound Ku70/80 and is able to bring DNA ends together despite its lack of kinase activity (60). In this capacity, the scid DNA-PKcs may serve as a scaffold for other proteins involved in CSR, and thereby enable 3H9sdVκ8sd scid B cells to class switch with close to normal efficiency.
NHEJ May Not Be the (Primary or Only) Mechanism for Repair of DNA Breaks Resulting from CSR.
Given the preceding discussion, we are led to question whether a protein other than DNA-PKcs may associate with DNA-bound Ku70/80 and provide the necessary kinase activity, or alternatively, whether NHEJ is the (sole) mechanism for joining DNA ends resulting from CSR. It has been reported that DNA lesions associated with the initiation of CSR are most frequent during the G1 phase of the cell cycle (61), which is consistent with when NHEJ is believed to occur (62, 63). But whether the observed CSR-associated DNA lesions in the G1 phase of the cell cycle signify repair of DNA double strand breaks by NHEJ is open to question. Repair of such lesions could possibly occur later and involve a replication-dependent mechanism (64–68) in which the intermediate products are nicked rather than fully cleaved DNA molecules. Even if all CSR-associated DNA nicks were to result in staggered DNA double strand breaks (4), the repair of these breaks may not necessarily involve NHEJ. This reservation is not only prompted by the results presented here, but also by results of early studies showing a high frequency of point mutations, insertions, and deletions in switch junctions (69, 70). The latter findings led to the proposal of an illegitimate priming model for CSR (69, 70) whereby DNA double strand breaks, followed by exonuclease digestion, result in a single (donor) strand end that anneals to a microhomologous sequence in the acceptor switch region. The acceptor switch region sequence then serves as a template for priming DNA synthesis. The reciprocal reaction also must occur to complete switch recombination for both strands of DNA. As pointed out by others (71, 72), the illegitimate priming model is a variant of single strand annealing, which can be considered a form of homologous recombination (7).
Homologous recombination has been postulated as the mechanism for repair of DNA breaks resulting from initiation of somatic hypermutation (SHM) in Ig variable region genes (73–76). Interestingly, there is evidence that some factors may be common to both SHM and CSR. For example, a recently discovered enzyme known as activation-induced cytidine deaminase is essential for both SHM and CSR (33). Moreover, SHM appears to involve error prone repair of DNA breaks (for review see reference 77), as originally postulated by Brenner and Milstein (78), and the same may be true of CSR, as switch junctions and surrounding regions often contain point mutations (66, 70). Further, in the absence of the mismatch repair enzyme, Msh2, the distribution of recombination breakpoints is altered in cells undergoing SHM (or CSR), such that most breakpoints occur at consensus motifs in the variable (or switch) regions (79, 80). Finally, both SHM and CSR can occur in the absence of DNA-PKcs activity (75 and this study). All of the aforementioned findings are consistent with some overlap in the machinery responsible for repair of DNA breaks resulting from initiation of SHM and CSR.
B Cell Anergy in 3H9sdV_κ_8sd Scid Mice Is Broken upon Reconstitution with T Cells.
The 3H9 and Vκ8 tgs code for anti-self antibody with specificity for single stranded DNA and transgenic B cells expressing this specificity (3H9Vκ8 B cells) appear to be inactive in the bone marrow at the pre-B to B transitional stage in nonautoimmune mouse strains (23, 37, 38). Consistent with such inactivity (anergy), both 3H9sdVκ8sd scid mice (this work) and 3H9sdVκ8 RAG−/− (37) mice are deficient in serum Ig. Also, 3H9Vκ8 B cells show limited ability in vitro to proliferate and secrete Ig in response to low doses of LPS (<10 μg/ml) or anti-IgM Fab(ab)′2 (<50 μg/ml) compared with nontransgenic B cells (23, 38). Significant proliferation of 3H9/Vκ8 B cells does occur, however, when these cells are stimulated with high doses of LPS (10–20 μg/ml) or anti-IgM (50 μg/ml; reference 37) or when their surface IgM is cross-linked in the presence of T cell help (38). Indeed, our results (Table III) suggest that B cells from 3H9sdVκ8sd scid and 3H9sdVκ8 scid/+ mice proliferate as well as (or better than) B cells from nontransgenic scid/+ mice in response to high doses of LPS (50 μg/ml) and IL4.
Thus, the ability of 3H9Vκ8 B cells to proliferate or respond to differentiation stimuli may depend on the strength of the stimuli and the microenvironment in which these cells arise. For example, as reported earlier (37), 3H9Vκ8 B cells that develop in the absence of T cells, as in 3H9sdVκ8 RAG−/− mice, may be partially activated. Two distinct populations of B cells were found in 3H9sdVκ8 RAG−/− mice: one lacked surface CD43 and expressed low IgM and high IgD (CD43-IgMlo IgDhi), the other expressed high CD43, high IgM, and low IgD (CD43hiIgMhi IgDlo), a phenotype associated with activated B cells (81). The latter cell population was not found in 3H9sdVκ8 RAG+/− mice (37). Full activation of the CD43hiIgMhi IgDlo B cell population may result if these B cells are provided T cell help. As shown here, when 3H9sdVκ8sd scid mice were provided with a source of T cells from JH−/− donors, the recipients generated T cells (Fig. 1 B) and expressed normal levels of serum IgG and IgA (Fig. 2). Moreover, all of the recipients expressed IgG1 and IgG2a of the tg allotype (unpublished data). What might be the mechanism for this apparent breakage in B cell tolerance? One possibility is that IgM-expressing B cells in the 3H9sdVκ8sd scid recipients serve as antigen-presenting cells for naive, autoreactive T cells from the JH−/− donor mice, and in turn, are T cell–activated to undergo IgM–IgG class switching. Regardless of the mechanism, it is clear that 3H9sdVκ8sd scid mice contain B cells that can be fully activated to undergo CSR in the presence of T cells and (self?) antigen.
Acknowledgments
The assistance of the following CORE facilities of the Fox Chase Cancer Center is gratefully acknowledged: Flow Cytometry and Cell Sorting Facility, Laboratory Animal Resources, and Histopathology Facility. We thank C. Congleton for help with mouse breeding, M. Weigert for providing mice with the 3H9sd and Vκ8sd tgs, and G. Taccioli for providing DNA-PKcs−/− mice. We also thank W. Dunnick, M. Gellert, R. Hardy, P. Nakajima, D. Roth, M. Wabl and M. Weigert for review of the manuscript and R. Diehl for assistance in typing the manuscript.
The work was supported by National Institutes of Health grants CA-04946 and CA-06927 and by a grant from the Commonwealth of Pennsylvania. M.G. Cotticelli was supported by the Greenwald fellowship.
M.G. Cotticelli's present address is Dept. of Pathology, University of Pennsylvania Medical School, Philadelphia, PA 19104.
N.R. Ruetsch's present address is Aventis Pharmaceuticals, Bridgewater, NJ 08807-0800.
Footnotes
*
Abbreviations used in this paper: A-MuLV, Abelson murine leukemia virus; CSR, class switch recombination; DNA-PK, DNA-dependent protein kinase; DNA-PKcs, DNA-PK catalytic subunit; I, intronic; NHEJ, nonhomologous end joining; RT-PCR, reverse transcriptase–PCR; S, switch; scid, severe combined immune deficiency; sd, site-directed; tg, transgene.
References
- 1.Tonegawa, S. 1983. Somatic generation of antibody diversity. Nature. 302:575–581. [DOI] [PubMed] [Google Scholar]
- 2.Stavnezer, J. 1996. Antibody class switching. Adv. Immunol. 61:79–146. [DOI] [PubMed] [Google Scholar]
- 3.Wuerffel, R.A., D. Jian, R.J. Thompson, and A.L. Kenter. 1997. Ig Sγ3 DNA-specific double strand breaks are induced in mitogen-activated B cells and are implicated in switch recombination. J. Immunol. 159:4139–4144. [PubMed] [Google Scholar]
- 4.Chen, X., K. Kinoshita, and T. Honjo. 2001. Variable deletion and duplication at recombination junction ends: implication for staggered double-strand cleavage in class-switch recombination. Proc. Natl. Acad. Sci. USA. 98:13860–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Honjo, T., K. Kinoshita, and M. Muramatsu. 2002. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu. Rev. Immunol. 20:165–196. [DOI] [PubMed] [Google Scholar]
- 6.Manis, J.P., M. Tian, and F.W. Alt. 2002. Mechanism and control of class-switch recombination. Trends Immunol. 23:31–39. [DOI] [PubMed] [Google Scholar]
- 7.Karran, P. 2000. DNA double strand break repair in mammalian cells. Curr. Opin. Genet. Dev. 10:144–150. [DOI] [PubMed] [Google Scholar]
- 8.Khanna, K.K., and S.P. Jackson. 2001. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27:247–254. [DOI] [PubMed] [Google Scholar]
- 9.Carter, T., I. Vancurova, I. Sun, W. Lou, and S. DeLeon. 1990. A DNA-activated protein kinase from HeLa cell nuclei. Mol. Cell. Biol. 10:6460–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lees-Miller, S.P., Y.-R. Chen, and C.W. Anderson. 1990. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol. Cell. Biol. 10:6472–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dvir, A., S.R. Peterson, M.W. Knuth, H. Lu, and W.S. Dynan. 1992. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl. Acad. Sci. USA. 89:11920–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottlieb, T.M., and S.P. Jackson. 1993. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 72:131–142. [DOI] [PubMed] [Google Scholar]
- 13.Manis, J.P., Y. Gu, R. Lansford, E. Sonoda, R. Ferrini, L. Davidson, K. Rajewsky, and F.W. Alt. 1998. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 187:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casellas, R., A. Nussenzweig, R. Wuerffel, R. Pelanda, A. Reichlin, H. Suh, X.-F. Qin, A. Kenter, K. Rajewsky, and M. Nussenzweig. 1998. Ku80 is required for immunoglobulin isotype switching. EMBO J. 17:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rolink, A., F. Melchers, and J. Andersson. 1996. The SCID but not the RAG-2 gene product is required for Sμ-Sɛ heavy chain class switching. Immunity. 5:319–330. [DOI] [PubMed] [Google Scholar]
- 16.Grawunder, U., N. Finnie, S. Jackson, B. Riwar, and R. Jessberger. 1996. Expression of DNA-dependent protein kinase holoenzyme upon induction of lymphocyte differentiation and V(D)J recombination. Eur. J. Biochem. 241:931–940. [DOI] [PubMed] [Google Scholar]
- 17.Beamish, H.J., R. Jessberger, E. Riballo, A. Priestley, T. Blunt, B. Kysela, and P.A. Jeggo. 2000. The C-terminal conserved domain of DNA-PKcs, missing in the SCID mouse, is required for kinase activity. Nucleic Acids Res. 28:1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manis, J.P., D. Dudley, L. Kaylor, and F.W. Alt. 2002. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 16:607–617. [DOI] [PubMed] [Google Scholar]
- 19.Kataoka, T., T. Miyata, and T. Honjo. 1981. Repetitive sequences in class-switch recombination regions of immunoglobulin heavy chain genes. Cell. 23:357–368. [DOI] [PubMed] [Google Scholar]
- 20.Nikaido, T., Y. Yamawaki-Kataoka, and T. Honjo. 1982. Nucleotide sequences of switch regions of immunoglobulin Cɛ and Cγ genes and their comparison. J. Biol. Chem. 257:7322–7329. [PubMed] [Google Scholar]
- 21.Szurek, P., J. Petrini, and W. Dunnick. 1985. Complete nucleotide sequence of the murine γ3 switch region and analysis of switch recombination sites in two γ3-expressing hybridomas. J. Immunol. 135:620–626. [PubMed] [Google Scholar]
- 22.Mowatt, M.R., and W. Dunnick. 1986. DNA sequence of the murine γ1 switch segment reveals novel structural elements. J. Immunol. 136:2674-2683. [PubMed] [Google Scholar]
- 23.Erikson, J., M.Z. Radic, S.A. Camper, R.R. Hardy, C. Carmack, and M. Weigert. 1991. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 349:331–334. [DOI] [PubMed] [Google Scholar]
- 24.Carmack, C.E., S.A. Camper, J.J. Mackle, W.U. Gerhard, and M.G. Weigert. 1991. Influence of a Vκ8 L chain transgene on endogenous rearrangements and the immune response to the HA(SB) determinant on influenza virus. J. Immunol. 147:2024–2033. [PubMed] [Google Scholar]
- 25.Chen, C., S. Nagy, E.L. Prak, and M. Weigert. 1995. b. Immunoglobulin heavy chain gene replacement: a mechanism of receptor editing. Immunity. 3:747–755. [DOI] [PubMed] [Google Scholar]
- 26.Prak, E.L., and M. Weigert. 1995. Light chain replacement: a model for antibody gene rearrangement. J. Exp. Med. 182:541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen, J., M. Trounstine, F.W. Alt, F. Young, C. Kurahara, J.F. Loring, and D. Huszar. 1993. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int. Immunol. 5:647–656. [DOI] [PubMed] [Google Scholar]
- 28.Taccioli, G.E., A.G. Amatucci, H.J. Beamish, D. Gell, X.H. Xiang, M.I. Torres Arzayus, A. Priestley, S.P. Jackson, A. Marshak Rothstein, P.A. Jeggo, and V.L.M. Herrera. 1998. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity. 9:355–366. [DOI] [PubMed] [Google Scholar]
- 29.Bosma, G.C., M. Fried, R.P. Custer, A. Carroll, D.M. Gibson, and M.J. Bosma. 1988. Evidence of functional lymphocytes in some (leaky) scid mice. J. Exp. Med. 167:1016–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang, Y., G.C. Bosma, and M.J. Bosma. 1995. Development of B cells in scid mice with immunoglobulin transgenes: implications for the control of V(D)J recombination. Immunity. 2:607–616. [DOI] [PubMed] [Google Scholar]
- 31.Hardy, R.R. 1986. Purification and coupling of fluorescent proteins for use in flow cytometry. Handbook of Experimental Immunology, 4th ed. D.M. Weir, L.A. Herzenberg, C.C. Blackwell, and L.A. Herzenberg, editors. Blackwell Scientific Publishers, Ltd., Edinburgh, UK. 31.1–31.12.
- 32.Kinoshita, K., M. Harigai, S. Fagarasan, M. Muramatsu, and T. Honjo. 2001. A hallmark of active class switch recombination: transcripts directed by I promoters on looped-out circular DNAs. Proc. Natl. Acad. Sci. USA. 98:12620–12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg, N., and D. Baltimore. 1976. A quantitative assay for transformation of bone marrow cells by Abelson murine leukemia virus. J. Exp. Med. 143:1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews, N.G., and D.V. Faller. 1991. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19:2499 (Abstr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woo, R.A., K.G. McLure, S.P. Lees-Miller, D.E. Rancourt, and P.W.K. Lee. 1998. DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature. 394:700–704. [DOI] [PubMed] [Google Scholar]
- 37.Xu, H., H. Li, E. Suri-Payer, R.R. Hardy, and M. Weigert. 1998. Regulation of anti-DNA B cells in recombination-activating gene-deficient mice. J. Exp. Med. 188:1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen, K.-A.T., L. Mandik, A. Bui, J. Kavaler, A. Norvell, J.G. Monroe, J.H. Roark, and J. Erikson. 1997. Characterization of anti-single-stranded DNA B cells in a non-autoimmune background. J. Immunol. 159:2633–2644. [PubMed] [Google Scholar]
- 39.Schlissel, M., A. Constantinescu, T. Morrow, M. Baxter, and A. Peng. 1993. Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′-phosphorylated, RAG-dependent, and cell cycle regulated. Genes Dev. 7:2520–2532. [DOI] [PubMed] [Google Scholar]
- 40.Li, S.C., P.B. Rothman, J. Zhang, C. Chan, D. Hirsh, and F.W. Alt. 1994. Expression of Iμ-Cγ hybrid germline transcripts subsequent to immunoglobulin heavy chain class switching. Int. Immunol. 6:491–497. [DOI] [PubMed] [Google Scholar]
- 41.Snapper, C.M., F.D. Finkelman, and W.E. Paul. 1988. Differential regulation of IgG1 and IgE synthesis by interleukin 4. J. Exp. Med. 167:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lebman, D.A., and R.L. Coffman. 1988. The effects of IL-4 and Il-5 on the IgA response by murine peyer's patch B cell populations. J. Immunol. 141:2050–2056. [PubMed] [Google Scholar]
- 43.Kirchgessner, C.U., C.K. Patil, J.W. Evans, C.A. Cuomo, L.M. Fried, T. Carter, M.A. Oettinger, and M.J. Brown. 1995. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science. 267:1178–1182. [DOI] [PubMed] [Google Scholar]
- 44.Blunt, T., N.J. Finnie, G.E. Taccioli, G.C. Smith, J. Demengeot, T.M. Gottlieb, R. Mizuta, A.J. Varghese, F.W. Alt, P.A. Jeggo, et al. 1995. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell. 80:813–823. [DOI] [PubMed] [Google Scholar]
- 45.Danska, J., D.P. Holland, S. Mariathasan, K.M. Williams, and C.J. Guidos. 1996. Biochemical and genetic defects in the DNA-dependent protein kinase in murine scid lymphocytes. Mol. Cell. Biol. 16:5507–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finnie, N.J., T.M. Gottlieb, T. Blunt, P.A. Jeggo, and S.P. Jackson. 1995. DNA-dependent protein kinase activity is absent in xrs-6 cells: Implications for site-specific recombination and DNA double-strand break repair. Proc. Natl. Acad. Sci. USA. 92:320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bosma, M.J., and A.M. Carroll. 1991. The scid mouse mutant: definition, characterization, and potential uses. Annu. Rev. Immunol. 9:323–350. [DOI] [PubMed] [Google Scholar]
- 48.Lieber, M.R., J.E. Hessie, S. Lewis, G.C. Bosma, K. Mizuuchi, M.J. Bosma, and M. Gellert. 1988. The defect in murine severe combined immune deficiency: joining of signal segments but not coding segments in V(D)J recombination. Cell. 55:7–16. [DOI] [PubMed] [Google Scholar]
- 49.Harrington, J., C.L. Hsieh, J. Gerton, G. Bosma, and M.R. Lieber. 1992. Analysis of the defect in DNA end joining in the murine scid mutation. Mol. Cell. Biol. 12:4758–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roth, D.B., J.P. Menetski, P.B. Nakajima, M.J. Bosma, and M. Gellert. 1992. b. V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell. 70:983–991. [DOI] [PubMed] [Google Scholar]
- 51.Roth, D.B., C. Zhu, and M. Gellert. 1993. Characterization of broken DNA molecules associated with V(D)J recombination. Proc. Natl. Acad. Sci. USA. 90:10788–10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramsden, D.A., and M. Gellert. 1995. Formation and resolution of double-strand break intermediates in V(D)J rearrangement. Genes Dev. 9:2409–2420. [DOI] [PubMed] [Google Scholar]
- 53.Nakajima, P.B., and M.J. Bosma. 2002. Variable diversity joining recombination: nonhairpin coding ends in thymocytes of SCID and wild-type mice. J. Immunol. 169:3094-3104. [DOI] [PubMed] [Google Scholar]
- 54.Lansford, R., J.P. Manis, E. Sonoda, K. Rajewsky, and F.W. Alt. 1998. Ig heavy chain class switching in rag-deficient mice. Int. Immunol. 10:325–332. [DOI] [PubMed] [Google Scholar]
- 55.Gao, Y., J. Chaudhuri, C. Zhu, L. Davidson, D.T. Weaver, and F.W. Alt. 1998. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity. 9:367–376. [DOI] [PubMed] [Google Scholar]
- 56.Bogue, M.A., C. Jhappan, and D.B. Roth. 1998. Analysis of variable (diversity) joining recombination in DNA-dependent protein kinase (DNA-PK)-deficient mice reveals DNA-PK-independent pathways for both signal and coding joint formation. Proc. Natl. Acad. Sci. USA. 95:15559–15564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siebenkotten, G., C. Esser, M. Wabl, and A. Radbruch. 1992. The murine IgG1/IgE class switch program. Eur. J. Immunol. 22:1827–1834. [DOI] [PubMed] [Google Scholar]
- 58.Mandler, R., F.D. Finkleman, A.D. Levine, and C.M. Snapper. 1993. Il-4 induction of IgE class switching by lipopolysaccharide-activated murine B cells occurs predominantly through sequential switching. J. Immunol. 150:407–418. [PubMed] [Google Scholar]
- 59.Blunt, T., D. Gell, M. Fox, G.E. Taccioli, A.R. Lehmann, S.P. Jackson, and P.A. Jeggo. 1996. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc. Natl. Acad. Sci. USA. 93:10285–10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeFazio, L.G., R.M. Stansel, and G. Chu. 2002. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 21:3192–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petersen, S., R. Casellas, B. Reina-San-Martin, H.T. Chen, M.J. Difilippantonio, P.C. Wilson, L. Hanitsch, A. Celeste, M. Muramatsu, D.R. Pilch, et al. 2001. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 414:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee, S.E., R.A. Mitchell, A. Cheng, and E.A. Hendrickson. 1997. Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol. Cell. Biol. 17:1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takata, M., S. Sasaki, E. Sonoda, C. Morrison, M. Hashimoto, H. Utsumi, Y. Yamaguchi-Iwai, A. Shinohara, and S. Takeda. 1998. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 17:5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Severinson-Gronowicz, E., C. Doss, and J. Schroder. 1979. Activation to IgG secretion by lipopolysaccharide requires several proliferation cycles. J. Immunol. 123:2057–2062. [PubMed] [Google Scholar]
- 65.Kenter, A.L., and J.V. Watson. 1987. Cell cycle kinetics model of LPS-stimulated spleen cells correlates switch region rearrangements with S phase. J. Immunol. Methods. 97:111–117. [DOI] [PubMed] [Google Scholar]
- 66.Dunnick, W., M. Wilson, and J. Stavnezer. 1989. Mutations, duplication, and deletion of recombined switch regions suggest a role for DNA replication in the immunoglobulin heavy chain switch. Mol. Cell. Biol. 9:1850–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lundgren, M., L. Strom, L.-O. Bergquist, S. Skog, T. Heiden, J. Stavnezer, and E. Severinson. 1995. Cell cycle regulation of immunoglobulin class switch recombination and germ-line transcription: potential role of Ets family members. Eur. J. Immunol. 25:2042–2051. [DOI] [PubMed] [Google Scholar]
- 68.Hodgkin, P.D., J.-H. Lee, and A. Bruce Lyons. 1996. B cell differentiation and isotype switching is related to division cycle number. J. Exp. Med. 184:277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dunnick, W., and J. Stavenzer. 1990. Copy choice mechanism of immunoglobulin heavy-chain switch recombination. Mol. Cell. Biol. 10:397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dunnick, W., G.Z. Hertz, L. Scappino, and C. Gritzmacher. 1993. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 21:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stavnezer, J. 1996. Antibody class switch. Adv. Immunol. 61:79–146. [DOI] [PubMed] [Google Scholar]
- 72.Papavasiliou, F.N., and D.G. Schatz. 2002. Somatic hypermutation of immunoglobulin genes: Merging mechanisms for genetic diversity. Cell. 109:s35–s44. [DOI] [PubMed] [Google Scholar]
- 73.Maizels, N. 1995. Somatic hypermutation: how many mechanisms diversify V region sequences. Cell. 83:9–12. [DOI] [PubMed] [Google Scholar]
- 74.Selsing, E., B. Xu, and D. Sigurdardottir. 1996. Gene conversion and homologous recombination in murine B cells. Semin. Immunol. 8:151–158. [DOI] [PubMed] [Google Scholar]
- 75.Bemark, M., J.E. Sale, H.-J. Kim, C. Berek, R.A. Cosgrove, and M.S. Neuberger. 2000. Somatic hypermutation in the absence of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) or recombination-activating gene (RAG)1 activity. J. Exp. Med. 192:1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Papavasiliou, F.N., and D.G. Schatz. 2000. Cell-cycle-related DNA double-strand breaks in somatic hypermutation of immunoglobulin genes. Nature. 408:216–221. [DOI] [PubMed] [Google Scholar]
- 77.Gearhart, P.J., and R.D. Wood. 2001. Emerging links between hypermutation of antibody genes and DNA polymerases. Nat. Rev. Immunol. 1:187–192. [DOI] [PubMed] [Google Scholar]
- 78.Brenner, S., and C. Milstein. 1966. Origin of antibody variation. Nature. 211:242–243. [PubMed] [Google Scholar]
- 79.Rada, C., M.R. Ehrenstein, M.S. Neuberger, and C. Milstein. 1998. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 9:135–141. [DOI] [PubMed] [Google Scholar]
- 80.Ehrenstein, M.R., and M.S. Neuberger. 1999. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. EMBO J. 18:3484–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wells, S.M., A.B. Kantor, and A.M. Stall. 1994. CD43 (S7) expression identifies peripheral B cell subsets. J. Immunol. 153:5503–5515. [PubMed] [Google Scholar]