The Balance between Sphingosine and Sphingosine-1-Phosphate Is Decisive for Mast Cell Activation after Fc∈ Receptor I Triggering (original) (raw)
Abstract
Over the last few years, sphingolipids have been identified as potent second messenger molecules modulating cell growth and activation. A newly emerging facet to this class of lipids suggests a picture where the balance between two counterregulatory lipids (as shown in the particular example of ceramide and sphingosine-1-phosphate in T lymphocyte apoptosis) determines the cell fate by setting the stage for various protein signaling cascades. Here, we provide a further example of such a decisive balance composed of the two lipids sphingosine and sphingosine-1-phosphate that determines the allergic responsiveness of mast cells. High intracellular concentrations of sphingosine act as a potent inhibitor of the immunoglobulin (Ig)E plus antigen–mediated leukotriene synthesis and cytokine production by preventing activation of the mitogen-activated protein kinase pathway. In contrast, high intracellular levels of sphingosine-1-phosphate, also secreted by allergically stimulated mast cells, activate the mitogen-activated protein kinase pathway, resulting in hexosaminidase and leukotriene release, or in combination with ionomycin, give cytokine production. Equivalent high concentrations of sphingosine-1-phosphate are dominant over sphingosine as they counteract its inhibitory potential. Therefore, it might be inferred that sphingosine-kinase is pivotal to the activation of signaling cascades initiated at the Fc∈ receptor I by modulating the balance of the counterregulatory lipids.
Keywords: mast cells, signal transduction, gene regulation
Phosphoinositides and corresponding lipid kinases comprise a signaling system of increasingly known importance for cell survival and proliferation. This is illustrated by the central role of phosphatidylinositol-3-kinase (PI-3K)1 in signaling cascades initiated at receptors such as the platelet-derived growth factor (PDGF) receptor, insulin receptor, CD28, and CD40. The phospholipase C–mediated generation of diacylglycerol, which is a prominent example of another lipid signaling system, leads to the activation of classical and novel protein kinase C (PKC) isoforms, mainly involved in differentiation and regulation of effector functions (participation in T cell receptor cycling, Fas expression, as well as IL-2 and IL-2 receptor expression 1 2 3). Besides these two well-established systems, another class of lipids, the sphingolipids, are emerging as components of the cellular lipid signaling machinery participating in cell growth and differentiation, oncogenic transformation, and immune recognition and responsiveness 4 5 6. Sphingosine (S) itself, the central component to this lipid mediator class, functions as a dual modulator of cellular responses. It acts as a potent inhibitor of PKCs, but recently activation of the retinoblastoma (Rb) protein and p21-activated kinases (PAKs) with subsequent triggering of the corresponding signaling pathways has been described 7 8. The seemingly conflicting data on activation and repression of the same signaling steps in different cell types by one sphingolipid are, in addition to cell type specificity and different costimuli, explained by the finding of a “lipid network.” For this hypothesis, it is not the mere presence of one sphingolipid that determines the outcome but rather its relative ratio to other members of this mediator class. This was first suggested for the intracellular balance of ceramide and sphingosine-1-phosphate (S1P) in the regulation of T lymphocyte apoptosis 9.
In the rat basophilic leukemia cell line, RBL, Fc∈RI cross-linking leads to S-kinase (SK) activation and conversion of S to S1P. This compound has been demonstrated to act as an alternative second messenger to inositol-1,4,5-trisphosphate (IP3). Inhibition of SK by the S analogue, d-l-threo-dihydrosphingosine, strongly suppresses the IgE plus antigen (IgE/Ag)-mediated calcium influx, but has no influence on the activation of the tyrosine kinase, syk 10. In the context of the lipid network hypothesis, S and S1P can be interpreted as two counterregulatory factors determining the allergic responsiveness in RBL cells, with SK acting as a kind of “permissive switch.”
Here, we show the effects of S and S1P on noninduced and IgE/Ag-induced CPII mouse mast cells as well as primary bone marrow–derived mouse mast cells (BMMCs) in an attempt to determine the influence of the SK activity on an allergic response. In CPII cells, the early phase of mast cell activation (degranulation) is insensitive to S application, whereas leukotriene synthesis and the transcriptional induction of cytokines are strongly inhibited by this lipid due to abrogation of the mitogen-activated protein kinase (MAPK) pathway. This effect was confirmed on BMMCs regarding the cytokine response. However, shifting the balance towards S1P leads to a restoration of the cellular response in both cell types, demonstrating that the ratio of these two lipids is decisive for the signal transduction events initiated at the Fc∈RI. This is based on the antagonistic activity of S and S1P as inhibitor and activator of the MAPK pathway, respectively. As a consequence of higher intracellular concentrations of S1P, AP1 transcription factors are induced in CPII cells, a response reminiscent of the AP1-mediated cell division of Swiss 3T3 fibroblasts after S1P treatment 11.
Materials and Methods
Transient transfections, gel shift analyses, hexosaminidase release assay, leukotriene assay, TNF-α ELISA, use of ionomycin and apigenin, Western blot analyses, preparation of radiolabeled probes, nuclear extracts, and electrophoretic mobility shift assays were done as described 12 13 14 15 16.
Materials.
S was purchased from Sigma Chemical Co. and dissolved in DMSO at a concentration of 10 mM. S1P was purchased from Calbiochem. For dissolving S1P, 2 μl morpholin was used to convert 1 mg S1P to its salt before MeOH was added to achieve a concentration of 10 mM.
Antibodies.
Antibodies for raf and MAPK kinase (MEK) 1 used in Western blot analyses were purchased from Transduction Laboratories. Antibodies directed against extracellular signal regulatory kinase (erk) 1,2, c-jun NH2-terminal kinase (jnk) 1,2, and p38 (phospho-specific and non–phospho-specific) come from New England Biolabs. Supershift antibodies directed against AP1 components come from Santa Cruz Biotechnologies.
BMMC Generation.
BMMCs were generated by the method of Rottem et al. 17. Fresh bone marrow cells were cultured in complete RPMI 1640 supplemented with 10 ng/ml IL-3 (PeproTech) and 100 ng/ml stem cell factor (SCF; PeproTech) for at least 2 wk. They were characterized as BMMCs by flow cytometry as CD45+, CD117+, CD9+ and positive for staining with FITC-labeled mouse IgE but were CD90.2−, CD4−, CD8−, CD45/B220−, CD11b−, CD11c−, Gr-1−, TER-119−, and MHC class II−. Purity was estimated at >95%. All antibodies were FITC, PE, or biotin labeled and purchased from PharMingen except for mouse IgE (clone SPE-7; Sigma Chemical Co.), which was directly labeled with FITC Cellite (Calbiochem) and purified over Sephadex G-25 (Amersham Pharmacia Biotech) before use. Cells were stimulated with 2 μg/ml murine IgE (Sigma Chemical Co.) and 100 ng/ml DNP-BSA (Calbiochem) for the time points indicated in the figure legends.
Immunoprecipitations/Kinase Assays.
The kinase assay for c-raf activity was done using the c-raf 1 immunoprecipitation kinase cascade assay kit (Upstate Biotechnology) according to the manufacturer's protocol. The MEK 1 kinase activity was determined using the MAPKK immunoprecipitation kinase cascade kit (Upstate Biotechnology). In both cases, cells were either left unstimulated or were stimulated for 7 min (raf) or 9 min (MEK) with and without S application. Reactions were separated by SDS-PAGE, then subjected to autoradiography.
Determination of Sphingolipids in Mast Cells.
4 × 105 mast cells were incubated with 10 μM S (Sigma Chemical Co.) containing 100 nM (2 μCi) 3H-S (Amersham Pharmacia Biotech) for up to 4 h. Subsequently, cells or supernatants were extracted with 2 vol CHCl3/methanol (1:2), sonicated, and again extracted with CHCl3/KCl (2:1). After lyophilization, lipids were dissolved in CHCl3 and analyzed by thin layer chromatography (_n_-butanol/CH3COOH/H2O [6:2:2]). After drying, the plate was subjected to autoradiography. For detection of Ser-containing lipids, 107 cells were incubated with 5 μCi 14C-Ser (Amersham Pharmacia Biotech) for 24 h. Cells were then either left unstimulated or were stimulated with IgE/Ag. Lipids were extracted and separated as described above. After chromatography, the plates were subjected to autoradiography.
SK Assay.
SK activity of mast cells was determined by adding 1 μM and 10 μM S (Sigma Chemical Co.) to cell lysates (from 106 cells) prepared by repeated freeze–thaw cycles in kinase buffer (50 mM Hepes, pH 7.4, 50 mM LiCl, 15 mM MgCl2, 15 mM CaCl2, 1 mM EDTA, 1 mM EGTA, 0.2 mM PMSF, 10 μM ATP). 5 μCi [γ-32P]ATP (Amersham Pharmacia Biotech) was added, and the reaction was incubated at room temperature for 1 h. Lipids were extracted and separated as described above.
Results
A Strong SK Activity Leads to a Rapid Conversion of S to S1P in CPII Mast Cells.
In an in vitro kinase reaction with exogenous S, a strong basal SK activity is detected in cellular extracts from nonactivated CPII cells, which is slightly induced after IgE/Ag activation, comparable to the recently reported data on RBL cells 10. Without the addition of S, no phosphorylation is observed, suggesting that the intracellular concentration of this lipid is normally low in CPII cells (Fig. 1 A). This is supported by the failure to detect S in lipid extractions of CPII cells by thin layer chromatography either after staining with fluram (data not shown; detection limit ∼1 × 10−15 g/cell; S content in other cells ∼3 × 10−14 g/cell) or by autoradiography after incubating cells with 14C-Ser, a precursor for cellular sphingolipids, for 24 h. However, other Ser-containing lipids are easily found by these methods (Fig. 1 B). Taken together, these findings suggest a permanent conversion of S to S1P. In vivo, this conversion is visualized by the addition of 100 nM 3H-S to CPII cells. Within 10 min of incubation, S is converted to S1P and a further degradation product (phosphoethanolamine; Fig. 1 C, left). Overloading the cellular machinery by applying 10 μM S in vivo leads to a delay of both the phosphorylation and degradation, resulting in considerable levels of internal S present up to 4 h in nonstimulated as well as IgE/Ag-stimulated cells (Fig. 1 C, middle and right).
Figure 1.


A strong SK activity mediates the rapid degradation of S in CPII mast cells. (A) Determination of SK activity of either nonstimulated (nst) or 2 min–stimulated (IgE/Ag) CPII cells. −, reactions without exogenous S. The position of S1P on the thin layer plate is indicated by an arrow (right). (B) Ser-containing lipids from nonstimulated (nst) or stimulated (IgE/Ag) CPII cells. The position of S, which served as a standard (Std), is indicated by an arrow (right). (C) S phosphorylation and degradation (100 nM or 10 μM as indicated) after incubating nonstimulated and stimulated cells (indicated at the bottom) for various time points (top). −, nonstimulated cells. Stimulated cells were preincubated with S for 1 h before stimulation. S served as a standard (Std); d, degradation product. The positions of S, S1P, and the degradation product are indicated by arrows (right).
High Levels of Intracellular S Prevent Leukotriene Synthesis and Cytokine Transcription.
To investigate if the accumulation of S affects the allergic responsiveness of CPII mast cells, readouts measuring the degranulation (hexosaminidase release assay), leukotriene synthesis (ELISA), and cytokine transcription (reporter gene assays) were performed. The IgE/Ag-mediated degranulation reaction was, if at all, only marginally inhibited by 10 μM S (Fig. 2 A). However, leukotriene synthesis, as well as the transcriptional activation of IL-5 and TNF-α that is usually seen in CPII cells after IgE/Ag activation, was dramatically reduced (Fig. 2B and Fig. c). This indicates that high intracellular concentrations of S specifically abrogate signaling cascades essential for the induction of these mediators.
Figure 2.


High levels of intracellular S strongly inhibit leukotriene synthesis and cytokine transcription. Cells were either left unstimulated (nst) or were pretreated with solvent before activation with IgE/Ag (IgE/Ag) or pretreated with S (IgE/Ag + S) for 1 h before activation with IgE/Ag. (A) Hexosaminidase release as indicated on the y-axis. Stimulation conditions are given on the x-axis. Values represent mean of quadruplicate experiments ± SD. (B) Leukotriene (LT) release as indicated on the y-axis (in pg/ml LT). Stimulation conditions are given on the x-axis. Values represent mean of quadruplicate experiments ± SD. (C) The transcriptional activation of IL-5 and TNF-α was determined in a reporter gene assay after transient transfections. pGL2 represents the parental vector. Corrected luciferase values are given on the y-axis; stimulation conditions and input DNA are indicated on the x-axis. Data represent mean ± SD of triplicate determinations.
S Is a Potent Inhibitor of the MAPK Pathways in IgE/Ag-stimulated CPII Cells.
PMA-dependent PKCs, the prime targets for inhibition by lysosphingolipids, are known to be dispensable for the induction of TNF-α in CPII cells after Fc∈RI triggering 16. The production of this cytokine and leukotriene synthesis strongly depend on the activation of the MAPK pathway, suggesting that this signaling cascade is a previously unrecognized target for S-mediated modulation 18. 1-h treatment with 10 μM S before IgE/Ag activation prevented the 50–100-fold enhancement of the raf and MEK kinase activity, measured in in vitro kinase reactions of immunoprecipitates using myelin basic protein (MBP) as a readout (Fig. 3A and Fig. B). Subsequently, the two IgE/Ag-responsive MAPKs in CPII cells, erk 1,2 and jnk 1, are hypophosphorylated, as measured in a Western blot analysis (pThr 202/pTyr 204 and pThr 183/pTyr 185; Fig. 3C and Fig. D). This shows that the inhibitory potential of S affects all MAPK pathways that are induced in mast cells after IgE/Ag stimulation 16.
Figure 3.

S inhibits the MAPK pathways. Cells were either left unstimulated (nst) or were pretreated with solvent before activation with IgE/Ag (IgE/Ag) or pretreated with S (IgE/Ag + S) for 1 h before activation with IgE/Ag. (A) Raf kinase activity was determined after immunoprecipitations. MBP was used as substrate (indicated by an arrow). Reactions were separated by SDS-PAGE. Western blots of the immunoprecipitate for normalization are shown below (raf IP, indicated by an arrow). (B) MEK 1 activity was determined after immunoprecipitations. MBP was used as a substrate (indicated by an arrow). Reactions were separated by SDS-PAGE. Western blots of the immunoprecipitate for normalization are shown below (MEK 1 IP, indicated by an arrow) (C) Western blots to investigate erk 1,2 activation/phosphorylation. Cells were activated for 10 min. erk 1,2 and phospho-erk 1,2 are indicated by arrows (right). (D) Western blots for jnk 1,2 activation/phosphorylation. Cells were stimulated for 10 min. jnk 1,2 and phospho-jnk 1 are indicated by arrows (right).
AP1-mediated Transcription Is Prevented by S.
The lack of MAPK activity results in a strongly reduced phosphorylation of the AP1 component, c-jun, one of the targets of jnk1 at the transcription factor level (Fig. 4 A). As a consequence, AP1 transcriptional activity (3 x TRE reporter gene plasmid) is reduced compared with stimulated cells without S treatment, as shown by transient transfections (Fig. 4 B). This explains the inhibition observed for the transcription of TNF-α, which strongly depends on AP1 serving as a cofactor for nuclear factor of activated T cells (NF-AT) at the κ3 site of the promoter in CPII cells 16.
Figure 4.

AP1 transcription is prevented by S. (A) Western blots for c-jun activation/phosphorylation were performed with lysates of either nonstimulated (nst), 1 h–stimulated (IgE/Ag), or 1 h S-pretreated cells before activation (IgE/Ag + S). c-jun and phospho-c-jun are indicated by arrows (right). (B) The transcriptional activation of an AP1-dependent reporter gene (3 x TRE) was measured in transient transfections. Corrected luciferase values are indicated on the y-axis, stimuli and input DNA on the x-axis. pGL2 represents the parental vector. Cells were activated for 3 h. Data represent mean ± SD of triplicate determinations.
The Effect of S Is Prevented by Equimolar Concentrations of S1P.
At this point, we speculated that the internal balance of S and S1P is a kind of permissive switch that might determine the potential of mast cells to respond to Fc∈RI triggering. In this hypothesis, it is not the overall concentration of S but rather the relative ratio of S to its derivative S1P that would be decisive for initiating a response. To test this, we incubated CPII cells with equimolar amounts of S and S1P (10 μM) for 1 h before IgE/Ag stimulation. Under these conditions, the transcriptional activation of TNF-α can be restored to ∼60% of IgE/Ag-induced levels (Fig. 5 A). The ability of S1P to decisively overcome the S-mediated inhibition is not due to the fact that less S is present in the cells (shown by uptake experiments with 3H-S), but rather suggests that S1P is an activation molecule, as already proven for other cell types (10 19 20 21; Fig. 5 B).
Figure 5.

S1P reverts S inhibition of TNF-α. (A) The transcriptional activation of a TNF-α reporter gene construct was determined in transient transfections. Corrected luciferase values are indicated on the y-axis, stimulation conditions on the x-axis. Cells were either left unstimulated (nst) or were pretreated with solvent, 10 μM S (S), or 10 μM S plus 10 μM S1P (S + S1P) before activation with IgE/Ag. Data represent mean ± SD of triplicate determinations. (B) Determination of S in stimulated cells (time points are given above the panels; −, nonstimulated cells) incubated either with S alone or S plus S1P as indicated at the top by thin layer chromatography. The positions of S, S1P, and degradation product (d) are indicated with arrows (right). S served as standard (Std).
Effects of S and S1P on BMMCs.
To confirm the general applicability of the hypothesis that the balance between S and S1P is decisive for activation of mast cells via the Fc∈RI, we generated BMMCs and repeated several key experiments. Addressing the fact that there was a lack of S in nonstimulated CPII cells, which we speculated to be a particular characteristic of our cell line, we investigated the composition of Ser-containing lipids in nonstimulated as well as 1-h IgE/Ag-stimulated BMMCs after incubation with 14C-Ser for 24 h (Fig. 6 A). In both cases, internal S is readily detectable with a slightly reduced concentration in induced cells. Complementary to this reduction, an in vitro kinase reaction demonstrates a strongly inducible, but judged from the exposure time and input concentration of exogenous S, much weaker SK activity in stimulated BMMCs compared with CPII cells (Fig. 6 B). This weaker SK activity also results in no detectable conversion of endogenous S in this experimental setup, and is further demonstrated by uptake experiments with 100 nM and 10 μM S shown in Fig. 6 C. Compared with CPII cells, BMMCs show a reduced phosphorylation rate and a delayed conversion of S to S1P and the degradation product (phosphoethanolamine), whether with a 100 nM or a 10 μM S application (compare Fig. 6 C with Fig. 1 C). However, in agreement with the inducible SK activity in these cells, there is a more rapid and pronounced phosphorylation of S seen in IgE/Ag-stimulated cells (Fig. 6 C). As in CPII cells, the high intracellular concentration of S, if 10 μM is applied, leads to a strong inhibition of the IgE/Ag-inducible production of TNF-α as measured in a corresponding ELISA. This effect is partially reversible by applying an equivalent amount of S1P, even though it is not as strong as that observed in the CPII cell line (Fig. 6 D). This is most likely due to the fact that BMMCs contain substantial intracellular concentrations of S, and that the constitutive and induced conversion to S1P is much slower in BMMCs compared with CPII cells. To conclusively demonstrate that in BMMCs S abrogates the MAPK pathway, Western blot analyses were performed to determine the phosphorylation status of erk 1,2 in both 10-min IgE/Ag-stimulated and S-pretreated IgE/Ag-stimulated cells. Comparable to CPII cells, phosphorylated erk 1,2 was detected in induced cells, whereas S pretreatment strongly prevents the kinase from being activated without affecting its constitutive expression (Fig. 6 E).
Figure 6.
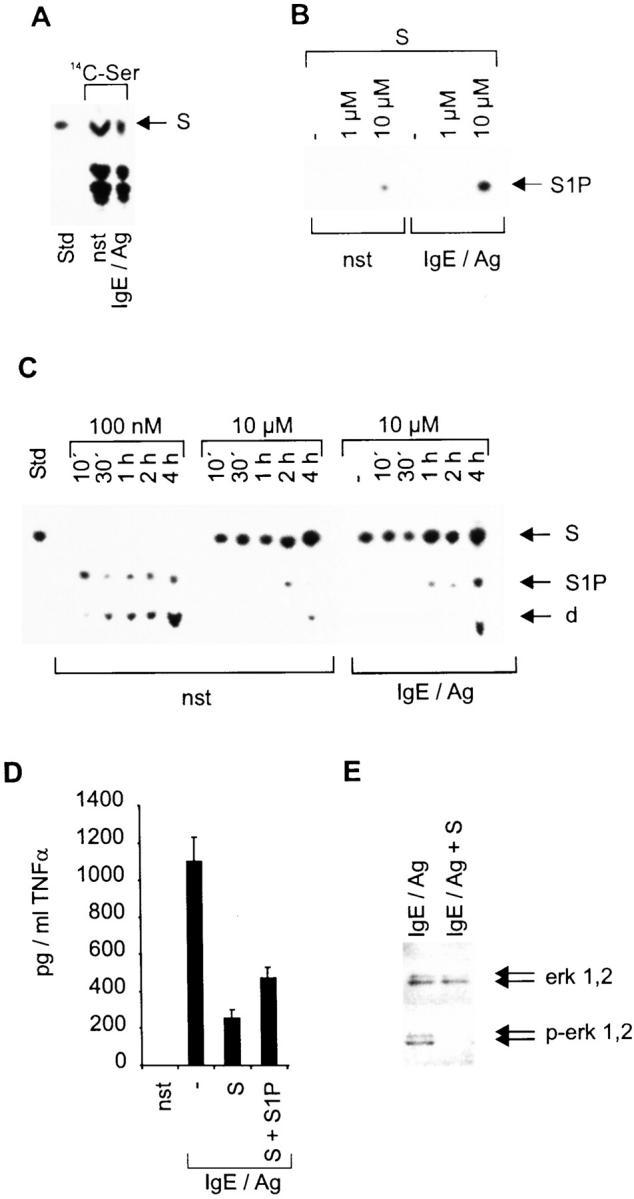
Effects of S and S1P on BMMCs. (A) Ser-containing lipids from nonstimulated (nst) or 1 h–stimulated (IgE/Ag) BMMCs. The position of S, which served as a standard (Std), is indicated by an arrow (right). (B) Determination of SK activity of either nonstimulated (nst) or 2 min–stimulated (IgE/Ag) BMMCs. −, reactions without exogenous S. The position of S1P on the thin layer chromatography plate is indicated by an arrow (right). (C) S phosphorylation and degradation (100 nM or 10 μM as indicated) after incubating nonstimulated and stimulated cells (indicated at the bottom) for various time points (top); −, nonstimulated cells. Stimulated cells were preincubated with S for 1 h before stimulation. S served as a standard (Std); d, degradation product. The positions of S, S1P, and the degradation product are indicated by arrows (right). (D) The secretion of TNF-α by BMMCs was determined by a corresponding ELISA. pg/ml TNF-α are indicated on the y-axis, stimulation conditions on the x-axis. Cells were either left unstimulated (nst) or were pretreated with solvent, 10 μM S (S), or 10 μM S plus 10 μM S1P (S + S1P) before activation with IgE/Ag for 6 h. Data represent mean ± SD of quadruplicate determinations. (E) Western blots to investigate erk 1,2 activation/phosphorylation. Cells were activated for 10 min. erk 1,2 and p-erk 1,2 are indicated by arrows (right).
S1P Leads to Mast Cell Degranulation, Stimulation of Leukotriene Synthesis, and Activation of the MAPK Pathways.
To address the molecular mechanism by which S1P overcomes the S-mediated inhibition, we again used CPII cells as a model system. In these cells, application of S1P resulted in a direct dose-dependent release of hexosaminidase and leukotriene synthesis (Fig. 7A and Fig. B). In addition, the transcriptional induction of TNF-α is induced if S1P is added together with ionomycin, but not with PMA (Fig. 7 C). Taken together, these data suggest that S1P acts contrary to S, mediating the activation of the MAPK pathways.
Figure 7.


S1P is an activator of CPII mast cells. (A) Hexosaminidase release assay with either nonstimulated (nst) or 1 h–stimulated cells. Hexosaminidase values are indicated on the y-axis, stimuli on the x-axis. Values represent mean of quadruplicate experiments ± SD. (B) Leukotriene (LT) release of nonstimulated (nst) or 4 h–stimulated cells. pg/ml LT are indicated on the y-axis, stimuli on the x-axis. Data represent mean of quadruplicate determinations ± SD. (C) TNF-α reporter gene assay. Cells were either left unstimulated (nst) or were pretreated with solvent before stimulation with IgE/Ag, or stimulated with 10 μM S1P, 100 ng/ml ionomycin, and 20 ng/ml PMA. Data represent mean ± SD of triplicate determinations. Luciferase values are indicated on the y-axis, stimuli on the x-axis.
To prove this hypothesis, Western blot analyses were performed measuring the phosphorylation of the MAPK pathway components MEK 1,2, erk 1,2, and jnk 1, which are required for TNF-α induction after IgE/Ag triggering 16. 5 min after S1P treatment, phosphorylation/activation of these kinases was detected (Fig. 8 A). This leads to the subsequent enhancement in the phosphorylation of c-jun and the assembly of several AP1 proteins on a consensus binding site as demonstrated by a supershift analysis (Fig. 8A and Fig. B). The composition of the complex is similar to the complex observed after IgE/Ag stimulation. In both settings, binding of fosB, c-fos, c-jun, and junD was observed 22. Therefore, a combination of S1P and Ca2+ ionophores allows the activation of AP1 and NF-AT, which synergistically mediates TNF-α transcription 16.
Figure 8.

S1P induces the MAPK pathway and AP1 transcription factors. (A) Western blot analysis with lysates of nonstimulated (nst) or 10 μM S1P–stimulated cells for the time points indicated. MEK 1,2, phospho-MEK 1,2, erk 1,2, p-erk 1,2, jnk 1,2, p-jnk 1, c-jun, and p-c-jun are indicated by arrows (right). (B) Supershift analysis on an AP1 consensus site as radiolabeled probe with nuclear extracts of either nonstimulated (nst) or 1 h S1P–stimulated cells as indicated at the top. f, free probe. Antibodies against AP1 transcription factors are given at the top. −, activated complex without antibody.
Allergically Activated Mast Cells Release S1P into the Supernatant.
Recent reports indicated that activated platelets secrete S1P 23. Therefore, we tested supernatants of nonstimulated and IgE/Ag-stimulated CPII cells for the presence of sphingolipids by labeling with 14C-Ser and 3H-S. In both settings, secretion of S1P by CPII cells was demonstrated after 2- and 4-h IgE/Ag triggering (Fig. 9A and Fig. B). The same results were obtained if supernatants of IgE/Ag-stimulated BMMCs labeled with 14C-Ser were tested (Fig. 9 C). Matching the lack of endogenous S in CPII cells, this lipid was also not found in the supernatants of allergically triggered cells. However, IgE/Ag-activated BMMCs do not release S into the supernatant, although it is easily detectable intracellularly (Fig. 9A and Fig. C). These findings, together with the delayed appearance of S1P in the supernatant, strongly suggest an active secretion of this mediator rather than its being a by-product of the degranulation reaction or the result of passive diffusion of this lipid. This implies that only an activating factor is released. To further highlight the physiological relevance for this secretion process, we tested BMMCs for their ability to respond to S1P. Similar to CPII cells (compare Fig. 7 A), there was a dose-dependent degranulation of primary mast cells as observed in a hexosaminidase release assay, suggesting that an immunecomplex-independent amplification process of early allergic reactions via released S1P could occur (Fig. 9 D).
Figure 9.

Activated mast cells secrete S1P. (A) Lipids detected in the supernatants of nonstimulated (nst) or stimulated (IgE/Ag) CPII cells that were fed with 14C-Ser for 24 h. After extraction, lipids were separated by thin layer chromatography. S and S1P served as standard (Std); the positions of S, S1P, and the degradation product (d) are indicated by arrows (right). (B) Sphingolipids detected in supernatants of nonstimulated or stimulated (IgE/Ag) CPII cells that were fed with 3H-S for 1 h before activation. Time points of activation are indicated at the top; control is the cellular content of sphingolipids. S and S1P served as standard. The positions of S, S1P, and the degradation product (d) are indicated by arrows (right). (C) Lipids detected in the supernatants of nonstimulated (nst) or stimulated (IgE/Ag) BMMCs that were fed with 14C-Ser for 24 h. After extraction, lipids were separated by thin layer chromatography. S and S1P served as standard (Std); the positions of S, S1P, and the degradation product (d) are indicated by arrows (right). (D) Hexosaminidase release assay with either nonstimulated (nst) or 1 h–stimulated BMMCs. Hexosaminidase values are indicated on the y-axis, stimuli on the x-axis. Values represent mean of quadruplicate experiments ± SD.
Discussion
The linkage of the Fc∈RI to SK in RBL cells and our data in the CPII mast cell system as well as primary cells are in line with the assumption that allergic responsiveness of these effector cells is under the control of the exact intracellular balance of S and S1P 10. In this hypothesis, SK would constitute a kind of permissive switch initiated at the Fc∈RI or being active constitutively, that generates an intracellular lipid milieu, allowing protein-based signaling cascades such as MAPKs and PKCs to fully activate these effector cells. However, an alternative explanation for our finding might be a receptor-mediated triggering of a so far unknown signaling cascade by S1P, thereby overcoming the intracellular-mediated inhibition by S. This hypothesis is supported by the recent finding that S1P binds to its receptor, endothelial differentiation gene 1 (EDG-1), and thereby extracellularly leads to the activation of MAPKs, a response also observed in CPII mouse mast cells after S1P treatment 24. Contrary to this receptor-mediated effect of S1P argues the extremely fast activation of all the MAPKs tested, which in comparison to an Fc∈RI activation is three times more rapid. In addition, CPII cells respond with a similar but slightly lower increase in the intracellular Ca2+ concentration than after Fc∈RI triggering (data not shown), which recently was shown to be independent of the presence of EDG-1 and mediated exclusively by an increase in intracellular S1P 25. Despite this increase of the intracellular Ca2+ concentration after S1P treatment, TNF-α transcription and secretion are only observed when ionomycin is added simultaneously as a further stimulus. The synergistic effects of S1P and ionomycin are explained by the dependence of TNF-α transcription on the activation of the transcription factors AP1 and NF-AT, which together form a complex at the κ3 site of this cytokine promoter 12. Ionomycin but not S1P provokes the required extracellular Ca2+ influx necessary for the activation of the Ca2+-dependent phosphatase calcineurin and the dephosphorylation and translocation of NF-AT to the nucleus. In contrast, the AP1 components are fully activated by S1P alone, which is in line with its activation of the MAPK pathway. A similar observation has been made for sphingosylphosphocholine and the activation of AP1 in Swiss 3T3 fibroblasts resulting in enhanced proliferation 26.
Initially, S was described as a potent inhibitor of PKC isozymes in vitro if applied at a concentration of ∼100 μM. However, in cell cultures, specific effects of this lipid are already observed at concentrations <10 μM, resulting in both activation and inhibition of particular cellular functions 27 28. The diverse spectrum of these reactions as well as the 10-fold lower concentration to provoke those reactions a priori suggest further targets besides PKCs for this lipid. This is illustrated further by the fact that related cell types such as Th1 and Th2 cells respond in an opposite manner to S. The synthesis of IFN-γ is strongly inhibited in Th1 cells, whereas IL-4 production is augmented by S in Th2 cells. An inverse relationship is seen in these two cell types regarding DNA synthesis, which is repressed by S in Th2 cells but not in Th1 cells 29. Mast cells, although similar to Th2 cells with respect to cytokine profile, react with a strong inhibition of their cytokine transcription and secretion, mimicking the inhibition of IFN-γ in Th1 cells 30. Taken together, these data suggest that there must be fundamental cell type/subtype–specific differences that cannot be explained by our current knowledge of common signaling pathways in cell types such as PKCs, MAPKs, PI-3K, and others. Different constitutive and inducible SK activities in the different cell types and the presence and concentration of further sphingolipids with counterregulatory potentials may be responsible for the diverse outcome after S treatment.
In allergically activated mouse mast cells, it was identified that MAPK pathways are one of the common “protein-based signaling cascades” that are sensitive to high intracellular concentrations of S. Based on this finding and the concentration of S applied, we favor the idea that many of the inhibitory effects of S described in the literature are due to this potential to inhibit MAPKs and not due to their PKC inhibitory potential, which is usually seen at much higher concentrations. Already at the level of raf, a clear abrogation of signaling is observed leading to subsequent hypophosphorylation of downstream kinases and diminished phosphorylation and expression of transcription factors such as c-jun. This suggests that the inhibitory event is relatively proximal to the receptor, whereas the finding that degranulation, which is PI-3-kinase dependent, is unaffected rules out a central block at the Fc∈RI receptor. This assumption is in agreement with the finding by J.P. Kinet and colleagues that the S analogue d-l-threo-dihydrosphingosine has no influence on syk activity 10.
The secretion of S1P by IgE/Ag-activated mast cells adds this lipid to a variety of mediators that are released during an allergic reaction. By using a variety of inhibitors of different signaling cascades, such as Gö6976, FK506, dimethoxyviridine, and 4′,5,7-trihydroflavone, to investigate which processes S1P secretion is dependent on, only the latter substance showed any effect. However, the same drug also lowers endogenous S1P levels, therefore suggesting an indirect inhibition of S1P secretion (data not shown). The ability of S1P to activate besides mast cells, monocytes and endothelial cells, and to mediate lymphocyte infiltration implies that it augments the allergic reaction on a broader scale 25 31 32.
However, the control of signaling processes by two or more antagonistic lipids as exemplified by our study with S and S1P is not restricted to Fc∈RI triggering, mast cells, or this lipid combination (S, S1P), as demonstrated by a number of recent reports. Fas-initiated signaling depends on the balance of ceramide and S1P in T lymphocytes 9. The same regulatory principle applies for the ceramide- and detachment-induced apoptosis that can be reverted by S1P in the monocytic cell line U937 20 33. In HL-60 cells, this reversion was demonstrated to be due to a shift in the activation of the two MAPKs erk 1,2 and jnk 1,2. Although ceramide mediates the phosphorylation of the latter, S1P not only stimulates erk 1,2 but also represses jnk 1,2 in this specific cell type 20. This difference from the situation in CPII cells, where both branches of MAPKs are activated by S1P, indicates that a particular lipid has no specifically assigned function per se but that its action strongly depends on the context.
Acknowledgments
We thank L. Machado and E. Liehl for critical reading of the manuscript.
Footnotes
1used in this paper: BMMC, bone marrow–derived mouse mast cell; erk, extracellular signal regulatory kinase; jnk, c-jun NH2-terminal kinase; MAP, mitogen-activated protein; MAPK, MAP kinase; MBP, myelin basic protein; MEK, MAPK kinase; NF-AT, nuclear factor of activated T cell(s); PI-3K, phosphatidylinositol-3-kinase; PKC, protein kinase C; RBL, rat basophilic leukemia; S, sphingosine; S1P, sphingosine-1-phosphate; SK, S-kinase
References
- Dietrich J., Backstrom T., Lauritsen J.P., Kastrup J., Christensen M.D., von Bulow F., Palmer E., Geisler C. The phosphorylation state of CD3gamma influences T cell responsiveness and controls T cell receptor cycling. J. Biol. Chem. 1998;273:24232–24238. doi: 10.1074/jbc.273.37.24232. [DOI] [PubMed] [Google Scholar]
- Wang R., Zhang L., Yin D., Mufson R.A., Shi Y. Protein kinase C regulates Fas (CD95/APO-1) expression. J. Immunol. 1998;161:2201–2207. [PubMed] [Google Scholar]
- Szamel M., Appel A., Schwinzer R., Resch K. Different protein kinase C isoenzymes regulate IL-2 receptor expression or IL-2 synthesis in human lymphocytes stimulated via the TCR. J. Immunol. 1998;160:2207–2214. [PubMed] [Google Scholar]
- Hakomori S. Bifunctional role of glycosphingolipids. Modulators for transmembrane signaling and mediators for cellular interactions. J. Biol. Chem. 1990;265:18713–18716. [PubMed] [Google Scholar]
- Laulederkind S.J., Bielawska A., Raghow R., Hannun Y.A., Ballou L.R. Ceramide induces interleukin 6 gene expression in human fibroblasts. J. Exp. Med. 1995;182:599–604. doi: 10.1084/jem.182.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder J.J., Crane H.M., Xia J., Liotta D.C., Merrill A.H., Jr. Disruption of sphingolipid metabolism and stimulation of DNA synthesis by fumonisin B1. A molecular mechanism for carcinogenesis associated with Fusarium moniliforme . J. Biol. Chem. 1994;269:3475–3481. [PubMed] [Google Scholar]
- Bokoch G.M., Reilly A.M., Daniels R.H., King C.C., Olivera A., Spiegel S., Knaus U.G. A GTP-ase independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. J. Biol. Chem. 1998;273:8137–8144. doi: 10.1074/jbc.273.14.8137. [DOI] [PubMed] [Google Scholar]
- Dbaibo G.S., Wolff R.A., Obeid L.M., Hannun Y.A. Activation of a retinoblastoma-dependent signaling pathway by sphingosine. J. Biochem. 1995;310:453–459. doi: 10.1042/bj3100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvillier O., Rosenthal D.S., Smulson M.E., Spiegel S. Sphingosine-1-phosphate inhibits activation of caspases that cleave poly(ADP-ribose)polymerase and lamins during Fas- and ceramide-mediated apoptosis in Jurkat T-lymphocytes. J. Biol. Chem. 1998;273:2910–2916. doi: 10.1074/jbc.273.5.2910. [DOI] [PubMed] [Google Scholar]
- Choi O.H., Kim J.H., Kinet J.P. Calcium mobilization via sphingosine kinase in signalling by the Fc epsilon RI antigen receptor. Nature. 1996;380:634–636. doi: 10.1038/380634a0. [DOI] [PubMed] [Google Scholar]
- Su Y., Rosenthal D., Smulson M., Spiegel S. Sphingosine 1-phosphate, a novel signaling molecule, stimulates DNA binding activity of AP-1 in quiescent Swiss 3T3 fibroblasts. J. Biol. Chem. 1994;269:16512–16517. [PubMed] [Google Scholar]
- Prieschl E.E., Pendl G.G., Elbe A., Serfling E., Harrer N.E., Stingl G., Baumruker T. Induction of the TNFα promoter in the murine dendritic cell line DC18 and the murine mast cell line CPII is differently regulated. J. Immunol. 1996;157:2645–2653. [PubMed] [Google Scholar]
- Pendl G.G., Prieschl E.E., Harrer N.E., Baumruker T. Effects of phosphatidylinositol-3-kinase inhibitors on degranulation and gene induction of allergically triggered mouse mast cells. Int. Arch. Allergy Immunol. 1997;112:392–399. doi: 10.1159/000237486. [DOI] [PubMed] [Google Scholar]
- Prieschl E.E., Novotny V., Csonga R., Jaksche D., Elbe-Bürger A., Thumb W., Auer M., Stingl G., Baumruker T. A novel splice variant of the transcription factor Nrf1 interacts with the TNFα promoter and stimulates transcription. Nucleic Acids Res. 1998;26:2291–2297. doi: 10.1093/nar/26.10.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieschl E.E., Gouilleux-Gruart V., Walker C., Harrer N.E., Baumruker T. An NFAT-like transcription factor on mast cells is involved in IL5 gene regulation after IgE plus Ag stimulation. J. Immunol. 1995;154:6112–6119. [PubMed] [Google Scholar]
- Csonga R., Prieschl E.E., Jaksche D., Novotny V., Baumruker T. Common and distinct signaling pathways mediate the induction of TNFα and IL-5 in IgE plus antigen stimulated mast cells. J. Immunol. 1998;160:273–283. [PubMed] [Google Scholar]
- Rottem M., Hull G., Metcalfe D.D. Demonstration of differential effects of cytokines on mast cells derived from murine bone marrow and peripheral blood mononuclear cells. Exp. Hematol. 1994;22:1147–1155. [PubMed] [Google Scholar]
- Hirasawa N., Santini F., Beaven M.A. Activation of the mitogen-activated protein kinase/cytosolic phospholipase A2 pathway in a rat mast cell line. Indications of different pathways for release of arachidonic acid and secretory granules. J. Immunol. 1995;154:5391–5402. [PubMed] [Google Scholar]
- Olivera A., Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–560. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- Cuvillier O., Pirianov G., Kleuser B., Vanek P.G., Coso O.A., Gutkind S., Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- Lee M.J., Van Brocklyn J.R., Thangada S., Liu C.H., Hand A.R., Menzeleev R., Spiegel S., Hla T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- Novotny V., Prieschl E.E., Csonga R., Fabjani G., Baumruker T. Nrf1 in a complex with fosB, c-jun, junD, and ATF2 forms the AP1 component at the TNFα promoter in stimulated mast cells. Nucleic Acids Res. 1998;26:5480–5485. doi: 10.1093/nar/26.23.5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatomi Y., Ruan F., Hakomori S., Igarashi Y. Sphingosine-1-phosphatea platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood. 1995;86:193–202. [PubMed] [Google Scholar]
- Lee M.J., Evans M., Hla T. The inducible G protein-coupled receptor edg-1 signals via the G(i)/mitogen-activated protein kinase pathway. J. Biol. Chem. 1996;271:11272–11279. doi: 10.1074/jbc.271.19.11272. [DOI] [PubMed] [Google Scholar]
- Van Brocklyn J.R., Lee M.J., Menzeleev R., Olivera A., Edsall L., Cuvillier O., Thomas D.M., Coopman P.J.P., Thangada S., Liu C.H. Dual actions of sphingosine-1-phosphateextracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J. Cell Biol. 1998;13:229–240. doi: 10.1083/jcb.142.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A., Bittman R., Schmidt R.R., Spiegel S. Structural requirements of sphingosylphosphocholine and sphingosine-1-phosphate for stimulation of activator protein-1 activity. Mol. Pharmacol. 1996;50:451–457. [PubMed] [Google Scholar]
- Hannun Y.A., Bell R.M. Lysosphingolipids inhibit protein kinase Cimplications for the sphingolipidoses. Science. 1987;235:670–674. doi: 10.1126/science.3101176. [DOI] [PubMed] [Google Scholar]
- Shayman J.A. Sphingolipidsthe role in intracellular signaling and renal growth. J. Am. Soc. Nephrol. 1996;2:171–182. doi: 10.1681/ASN.V72171. [DOI] [PubMed] [Google Scholar]
- Tokura Y., Wakita H., Yagi H., Nishimura K., Furukawa F., Takigawa M. Th2 suppressor cells are more susceptible to sphingosine than Th1 cells in murine contact photosensitivity. J. Invest. Dermatol. 1996;107:34–40. doi: 10.1111/1523-1747.ep12297849. [DOI] [PubMed] [Google Scholar]
- Burd P.R., Rogers H.W., Gordon J.R., Martin C.A., Jayaraman S., Wilson S.D., Dvorak A.M., Galli S.J., Dorf M.E. Interleukin 3–dependent and –independent mast cells stimulated with IgE and antigen express multiple cytokines. J. Exp. Med. 1989;170:245–257. doi: 10.1084/jem.170.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatrov V.A., Lehmann V., Chouaib S. Sphingosine-1-phosphate mobilizes intracellular calcium and activates transcription factor NF-kappa B in U937 cells. Biochem. Biophys. Res. Commun. 1997;234:121–124. doi: 10.1006/bbrc.1997.6598. [DOI] [PubMed] [Google Scholar]
- Stam J.C., Michiels F., Kammen R.A., Moolenaar W.H., Collard J.G. Invasion of T-lymphoma cellscooperation between Rho family GTPases and lysophospholipid receptor signaling. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:4066–4074. doi: 10.1093/emboj/17.14.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K., Nakamura H., Oda T., Hirano T., Shimizu N., Utiyama H. Involvement of Mac-1-mediated adherence and sphingosine 1-phosphate in survival of phorbol ester-treated U937 cells. Biochem. Biophys. Res. Commun. 1998;244:745–750. doi: 10.1006/bbrc.1998.8328. [DOI] [PubMed] [Google Scholar]