Autoantigen-Specific B Cell Activation in FAS-Deficient Rheumatoid Factor Immunoglobulin Transgenic Mice (original) (raw)
Abstract
In systemic autoimmune disease, self-tolerance fails, leading to autoantibody production. A central issue in immunology is to understand the origins of activated self-reactive B cells. We have used immunoglobulin (Ig) transgenic mice to investigate the regulation of autoreactive B cells with specificity for self-IgG2a (the rheumatoid factor [RF] specificity) to understand how normal mice regulate RF autoantibodies and how this fails in autoimmune mice. We previously showed that normal mice do not tolerize the AM14 RF clone, nor do they appear to activate it. Here we show that in Fas-deficient autoimmune mice, the picture is quite different. RF B cells are activated to divide and secrete, but only when the autoantigen is present. Thus, B cells that are ignored rather than anergized in normal mice can be stimulated to produce autoantibody in Fas-deficient mice. This demonstrates a novel developmental step at which intact Fas–Fas ligand signaling is required to regulate B cells in order to prevent autoimmunity. These data also establish the relevance of ignorant self-specific B cells to autoantibody production in disease and prove that in the case of the RF specificity, the nominal autoantigen IgG2a is the driving autoantigen in vivo.
Keywords: autoimmunity, systemic lupus, B cell tolerance, autoantibody, IgG
Acharacteristic of systemic autoimmune diseases is the production of high-titer autoantibodies (autoAbs)1 to a variety of self-constituents 1. These autoAbs are important diagnostic markers of disease, and their patterns are specific for particular autoimmune diseases 1. They are also important because autoAbs can be pathogenic under certain circumstances 2 3 4. The pathogenic consequences of activated autoreactive B cells are not limited to autoAb production, as B cells also promote T cell activation 5.
Thus, it has been of great interest to understand the origins of autoreactive B cells in autoimmune animals and, conversely, how they are controlled in normal animals. It is possible that intrinsic B cell defects 6 7 8 leading to B cell hyperactivity 9 10 account for the production of autoAbs. In this view, autoAbs are the result of nonspecific B cell activation. On the other hand, an early clue to an important role of autoantigen (autoAg) in the genesis of such B cells in autoimmune animals came from the study of autoAb hybridomas. These were found to be somatically mutated and clonally related 11 12 13 14 15 16 17 18 19. In many cases, the somatic mutations had high ratios of replacement to silent mutations in the CDRs, and in some instances, mutations led to higher affinity for the nominal autoAg. These data were interpreted as evidence that autoAg played a critical role in driving the expansion and selection of autoreactive B cells. These interpretations were only indirect; the analysis of autoAb hybridomas could not provide direct proof that autoAg drives autoreactive B cells, and this issue remains controversial 20.
A second issue is at what stage tolerance must fail in order to allow for the developmental progression of self-reactive B cells. Transgenic (Tgic) mice have been invaluable tools in efforts to address this question. The earliest models, using model antigens such as hen egg lysozyme (HEL) or class I, revealed deletion, receptor editing, and anergy as basic mechanisms of B cell self-tolerance 21 22 23 24. When the anti-HEL or anti–class I models were crossed onto the Fas-deficient background, tolerance was generally found to be intact 25 26. Studies of model autoAgs do not permit a clear extrapolation to the situation in disease. This is because particular autoAgs, such as DNA, chromatin, or self-IgG are targets in autoimmune diseases 1; tolerance to many other autoAgs remains intact even in systemic autoimmune disease. Why these autoAgs are preferred targets is still unclear. Nonetheless, the regulation of autoreactive B cells with disease-related specificities must be unique and could not be readily predicted by the behavior of B cells specific for model autoAgs that are not actual disease targets.
Recognizing this, several groups have extensively studied Tgic mice expressing anti–single-stranded (ss)DNA and/or –double-stranded (ds)DNA 27 28 29 30 31 32 33 34 35 36 37 38. Such B cells may be regulated by receptor editing, deletion, and anergy, possibly depending on the fine specificity of the cell. In contrast to the artificial autoAg systems, anti-DNA B cells that use a 3H9 H chain transgene (Tg) are activated in Tgic mice on the MRL.Faslpr background 39. This is an extremely important observation because it demonstrates that in an autoimmune mouse, B cells with disease-related specificities may indeed be regulated differently from B cells specific for arbitrary “self” antigens. However, it is not yet clear how these activated DNA-specific B cells arise, whether by defeating central tolerance, anergy, or both.
The regulation of rheumatoid factors (RFs), another typical yet less-studied autoAb, may again be different. Our group has investigated a model based on an RF, AM14, that was originally isolated from an MRL.Faslpr mouse 40. Because AM14 binds only to IgG2a of the “a” allotype (IgG2aa), we have the opportunity to study the Tgic B cells in the presence and absence of autoAg in both normal and autoimmune-prone backgrounds. This capability is similar to the design of the model autoAg systems but is not possible in other Tgics expressing authentic autoAbs. We have previously shown that AM14 B cells are clonally ignorant. They are not tolerized by deletion, editing, or anergy in normal BALB/c mice, nor do they appear activated in the absence of intentional immunization 41.
We suggested based on these results that the precursors of autoAb-secreting cells were not necessarily tolerized in normal animals and became selectively activated in autoimmune animals due to increased propensity to activate the otherwise quiescent cells and/or a failure to downregulate them once activated. This hypothesis, inferred from the phenotype of normal animals, has never been tested in autoimmune mice. It remained possible that such cells would also be ignored in the autoimmune-prone animal. Alternatively, RF B cells might be activated in the autoimmune-prone background regardless of whether autoAg is present.
To investigate these possibilities, we crossed the separate H and L chain Tgs that comprise the AM14 RF specificity onto both the C57B6.MRL.Faslpr (B6/lpr: IgHb, Ag−) and B6/lpr/IgHa background. B6/lpr mice produce large numbers of autoAbs, including RF and antichromatin and have a mild autoimmune disease 7 42 43 44, owing chiefly to their Fas deficiency but also to some B6-derived background genes 45. The B6/lpr strain was a particularly attractive model, as congenic strains were available that both had B6/lpr/IgHa and lacked the autoAg 7. We studied the activation state of RF B cells and T cells in vivo in age-matched sets of congenic mice. These studies have provided insights into the role of autoAg in driving B cell autoimmunity, the identity of the RF Ag, where tolerance breaks down to permit autoAb production, and the role of Fas in this process.
Materials and Methods
Mice.
The following three strains of mice were constructed from our BALB/c-based Tgic lines: AM14Vh/B6/lpr (H chain Tgics), AM14Vκ/B6lpr, and AM14Vκ/B6/lpr/IgHa (L chain Tgics). These were derived by continuous backcrossing to either B6/lpr (originally obtained from The Jackson Laboratory) or B6/lpr/IgHa mice 7 (a gift of Dr. Robert Eisenberg, University of Pennsylvania, Philadelphia, PA). B6/lpr and B6/lpr/IgHa mice were also maintained by intercrossing at Yale University. At each generation, Tgic mice were identified by PCR (see below) for breeding to the next generation. At BC1, mice were typed for homozygosity for the Faslpr mutation by PCR 5, which was confirmed at BC2. The IgHa genotype was also confirmed at BC1 by an allele-specific PCR assay for IgG2aa versus IgG2ab. From BC4 (97% B6 genes) and beyond, AM14Vh/B6/lpr mice were crossed with AM14Vl/B6/lpr mice to create Ag− double-Tgic controls (HLb mice) or to AM14Vh/B6/lpr/IgHa mice to create Ag+ double-Tgic experimental mice (HLab). All other transgenotypes (H, H chain only; L, L chain only; and N, non-Tgic) were also obtained in these crosses and were analyzed as additional controls (see Results). Age-matched IgHb or IgHab mice 40 that were wild type at the Fas locus were available on the BALB/c background and were analyzed as controls. All mice were housed in the same room in our specific pathogen–free barrier colony.
PCR Genotyping.
PCR to genotype for H and L Tgs, IgH genotype, and the lpr mutation was performed as described 40. PCR to genotype the IgG2a locus was performed as previously described 46.
Antibodies.
The selection, preparation, and labeling of antibodies was as described 40 41. Anti-CD3–biotin was obtained from PharMingen.
Cell Isolation and FACS™ Analysis.
These were performed essentially as described 41 with the following modifications. Spleens were harvested, weighed, and then divided, with a portion being quick-frozen in OCT for later immunohistochemical analysis. The other portion was weighed again and then processed into a single-cell suspension. Red cells were then lysed with ammonium chloride/Tris solution. Cell number was determined by counting in a hemocytometer, and a corrected total number of cells in the spleen was derived by considering the fraction by weight of the total spleen that was used to create the cell suspension. In most experiments, cells were preincubated with saturating concentrations of 2.4G2 (rat anti–mouse FcR) to block nonspecific binding. Stained cells were analyzed on a FACSCalibur™ (Becton Dickinson Immunocytometry Systems). When possible, live gating using propidium iodide was used to exclude dead cells. When four labeled mAbs were used, forward and side scatter profiles were used to exclude most dead cells and RBCs. At least 30,000 events were collected for two- and three-color analysis, and 50,000 events were collected for four-color analysis.
ELISA and Enzyme-linked Immunospot Assay.
The assays were performed as described 41.
Statistics.
Data were not distributed normally, mandating the use of Mann-Whitney nonparametric tests (two-tailed) to compare groups. Tests were performed using StatView 4.5 (Abacus Concepts, Inc.).
Results
Protocol.
Age-matched cohorts of AM14 Tgic mice were established on the B6/lpr (IgHb) and B6/lpr/IgHa backgrounds. As these were generated by intercrossing H and L single-Tgic mice, all possible genotypes were created, among which H, HL, and N were extensively studied. In addition, similar age-matched cohorts were generated on the BALB/c (IgHa) and congenic CB.17 (IgHb) backgrounds. These strains were available as nonautoimmune, Fas-sufficient controls. Mice were allowed to age to 4–7 mo to allow spontaneous autoimmunity to develop, at which point they were killed and analyzed as described below.
Spleen Cell Number.
The number of total splenocytes was greater in the HLab Tgics (which had the AM14 autoAg) compared with the corresponding HLb Tgic mice, which lacked the autoAg (Table ; P = 0.0003). Interestingly, a similar difference was observed in the H chain Tgic mice (P = 0.0017). This was not due to differences between B6/lpr and B6/lpr/IgHa per se, as no such difference was seen in the N controls (Table ; P = 0.53). Notably, this difference was also not observed in the comparison between BALB/c and CB.17 HL Tgics of similar age (P = 0.76).
Table 1.
Summary of Total and RF Id+ B Cells in Spleen
| Transgene type | IgH allotype | Fas genotype | Total cells | P value | Idiotype-positive cells | P value |
|---|---|---|---|---|---|---|
| ×10−7 | ×10−5 | |||||
| HL | ab | lpr/lpr | 6.2 ± 0.17 | 0.0003 | 43.3 ± 1.20 | 0.0016 |
| HL | bb | lpr/lpr | 2.8 ± 0.08 | 12.8 ± 0.39 | ||
| H | ab | lpr/lpr | 8.8 ± 0.28 | 0.0017 | 43.1 ± 3.15 | 0.0076 |
| H | bb | lpr/lpr | 4.1 ± 0.16 | 6.4 ± 0.38 | ||
| N | ab | lpr/lpr | 10.4 ± 0.38 | 0.53 | ND | ND |
| N | bb | lpr/lpr | 12.9 ± 0.48 | ND | ||
| HL | ab | +/+ | 5.4 ± 0.15 | 0.76 | 164.2 ± 5.10 | 0.70 |
| HL | bb | +/+ | 5.5 ± 0.23 | 157.0 ± 5.91 |
Number and Percentage of AM14 Id Cells.
Percentages (Fig. 1) and numbers (Table ) of AM14 Id+ B cells were greater in the HLab Tgic mice compared with HLb mice. Again, H chain Tgic mice were similar in this regard. Indeed, many Hab mice had substantial percentages of Id+ B cells, whereas this was only rarely observed among Hb mice. Such cells are detectable only at low frequency in young (reference 40) or old BALB/c or CB.17 H Tgics (not shown). AM14 Id+ cells in Hab Tgics presumably represent cells with endogenous Vκ8 L chains that are identical to or resemble the germline-encoded Tgic Vκ8. This interpretation is supported by the fact that among LPS hybridomas isolated from BALB/c H chain Tgics, both of the Id+ cell lines that were isolated had the same endogenously derived Vκ8 sequence as the Tg L chain (Shlomchik, M., unpublished data). If indeed these Id+ cells in H-only mice reconstitute the RF specificity of the original AM14 HL pair, finding higher percentages in the Hab mice compared with the Hb mice would be consistent with an active, autoAg-driven process in which rare RF B cells are substantially expanded. RF ELISpot data (see below) is also consistent with this interpretation; however, confirmation of it will require isolation of the cells from Hab mice and sequencing of their L chains.
Figure 1.

Increased numbers of RF Id+ cells in spleens of Tgic mice with AutoAg. The y-axis shows on a logarithmic scale the calculated numbers of Id+ cells based on total cell counts and the percentage of Id+ cells determined by FACS™ analysis. Each diamond is an individual mouse, and the horizontal bar is the median. Mouse genotypes are shown on the category axis as follows: HL, both heavy and light Tgs; H, heavy Tg only; ab, heterozygous for IgHa and IgHb allotypes (i.e., autoAg present); bb, homozygous for the IgHb allotype (autoAg absent). Left panel, B6/lpr-derived strains; right panel, BALB/c-derived strains. P values for comparisons between each autoAg-positive versus -negative group are shown above the graphs. The numbers of mice analyzed are: 41 HLab, 31 HLb, 17 Hab, 17 Hb, 23 BALB/c HLab, and 19 CB.17 HLb.
Activation of AM14 Id Cells.
Larger numbers of AM14 Id+ cells in mice that express the autoAg (i.e., when AM14 is an autoAb) suggests that spontaneous activation may be occurring in this setting. This is indeed the case, as demonstrated by expression of CD44 47 48 and by the presence of Id+ ELISpots, which reflect differentiation into plasma cells. Fig. 2 shows CD44 expression in AM14 HL and H Tgics of both IgHa and IgHb allotypes. Many of the IgHa mice had a substantial fraction of CD44hi/Id+ cells, a phenotype only rarely observed in IgHb mice. These data are summarized over the entire cohort in Fig. 2 B as the ratio of CD44hi/CD44lo Id+ cells. There is a statistically significant difference in this measure between the IgHa and IgHb mice of both HL (P = 0.0009) and H (P = 0.0007) genotypes. A similar comparison of IgHa and IgHb mice on the BALB/c background did not reveal any differences, suggesting that accumulation of spontaneously activated RF B cells is dependent on Fas deficiency.
Figure 2.


Increased fraction of CD44hi Id+ B cells in spleens of Tgic mice with autoAg. (A) Representative FACS™ data from typical mice from a single experiment. Staining with anti–Id 4-44 is shown on the y-axis and anti-CD44 is on the x-axis. 4% contour plots are depicted. The percentage of total lymphocytes in the upper quadrants is shown. Left panels show HL+/+ mice that are wild-type at Fas (BALB/c-derived strains). Center and right panels are from B6/lpr-derived strains, with the center panels from HL Tgics and the right from H Tgics. As indicated, the top row is from mice that express the autoAg (IgHab), and the bottom row is from mice that lack the autoAg (IgHb). (B) Summary of FACS™ data as represented in A for all analyzed mice. The y-axis shows on a logarithmic scale the ratio of CD44hi (top right quadrant in A) to CD44lo (top left quadrant in A). Each diamond is an individual mouse, and the horizontal bar is the mean. The layout of the figure is otherwise as in Fig. 1. The numbers of mice analyzed are: 38 HLab, 29 HLb, 17 Hab, 16 Hb, 22 BALB/c HLab, and 19 CB.17 HLb.
Another measure of activation is differentiation into high rate Ab-secreting cells, which we measured by ELISpot. Similar to the CD44 data, a substantial proportion of both HLab and Hab mice had high numbers of Id+ ELISpots (Fig. 3). The median difference between IgHa and IgHb mice was 16-fold for HL Tgics (P < 0.0001) and 407-fold for H Tgics (P = 0.002). Again, there were no significant differences between BALB/c and CB.17 mice. To directly demonstrate this, on a subset of H chain mice, we measured RF ELISpots in both IgHa and IgHb B6/lpr mice. RF spots were much higher in most Hab than in Hb mice (P = 0.009). These findings directly demonstrate that in Ha mice, concurrent with the expansion and differentiation of Id+ cells, RF B cells are also expanded. In combination with our other observations, this suggests that most of the Id+ B cells are RF B cells as well.
Figure 3.

Increased numbers of Id+ ELISpots in spleens of Tgic mice with autoAg. The y-axis shows on a logarithmic scale the calculated numbers of Id+ ELISpots based on total cell counts and the number of Id+ ELISpots determined on a measured number of cells as described in Materials and Methods. The layout of the figure is otherwise as in Fig. 1. The numbers of mice analyzed are: 38 HLab, 29 HLb, 16 Hab, 16 Hb, 22 BALB/c HLab, and 19 CB.17 HLb.
Splenic Architecture.
To confirm the identification of AM14 Id+ B cells and antibody-forming cells (AFCs) by FACS™ and ELISpot and to identify their locations in vivo, frozen sections of spleens from representative Fas-deficient animals (HL or H) were prepared and stained for CD3 and Id expression (Fig. 4A–D). Spleens from age-matched HLab and HLb Fas-sufficient mice were similarly prepared as controls (Fig. 4E and Fig. F). Control mice of either allotype had normal splenic architecture, with most AM14 Id+ cells in B cell follicles and few AFCs in the red pulp. In contrast, large numbers of cells staining darkly with the 4-44 anti-Id were detected in Fas-deficient IgHa mice of both HL and H genotypes (Fig. 4A and Fig. C). Such cells were prominent in the red pulp, a site known to harbor plasma cells early after immunization in normal mice 49. These putative AFCs were more prominent in mice that also had large numbers of ELISpots (data not shown). In addition, such cells were also seen at the border of the T and B cell zones. In contrast to IgHa HL and H mice, darkly staining cells were rare to absent in all IgHb mice examined. In mice of both allotypes, cells staining moderately for the AM14 Id were seen scattered within the T cell zones, a site where B cells are rarely observed in normal mice (Fig. 4E and Fig. F). Others who have examined the splenic architecture of Fas-deficient mice have also noted the presence of B cells scattered in the T zones 50 51. Our data suggest that such localization is not necessarily dependent on B cell receptor engagement, as it was equally seen in the presence or absence of Ag. Finally, residual B cell zones, located at the edges of the T zones, containing lightly staining Id+ cells were noted in all IgHa mice. Some IgHb mice virtually lacked these B zones (Fig. 4 B), whereas in others they appeared less well developed (not shown). This immunohistological phenotype may underlie the observation that overall, IgHb mice have fewer AM14 Id+ B cells as determined by FACS™.
Figure 4.
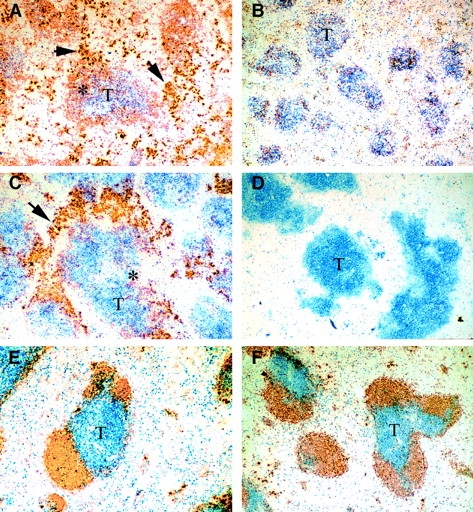
Histologic analysis of spleens. Frozen sections of spleens from representative mice were stained with anti-CD3 (blue) and 4-44 anti-Id (gold). A, C, and E are from IgHab mice, and B, D, and F are IgHb. A–D, B6/lpr mice; E and F, BALB/c background mice. A, B, E, and F are HL mice, whereas C and D show spleens from H-only Tgic mice. T cell zones are indicated by T; foci of darkly staining Id+ plasma cells are shown by arrows. Asterisks indicate T–B cell borders in which Id19 B cells are mixing with adjacent T cells. HLab (A) and Hab (C) are remarkable for large numbers of Id19 plasma cells as well as lighter-staining Id19 cells, both of which are absent in the IgHb congenics (B and D). Fas-sufficient mice (E and F) show normal histology, with well-defined T and B zones with most Id+ cells in the follicles and few plasma cells. There are no remarkable differences between the allotype congenics in E and F.
T Cell Activation.
B cells play a key role in the activation of T cells in MRL/lpr mice 5 52. Many activated T cells in Fas-deficient mice express the B220 antigen 53. This is an activation marker in normal mice 54, and the weight of data suggests that in Fas-deficient mice, B220+ T cells represent accumulating cells that had been activated and, in the presence of Fas, would have been eliminated after activation. Thus, the frequency of such T cells is a measure of the extent of T cell activation. In the course of our analysis of the B6/lpr cohorts, we determined the percentages of B220+ T cells as demonstrated in Fig. 5. The extent of T cell activation, as measured by B220+ T cell percentages, depends on whether the B cell autoAg is present. This was true in both the HL and H Tgics, but the IgH genotype did not have an effect on B220+ T cell numbers in N mice. Similar conclusions were reached based on B220+ T cell numbers (data not shown), which is not surprising because HLab mice also have greater numbers of splenocytes than HLb mice (Table ).
Figure 5.

Increased percentage of B220+ T cells in spleens of Tgic mice with the autoAg. B220+ T cells were identified by FACS™ analysis as described in Materials and Methods. The y-axis shows percentages from individual mice. Each diamond is an individual mouse, and the horizontal bar is the mean. The layout of the figure is otherwise as in Fig. 1. Only B6/lpr mice are shown in this figure, as the percentages in the BALB/c-derived strains were negligible. The numbers of mice analyzed are: 39 HLab, 29 HLb, 18 Hab, 17 Hb, 18 Nab, and 19 Nb.
Discussion
The origin of autoreactive B cells has been a question of great interest: are these cells that aberrantly escape central tolerance, are abnormally rescued from an anergic state, or are derived from clonally ignorant B cells whose activation or cell death is improperly regulated? Although several of these pathways may operate—depending on the autoAg, B cell affinity, and stage of disease—it is important to demonstrate experimentally which pathways are possible and thereby have appropriate models for further study. Here, we show that RF B cells that are clonally ignorant in BALB/c mice are no longer ignorant but rather are stimulated in the presence of their autoAg in the B6/lpr genetic background. We were able to demonstrate both increased numbers of autoreactive B cells and differentiation of these B cells into AFCs in mice that were genetically capable of expressing the autoAg, compared with congenic mice that lacked the autoAg.
The existence of low-affinity “natural” autoantibodies in normal individuals has been known for a long time 55 56. These B cells are likely to be clonally ignorant, as suggested by the phenotype of AM14 B cells in normal mice. What has remained controversial is whether such B cells had any relevance or relationship to pathologic autoantibodies produced in systemic autoimmune disease 56 57. Our results indicate that clonally ignorant cells can indeed be relevant precursors for pathologic autoantibodies.
Evidence that Proliferation and Differentiation of Autoreactive B Cells In Vivo Depends on AutoAg.
A second issue that our data addresses is the role of autoAg-stimulated versus nonspecific B cell activation in the induction of autoAbs. Several studies had shown that antibodies to a wide variety of antigens can be detected in autoimmune animals, suggesting that polyclonal activation was at work 58 59 60. On the other hand, the oligoclonal nature of B cell hybridomas specific for IgG, DNA, nucleosomes, or Sm isolated from autoimmune animals suggested an antigen-driven process 11 13 14 15 16 17 18 19. Notably, a nonrandom pattern of somatic mutations and the presence of certain mutations that increased the affinity for Ag suggested that Ag was playing a role in driving at least some autoimmune responses. These conflicting interpretations were based on indirect inferences. Moreover, neither argument could establish whether autoAg was required or was simply altering or exacerbating an underlying polyclonal process.
The data in this report provide more direct evidence that autoAg is required for the activation of autoreactive B cells. In the presence of autoAg, substantial accumulations of RF plasma cells were observed in the spleens of Tgic mice compared with those mice that lacked the autoAg. Remarkably, only in the presence of the autoAg did H chain Tgic mice efficiently select rare endogenous L chains that reconstruct both the idiotype and RF specificity of the original HL pair that comprised AM14. This phenomenon provides an additional strong argument for the role of autoAg in driving RF B cells.
These conclusions are based on direct ELISpot data and splenic histology; we did not systematically measure and do not present serum RF data that are at best an indirect measurement of B cell activation and differentiation. Serum RF data are widely gathered in humans, where such direct measurements as ELISpot are not feasible. In our system, serum RF levels would be influenced by Ag competition and increased clearance rates of immune complexes as well as by competition by IgG2a naturally occurring in serum. These confounding features would be present in IgHa mice but not in IgHb mice, precluding any meaningful comparison. Furthermore, it would be difficult in such assays to distinguish RF secreted by B cells expressing endogenous Ig genes. ELISpot and histology assays are not subject to these concerns.
The Nominal AutoAg IgG2a Is the In Vivo Antigen Driving RF B Cells.
These observations raise the related issue of the identification of the true autoAg. It has been controversial whether the nominal Ags assayed in vitro as targets of autoAbs are actually those that drive autoreactive B cell clones in vivo 61 62. Perhaps the strongest evidence in favor of nominal Ags as actual Ags came from a single study in which the clonality and specificity were simultaneously determined for a large number of hybridomas isolated from a single autoimmune mouse 63. Nearly all of the expanded clones could be assigned to one of a relative few nominal autoAb specificities, again suggesting that the nominal antigens were driving most of the clonal expansion. As congenic strains were compared in this study, the only important difference between the two strains is almost certainly the IgH locus allotype and, in particular, the presence or absence of IgG2aa, which is the only ligand for AM14 that is encoded by the IgH locus (Shlomchik, M., unpublished data). Thus, IgG2aa is sufficient in vivo to drive B cell clonal expansion and to recapitulate the original expansion that must have occurred in the MRL/lpr mouse from which AM14 was isolated as a hybridoma. No other autoAg on the B6/lpr (IgHb) background was capable of driving this activation.
T Cell Activation Is Promoted by the RF B Cell Antigen.
A surprising observation was that T cell activation, as reflected by the accumulation of B220+ T cells, was affected by the presence of the B cell Ag. This means that the extent of B cell activation of one particular clone was sufficient to affect the activation of polyclonal T cells. The mechanism for this is unclear but intriguing. It could be an indirect effect due to cytokines produced by B cells 64 65 66 67. More likely, it is a direct effect of T–B cell collaboration. Recently, our group has shown that the absence of B cells affects T cell activation 5. Sobel and colleagues also showed in mixed BM chimeras that Fas-deficient B cells are largely responsible for promoting T cell activation 52 and that cognate interactions are required for autoAb elicitation 68. Our recent data 69 demonstrate that this effect on T cell activation is independent of secreted antibody, again arguing for a direct, cognate interaction. To explain the effect of the B cell Ag on T cell activation we observed here, one would have to suppose that AM14 B cells could activate a substantial enough fraction of T cells to make this pathway apparent by bulk FACS™ analysis. It is unlikely that there is a high frequency of T cells specific for self-IgG2a, the AM14 antigen. However, RF B cells can take up immune complexes and present a wide variety of autoAgs, provided they were complexed with self-IgG 70. This is expected to be the case in B6/lpr mice, which produce a variety of IgG2a autoAbs, including antichromatin 68. Indeed, this feature of RF B cells—the ability to present many autoAgs to T cells and thus garner T cell help—may explain why RF is a predominant specificity in Fas-deficient mice 63 as well as in several autoimmune diseases. It is worth noting that although the origin of B220+ T cells in lpr mice is unclear, most “double-negative” T cells arise from a CD8+ T cell precursor 71 72. If the generation of B220+ T cells is related to the same process, then an effect of B cells on CD8+ T cells may be playing a role. In this regard, activated and memory phenotype CD8+ T cells fail to accumulate in MRL/lpr mice in the absence of B cells 5. Further work will be required to expand on this unexpected observation and to identify the T cells promoted by RF B cells and the autoAgs that are recognized.
Implications for the Role of Fas/FasL in Regulating Autoimmunity.
Fas deficiency is the major determinant of autoAb production in B6/lpr mice 44 45. Because of constraints and logistics of animal breeding as well as the desire to compare results to those previously obtained on the BALB/c background 40 41, we have only been able to compare Fas-deficient mice on the B6 background to Fas-sufficient mice on the BALB/c background. Thus, we cannot formally rule out a role for background genes in the B6 strain. However, this seems very unlikely, as neither strain background is associated with autoimmunity or RF production and especially because many of the phenotypes we show—high numbers of RF ELISpots, activated B cells at the T–B interface and in the red pulp, and accumulation of B220+ T cells—are distinctive to the Fas-deficient phenotype. With this caveat in mind, our results bear on how Fas normally prevents autoAb production. Fas is expressed throughout B cell ontogeny, including in developing B cells, newly anergized B cells, newly activated B cells, germinal center (GC) B cells, and plasma cells 73 74 75 76 77 78 79. In principle, Fas deficiency could be critical at multiple stages of B cell development and tolerance in promoting autoimmunity. Our data demonstrate that Fas deficiency need not act at early stages of tolerance (editing, deletion, and anergy) to promote autoreactivity. In the presence of functional Fas, AM14 B cells develop beyond these stages and are quiescent in the presence or absence of antigen. Fas must therefore play an important role in the regulation of RF B cells during or after autoAg-specific activation, because when Fas is deficient, these B cells expand, persist, and make autoAb, but only in the presence of autoAg. It is also possible that Fas is acting more indirectly, e.g., by causing increased IgG2a autoAg levels. In our case, this is doubtful, as the average serum IgG2a level in a small group of 4–5-mo-old HLab mice is 40 ± 19 μg/ml (± 1 SD), levels that are similar to those in BALB/c mice. More work needs to be done to pinpoint at which stage(s) after the initial Ag activation event Fas is playing a role. It is notable in this regard that although Fas is expressed in GC B cells, GC reactions appear relatively normal in Fas-deficient mice 80 81. We observed that RF Tgic B6/lpr mice accumulated large numbers of RF AFCs with few GCs (data not shown) and had a relative B lymphopenia. This raises the possibility that Fas on plasma cells is playing at least one critical role.
Fas deficiency is likely not the cause of human systemic autoimmunity. However, given the central role of Fas/FasL and possibly other homologues in the TNFR/TNF family in immune system homeostasis, it does seem likely that subtle defects in apoptotic pathways could underlie human disease 82 83. In this regard, pure Fas deficiency may be an excellent model for understanding the in vivo pathogenic mechanisms of such deficiencies. The overall impact of this idea will only be known when genes that promote lupus are identified 84 85 and the Fas and Fas-related signaling pathways are better elucidated.
AM14 B6/lpr Mice Have a Novel Phenotype Compared with Other AutoAb Tgic Mice.
Previous studies had investigated how autoimmune-prone genetic backgrounds affected the regulation of B cells that would normally be tolerant rather than ignorant. In the case of mice that edited or deleted B cells specific for model self-antigens such as Class I or HEL, it was generally found that regulation was intact even in autoimmune-prone backgrounds 25 26. These data suggested that Fas and Fas/MRL defects did not grossly impair central tolerance at this level for these autoAgs.
Regulation of B cells specific for either HEL or DNA that are anergic in normal mice has also been investigated on the B6/lpr or MRL/lpr backgrounds. In the case of HEL, induction of anergy was essentially intact 25. In related experiments, Rathmell et al. transferred anergic anti-HEL B cells along with activated CD4+ T cells and demonstrated that anergic cells are sensitive to elimination; this did not occur when the T cells were deficient in FasL, indicating a role for this pathway in the elimination of anergic cells when T cell help was also being delivered 86. This remains a potential pathway by which autoreactive B cells could escape regulation, although unmanipulated Fas- or FasL-deficient mice did not show gross defects in self-tolerance, as discussed above. The situation is different in our model, as the B cells are not anergic and should be capable of rescue by surface Ig cross-linking 87; thus, the Fas pathway must function during additional regulatory steps (see below).
The situation is more complex for anti-DNA B cells; their fate may depend on the specificity/affinity of the DNA-specific B cell. In normal mice, anti-ssDNA B cells appear anergic, albeit with a somewhat different phenotype from the anti-HEL anergic B cells 27 33. Some dsDNA-specific B cells may be subject to receptor editing/deletion 28 29 30 88, but others may persist in the periphery localized at the T–B interface, where they turn over rapidly 32 34. In autoimmune MRL/lpr mice Tgic for the 3H9 H chain, which can generate a variety of anti-DNA depending on the endogenous L chain 89, anti-DNA is seen in the serum and hybridomas secreting antinuclear Abs with homogenous nuclear staining are readily detected 31 39. These autoAbs may arise through several pathways; they may result from loss of central tolerance, though recent detection of anti-dsDNA B cells in BALB/c spleen by Roark et al. 34 suggests that rescue from anergy may also be possible. Recent work by Weigert and colleagues on anti-ssDNA site-directed Tgic mice also suggests that rescue from anergy in MRL/lpr mice can occur 90. As there cannot be an antigen-free anti-DNA Tgic, determination of the ontogeny of the autoreactive B cells and, in particular, proving that this is a specific process is less straightforward than in the RF system. In any case, the anti-DNA models represent a different scenario from ours in that the precursors of anti-DNA Abs are thought to escape from either deletion or anergy, whereas in the case of AM14, ignorant B cells are being positively selected by Ag.
Conclusion.
We have shown that B cells that would be ignored in a normal mouse are driven to activation, expansion, and secretion in an autoimmune-prone Fas-deficient mouse. This process is specific and requires the presence of autoAg as well as the Fas defect. Because this is the only autoAb Tgic system, to our knowledge, that demonstrates Ag-specific activation of defined, nontolerant mature B cells, it provides a unique opportunity to study the temporal and spatial course of the initiating events of Fas-deficient B cell autoimmunity and perhaps ultimately other scenarios of B cell autoimmunity. It may also shed light on the induction of autoreactive T cells and may be a system for their generation and study in vitro and in vivo.
Acknowledgments
We thank Hong Zou and Heather Schweiger for expert technical assistance. We thank Owen Chan, Joe Craft, Ann Haberman, Charles A. Janeway, Jr., Mark Mamula, and Martin Weigert for comments on the manuscript.
This work was supported by National Institutes of Health grant P01 AI36529.
Footnotes
1used in this paper: AFCs, antibody-forming cells; ds, double-stranded; ELISpot, enzyme-linked immunospot assay; GC, germinal center; HEL, hen egg lysozyme; N, nontransgenic; RF, rheumatoid factor; ss, single-stranded; Tg, transgene; Tgic, transgenic
References
- Tan E.M. Antinuclear antibodiesdiagnostic markers for autoimmune diseases and probes for cell biology. Adv. Immunol. 1989;44:93–151. doi: 10.1016/s0065-2776(08)60641-0. [DOI] [PubMed] [Google Scholar]
- Madaio M.P., Carlson J., Cataldo J., Ucci A., Migliorini P., Pankewycz O. Murine monoclonal anti-DNA antibodies bind directly to glomerular antigens and form immune deposits. J. Immunol. 1987;138:2883–2889. [PubMed] [Google Scholar]
- Raz E., Brezis M., Rosenmann E., Eilat D. Anti-DNA antibodies bind directly to renal antigens and induce kidney dysfunction in the isolated perfused rat kidney. J. Immunol. 1989;142:3076–3082. [PubMed] [Google Scholar]
- Lefkowith J.B., Gilkeson G.S. Nephritogenic autoantibodies in lupus. Arthritis Rheum. 1996;39:894–903. doi: 10.1002/art.1780390605. [DOI] [PubMed] [Google Scholar]
- Chan O., Shlomchik M.J. A new role for B cells in systemic autoimmunityB cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J. Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- Nemazee D., Guiet C., Buerki K., Marshak-Rothstein A. B lymphocytes from the autoimmune-prone mouse strain MRL/lpr manifest an intrinsic defect in tetraparental MRL/lpr DBA/2 chimeras. J. Immunol. 1991;147:2536–2539. [PubMed] [Google Scholar]
- Sobel E.S., Katagiri T., Katagiri K., Morris S.C., Cohen P.L., Eisenberg R.A. An intrinsic B cell defect is required for the production of autoantibodies in the lpr model of murine systemic autoimmunity. J. Exp. Med. 1991;173:1441–1449. doi: 10.1084/jem.173.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakkanaiah V.N., Sobel E.S., MacDonald G.C., Cheek R.L., Cohen P.L., Eisenberg R.A. B cell genotype determines the fine specificity of autoantibody in lpr mice. J. Immunol. 1997;159:1027–1035. [PubMed] [Google Scholar]
- Klinman D.M., Steinberg A.D. Proliferation of anti-DNA-producing NZB B cells in a non-autoimmune environment. J. Immunol. 1986;137:69–75. [PubMed] [Google Scholar]
- Eastcott J.W., Schwartz R.S., Datta S.K. Genetic analysis of the inheritance of B cell hyperactivity in relation to the development of autoantibodies and glomerulonephritis in NZB × SWR crosses. J. Immunol. 1983;131:2232–2239. [PubMed] [Google Scholar]
- Shlomchik M.J., Marshak-Rothstein A., Wolfowicz C.B., Rothstein T.L., Weigert M.G. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805–811. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- Shlomchik M.J., Aucoin A.H., Pisetsky D.S., Weigert M.G. Structure and function of anti-DNA antibodies derived from a single autoimmune mouse. Proc. Natl. Acad. Sci. USA. 1987;84:9150–9154. doi: 10.1073/pnas.84.24.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomchik M., Mascelli M.A., Shan H., Radic M.Z., Pisetsky D., Marshak-Rothstein A., Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J. Exp. Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion T.N., Bothwell A.L.M., Briles D.E., Janeway C.A., Jr. IgG anti-DNA antoantibodies within an individual autoimmune mouse are the products of clonal selection. J. Immunol. 1989;142:4269–4274. [PubMed] [Google Scholar]
- Bloom D.D., Davignon J.-L., Retter M.W., Shlomchik M.J., Pisetsky D.S., Cohen P.L., Eisenberg R.A., Clarke S.H. V region gene analysis of anti-Sm hybridomas from MRL/Mp-lpr/lpr. J. Immunol. 1993;150:1591–1610. [PubMed] [Google Scholar]
- Losman M.J., Fasy T.M., Novick K.E., Monestier M. Monoclonal autoantibodies to subnucleosomes from a MRL/Mp(-)+/+ mouseoligoclonality of the antibody response and recognition of a determinant composed for histones H2A, H2B, and DNA. J. Immunol. 1992;148:1561–1569. [PubMed] [Google Scholar]
- Randen I., Brown D., Thompson K.M., Hughes-Jones N., Pascual V., Victor K., Capra J.D., Forre O., Natvig J.B. Clonally related IgM rheumatoid factors undergo affinity maturation in the rheumatoid synovial tissue. J. Immunol. 1992;148:3296–3301. [PubMed] [Google Scholar]
- van Es J.H., Gmelig-Meyling F.H., van De Akker W.R., Aanstoot H., Derksen R.H., Logtenberg T. Somatic mutations in the variable regions of a human IgG anti-double-stranded DNA autoantibody suggest a role for antigen in the induction of systemic lupus erythematosus. J. Exp. Med. 1991;173:461–470. doi: 10.1084/jem.173.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler T.H., Fehr H., Kalden J.R. Analysis of immunoglobulin variable region genes from human IgG anti-DNA hybridomas. Eur. J. Immunol. 1992;22:1719–1728. doi: 10.1002/eji.1830220709. [DOI] [PubMed] [Google Scholar]
- Dunn-Walters D.K., Spencer J. Strong intrinsic biases towards mutation and conservation of bases in human IgVh genes during somatic hypermutation prevent statistical analysis of antigen selection. Immunology. 1998;95:339–345. doi: 10.1046/j.1365-2567.1998.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemazee D.A., Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class-I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- Hartley S.B., Crosbie J., Brink R.A., Kantor A.B., Basten A., Goodnow C.C. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- Tiegs S.L., Russell D.M., Nemazee D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley S.B., Cooke M.P., Fulcher D.A., Harris A.W., Cory S., Basten A., Goodnow C.C. Elimination of self-reactive B lymphocytes proceeds in two stagesarrested development and cell death. Cell. 1993;72:325–335. doi: 10.1016/0092-8674(93)90111-3. [DOI] [PubMed] [Google Scholar]
- Rathmell J.C., Goodnow C.C. Effects of the lpr mutation on elimination and inactivation of self-reactive B cells. J. Immunol. 1994;153:2831–2842. [PubMed] [Google Scholar]
- Rubio C.F., Kench J., Russell D.M., Yawger R., Nemazee D. Analysis of central B cell tolerance in autoimmune-prone MRL/lpr mice bearing autoantibody transgenes. J. Immunol. 1996;157:65–71. [PubMed] [Google Scholar]
- Erikson J., Radic M.Z., Camper S.A., Hardy R.R., Weigert M.G. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 1991;349:331–334. doi: 10.1038/349331a0. [DOI] [PubMed] [Google Scholar]
- Gay D., Saunders T., Camper S., Weigert M. Receptor editingan approach by autoreactive B cells to escape tolerance. J. Exp. Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Radic M.Z., Erikson J., Camper S.A., Litwin S., Hardy R.R., Weigert M. Deletion and editing of B cells that express antibodies to DNA. J. Immunol. 1994;152:1970–1982. [PubMed] [Google Scholar]
- Xu H., Li H., Suri-Payer E., Hardy R.R., Weigert M. Regulation of anti-DNA B cells in recombination-activating gene-deficient mice. J. Exp. Med. 1998;188:1247–1254. doi: 10.1084/jem.188.7.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roark J.H., Kuntz C.L., Nguyen K.A., Mandik L., Cattermole M., Erikson J. B cell selection and allelic exclusion of an anti-DNA Ig transgene in MRL-lpr/lpr mice. J. Immunol. 1995;154:4444–4455. [PubMed] [Google Scholar]
- Mandik-Nayak L., Bui A., Noorchashm H., Eaton A., Erikson J. Regulation of anti–double-stranded DNA B cells in nonautoimmune micelocalization to the T–B interface of the splenic follicle. J. Exp. Med. 1997;186:1257–1267. doi: 10.1084/jem.186.8.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K.A., Mandik L., Bui A., Kavaler J., Norvell A., Monroe J.G., Roark J.H., Erikson J. Characterization of anti-single-stranded DNA B cells in a non-autoimmune background. J. Immunol. 1997;159:2633–2644. [PubMed] [Google Scholar]
- Roark J.H., Bui A., Nguyen K.-A.T., Mandik L., Erikson J. Persistence of functionally compromised anti-double-stranded DNA B cells in the periphery of non-autoimmune mice. Int. Immunol. 1997;9:1615–1629. doi: 10.1093/intimm/9.11.1615. [DOI] [PubMed] [Google Scholar]
- Offen D., Spatz L., Escowitz H., Factor S., Diamond B. Induction of tolerance to an IgG autoantibody. Proc. Natl. Acad. Sci. USA. 1992;89:8332–8336. doi: 10.1073/pnas.89.17.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev A., Spatz L., Ray S., Diamond B. Lack of allelic exclusion permits autoreactive B cells to escape deletion. J. Immunol. 1994;153:3551–3556. [PubMed] [Google Scholar]
- Tsao B.P., Ohnishi K., Cheoutre H., Mitchell B., Teitell M., Mixter P., Kronenberg M., Hahn B. Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. J. Immunol. 1992;149:350–358. [PubMed] [Google Scholar]
- Pewzner-Jung Y., Friedmann D., Sonoda E., Jung S., Rajewsky K., Eilat D. B cell deletion, anergy, and receptor editing in “knock in” mice targeted with a germline-encoded or somatically mutated anti-DNA heavy chain. J. Immunol. 1998;161:4634–4645. [PubMed] [Google Scholar]
- Roark J.H., Kuntz C.L., Nguyen K.A., Caton A.J., Erikson J. Breakdown of B cell tolerance in a mouse model of systemic lupus erythematosus. J. Exp. Med. 1995;181:1157–1167. doi: 10.1084/jem.181.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomchik M.J., Zharhary D., Camper S., Saunders T., Weigert M. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int. Immunol. 1993;5:1329–1341. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- Hannum L.G., Ni D., Haberman A.M., Weigert M.G., Shlomchik M.J. A disease-related RF autoantibody is not tolerized in a normal mouseimplications for the origins of autoantibodies in autoimmune disease. J. Exp. Med. 1996;184:1269–1278. doi: 10.1084/jem.184.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren R.W., Sailstad D.M., Pisetsky D.S. Monoclonal rheumatoid factors from B6-lpr/lpr mice. Clin. Exp. Immunol. 1984;58:731–736. [PMC free article] [PubMed] [Google Scholar]
- Izui S., Kelley V.E., Masuda K., Yoshida H., Roths J.B., Murphy E.D. Induction of various autoantibodies by mutant gene lpr in several strains of mice. J. Immunol. 1984;133:227–233. [PubMed] [Google Scholar]
- Pisetsky D.S., Caster S.A., Roths J.B., Murphy E.D. lpr gene control of the anti-DNA antibody response. J. Immunol. 1982;128:2322–2325. [PubMed] [Google Scholar]
- Vidal S., Kono D.H., Theofilopoulos A.N. Loci predisposing to autoimmunity in MRL-Faslpr and C57BL/6-Faslpr mice. J. Clin. Invest. 1998;101:696–702. doi: 10.1172/JCI1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Shlomchik M.J. High affinity rheumatoid factor transgenic B cells are eliminated in normal mice. J. Immunol. 1997;159:1125–1134. [PubMed] [Google Scholar]
- Murakami S., Miyake K., June C.H., Kincade P.W., Hodes R.J. IL-5 induces a Pgp-1 (CD44) bright B cell subpopulation that is highly enriched in proliferative and Ig secretory activity and binds to hyaluronate. J. Immunol. 1990;145:3618–3627. [PubMed] [Google Scholar]
- Camp R.L., Kraus T.A., Birkeland M.L., Pure E. High levels of CD44 expression distinguish virgin from antigen-primed B cells. J. Exp. Med. 1991;173:763–766. doi: 10.1084/jem.173.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.J., Zhang J., Lane P.J., Chan E.Y., MacLennan I.C. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur. J. Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- Lieberum B., Hartmann K.U. Successive changes of the cellular composition in lymphoid organs of MRL-Mp/lpr/lpr mice during the development of lymphoproliferative disease as investigated in cryosections. Clin. Immunol. Immunopathol. 1988;46:421–431. doi: 10.1016/0090-1229(88)90061-x. [DOI] [PubMed] [Google Scholar]
- Jacobson B.A., Panka D.J., Nguyen K.A., Erikson J., Abbas A.K., Marshak-Rothstein A. Anatomy of autoantibody productiondominant localization of antibody-producing cells to T cell zones in Fas-deficient mice. Immunity. 1995;3:509–519. doi: 10.1016/1074-7613(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Sobel E.S., Kakkanaiah N., Schiffenbauer J., Reap E.A., Cohen P.L., Eisenberg R.A. Novel immunoregulatory B cell pathways revealed by lpr−+ mixed chimeras. J. Immunol. 1998;160:1497–1503. [PubMed] [Google Scholar]
- Davidson W.F., Dumont F.J., Bedigian H.G., Fowlkes B.J., Morse H.C.I. Phenotypic, functional, and molecular genetic comparisons of the abnormal lymphoid cells of C3H-lpr/lpr and C3H-gld/gld mice. J. Immunol. 1986;136:4075–4084. [PubMed] [Google Scholar]
- Renno T., Attinger A., Rimoldi D., Hahne M., Tschopp J., MacDonald H.R. Expression of B220 on activated T cell blasts precedes apoptosis. Eur. J. Immunol. 1998;28:540–547. doi: 10.1002/(SICI)1521-4141(199802)28:02<540::AID-IMMU540>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Casali P., Notkins A.L. CD5+ B lymphocytes, polyreactive antibodies and the human B-cell repertoire. Immunol. Today. 1989;10:364–368. doi: 10.1016/0167-5699(89)90268-5. [DOI] [PubMed] [Google Scholar]
- Coutinho A., Kazatchkine M.D., Avrameas S. Natural autoantibodies. Curr. Opin. Immunol. 1995;7:812–818. doi: 10.1016/0952-7915(95)80053-0. [DOI] [PubMed] [Google Scholar]
- Hentati B., Ternynck T., Avrameas S., Payelle-Brogard B. Comparison of natural antibodies to autoantibodies arising during lupus in (NZB × NZW)F1 mice. J. Autoimmun. 1991;4:341–356. doi: 10.1016/0896-8411(91)90029-c. [DOI] [PubMed] [Google Scholar]
- Klinman D.M., Steinberg A.D. Systemic autoimmune disease arises from polyclonal B cell activation. J. Exp. Med. 1987;165:1755–1760. doi: 10.1084/jem.165.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman D.M., Eisenberg R.A., Steinberg A.D. Development of the autoimmune B cell repertoire in MRL-lpr/lpr mice. J. Immunol. 1990;144:506–511. [PubMed] [Google Scholar]
- Klinman D. Similarities in B cell repertoire development between autoimmune and aging normal mice. J. Immunol. 1992;148:1353–1358. [PubMed] [Google Scholar]
- Ishigatsubo Y., Igarashi T., Ohno S., Ueda A., Okubo T., Klinman D.M. Cross-reactivity of IgM- and IgG-secreting B cells in autoimmune mice. Arthritis Rheum. 1993;36:1003–1006. doi: 10.1002/art.1780360718. [DOI] [PubMed] [Google Scholar]
- Shoenfeld Y., George J. Induction of autoimmunity. A role for the idiotypic network. Ann. NY Acad. Sci. 1997;815:342–349. doi: 10.1111/j.1749-6632.1997.tb52080.x. [DOI] [PubMed] [Google Scholar]
- Shan H., Shlomchik M.J., Marshak-Rothstein A., Pisetsky D.S., Litwin S., Weigert M.G. The mechanism of autoantibody production in an autoimmune MRL/lpr mouse. J. Immunol. 1994;153:5104–5120. [PubMed] [Google Scholar]
- Ware C.F., Crowe P.D., Grayson M.H., Androlewicz M.J., Browning J.L. Expression of surface lymphotoxin and tumor necrosis factor on activated T, B, and natural killer cells. J. Immunol. 1992;149:3881–3888. [PubMed] [Google Scholar]
- Spencer N.F.L., Daynes R.A. IL-12 directly stimulates expression of IL-10 by CD5+ B cells and IL-6 by both CD5+ and CD5− B cellspossible involvement in age-associated cytokine dysregulation. Int. Immunol. 1997;9:745–754. doi: 10.1093/intimm/9.5.745. [DOI] [PubMed] [Google Scholar]
- Fu Y.X., Huang G., Wang Y., Chaplin D.D. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin α–dependent fashion. J. Exp. Med. 1998;187:1009–1018. doi: 10.1084/jem.187.7.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V., Famiglietti S., Lacaud G., Lang P., Rider J.E., Kay B.K., Cambier J.C., Nemazee D. Antigens varying in affinity for the B cell receptor induce differential B lymphocyte responses. J. Exp. Med. 1998;188:1453–1464. doi: 10.1084/jem.188.8.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel E.S., Kakkanaiah V.N., Kakkanaiah M., Cheek R.L., Cohen P.L., Eisenberg R.A. T-B collaboration for autoantibody production in lpr mice is cognate and MHC-restricted. J. Immunol. 1994;152:6011–6016. [PubMed] [Google Scholar]
- Chan O.T., Hannum L.G., Haberman A.M., Madaio M.P., Shlomchik M.J. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosnek E., Lanzavecchia A. Efficient and selective presentation of antigen–antibody complexes by rheumatoid factor B cells. J. Exp. Med. 1991;173:487–489. doi: 10.1084/jem.173.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado M.A., Eisenberg R.A., Roper E., Cohen P.L., Kotzin B.L. Greatly reduced lymphoproliferation in lpr mice lacking major histocompatibility complex class I. J. Exp. Med. 1995;181:641–648. doi: 10.1084/jem.181.2.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohteki T., Iwamoto M., Izui S., Macdonald H.R. Reduced development of CD4−8−B220+ T cells but normal autoantibody production in lpr/lpr mice lacking major histocompatibility complex class I molecules. Eur. J. Immunol. 1995;25:37–41. doi: 10.1002/eji.1830250108. [DOI] [PubMed] [Google Scholar]
- Daniel P.T., Krammer P.H. Activation induces sensitivity toward APO-1 (CD95)-mediated apoptosis in human B cells. J. Immunol. 1994;152:5624–5632. [PubMed] [Google Scholar]
- Onel K.B., Tucek-Szabo C.L., Ashany D., Lacy E., Nikolic-Zugic J., Elkon K.B. Expression and function of the murine CD95/FasR/APO-1 receptor in relation to B cell ontogeny. Eur. J. Immunol. 1995;25:2940–2947. doi: 10.1002/eji.1830251034. [DOI] [PubMed] [Google Scholar]
- Mandik L., Nguyen K.-A., Erikson J. Fas receptor expression on B-lineage cells. Eur. J. Immunol. 1995;25:3148–3154. doi: 10.1002/eji.1830251124. [DOI] [PubMed] [Google Scholar]
- Watanabe D., Suda T., Nagata S. Expression of Fas in B cells of the mouse germinal center and Fas-dependent killing of activated B cells. Int. Immunol. 1995;7:1949–1956. doi: 10.1093/intimm/7.12.1949. [DOI] [PubMed] [Google Scholar]
- Wang J., Taniuchi I., Maekawa Y., Howard M., Cooper M.D., Watanabe T. Expression and function of Fas antigen on activated murine B cells. Eur. J. Immunol. 1996;26:92–96. doi: 10.1002/eji.1830260114. [DOI] [PubMed] [Google Scholar]
- Nishiuchi R., Yoshino T., Matsuo Y., Sakuma I., Cao L., Seino Y., Takahashi K., Akagi T. The Fas antigen is detected on immature B cells and the representative cell lines show Fas-mediated apoptosis. Br. J. Haematol. 1996;92:302–307. doi: 10.1046/j.1365-2141.1996.d01-1463.x. [DOI] [PubMed] [Google Scholar]
- Spets H., Georgii-Hemming P., Silajason J., Nilsson K., Jernberg-Wiklund H. Fas/APO-1 (CD95)-mediated apoptosis is activated by interferon-γ and interferon-α in interleukin-6 (IL-6)-dependent and IL-6-independent multiple myeloma cell lines. Blood. 1998;92:2914–2923. [PubMed] [Google Scholar]
- Smith K.G., Nossal G.J., Tarlinton D.M. Fas is highly expressed in the germinal center but is not required for regulation of the B-cell response to antigen. Proc. Natl. Acad. Sci. USA. 1995;92:11628–11632. doi: 10.1073/pnas.92.25.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Zheng B., Dal Porto J., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. IV. Affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J. Exp. Med. 1995;182:1635–1644. doi: 10.1084/jem.182.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Edwards C.K.I., Yang P., Wang Z., Bluethmann H., Mountz J.D. Greatly accelerated lymphadenopathy and autoimmune disease in lpr mice lacking tumor necrosis factor receptor 1. J. Immunol. 1996;156:2661–2665. [PubMed] [Google Scholar]
- Mountz J.D., Wu J., Cheng J., Zhou T. Autoimmune disease-a problem of defective apoptosis. Arthritis Rheum. 1994;37:1415–1420. doi: 10.1002/art.1780371002. [DOI] [PubMed] [Google Scholar]
- Kotzin B.L. Susceptibility loci for lupusa guiding light from murine models? J. Clin. Invest. 1997;99:557–558. doi: 10.1172/JCI119194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan C., Alas E., Morel L., Yang P., Wakeland E.K. Genetic dissection of SLE pathogenesis. J. Clin. Invest. 1998;101:1362–1372. doi: 10.1172/JCI728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell J.C., Cooke M.P., Ho W.Y., Grein J., Townsend S.E., Davis M.M., Goodnow C.C. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376:181–184. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- Rothstein T.L., Wang J.K., Panka D.J., Foote L.C., Wang Z., Stanger B., Cui H., Ju S.T., Marshak-Rothstein A. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 1995;374:163–165. doi: 10.1038/374163a0. [DOI] [PubMed] [Google Scholar]
- Chen C., Nagy Z., Radic M.Z., Hardy R.R., Huszar D., Camper S.A., Weigert M. The site and stage of anti-DNA B-cell deletion. Nature. 1995;373:252–255. doi: 10.1038/373252a0. [DOI] [PubMed] [Google Scholar]
- Radic M.Z., Mascelli M.A., Erikson J., Shan H., Weigert M. Ig H and L chain contributions to autoimmune specificities. J. Immunol. 1991;146:176–182. [PubMed] [Google Scholar]
- Brard F., Shannon M., Prak E.L., Litwin S., Weigert M. Somatic mutation and light chain rearrangement generate autoimmunity in anti–single-stranded DNA transgenic MRL/lpr mice. J. Exp. Med. 1999;190:691–704. doi: 10.1084/jem.190.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]