Selection at Multiple Checkpoints Focuses VH12 B Cell Differentiation toward a Single B-1 Cell Specificity (original) (raw)
Abstract
Phosphatidyl choline (PtC)-specific B cells segregate to the B-1 subset, where they comprise up to 10% of the B-1 repertoire. About half express VH12 and Vκ4/5H and are restricted in VHCDR3. We have previously reported that anti-PtC VHCDR3 is enriched among VH12-expressing cells by selective elimination of pre-B cells. We report here a bias for Vκ4/5H expression among VH12-expressing B cells, even among those that do not bind PtC and are not B-1. This is due in part to an inability of VH12 to associate with many light (L) chains but must also be due to a selective advantage in survival or clonal expansion in the periphery for Vκ4/5H-expressing cells. Thus, the bias for Vκ4/5H expression is independent of PtC binding, and, as segregation to B-1 occurs after Ig gene expression, it precedes segregation to the B-1 subset. In 6-1 mice, splenic B-1 cells reside in follicles but segregate to follicles distinct from those that contain B-2 cells. These data indicate that selection at multiple developmental checkpoints ensures the co-expression of an anti-PtC VHCDR3 and L chain in a high frequency of VH12 B cells. This focus toward specificity for PtC facilitates the development of a large anti-PtC B-1 repertoire.
Keywords: B-0 cells, B-1 cells, follicles, phosphatidyl choline, heavy and light chain association
The B-1 cells comprise a distinct subset of the B cell repertoire in normal mice. For example, B-1 cells are the predominant B cell type in the peritoneum, are infrequent in the spleen, and are generally absent from lymph nodes and bone marrow, whereas conventional B-2 cells are the predominant B cell type in both spleen and lymph node 1 2. In addition, whereas most conventional B cells are small, resting, naive B cells, B-1 cells have characteristics of activated cells; they are larger and more granular 2, they express activated signal transducer and activator of transcription 3 (STAT-3) in the peritoneum 3, and in both the spleen and peritoneum they are resistant to tolerance induction by anti-Ig 4. They also express VH and Vκ repertoires that differ considerably from those of conventional B cells 5. These and other differences suggest that conventional and B-1 cells have different roles in the immune system.
An intriguing feature of B-1 cells is their unusual repertoire. High frequencies of B-1 cells are polyreactive and autoreactive. These include B cells specific for single-stranded DNA, IgG (rheumatoid factor), and phosphatidyl choline (PtC),1 a common membrane phospholipid 6 7 8 9, all of which are rare among B-2 cells 8 10. The B-1 subset also includes B cells specific for bacterial carbohydrate antigens and phosphoryl choline 11 12, prompting suggestions that B-1 cells are involved in T cell–independent responses to common bacterial antigens.
How B cells of certain specificities segregate to one subset or the other is unknown but is likely to be a function of B-1 and B-2 cell origins. One hypothesis (the lineage hypothesis) to explain B-1 and B-2 cell origins posits that they derive from stem cells committed to producing only B cells of one subset or the other 13 14 15. It is based primarily on cell transfer experiments indicating that bone marrow and fetal liver are differentially able to restore the B-1 cell subset in lethally irradiated mice. We and others 16 17 have proposed the induced differentiation hypothesis that posits a single B cell lineage and that, upon exposure to certain antigens, B-2 cells differentiate to B-1 cells. To reflect the ability of B-2 cells to differentiate to B-1, the former have been referred to as B-0. Thus, B-2 cells of the lineage hypothesis and B-0 cells of the induced differentiation hypothesis are equivalent and in this report are referred to as B-0. This hypothesis was prompted by the observation that anti-IgM and IL-6 can induce splenic conventional B cells to acquire a B-1 cell phenotype in vitro 16. It is supported by our observations using transgenic (Tg) mice that PtC-specific B cells segregate to the B-1 subset after Ig gene rearrangement and by the finding that these Tg B cells are B-0 under circumstances in which signals initiated by the B cell receptor (BCR) are blocked 10 18.
To understand segregation to the B-1 subset, we have followed the differentiation of PtC-specific B cells, a population that accounts for 5–10% of the normal B-1 repertoire 8. They express predominantly one of two VH/Vκ combinations, VH12/Vκ4/5H and VH11/Vκ9, and are restricted in VH CDR3 19. Among VH12 anti-PtC B cells, the CDR3 is made up of 10 amino acids with an invariant glycine in the fourth position and a tyrosine encoded by JH1 in the fifth position, a motif designated 10/G4. These restrictions indicate that antigen-driven clonal expansion is responsible for the large number of B-1 cells of this specificity in normal mice 8.
The 10/G4 CDR3 is enriched at the pre-B cell stage 20. This occurs by the elimination of most VH12 pre-BII cells that do not have a 10/G4 CDR3 sequence (non-10/G4) during the transition from pre-BI to pre-BII. As this coincides with the expression of the pre-BCR, we have proposed a positive selection mechanism to account for this differential survival. 10/G4 pre-BCRs bind ligand and transduce a signal to turn off ongoing programmed cell death, whereas non-10/G4 pre-B cells are unable to bind ligand and fail to turn off programmed cell death. Whatever the mechanism, selection at the pre-B cell stage enriches for 10/G4 B cells that will ultimately contribute to the PtC-specific B-1 repertoire.
We demonstrate in this report that splenic VH12 B cells expressing Vκ4/5H are enriched, even among non-PtC–binding VH12 B cells. This, together with the enrichment for 10/G4 VH12 rearrangements at the pre-BII cell stage, ensures that a high proportion of VH12-expressing B cells bind PtC and differentiate to B-1. We suggest that this unprecedented developmental selection has evolved to ensure that a high percentage of VH12 B cells bind PtC and are present in every individual, underscoring their importance to the survival of the individual.
Materials and Methods
Mice.
VH12 Tg mice (6-1) were previously generated 10 and maintained in our pathogen-free animal facility at the University of North Carolina (UNC) by backcrossing to C.B17 mice. Offspring carrying the transgene were identified by PCR of tail genomic DNA as previously described 10. Mice homozygous for the deletion of the κ locus were provided by GenPharm International 21 and bred to 6-1 mice to obtain 6-1/κ−/− mice.
Sorting and Vκ Repertoire Determination.
The κ L chain repertoire was determined for subsets of PtC-binding and -nonbinding lymphocytes from 6-1 mice. Splenic lymphocytes were stained with anti-B220–PE antibodies and liposome-encapsulated carboxyfluorescein, and the B220+ cells that were PtCbri, PtCint, and PtCneg were sorted on a MoFlo high speed sorter (Cytomation, Inc.). Total RNA was extracted from ∼106 sorted cells using TRIzol (Life Technologies, Inc.) according to the manufacturer's protocol. The RNA was subjected to RT-PCR using 5′ RACE (rapid amplification of cDNA ends; version 2.0; Life Technologies, Inc.). For reverse transcription, we used a GSP1 custom primer, GGGGTAGAAGTTGTT, that anneals to the Cκ encoding sequence ∼80 bases 3′ of the J–Cκ junction. For the PCR, we used the 5′ RACE anchor primer and a custom GSP2 primer, CAUCAUCAUCAUCTGAGGCACCTCCAGATGTTA, that anneals to a region of the Cκ sequence 50 bases from the J–C junction. 40 cycles of amplification were performed. The conditions for the PCR were 94°C for 1 min, annealing at 55°C for 1 min, and primer extension at 72°C for 1.5 min. The final extension was at 72°C for 5 min. The amplification product was cloned into the pAMP1 vector using the CloneAmp® pAMP1system (Life Technologies, Inc.) according to the manufacturer's protocol. Plasmid DNA was isolated from randomly picked colonies and sequenced using the Sequenase II kit (Stratagene, Inc.) or the UNC Automated DNA Sequence Facility. The oligonucleotide used to prime sequencing was GGCTCTGACTAGATCTGCAAGAGAT. Sequence comparisons and analysis used DNASIS 2.5 software and the BLAST (Basic Local Alignment Search Tool) sequence search facility (GenBank).
For the sorting of B cells for adoptive transfer, spleen cells from 6-1 mice were stained for B220 and CD23 and sorted for B220+CD23− cells. Sorting was done using the MoFlo high speed sorter (Cytomation, Inc.). Approximately 3–5 × 106 cells were transferred intravenously to unirradiated C.B17 mice. Spleens were taken after 24 h and after 7 d for sectioning and analysis by immunofluorescence microscopy as described below.
Flow Cytometry and Immunofluorescence Microscopy.
The antibodies used for flow cytometry and the method for staining were as previously described 10 18. The cells were analyzed using a FACScan™ (Becton Dickinson) with hardware interface and acquisition and analysis software from Cytomation, Inc. All data represent cells that fall within the lymphocyte gate determined by forward and 90° light scatter. All contour plots are 5% probability.
For immunofluorescence microscopy, spleens were imbedded in TBS compound (Triangle Biomedical Sciences) and flash frozen in liquid nitrogen and 2-methylbutane. Frozen sections were air dried and fixed in acetone for 2 min and subsequently washed with 1× PBS. Blocking was performed for 1 h at room temperature with normal rat and mouse serum. The first step staining was performed for 1 h at room temperature with anti-IgMa (or -IgMb)–FITC and anti-CD23–biotin. After incubation, sections were washed two times with 1× PBS and then stained with anti-CD3–PE and streptavidin–Cy5, washed with PBS, and mounted in Fluoromount G (Southern Biotechnology Associates, Inc.). Slides were examined with a Leica TCS-NT confocal laser scanning microscope (Leica Inc.) equipped with argon and helium-neon lasers. Photomultiplier tube voltages and laser powers were set to eliminate the background signal given by the IgG isotype and streptavidin–Cy5-only controls. Images from three different fluorescent channels were recorded simultaneously. Image processing was performed with the Leica TCS-NT proprietary software and Adobe Photoshop (Adobe Systems, Inc.).
H and L Chain Association.
Gene transfections were done as described previously 22. In brief, μ expression vectors were transfected into L chain–only hybridoma cell lines. These hybridoma lines were J558L (λ), 4A9 (VκRF), 2-12 (Vκ31), 1E5 (Vκ8), D35.2 (Vκ8), and CH12.2b4 (Vκ10) 22. An H chain loss mutant of the anti-Sm hybridoma 1-8C2 23, provided by M. Borrero (UNC), was used to test association with Vκ1A L chains. Vκ4/5H and Vκ21C L chain–only cell lines were not available. Therefore, we cotransfected into P3-X63-Ag8.653 myeloma cells with the 10/G4 or 2-12 expression constructs with L chain expression vectors containing Vκ4/5H or Vκ21C rearrangements as described 22. To test whether a complete Ig molecule was formed, supernatant was subjected to ELISA using microtiter plates coated with polyclonal goat anti–mouse μ (Southern Biotechnology Associates, Inc.) and alkaline phosphatase–labeled polyclonal goat anti–mouse κ (Southern Biotechnology Associates, Inc.) to develop the reaction. In those cases where Ig secretion was not detected, the production of H and L chains was confirmed by ELISA using cell lysates and the polyclonal goat anti–mouse μ– or polyclonal goat anti–mouse κ-coated plates as above. The former were developed with phosphatase-labeled polyclonal goat anti–mouse μ to detect H chain, and the latter were developed with phosphatase-labeled goat anti–mouse κ to detect L chain. OD readings were determined with an automated plate reader (Emax; Molecular Devices).
Results
The Phenotype of the Splenic B Cell Subpopulations in 6-1 Mice.
We have previously described the PtC-specific B-1 cells of VH12 Tg (6-1) mice 10. As a result of antigen-driven clonal expansion, the number of these cells is considerable, comprising 60–90% of the splenic B cell population in adults 10. They can be detected by staining with a liposome probe that contains PtC as a membrane component and that encapsulates carboxyfluorescein 8. These cells stain brightly with liposomes (PtCbri) and, as described previously 10 18, are B220lowIgMhighCD23−, and most express CD5 and CD43 (Fig. 1).
Figure 1.
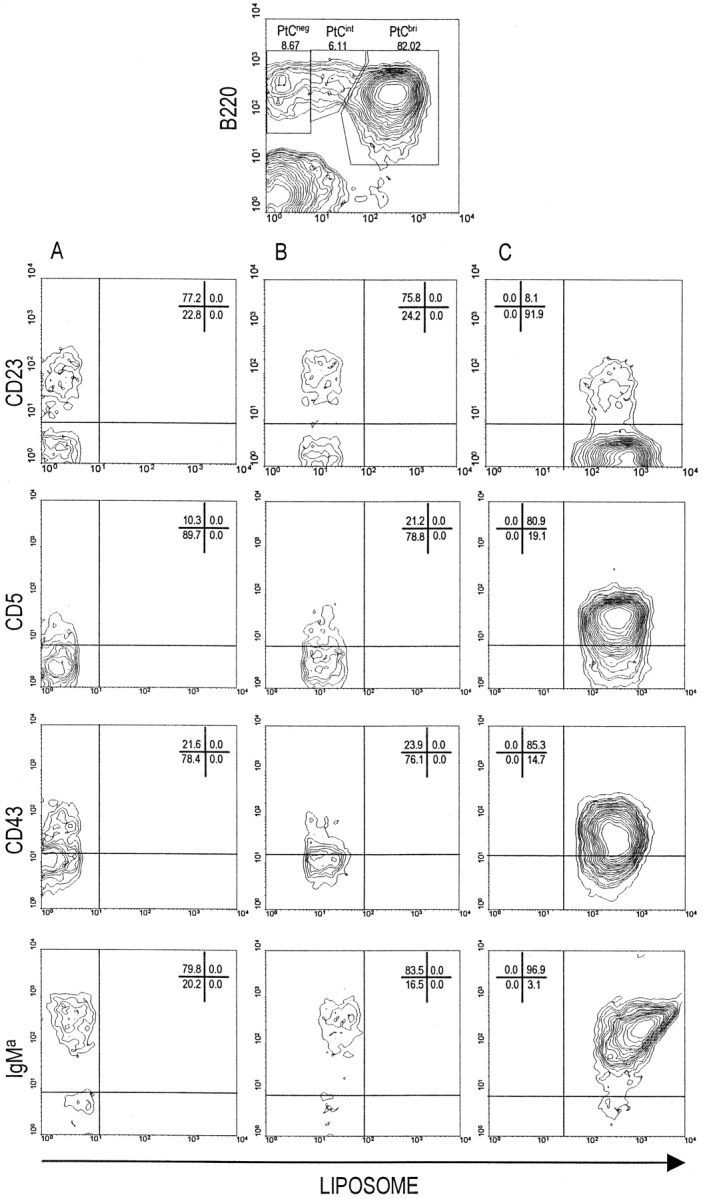
The phenotype of PtCneg, PtCint, and PtCbri cells of 6-1 mice. 6-1 splenic B cells were stained for liposome binding, B220, and CD23, CD5, CD43, or IgMa. Shown is the histogram for B220 versus liposomes, and the gates used to identify the three populations of B cells are shown at the top of the figure. In columns A, B, and C is the phenotypic analysis of PtCneg, PtCint, and PtCbri cells, respectively. For each mouse, 7–8 × 107 spleen cells were stained and 50,000 cells per sample were acquired on the flow cytometer.
Based on the liposome staining, there are at least two other B cell populations in 6-1 mice (Fig. 1), cells that do not stain with liposomes (PtCneg) and cells that have an intermediate level of liposome staining (PtCint). The PtCneg and PtCint populations are about equal in size and together account for 10–30% of splenic B cells. Most cells of both populations are B-0, i.e., CD23+CD5−CD43−B220high (Fig. 1). Some cells of both populations are CD23−. This population is likely to include immature B cells but may also include B-1 cells, as the PtCneg and PtCint populations appear to include cells that express CD43. Inasmuch as there are essentially no 6-1 splenic B cells that express IgMb 10, we attribute the differences in liposome binding and segregation to the B-0 or B-1 subsets to differences in the L chain.
L Chain Gene Use by 6-1 Splenic B Cells.
The Vκ repertoire of 6-1 splenic B cells was determined independently for sorted PtCneg, PtCint, and PtCbri B cell populations. mRNA was isolated from each population and used in a κ-specific RT-PCR. cDNA clones were generated, and randomly selected clones were sequenced to identify the Vκ and Jκ gene segments. Sequence analysis indicates remarkable similarities in the Vκ repertoire among these populations.
All but two of the cDNA clones from the PtCbri B-1 cells and three of the clones from the PtCint B-0 cells are of the identical Vκ gene (designated Vκ4/5H; reference 24) (Fig. 2C and Fig. D). This is the same Vκ gene expressed by PtC-specific lymphomas and peritoneal B-1 cell hybridomas 19. In contrast, the Vκ genes cloned from the PtCneg population are more heterogeneous (Fig. 2 A). 57% of the repertoire consists of non-Vκ4/5 genes from 12 Vκ groups. The remaining 43% are Vκ4/5 genes, and of these, 68% are Vκ4/5H (Fig. 2 B). Combining the PtCneg and PtCint B-0 populations, we estimate that 66% of B-0 cells use a member of the Vκ4/5 group and 59% use Vκ4/5H. Such a bias among B-0 cells is not evident in non-Tg littermate mice, as only two (6%) of the B-0 sequences express a Vκ4/5 gene, neither of which are Vκ4/5H (Fig. 3). Thus, Vκ4/5H dominates both the B-0 (PtCneg and PtCint) and B-1 subsets in 6-1 mice as a consequence of VH12 expression.
Figure 2.

Vκ use by PtCneg, PtCint, and PtCbri cells from 6-1 mice. Approximately 106 splenic B cells were sorted from 6-1 mice as indicated in the histogram (B220 versus liposomes) and mRNA isolated separately from each subset. RT-PCR was performed to amplify the expressed rearranged Vκ genes and individual clones sequenced. The percent of clones corresponding to the indicated Vκ groups is indicated in the bar graphs at the top, and the total number of sequences is given in parentheses at the top of each graph. (A) Vκ use by PtCneg B cells. (B) The breakdown of Vκ4/5 gene use by PtCneg cells into Vκ4/5H and other members of this family shown in A. The total numbers of sequences corresponding to the Vκ4/5 and non-Vκ4/5 subsets are given in parentheses. (C) Vκ use by PtCint B cells. (D) Vκ use by PtCbri B-1 cells. All Vκ identifications are Vκ groups except Vκ4/5H, which denotes a specific Vκ4/5 gene.
Figure 3.

Vκ use by B-0 cells from non-Tg 6-1 littermates. CD23+B220+ cells were sorted as indicated by the gates shown in the histogram. As for the analysis in Fig. 2, mRNA was isolated from ∼106 sorted cells for RT-PCR of expressed Vκ genes. Individual clones were sequenced and the Vκ groups identified. The representation of individual Vκ groups is presented as the percentage of total 32 Vκ sequences determined.
These three populations of B cells in 6-1 mice are distinct in Jκ use. The PtCbri B-1 cells use predominantly Jκ2 and Jκ4 (Fig. 4 A), as is true of PtC-specific lymphomas and hybridomas 19. This bias undoubtedly reflects selection for this specificity and expansion in the B-1 subset. PtCint B-0 cells use Jκ2 almost exclusively. We attribute this to the fact that a tyrosine at the Vκ–Jκ junction, which is encoded by Jκ2, results in PtCint binding. Vκ4/5H-expressing PtCneg B-0 cells use Jκ2, -4, and -5, with Jκ5 used at almost twice the frequency as Jκ2 (Fig. 4 A). Notably, almost none of the Vκ4/5H-expressing B cells from any of these populations use Jκ1. This contrasts with the predominant use of Jκ1 and Jκ2 by non-Tg B-0 cells (Fig. 4 C) and even by non-Vκ4/5 genes from the PtCneg B-0 cells (Fig. 4 B).
Figure 4.



Jκ use by 6-1 and non-Tg littermate mice. (A) Jκ use by Vκ4/5H rearrangements from the PtCneg, PtCint, and PtCbri populations of 6-1 mice. (B) Jκ use by non-Vκ4/5 rearrangements from the PtCneg population of 6-1 mice. (C) Jκ use by B-0 (CD23+B220+) cells of non-Tg littermates. The total numbers of sequences analyzed in each group are given in Fig. 2 and Fig. 3.
Vκ4/5H L Chains Segregate According to the Vκ–Jκ Junctional Sequence.
The Vκ4/5H L chains of each population have a characteristic CDR3 sequence. Most (67%) of the Vκ4/5H rearrangements from PtCbri B-1 cells encode a charged amino acid (primarily arginine) at the Vκ–Jκ junction, position 96 (R96) 25, although several have the nonpolar residues leucine and phenylalanine at this position (Fig. 5). PtC-specific hybridoma and lymphoma antibodies have R96 or L96 19, confirming that anti-PtC B cells use these junctions.
Figure 5.

CDR3 sequences of Vκ4/5H rearrangements from PtCneg, PtCint, and PtCbri populations from 6-1 mice. The PtCneg sequences are presented as two groups. The top group is sequences from Vκ4/5H rearrangements, and the bottom group is from other Vκ4/5 gene rearrangements (non-H). The sequences from non-Vk4/5 are quite diverse and not shown. Arrow indicates position 96 at the Vκ–Jκ junction.
PtCint rearrangements are the most homogeneous in their encoded CDR3. Nearly all (∼90%) encode Y96. Jκ2 is the only Jκ that encodes Y96. That this residue can confer binding to PtC is indicated by the anti-PtC lymphoma CH32 that has Y96 19. The presence of Y96, along with the lower IgM levels inherent to B-0 cells, offers an explanation for the weak liposome staining by these cells.
The non-Vκ4/5H CDR3 sequences of the PtCneg population are quite heterogeneous in length and sequence (data not shown), reflecting the fact that most of the L chain CDR3 is encoded by Vκ. The CDR3 regions of Vκ4/5H L chains commonly have a nonpolar, noncharged amino acid at position 96 and in some cases are one or two amino acids shorter than the majority of those in the PtCint and PtCbri populations.
Paradoxically, many Vκ4/5H junctional sequences from PtCneg cells are identical to sequences from PtCbri cells. Four sequences encode L96 (and are nine amino acids in length), seven encode F96, and four encode R96. The presence of rearrangements compatible with PtC binding in this population are unlikely to be due to a contamination from other populations during the sort for the following reasons: sort contamination could come from two sources, the inability to resolve cells from neighboring populations and machine error, in which an incorrect cell not necessarily from a neighboring population is sorted. In the case of the former, PtCint cells would most likely contaminate PtCneg cells. But this is not the case, as the potential contaminants in the PtCneg population do not encode Y96. In the case of machine error, the most common contaminant would be from the PtCbri population, as it is the largest population. We rule this out as an explanation for two reasons. First, by this scenario, both the PtCneg and PtCint populations should be contaminated with PtCbri cells, but the PtCint population is almost completely lacking in sequences that could derive from the PtCbri population. Second, the potential PtC-specific sequences in the PtCneg population are not representative of the PtCbri population; the former are predominantly L96 and F96, whereas the latter are predominantly R96. Thus, the potential PtC-specific sequences in the PtCneg population are not contaminants. We calculate from these data that ∼18% of the PtCneg repertoire, which would amount to 12% of the total B-0 repertoire, could be PtC binding.
The Expressed Vκ Repertoire Is Limited by the Inability of the VH12 H Chain to Pair with Many L Chains.
One possible explanation for the restricted Vκ repertoire expressed by VH12 B cells is that VH12 is unable to associate with all L chains. To test this possibility, we transfected a 10/G4 VH12–D–JH1 expression construct into several L chain–only-expressing cell lines, or cotransfected it with different L chain expression constructs into a nonexpressing cell line, and assayed for secretion of IgM. This rearrangement is identical to that used to generate 6-1 mice. As shown in Table , we were unable to detect secreted antibody with λ1 and 5 of the 8 κ chains. In those cases in which secreted antibody was not detected, we could detect cytoplasmic H and L chains. Thus, most L chains failed to associate with VH12 and were considered nonpermissive.
Table 1.
VH12 Antibody Secretion
| VH12 | 2-12H | |
|---|---|---|
| λ1 | — | + |
| VκRF | — | + |
| Vκ4/5H | + | + |
| Vκ31 | — | + |
| Vκ8 | — | + |
| Vκ8 | — | + |
| Vκ1A | + | + |
| Vκ10 | + | + |
| Vκ21C | — | + |
The inability of VH12 to associate with λ1 chains was confirmed in vivo by generating 6-1 mice that lacked an intact κ locus (6-1/κ−/−). The B cells in these mice can only use λ chains. As seen in Fig. 6B cells are rare in both the spleens and bone marrow of these mice, indicating that λ1 is nonpermissive in vivo, as are probably most other λ chains, leading to cell death in the bone marrow. The few B cells present in the spleen use λ L chains and appear to be immature, i.e., CD23 −CD5− CD43 − (data not shown). Thus, the limited ability of VH12 to associate with L chains affects B cell production.
Figure 6.

FACS™ analysis of spleen and bone marrow cells from 6-1/κ−/− mice. The B cell populations of 6-1/κ−/− (bold line) and 6-1 (dotted line) spleen and bone marrow were compared using the pan-B cell marker CD19.
B-1 Cells Reside in Splenic Follicles in 6-1 Mice.
6-1 B-0 and B-1 cells are distinct in specificity and Vκ gene use. To determine whether there is also a histological distinction between B-0 and B-1 cells within the spleen, we prepared splenic sections from 6-1 and control mice for histological comparison. Spleen morphology in 6-1 mice, as shown by hematoxylin and eosin staining, does not appear to be different from non-Tg control littermates (data not shown). In the white pulp, follicular structures are associated with periarteriolar lymphatic sheaths (PALS) and are separated from the red pulp by lymphatic sinuses. Considering that 60–70% of the splenic B cells in 6-1 mice are B-1, it is likely that most if not all B-1 cells are in follicles.
To determine if B-1 cells occupy splenic follicles, we analyzed spleen sections from 6-1 and non-Tg littermate mice by confocal scanning laser microscopy. Sections were stained with antibodies specific for IgMa or IgMb and CD3 to identify B and T cells, respectively. To discriminate between B-0 and B-1 cells, we used antibodies to CD23, as CD23 is present on B-0 cells but not B-1 cells. As can be seen in Fig. 7 B, 6-1 mice have T cell–rich PALS that are adjacent to B cell–rich follicles. In addition, these follicles have identifiable marginal zones. Most follicles have B cells that do not stain for CD23 (Fig. 7 B), indicating that they contain B-1 cells, whereas follicular B cells from control non-Tg spleen sections stained simultaneously demonstrate unambiguous expression of CD23 (Fig. 7 A). Thus, B-1 cells occupy splenic follicles in 6-1 mice. CD23+ B-0 cells can be seen in splenic sections of 6-1 mice, but, surprisingly, they are concentrated to a subset of follicles rather than intermixed with B-1 cells in all follicles (Fig. 7 C).
Figure 7.
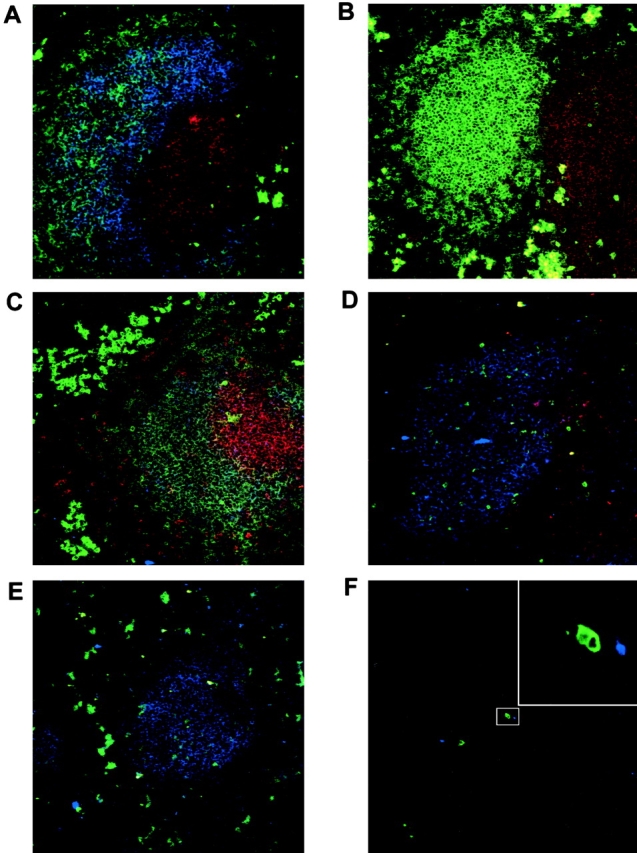
Histological analyses of B-1 cells in 6-1 mice. All photographs are of stains using anti-IgMa–FITC (green) (except A, which uses IgMb–FITC), anti-CD3-PE (red), and anti-CD23–streptavidin–Cy5 (blue) as described in Materials and Methods. (A) Non-Tg littermate follicle and PALS; (B and C) 6-1 follicles and PALS; (D and E) littermate follicles 24 h or 7 d, respectively, after transfer of 7 × 106 6-1 B-1 cells; (F and inset) isolation on IgMa cells in the red pulp 7 d after transfer. Magnification is 200 except for the inset in F, which is at 2,000.
The ability of B cells to occupy follicles can be affected by the presence of B cells of diverse specificity 26. To determine whether PtC-specific B-1 cells enter follicles in the presence of a majority of heterogeneous B-0 cells, we adoptively transferred sorted B220+ CD23 − B-1 cells from 6-1 mice to non-Tg littermates. At 24 h and 7 d after transfer, mice were killed and spleens taken for histological and flow cytometry analyses. At the time mice were killed, essentially all of the recovered transferred cells were PtCbri B-1 cells (CD23 −CD5+CD43+) (data not shown). As can be seen in Fig. 7D and Fig. E, IgMa B cells are visible in follicles and PALS at both time points. On day 7, numerous IgMa bright cells showing cytoplasmic IgM staining are present in the red pulp (Fig. 7 E). Similar cells are seen in 6-1 mice (Fig. 7B and Fig. C). These cells were not seen at 24 h, suggesting that some B-1 cells have differentiated to plasmablasts by 7 d after transfer and have migrated to the red pulp. Many of these cells are present in clusters of two to five cells (Fig. 7 F), suggesting recent cell division in the red pulp.
Discussion
The role played by the Ig receptor specificity in the induction of the B cell developmental program is a well established immunological paradigm. Our data show here that the specificity of the cell surface Ig receptor is not only important in the developmental steps taken among conventional B-0 cells but also in the segregation of cells to the B-0 and B-1 subsets.
Segregation to B-1.
Our previous analysis of anti-PtC Tg mice supports the idea of an antigen-driven differentiation of cells from B-0 to B-1. This was suggested by the observation that segregation of PtC-specific B cells to the B-1 subset is intact in 6-1 mice and in 6-1/Vκ4/5 double-Tg mice in which essentially all developing B cells are PtC specific 10. These data argue that the mechanism of segregation operates after Ig gene rearrangement. The differentiation of B-1 cells was more directly addressed by combining the VH12 and Vκ4 transgenes with the xid mutation 20. The xid mutation is a loss of function mutation in the gene for Bruton's tyrosine kinase 27 28 29 30 that causes a disruption in BCR signaling. Among other deficiencies, xid mice have few B-1 cells 1. VH12/Vκ4 double-Tg mice with the xid mutation exhibit a significant deficiency in B-1 cell development as expected 20. However, the majority of splenic PtC-specific B cells are B-0, not B-1, revealing the existence of a differentiative pathway from B-0 to B-1 that is dependent on signals initiated by the BCR. We have recently demonstrated this differentiative pathway in anti-PtC Tg, non-xid mice by manipulation of PtC-specific cells that are at intermediate differentiative stages in this pathway (Arnold, L.W., S.K. McCray, C. Tatu, and S.H. Clarke, manuscript submitted for publication).
Viewed in this context, we interpret the segregation of 6-1 B cells to be based on their ability to bind PtC. All newly differentiated B cells from the adult bone marrow are B-0. However, those that bind PtC with high affinity (PtCbri) are induced to become B-1, whereas those that bind PtC weakly or not at all (PtCint and PtCneg, respectively) are not signaled sufficiently and remain B-0. Among 6-1 cells that express a 10/G4 VH12 H chain and Vκ4/5H L chain, the ability to bind PtC is dependent on the amino acid at the Vκ–Jκ junction, position 96. These data therefore disprove any notion that VH or Vκ gene expression plays a role in segregation and demonstrate that the level of PtC binding determines differentiation to B-1. This is further evidence that segregation to B-1 occurs after Ig gene rearrangement.
An unexpected finding from this analysis was the presence of Vκ4/5H rearrangements in the PtCneg population that are identical to some in the PtCbri B-1 population. Because IgM− cells exist among the PtCneg population, it is plausible that these rearrangements derive from PtCbri cells that have lost surface IgM (Fig. 1) and are therefore sorted with the PtCneg population. IgM− cells are ∼20% of the PtCneg population, similar to the 18% estimate made from the sequence analysis. Loss of surface Ig can occur in B cells undergoing cell division. For example, rapidly dividing germinal center centroblasts do not express surface Ig 31. A similar downregulation may occur in dividing B-1 cells or in cells differentiating to B-1. Alternatively, these cells could be plasmablasts that have lost surface IgM, such as the cells seen in the red pulp in 6-1 mice and in normal mice after adoptive transfer of PtCbri B-1 cells (Fig. 7).
Upon differentiation in 6-1 mice, PtC-specific B-1 cells reside in splenic follicles and in fact occupy most splenic follicles, as they are in the majority. However, it is interesting that B-0 and B-1 cells segregate to different follicles. Whether this occurs in non-Tg mice is unknown. B-1 cells are not excluded from entry into a follicle composed mostly of B-0 cells as are other autoreactive B cells 26, indicating that exclusion from B-0 follicles is not the basis for segregation. Perhaps B-0 cells are excluded from B-1 follicles, or this segregation reflects competition between B-0 and B-1 cells during the time of follicle formation.
Some adoptively transferred B-1 cells have moved into the red pulp by 7 d after transfer and differentiated to plasmablasts. That not all B-1 cells differentiate to plasmablasts in 6-1 mice or in non-Tg mice that have received B-1 cell transfers suggests that migration to the red pulp and differentiation to a plasmablast are regulated independently from differentiation to B-1, although both presumably require antigen. This transition could be a controlling checkpoint for the secretion of natural IgM, as B-1 cells are considered the major source of this antibody 1 9 12.
The Bias in Vκ Repertoire Precedes Segregation to B-1 and Is Independent of PtC Binding.
Although it was anticipated that Vκ4/5H would dominate the B-1 PtCbri population, the dominance of Vκ4/5H in the B-0 subset was a surprise. As many as 59% of B-0 cells in these mice use Vκ4/5H. Even though some Vκ4/5H-expressing B cells may be PtC-specific B-1 cells that have lost surface Ig, this only decreases the proportion to 51%. Thus, extraordinarily high frequencies of B-0 cells express Vκ4/5H, indicating that the bias in Vκ expression is independent of PtC binding. As the segregation to B-1 occurs after expression of H and L chains 10, this bias must also precede segregation to the B-1 subset.
The inability of VH12 to associate with many L chains is no doubt a contributing factor, as it limits Vκ repertoire expressed by VH12 B cells. The fact that most L chains are nonpermissive in 6-1 mice (Fig. 6), provides an explanation for why 6-1 mice develop 10% as many B-0 cells as non-Tg littermates 18. The basis for the inability of VH12 to associate with most κ and λ chains is unknown. It is well established that V region structures influence L chain association 32 33 34 and that this involves both framework regions and CDRs 32 33 34 35 36. A second VH12 H chain, differing only in CDR3 from the 10/G4 VH12 used here, is similarly deficient in ability to associate with L chains (Ye, J., H. Wang, L.W. Arnold, and S.H. Clarke, manuscript in preparation), implicating the VH-encoded segment rather than CDR3 in this inability. As this VH is unmutated, this characteristic must be evolutionarily selected, possibly to limit the diversity of VH12 B cells.
Although the large number of nonpermissive L chains would account for the small B-0 population in 6-1 mice, it would not by itself account for the dominance of Vκ4/5H in B-0, as there are multiple permissive L chains (Fig. 2 A and Table ). Favor for Vκ4/5H could be achieved by a higher rearrangement frequency for this gene than for others. Although there is evidence that Vκ4/5 genes preferentially rearrange 37, there is no apparent Vκ4/5 bias among B-0 cells from non-Tg littermates. We therefore propose that VH12 B cells that express Vκ4/5H are favored over others for survival or clonal expansion. In this context, we have recently observed that the B cells in 6-1/Vκ1A double-Tg mice are predominantly immature, suggesting that not all VH12 B cells expressing a permissive L chain have an equal ability to contribute to the long-lived mature repertoire. We are currently testing this hypothesis.
The pattern of Jκ use among B-0 cells in 6-1 mice provides clues to the mechanism behind Vκ4/5H dominance. In normal mice, ∼80% of Vκ rearrangements in B-0 cells are to Jκ1 and Jκ2 38, as seen in our non-Tg control mice (Fig. 4 C). But 6-1 B-0 cells show a different pattern of Jκ use depending on whether or not they express Vκ4/5H. Vκ4/5H rearrangements are skewed to downstream Jκ gene segments and are rarely to Jκ1 (Fig. 4 A). In contrast, non-Vκ4/5 rearrangements are significantly less biased to downstream Jκs, and ∼35% are to Jκ1 (Fig. 4 B). The absence of rearrangements to Jκ1 in Vκ4/5H-expressing B-0 cells could be due to an inability of this gene to efficiently rearrange to Jκ1. Although we cannot exclude this possibility, we think it unlikely because we identified several rearrangements to Jκ1 (Fig. 4), and we know of no precedent for such a molecular defect. An alternative possibility is that Vκ4/5H-Jκ1 rearrangements cannot associate with VH12. Junctional amino acids affect association 35, and W96, unique to Jκ1, is not seen among the few Vκ4/5H-Jκ1 rearrangements identified and is present in only one Vκ4/5 (non-H) rearrangement (Fig. 5). However, rearrangements that delete the first codon of Jκ1 are common and would yield Jκ regions identical to those encoded by Jκ2, as Jκ1 and Jκ2 are identical in amino acid sequence after the first amino acid. In fact, many of the rearrangements with R96 are the result of a rearrangement to Jκ2 that includes the last codon of Vκ4/5H and deletes the first codon of Jκ2 19. The identical L chain could be generated by rearrangement to Jκ1. Thus, Jκ1 use should be seen among the PtCbri B-1 population, if not the PtCneg B-0 population, even if W96 disrupts association with VH12. That it is not argues against the idea that Jκ1 rearrangements are not represented because they disrupt association with VH12.
The more likely explanation is that Vκ4/5H expression is the result of secondary rearrangement. The occurrence of secondary rearrangement to delete a primary rearrangement is well established and would result in a bias for the use of downstream Jκ gene segments 39 40 41 42. Secondary rearrangement could occur in VH12 pre-B cells to replace primary rearrangements that encode nonpermissive L chains. As a majority of L chains appear to be nonpermissive with VH12 H chains, secondary rearrangement may occur in a high proportion of these cells. Unless replaced, expression of a nonpermissive L chain will result in cell death, as seen in 6-1/κ−/− mice (Fig. 6). A prediction of this hypothesis is that nonpermissive L chains are unable to mediate allelic exclusion. This was argued by others based on analyses of murine plasmacytomas that produce multiple L chains, of which only one was able to pair with the expressed H chain 43 44, and based on an analysis of κ Tg mice 45. We therefore suggest that Vκ4/5H rearrangements are predominantly secondary to primary rearrangements that encode nonpermissive L chains. Thus, secondary rearrangement probably contributes to the dominance of Vκ4/5H by VH12 B-0 cells. As secondary rearrangement occurs in the bone marrow before commitment to B-1, the anti-PtC B-1 repertoire must be dependent on secondary Vκ4/5H rearrangements as well, accounting for the absence of Jκ1 rearrangements from the B-1 repertoire. Why Vκ4/5H rearrangements are more skewed to downstream Jκs than non-Vκ4/5 rearrangements, implying that the former are more often secondary rearrangements than the latter, is still unresolved. This may be related to the suspected advantage that Vκ4/5H-expressing B cells have in entry into the long-lived mature B-0 repertoire and may involve additional Vκ rearrangement in transition to a mature B cell. We are currently testing this possibility.
Selection at Multiple Checkpoints Focuses the Specificity of VH12 B Cells to PtC.
These data reveal yet another checkpoint in B cell development that imposes a stringent limitation on VH12 B cell repertoire diversity. The first occurs at the transition from pre-BI to pre-BII, where the length and sequence of VH12 CDR3 are selected 20. Pre-B cells with 10/G4 rearrangements support pre-B cell differentiation, whereas most non-10/G4 rearrangements cannot, resulting in an enrichment of pre-B cells with 10/G4 rearrangements 20. The data reported here document a checkpoint after L chain gene rearrangement, during the transition to an immature B cell. The Vκ repertoire is limited at this stage due to an inability of most L chains to associate with VH12, resulting in a much smaller than normal B-0 subset. Because this cannot account for the dominance of Vκ4/5H among B-0 cells, we suggest an additional selective mechanism operating at a later stage that favors VH12/Vκ4/5H-expressing cells, possibly during the transition from an immature to a mature B cell. This process generates a pool of B-0 cells from which cells with the PtCbri phenotype are selected by antigen for entry into the B-1 repertoire and clonal expansion. As 10/G4 VH12 H chains and Vκ4/5H L chains are critical for the PtCbri phenotype, we suggest that there has been evolutionary pressure to develop a VH12 B-0 repertoire enriched in PtCbri cells. This would promote the production of a large number of PtC-specific B-1 cells. The evolutionary selection for the development of anti-PtC B cells complements an earlier finding by Booker and Haughton 46 that the VH12 and VH11 (also encoding anti-PtC antibodies) genes are evolutionarily more conserved than other VH genes.
In spite of sustained research efforts, the function of B-1 lymphocytes remains elusive. Their characterization in many vertebrate species suggests a strong phylogenetic conservation and a fundamental homeostatic or protective role. A limited repertoire of antigen specificities and the expression of low-affinity Ig receptors could indicate that these cells are a “first line” immune defense against bacterial organisms 47. A recent report by Boes et al. 48 showing that anti-PtC antibodies are protective in acute peritonitis provides direct evidence that anti-PtC antibodies are important in immediate protection against bacterial infections. Perhaps the physiological target of these antibodies is unlikely to change over time and is shared by a large number of organisms, and therefore the development of a response to this antigen could be evolutionarily selected to provide immediate protection before a T cell–dependent high-affinity response can develop. For the same reasons, B-1 cells could also have a role in the “scavenging” of senescent or apoptotic cells resulting from physiological or pathological events; these cells can express self-antigens on their surfaces (e.g., PtC, nuclear antigens, DNA) 49 50, toward which the B-1 Ig repertoire is oriented. As these autoantigens do not vary their epitopes in time, mechanisms to eliminate them could also be evolutionary selected. Such a strong survival value could have the 10/G4 VH12 H chain gene and its L chain partner, Vκ4/5H, expressed as a sine qua non component of the B-1 repertoire in mice and other species.
Acknowledgments
We gratefully acknowledge the Microscopy Facility, the DNA Sequencing Facility, and the Flow Cytometry Facility at the University of North Carolina for their assistance with this work. We are also indebted to Garnett Kelsoe and Biao Zheng for their generous assistance with histological analysis.
This work was supported by National Institutes of Health grants AI29576 and AI43587, grant 79017 from the American Cancer Society, and a grant from the Arthritis Foundation.
Footnotes
1used in this paper: BCR, B cell receptor; PALS, periarteriolar lymphatic sheaths; PtC, phosphatidyl choline; Tg, transgenic
References
- Hayakawa K., Hardy R.R., Parks D.R., Herzenberg L.A. The “Ly-1 B” cell subpopulation in normal, immunodefective, and autoimmune mice. J. Exp. Med. 1983;157:202–218. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K., Hardy R.R., Herzenberg L.A. Peritoneal Ly-1 B cellsgenetic control, autoantibody production, increased lambda light chain expression. Eur. J. Immunol. 1986;16:450–456. doi: 10.1002/eji.1830160423. [DOI] [PubMed] [Google Scholar]
- Karras J.G., Wang Z., Huo L., Howard R.G., Frank D.A., Rothstein T.L. Signal transducer and activator of transcription-3 (STAT-3) is constitutively activated in normal, self-renewing B-1 cells but only inducibly expressed in conventional B lymphocytes. J. Exp. Med. 1997;185:1035–1042. doi: 10.1084/jem.185.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou L.-B., Colosia A., Corley R.B., Clarke S.H., Scott D.W. Differential susceptibility to tolerance induction in vitro of splenic B cells from several transgenic mouse linesrole of B-1 cells. J. Immunol. 1995;154:6262–6274. [PubMed] [Google Scholar]
- Lalor P.A., Morahan G. The peritoneal Ly-1 (CD5) B cell repertoire is unique among murine B cell repertoires. Eur. J. Immunol. 1990;20:485–492. doi: 10.1002/eji.1830200305. [DOI] [PubMed] [Google Scholar]
- Casali P., Burastero S.E., Nakamura M., Inghirami G., Notkins A.L. Human lymphocytes making rheumatoid factor and antibody to ssDNA belong to the Leu-1+ B-cell subset. Science. 1987;236:77–81. doi: 10.1126/science.3105056. [DOI] [PubMed] [Google Scholar]
- Hardy R.R., Hayakawa K., Shimizu M., Yamasaki K., Kishimoto T. Rheumatoid factor secretion from human Leu-1+ B cells. Science. 1987;236:81–83. doi: 10.1126/science.3105057. [DOI] [PubMed] [Google Scholar]
- Mercolino T.J., Arnold L.W., Hawkins L.A., Haughton G. Normal mouse peritoneum contains a large number of Ly-1+ (CD5) B cells that recognize phosphatidyl choline. Relationship to cells that secrete hemolytic antibody specific for autologous erythrocytes. J. Exp. Med. 1988;168:687–698. doi: 10.1084/jem.168.2.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K., Hardy R.R., Honda M., Herzenberg L.A., Steinberg A.D., Herzenberg L.A. Ly-1 B cellsfunctionally distinct lymphocytes that secrete IgM autoantibodies. Proc. Natl. Acad. Sci. USA. 1984;81:2494–2498. doi: 10.1073/pnas.81.8.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold L.W., Pennell C.A., McCray S.K., Clarke S.H. Development of B-1 cellssegregation of phosphatidyl choline-specific B cells to the B-1 population occurs after immunoglobulin gene expression. J. Exp. Med. 1994;179:1585–1595. doi: 10.1084/jem.179.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masmoudi H., Mota-Santos S., Huetz F., Coutinho A., Casenave P.A. All T15 Id-positive antibodies (but not the majority of VHT15+ antibodies) are produced by peritoneal CD5 B lymphocytes. Int. Immunol. 1990;2:515–520. doi: 10.1093/intimm/2.6.515. [DOI] [PubMed] [Google Scholar]
- Forster I., Rajewsky K. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic newborn mice. Eur. J. Immunol. 1987;17:521–528. doi: 10.1002/eji.1830170414. [DOI] [PubMed] [Google Scholar]
- Hayakawa K., Hardy R.R., Herzenberg L.A., Herzenberg L.A. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J. Exp. Med. 1985;161:1554–1568. doi: 10.1084/jem.161.6.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy R.R., Hayakawa K. A developmental switch in B lymphopoiesis. Proc. Natl. Acad. Sci. USA. 1991;88:11550–11554. doi: 10.1073/pnas.88.24.11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor A.B., Herzenberg L.A. Origin of murine B cell lineages. Annu. Rev. Immunol. 1993;11:501–538. doi: 10.1146/annurev.iy.11.040193.002441. [DOI] [PubMed] [Google Scholar]
- Cong Y.Z., Rabin E., Wortis H.H. Treatment of murine CD5-B cells with anti-Ig, but not LPS, induces surface CD5two B cell activation pathways. Int. Immunol. 1991;3:467–476. doi: 10.1093/intimm/3.5.467. [DOI] [PubMed] [Google Scholar]
- Haughton G., Arnold L.W., Whitmore A.C., Clarke S.H. B1 cells are made, not born. Immunol. Today. 1993;14:84–87. doi: 10.1016/0167-5699(93)90064-R. [DOI] [PubMed] [Google Scholar]
- Clarke S.H., Arnold L.W. B-1 cell developmentevidence for an uncommitted immunoglobulin (Ig)M+ B cell precursor in B-1 cell differentiation. J. Exp. Med. 1998;187:1325–1334. doi: 10.1084/jem.187.8.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennell C.A., Mercolino T.J., Grdina T.A., Arnold L.W., Haughton G., Clarke S.H. Biased immunoglobulin variable region gene expression by Ly-1 B cells due to clonal selection. Eur. J. Immunol. 1989;19:1289–1295. doi: 10.1002/eji.1830190721. [DOI] [PubMed] [Google Scholar]
- Ye J., McCray S.K., Clarke S.H. The transition of pre-BI to pre-BII cells is dependent on the structure of the μ/surrogate L chain receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:1524–1533. [PMC free article] [PubMed] [Google Scholar]
- Chen J., Trounstine M., Kurahara C., Young F., Kuo C.C., Xu Y., Lorine J.F., Alt F.W., Huszar D. B cell development in mice that lack one or both immunoglobulin kappa light chain genes. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:821–830. doi: 10.1002/j.1460-2075.1993.tb05722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retter M.W., Eisenberg R.A., Cohen P.L., Clarke S.H. Sm and DNA binding by dual reactive B cells requires distinct VH, Vk, and VHCDR3 structures. J. Immunol. 1995;155:2248–2257. [PubMed] [Google Scholar]
- Bloom D.D., Davignon J.-L., Retter M.W., Shlomchick M.J., Pisetsky D.S., Cohen P.L., Eisenberg R.A., Clarke S.H. V region gene analysis of anti-Sm hybridomas from MRL/Mp-lpr/lpr mice. J. Immunol. 1993;150:1591–1610. [PubMed] [Google Scholar]
- Ibrahim S.M., Weigert M., Basu C., Erikson J., Radic M.Z. Light chain contribution to specificity in anti-DNA antibodies. J. Immunol. 1995;155:3223–3233. [PubMed] [Google Scholar]
- Kabat E.A., Wu T.T., Perry H.M., Gottesman K.S., Foeller C. Sequences of Proteins of Immunological Interest 1991. US Department of Health and Human Services; Washington, DC: pp. 151–259 [Google Scholar]
- Cyster J.G., Harley S.B., Goodnow C.C. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 1994;371:389–395. doi: 10.1038/371389a0. [DOI] [PubMed] [Google Scholar]
- Thomas J.D., Sideras P., Smith C.I., Vorechovsky I., Chapman V., Paul W.E. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- Rawlings D.J., Saffran D.C., Tsukada S., Largaespada D.A., Grimaldi J., Cohen L., Mohr R.N., Bazan J.F., Howard M., Copeland N.G. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- Khan W.N., Alt F.W., Gerstein R.M., Malynn B.A., Larsson I., Rathbun G., Davidson L., Muller S., Kantor A.B., Herzenberg L.A. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Kerner J.D., Appleby M.W., Mohr R.N., Chien S., Rawlings D.J., Maliszewski C.R., Witte O.N., Perlmutter R.M. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- Liu Y.-L., Zhang J., Lane P.J.L., Chan E.Y.-T., MacLennan I.C.M. Sites of specific B cell activation in primary and secondary responses to T-cell-dependent and T-cell independent antigens. Eur. J. Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- Grey H.M., Mannik M. Specificity of recombination of H and L chains from human γG-myeloma proteins. J. Exp. Med. 1965;122:619–632. doi: 10.1084/jem.122.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de-Preval C., Fougereau M. Specific interaction between VH and VL regions of human monoclonal immunoglobulins. J. Mol. Biol. 1976;102:657–678. doi: 10.1016/0022-2836(76)90340-5. [DOI] [PubMed] [Google Scholar]
- Hamel P.A., Klein M.H., Dorrington K.J. The role of the VL and VH-encoded segments in the preferential reassociation of immunoglobulin subunits. Mol. Immunol. 1986;23:503–510. doi: 10.1016/0161-5890(86)90113-6. [DOI] [PubMed] [Google Scholar]
- Hamel P.A., Isenman D.E., Klein M.H., Luedtke R., Dorrington K. Structural basis for the preferential association of autologous immunoglobulin subunitsrole of the J region of the light chain. Mol. Immunol. 1984;21:277–283. doi: 10.1016/0161-5890(84)90098-1. [DOI] [PubMed] [Google Scholar]
- Chen C., Martin T.M., Stevens S., Rittenberg M.B. Defective secretion of an immunoglobulin caused by mutations in the heavy chain complementarity determining region 2. J. Exp. Med. 1994;180:577–586. doi: 10.1084/jem.180.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalled S.L., Brodeur P.H. Preferential rearrangement of Vκ4 gene segments in pre-B cell lines. J. Exp. Med. 1990;172:559–566. doi: 10.1084/jem.172.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D.L., Coleclough C. Different joining region J segments of the murine κ immunoglobulin light chain locus are used at markedly different frequencies. Proc. Natl. Acad. Sci. USA. 1984;81:4756–4760. doi: 10.1073/pnas.81.15.4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S.H., McCray S. A shared κ reciprocal fragment and a high frequency of secondary rearrangements among influenza hemagglutinin specific B cell hybridomas. J. Immunol. 1991;146:343–349. [PubMed] [Google Scholar]
- Gay D., Saunders T., Camper S., Weigert M. Receptor editingan approach by autoreactive B cells to escape tolerance. J. Exp. Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiegs S.L., Russell D.M., Nemazee D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Dillon S.R., Zheng B., Shimoda M., Schlissel M.S., Kelsoe G. V(D)J recombinase activity in a subset of germinal center B lymphocytes. Science. 1997;278:301–305. doi: 10.1126/science.278.5336.301. [DOI] [PubMed] [Google Scholar]
- Kwan S.-P., Max E., Seidman J.G., Leder P., Scharff M.A. Two kappa immunoglobulin genes are expressed in the myeloma S107. Cell. 1981;26:57–66. doi: 10.1016/0092-8674(81)90033-7. [DOI] [PubMed] [Google Scholar]
- Bernard O., Gough N.M., Adams J.M. Plasmacytomas with more than immunoglobulin κ mRNAimplications for allelic exclusion. Proc. Natl. Acad. Sci. USA. 1981;78:5812–5816. doi: 10.1073/pnas.78.9.5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K.A., Brinster R.L., Storb U. Allelic exclusion and control of endogenous immunoglobulin gene rearrangement in κ transgenic mice. Nature. 1984;312:517–520. doi: 10.1038/312517a0. [DOI] [PubMed] [Google Scholar]
- Booker J.K., Haughton G. Mechanisms that limit diversity of autoantibodies. II. Evolutionary conservation of the Ig variable region genes which encode naturally occurring autoantibodies. Int. Immunol. 1994;6:1427–1436. doi: 10.1093/intimm/6.9.1427. [DOI] [PubMed] [Google Scholar]
- Herzenberg L.A., Herzenberg L.A. Toward a layered immune system. Cell. 1989;59:953–954. doi: 10.1016/0092-8674(89)90748-4. [DOI] [PubMed] [Google Scholar]
- Boes M., Prodeus A.P., Schmidt T., Carroll M.C., Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J. Exp. Med. 1998;188:2381–2386. doi: 10.1084/jem.188.12.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L.A., Anhalt G., Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen A., Casciola-Rosen L., Ahearn J. Novel packages of viral and self-antigens are generated during apoptosis. J. Exp. Med. 1995;181:1557–1561. doi: 10.1084/jem.181.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli-Marotto S., Retter M.W., Gee R., Mamula M.J., Clarke S.H. Autoreactive B cell regulationperipheral induction of developmental arrest by lupus-associated autoantigens. Immunity. 1998;8:209–219. doi: 10.1016/s1074-7613(00)80473-2. [DOI] [PubMed] [Google Scholar]