Development of Chronic Inflammatory Arthropathy Resembling Rheumatoid Arthritis in Interleukin 1 Receptor Antagonist–Deficient Mice (original) (raw)
Abstract
Interleukin (IL)-1 is a proinflammatory cytokine that plays important roles in inflammation, host defense, and the neuro-immuno-endocrine network. IL-1 receptor antagonist (ra) is an endogenous inhibitor of IL-1 and is supposed to regulate IL-1 activity. However, its pathophysiological roles in a body remain largely unknown. To elucidate the roles of IL-1ra, IL-1ra–deficient mice were produced by gene targeting, and pathology was analyzed on different genetic backgrounds. We found that all of the mice on a BALB/cA background, but not those on a C57BL/6J background, spontaneously developed chronic inflammatory polyarthropathy. Histopathology showed marked synovial and periarticular inflammation, with articular erosion caused by invasion of granulation tissues closely resembling that of rheumatoid arthritis in humans. Moreover, elevated levels of antibodies against immunoglobulins, type II collagen, and double-stranded DNA were detected in these mice, suggesting development of autoimmunity. Proinflammatory cytokines such as IL-1β, IL-6, and tumor necrosis factor α were overexpressed in the joints, indicating regulatory roles of IL-1ra in the cytokine network. We thus show that IL-1ra gene deficiency causes autoimmunity and joint-specific inflammation and suggest that IL-1ra is important in maintaining homeostasis of the immune system. Possible involvement of IL-1ra gene deficiency in RA will be discussed.
Keywords: IL-1 receptor antagonist, rheumatoid arthritis, autoimmunity, cytokine, animal model
Introduction
Rheumatoid arthritis (RA) is a systemic, chronic, inflammatory disorder expressed most commonly in the joints. Approximately 1% of adults worldwide are estimated to be affected with this disease and suffer from substantial pain and disability 1. Although various factors, including genetic factors, environmental factors, and infectious agents, have been suggested as causes of the disease 2, so far the etiopathogenesis has not been elucidated completely. Patients often produce autoantibodies against various self-substances such as IgG (rheumatoid factor [RF]), type II collagen (IIC), and nuclear antigens, suggesting an autoimmune nature of the disease 1. Moreover, various proinflammatory cytokines, including IL-1, IL-6, and TNF-α, are overexpressed in the joints of RA patients 2. As these cytokines can induce inflammation, promote synovial cell growth, and induce differentiation of osteoclasts, it is suspected that they may play an important role in the development of the disease 2. IL-1 seems especially crucial, because injection of IL-1 into normal rabbit joints causes severe arthritis 3, and injection of anti–IL-1 antibody or IL-1 receptor antagonist (ra) into affected joints ameliorates the disease 4 5 6 7.
IL-1 is a major mediator of inflammation and is produced by various types of cells, including macrophages, monocytes, and synovial lining cells 8. Three gene products, IL-1α, IL-1β, and IL-1ra, are known as the members of the IL-1 family that bind to IL-1Rs. IL-1α and IL-1β exert similar biological activities through the IL-1 type I receptor (IL-1RI; reference 9), whereas IL-1ra is a naturally occurring inhibitor of IL-1 and competes for the receptor 10 11. IL-1 signaling is also regulated by the IL-1 type II receptor (IL-1RII), a decoy receptor 12. Four isoforms of IL-1ra protein are synthesized by alternative splicing from a single gene; one is a secreted form with a signal polypeptide, and the other three exist intracellularly 13 14 15 16. All four isoforms can inhibit IL-1 activities. IL-1ra production is induced by a number of other cytokines, viral products, and acute phase proteins and is augmented in patients with autoimmune and inflammatory diseases, suggesting that this cytokine may play regulatory roles in those diseases 17.
It has been shown that IL-1 can induce many other genes. For example, we have shown that IL-1β induces IL-1α, and vice versa 18. IL-1 also induces IL-6 and cyclooxygenase (COX)-2 19. These factors, together with IL-1 or in a cascade, exhibit pleiotropic effects on an animal, including induction of inflammation and acute phase responses, host defense against bacterial and viral infection, activation of the immune system including thymocyte maturation and Th2 cell proliferation, and enhancement of bone metabolism by activating osteoclasts to secrete metalloproteases 19 20. Recently, we showed that IL-1 is also involved in fever development and activation of the hypothalamic-pituitary-adrenal axis 18. However, the role of IL-1 as well as IL-1ra in normal physiology and diseases has not been completely elucidated. Thus, we have recently produced mice deficient in genes for IL-1α, IL-1β, and IL-1ra using 129 mouse–derived embryonic stem cells to assess roles of these cytokines in a living animal 18. These mice showed no apparent abnormalities, except that IL–1ra deficient (IL-1ra−/−) mice showed some growth retardation.
We backcrossed these mice to several lines of inbred mice of different genetic backgrounds to see the difference of inflammatory reactions among strains. Interestingly, we found that IL-1ra−/− mice on a BALB/cA background spontaneously developed chronic inflammatory arthropathy that closely resembled RA in humans. Overexpression of proinflammatory cytokine genes was observed in the joints of these mice before onset of the disease. Elevation of serum Ig levels and autoantibody production was observed in these mice, suggesting development of autoimmunity. We will discuss the importance of IL-1ra in the homeostasis of the immune system and its possible involvement in human autoimmune diseases.
Materials and Methods
Production of IL-1ra−/− Mice.
IL-1ra−/− mice were produced by replacing the entire exons of the secreted form with the neo gene by homologous recombination as described 18. In these mutant mice, all four isoforms of the IL-1ra were destroyed. These mice were backcrossed to either BALB/cA or C57BL/6J strain mice for five to eight generations. Then, heterozygous mice were intercrossed with each other to obtain homozygous mutant mice. Mice were kept under specific pathogen–free conditions in an environmentally controlled clean room at the Laboratory Animal Research Center, Institute of Medical Science, University of Tokyo. The experiments were carried out according to the institutional ethical guidelines for animal experiments and safety guidelines for gene manipulation experiments.
Clinical Evaluation.
Incidence of arthritis was judged macroscopically. Each joint was examined weekly for swelling and redness. The severity of arthritis was graded on a scale of 0–3 for each paw for degree of redness and swelling. Grade 0 = normal, grade 1 = light swelling of the joint and/or redness of the footpad, grade 2 = obvious swelling of the joint, and grade 3 = severe swelling and fixation of the joint. A severity score was calculated for the four limbs (maximum 12 points for individual mice).
Histological Examination.
Joints were fixed in 10% phosphate-buffered formalin, decalcified in 10% EDTA-4Na, and embedded in paraffin. Sections (4 μm) were stained with hematoxylin/eosin.
FACS® Analysis.
Single-cell suspensions from thymi, spleens, and LNs were prepared, and 106 cells were pretreated with FcR-blocking antibody and stained with the following mAbs for 45 min. Cells were washed with FACS solution (HBSS/20% FCS) and analyzed with a FACScan™ cytometer using the LYSIS II™ program (Becton Dickinson). mAbs used for the staining were anti-CD45R/B220–PE (RA3-6B2), rat anti–CD3-∈–FITC (145-2C11), anti-CD8a–FITC (Ly-2), anti-CD4–PE, anti-CD44–PE, and anti-CD25–PE (PharMingen).
Antibody Titration.
Serum levels of IgG, IgM, and IgE were determined by ELISA as previously described 21. In brief, polyvinyl microtiter plates (Falcon MicroTest III; Becton Dickinson) were coated with 50 μl of rabbit anti–mouse IgG (8.7 μg/ml; DAKO Corp.), anti–mouse IgM (2 μg/ml; Zymed Labs., Inc.), or anti–mouse IgE (2 μg/ml; PharMingen) in PBS. For autoantibody titration, plates were coated with 50 μl of heat-denatured rabbit IgG (50 μg/ml) or bovine IIC (20 μg/ml) in TBS (25 mM Tris/HCl and 140 mM NaCl, pH 7.4). For the DNA, 50 μl of double-stranded (ds)DNA (1 μg/ml) in TBS was placed in wells of microtiter plates that had been coated with poly-l-lysine and dried overnight at 37°C. The plates were blocked with 1% skim milk (Coop)/5 mM EDTA/0.02% NaN3/TBS (blocking buffer) for 1 h at room temperature. Mouse serum was diluted by blocking buffer and added to each well. Alkaline phosphatase–conjugated goat anti–mouse IgG antibody (Zymed Labs., Inc.) or anti–mouse IgM antibody (Zymed Labs., Inc.) in blocking buffer was added as the secondary antibody. After washing with Tween 20/TBS, 100 μl of 1 mg/ml _p_-nitrophenylphosphate (Sigma Chemical Co.) in 50 mM NaHCO3/5 mM MgCl2, pH 9.5, was added, and the absorbance at 415 nm was measured by ELISA microreader (MTP-120; Colona). IgG and IgM concentrations were determined using mouse standard serum NOR-02/93 (Nordic Immunology), purified mouse myeloma IgM (Zymed Labs., Inc.), and purified mouse IgE (PharMingen), respectively, as the standards.
Northern Blot Hybridization Analysis.
Total RNA was isolated from the joints by an acid guanidium thiocyanate-phenol-chloroform extraction method, and poly (A)+ RNA was purified using the QuickPrep Micro mRNA Purification Kit (Pharmacia). Northern blot hybridization was performed as described 18. Probes for IL-1ra, IL-1α, IL-1β, IL-6, TNF-α, COX-2, and β-actin were described previously 18. The intensity of the bands on the autoradiogram was estimated by the BAS 2000 system (Fuji Photo Film Co.).
Mouse IL-1RI and IL-1RII probes were prepared by RT-PCR using RNA preparation from the mouse spleen. The PCR primers were 5′-TCGCAAGTGTCCTCTTACTCC-3′ and 5′-GTGGTAAGTGTGTTGCTGCC-3′ for IL-1RI and 5′-TGGACTCTTCTCAGCTGATCC-3′ and 5′-TGATCAGAGACAGAGGTGCC-3′ for IL-1RII. PCR cycles were 94°C for 1 min, 59°C for 1 min, and 72°C for 2 min (40 cycles).
Results
Development of Arthropathy in IL-1ra−/− Mice.
IL-1ra−/− mice were produced as described 18 and backcrossed to either BALB/cA or C57BL/6J mice. We noticed that, although (129 × C57BL/6J) F1 hybrid mice looked normal, IL-1ra−/− mice after backcrossing five generations to BALB/cA mice spontaneously developed chronic inflammatory arthropathy. In those mice, swelling and redness were observed in multiple joints. These symptoms were most remarkable at the ankle joint of the hindlimbs (Fig. 1, a and b) and appeared less frequently in the finger joints. The joints of the forelimbs also developed arthropathy, although the incidence and severity were low. Histological analysis of the ankle joints showed marked synovial and periarticular inflammation, with articular erosion caused by invasion of granulation tissues (Fig. 1c and Fig. d). Proliferation of synovial lining cells was remarkable, and invasion of inflammatory cells, including lymphocytes and neutrophils, was seen forming pannus (Fig. 1 d). Invasion of inflammatory cells mainly consisting of neutrophils was observed in the synovial space. Fibrin clots were also seen in this space (Fig. 1 e). Bone erosion was also remarkable, and the bone matrix was replaced with fibroblastic cells with marked activation of osteoclasts (Fig. 1 f). These histological findings were similar to those for RA in humans. Other histopathological abnormalities were not found in IL-1ra−/− mice, except for smaller body size 18.
Figure 1.
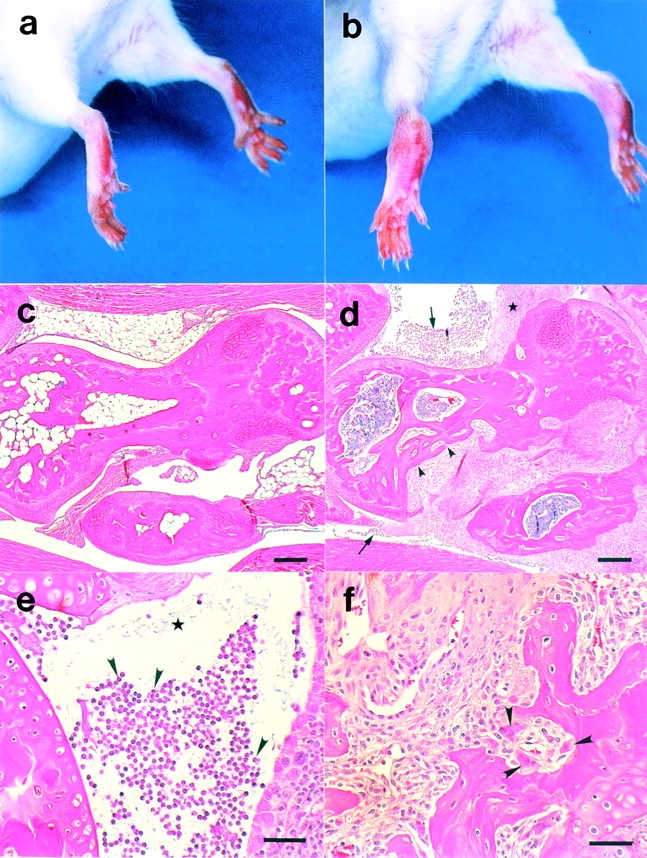
Histopathology of the ankle joints of BALB/cA IL-1ra−/− mice. The ankles of a normal IL-1ra+/+ mouse (a) and an affected IL-1ra−/− mouse (b) at 16 wk of age. Swelling and redness of the joints was observed in the IL-1ra−/− mouse. Microscopic observation of the joints of an IL-1ra+/+ mouse (c) and an IL-1ra−/− mouse (d), showing erosive destruction of the bone in the IL-1ra−/− mouse (arrowheads). Infiltration of inflammatory cells (arrows) and proliferation of the lining cells of the synovial membrane (★) were remarkable. (e) Neutrophil infiltration in the articular cavity (arrowheads) and deposition of fibrin clots (★). (f) Pannus formation; note the activation of osteoclasts (arrowheads). Scale bars: c and d, 100 μm; e and f, 50 μm.
The early sign of arthritis could be detected in IL-1ra−/− mice of the BALB/cA background as early as 5 wk of age. The incidence gradually increased, and >80% of mice became arthritic before 8 wk of age (Fig. 2 a). By 13 wk of age, all of the IL-1ra−/− mice developed arthritis, whereas none of the wild-type (IL-1ra+/+) or heterozygous (IL-1ra+/−) littermates did (data not shown). Arthritic severity score was also increased gradually in IL-1ra−/− mice (Fig. 2 b). Incidence and severity scores of arthritis were not significantly different between males and females (Fig. 2, a and b). It was thus shown that deficiency in the IL-1ra gene causes arthritis.
Figure 2.

Incidence of arthritis in BALB/cA IL-1ra−/− mice. (a) Incidence of arthritis in IL-1ra−/− mice. Development of arthritis was inspected weekly, and the percentage of arthritic mice (grade ≥1) is shown. (b) Arthritic severity score of IL-1ra−/− mice. The average scores of the mice with or without lesions are shown. Male mice: ▪, n = 10; female mice: •, n = 20. Average ± SEM is indicated. w, wk.
In contrast, none of the IL-1ra−/− mice on a C57BL/6J background developed the disease at all at 16 wk of age, when all of the BALB/cA IL-1ra−/− mice did (Table ). In older mice, arthritis developed at a low incidence. These observations indicate that a genetic factor(s) other than IL-1ra is involved in the development of this arthritis.
Table 1.
Incidence of Arthritis in IL-1ra−/− Mice in Different Genetic Backgrounds
| Age | BALB/cA | C57BL/6J | 129 × C57BL/6J |
|---|---|---|---|
| wk | |||
| 8 | 24/30 (80) | 0/56 (0) | 0/114 (0) |
| 16 | 56/56 (100) | 0/56 (0) | 0/114 (0) |
| 24 | ND | 1/13 (8) | 0/17 (0) |
| 32 | ND | 2/13 (15) | 0/2 (0) |
| 48 | 2/2 (100) | 2/7 (29) | ND |
Development of Autoimmunity in IL-1ra−/− Mice.
Histological analyses revealed that lymphoid tissues such as spleens and lymph nodes were enlarged in these mice; average spleen weight was 1.5-fold and that of lymph nodes was 1.8-fold heavier than in wild-type littermates at 16 wk of age (P < 0.001, t test), suggesting an activation of the immune system in arthritic IL-1ra−/− mice. However, the proportion of CD44+ or CD25+ T cell population as well as the T and B cell ratio and CD4+ and CD8+ T cell ratio was not significantly altered in IL-1ra−/− compared with IL-1ra+/+ mice (data not shown). These observations indicate that IL-1ra deficiency does not significantly affect T cell and B cell composition in normal physiology.
We next investigated Ig levels in the serum. The IgM concentration was not changed in these mice (Fig. 3 a). In contrast, total IgG and IgE levels were elevated two- to threefold at 16 wk of age (Fig. 3 a). Among the IgG subclasses, the level of IgG1, the major component of serum IgG, was significantly elevated, whereas IgG2a and IgG2b levels were not changed and the IgG3 level was slightly reduced compared with IL-1ra+/+ mice (Fig. 3 b).
Figure 3.

Antibody production in IL-1ra−/− mice. Serum antibody levels in IL-1ra−/− mice (−/−) and their wild-type littermates (+/+) were determined by ELISA at 16 wk of age. (a) Levels of total IgM, IgG, and IgE in BALB/cA background mice. (b) Levels of IgG subclasses in BALB/cA background mice. (c) Levels of autoantibodies against IgG (RF) of IgG class and IgM class, IIC, and dsDNA in BALB/cA background mice. (d) Autoantibody levels in C57BL/6J background mice. The serum dilution in c and d was 1/25. Autoantibody levels were expressed as the relative absorbance of the ELISA. Average ± SD of each genotype is shown. Open bars: wild-type mice; n = 13 (a), n = 11 (b), n = 9 (c), and n = 8 (d). Shaded bars: IL-1ra−/− mice; n = 14 (a), n = 12 (b), n = 10 (c), and n = 15 (d). Statistical significance was calculated by Student's t test. *P < 0.0001; ‡ P < 0.005; § P < 0.05.
As involvement of autoimmunity was suggested in the development of RA, we next measured autoantibody levels relevant to RA in the sera of these mice. As shown in Fig. 3 c, antibody levels against IgG (IgG class RF), IIC, and dsDNA were significantly elevated in IL-1ra−/− BALB/cA mice at 16 wk of age compared with IL-1ra+/+ and IL-1ra+/− littermates, whereas IgM class RF was not significantly elevated in these mice. On the other hand, autoantibody levels were not significantly elevated in IL-1ra−/− mice on the C57BL/6J background in which no arthritis developed (Fig. 3 d). Autoantibody levels, however, did not necessarily correlate to the severity score of arthritis, and even BALB/cA IL-1ra−/− mice with low antibody levels developed arthritis at 16 wk of age. These observations suggest that IL-1ra deficiency causes autoimmunity in mice with specific genetic backgrounds.
Augmented Expression of Inflammatory Mediators in the Joints of IL-1ra−/− Mice.
We then investigated the pathogenesis of arthritis and autoimmunity. It was conceivable that the deficiency of IL-1ra could exaggerate IL-1 signaling, because IL-1ra is a competitive inhibitor of IL-1α and IL-1β. As IL-1 is known to induce various inflammatory cytokines, we analyzed expression of inflammatory mediators in the joints of IL-1ra−/− mice to examine whether IL-1 signaling is augmented in these mice. As shown in Fig. 4, the IL-1β mRNA level was increased 10-fold in arthritic joints at 16 wk of age (grade = 6) compared with IL-1ra+/+ mice. The expression levels of IL-6 and TNF-α were also augmented in the arthritic joints, although the augmentation of TNF-α was less intensive (IL-6, 10–26-fold; TNF-α, 1.2–1.5-fold). On the other hand, the expression level of IL-1α was rather suppressed (1/3–1/2). The COX-2 mRNA level was elevated 2.5–3.5-fold in 16-wk-old arthritic mice, coinciding with inflammation in the joints. The expression levels of both IL-1RI and IL-1RII were also increased twofold at 16 wk of age.
Figure 4.


Augmentation of IL-1β, IL-6, TNF-α, COX-2, and IL-1R mRNA expression in the joints of BALB/cA IL-1ra−/− mice. Total RNA was isolated from the joints, and poly (A)+ RNA was purified. Expression of the genes for IL-1ra, IL-1α, IL-1β, IL-6, TNF-α, IL-1RI, IL-1RII, and COX-2 was examined by Northern blot hybridization analysis. (a) Lanes 1 and 2 and 5–8, IL-1ra+/+ mice; lanes 3 and 4 and 9–12, IL-1ra−/− mice. Lanes 1 and 2 and 5–12, nonarthritic mice; lanes 3 and 4, arthritic mice (severity score = 6). Ages of the mice are indicated at top. The results were reproducible in three independent experiments. (b) Densitometric analysis of mRNA expression levels. The radioactivity for a band shown in panel a was measured by a BAS 2000 system and normalized by that of β-actin; the radioactivities of the mutant mice relative to those of age-matched wild-type mice are shown. White bar, wild-type mouse; black bar, arthritic IL-1ra−/− mouse; gray bar, nonarthritic IL-1ra−/− mouse. w, wk.
Interestingly, the augmentation of these inflammatory mediator genes was also observed in the joints before onset of the disease (Fig. 4, a and b). The expression levels of IL-1β were elevated 2–3-fold in the nonarthritic joints at 4 wk and 1.5–2-fold at 10–12-wk of age, respectively. The IL-6 and TNF-α expression levels were also elevated 2–4- and 1.5–2-fold at 10–12 wk of age, respectively. It was thus shown that IL-1ra deficiency causes augmentation of proinflammatory cytokines and inflammatory mediators even before onset of the disease, suggesting that the low level of IL-1 that is expressed under normal conditions induces the inflammatory genes in these mutant mice.
Discussion
In this report, we have shown that deficiency of the IL-1ra gene causes autoimmunity and arthritis, emphasizing the importance of IL-1/IL-1ra balance in maintaining normal physiology of the joints and homeostasis of the immune system. We found that the expression of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α was augmented in IL-1ra−/− mouse joints compared with wild-type mice. In this regard, it is noteworthy that low levels of IL-1α and IL-1β were expressed constitutively in the normal joints without any stimulation (Fig. 4). Thus, the fact that proinflammatory cytokine expression was enhanced in IL-1ra−/− mice suggests that IL-1 activity is suppressed by IL-1ra under physiological conditions, and disruption of the IL-1ra gene exaggerates the action of the basal level IL-1. This excess IL-1 signal, as well as overproduced proinflammatory cytokines, is considered to cause inflammation by enhancing infiltration of inflammatory cells into synovial tissues, increasing permeability of blood vessels, activating synovial lining cells to produce collagenases and metalloproteases, and activating osteoclasts to destruct the bony structure 2.
It is known that these cytokines can also activate the immune system by enhancing recruitment of immune cells, activating both T cells and B cells, and enhancing antigen presentation 19 20. This cytokine overproduction may also explain why autoimmunity developed in these mice. This autoimmune reaction against self-IgGs and synovial components may also be involved in the development of arthritis 21. Consistent with this notion, we found that IL-1ra−/− mice on the C57BL/6J background developed arthritis at a high incidence when these mice were immunized with IIC, suggesting hyperresponsiveness of the immune system in IL-1ra−/− mice (Saijo, S., R. Horai, and Y. Iwakura, unpublished observation). We also observed earlier onset and more severe phenotype of the disease on a DBA/1 background (Saijo, S., and Y. Iwakura, unpublished observation). Recently, Ma et al. reported a similar observation 22. These observations indicate that IL-1ra plays an important role in the regulation of the immune system.
The incidence of arthritis was high only in IL-1ra−/− mice on the BALB/cA background, and incidence in the C57BL/6J or 129 × C57BL/6J hybrid background mice was low. These observations indicate that susceptibility to arthritis differs among strains, suggesting that genes other than IL-1ra are involved in the development of arteritis. In this regard, Nicklin et al. recently found that arthritis developed in IL-1ra−/− mice on the 129 × MF1 Swiss albino hybrid background, although they did not observe any joint abnormality in those mice 22a. Thus, IL-1ra−/− mice seem to develop tissue specific inflammation depending on their genetic backgrounds. These additional pathogenic genes are now under investigation.
So far, several lines of evidence have suggested that aberration of IL-1α, IL-1β, and IL-1ra genes, which form a gene cluster on the long arm of chromosome 2, are involved in inflammatory and autoimmune diseases in humans. For example, IL-1α polymorphism was detected in humans, and a specific variant was suggested to be associated with juvenile RA 23. Epidemiological studies have also invoked a possibility that IL-1ra is involved in the epithelial cell manifestations of diseases such as ulcerative colitis, systemic lupus erythematosus, psoriasis, lichen sclerosus, alopecia areata, and severity of Sjögren's syndrome, as these conditions are associated with a particular type of IL-1ra gene polymorphism 24 25 26 27 28 29. In addition, imbalance between IL-1ra and IL-1β production was observed in RA patients 30 31. RA patients exhibited a lower ratio of IL-1ra/IL-1β in plasma, even at baseline, in comparison with osteoarthritis or osteomyelitis patients 32. This observation suggests that IL-1ra production may be relatively deficient or inadequate in RA patients. Thus, it is tempting to speculate that IL-1ra gene deficiency is also involved in the development of arthritis in humans. However, so far IL-1ra gene polymorphism has not been reported in RA in humans. We are now examining this possibility in RA patients with familial history.
Various animal disease models have been developed to elucidate the ethiopathogenesis of RA in humans. It is well known that antigens cross-reactive with synovial components, such as IIC, and mycobacteria and streptococcal cell wall components induce arthritis in animals 33 34 35. MRL-lpr/lpr mice, which have a mutation in the fas gene, develop arthritis spontaneously due to development of autoimmunity 36. Besides these models, we recently developed an HTLV-I transgenic mouse model in which autoimmunity was caused by the action of the tax gene 21 37. In this report, we have shown a novel RA model, the BALB/cA IL-1ra−/− mouse. As the joint pathology and the immunological status in this model closely resembles that of RA patients and the penetrance is complete, these mice should also provide another useful model for RA in humans.
Acknowledgments
We thank Drs. C. Morimoto and Y. Kanai for critical reading of the manuscript, Drs. A. Matsuzawa and T. Yasuda for kind technical advice in determination of Ig levels, and Drs. M. Kotani and T. Sakatani for discussion and kind help with experiments. We thank Drs. T. Sudo, S. Yamamoto, T. Yokota, and T. Akiyama for mouse cDNA probes. We also thank all of the members of our labs for their kind discussion and help in animal care.
This work was supported by grants from the Ministry of Education, Culture, Sport, and Science of Japan, The Ministry of Health and Welfare of Japan, Core Research for Evolutional Science and Technology (CREST), the Japan Society for the Promotion of Science, the Pioneering Research Project in Biotechnology, and the Naito Foundation.
Footnotes
Abbreviations used in this paper: COX, cyclooxygenase; IIC, type II collagen; ra, receptor antagonist; RA, rheumatoid arthritis; RF, rheumatoid factor.
References
- Firestein, G.S., and N.J. Zvaifler. 1992. Rheumatoid arthritis: a disease of disordered immunity. In Inflammation: Basic Principles and Clinical Correlates, 2nd ed. Raven Press, Ltd., New York. 959–975.
- Feldmann M., Brennan F.M., Maini R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- Pettipher E.R., Higgs G.A., Henderson B. Interleukin 1 induces leukocyte infiltration and cartilage proteoglycan degradation in the synovial joint. Proc. Natl. Acad. Sci. USA. 1986;83:8749–8753. doi: 10.1073/pnas.83.22.8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinova-Mutafchieva L., Williams R.O., Mason L.J., Mauri C., Feldmann M., Maini R.N. Dynamics of proinflammatory cytokine expression in the joints of mice with collagen-induced arthritis (CIA) Clin. Exp. Immunol. 1997;107:507–512. doi: 10.1046/j.1365-2249.1997.2901181.x. [DOI] [PubMed] [Google Scholar]
- van den Berg W.B., Joosten L.A., Helsen M., van de Loo F.A. Amelioration of established murine collagen-induced arthritis with anti-IL-1 treatment. Clin. Exp. Immunol. 1994;95:237–243. [Google Scholar]
- Joosten L.A., Helsen M.M., van de Loo F.A., van den Berg W.B. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNF-α, anti-IL-1α/β, and IL-1Ra. Arthritis Rheum. 1996;39:797–809. doi: 10.1002/art.1780390513. [DOI] [PubMed] [Google Scholar]
- Makarov S.S., Olsen J.C., Johnston W.N., Anderle S.K., Brown R.R., Baldwin A.S., Jr., Haskill J.S., Schwab J.H. Suppression of experimental arthritis by gene transfer of interleukin 1 receptor antagonist cDNA. Proc. Nat. Acad. Sci. USA. 1996;93:402–406. doi: 10.1073/pnas.93.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocci M.J., Schmidt J.A. Interleukin-1structure and function. In: Remick D.G., Friedland J.S., editors. Cytokines in Health and Disease. 2nd ed. Marcel Dekker, Inc.; New York: 1997. pp. 1–27. [Google Scholar]
- Sims J.E., Gayle M.A., Slack J.L., Alderson M.R., Bird T.A., Giri J.G., Colotta F., Re F., Mantovani A., Shanebeck K. Interleukin 1 signaling occurs exclusively via the type I receptor. Proc. Natl. Acad. Sci. USA. 1993;90:6155–6159. doi: 10.1073/pnas.90.13.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter D.B., Deibel M.R., Jr., Dunn C.J., Tomich C.-S.C., Laborde A.L., Slightom J.L., Berger A.E., Bienkowski M.J., Sun F.F., McEwan R.N. Purification, cloning, expression and biological characterization of an interleukin-1 receptor antagonist protein. Nature. 1990;344:633–638. doi: 10.1038/344633a0. [DOI] [PubMed] [Google Scholar]
- Hannum C.H., Wilcox C.J., Arend W.P., Joslin F.G., Dripps D.J., Heimdal P.L., Armes L.G., Sommer A., Eisenberg S.P., Thompson R.C. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature. 1990;343:336–340. doi: 10.1038/343336a0. [DOI] [PubMed] [Google Scholar]
- Colotta F., Re F., Muzio M., Bertini R., Polentarutti N., Sironi M., Giri J.G., Dower S.K., Sims J.E., Mantovani A. Interleukin-1 type II receptora decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- Haskill S., Martin G., Van Le L., Morris J., Peace A., Bigler C.F., Jaffe G.J., Hammerberg C., Sporn S.A., Fong S. cDNA cloning of an intracellular form of the human interleukin 1 receptor antagonist associated with epithelium. Proc. Natl. Acad. Sci. USA. 1991;88:3681–3685. doi: 10.1073/pnas.88.9.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M., Polentarutti N., Sironi M., Poli G., De Gioia L., Introna M., Mantovani A., Colotta F. Cloning and characterization of a new isoform of the interleukin 1 receptor antagonist. J. Exp. Med. 1995;182:623–628. doi: 10.1084/jem.182.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyak M., Smith M.F., Jr., Abel A.A., Hance K.R., Arend W.P. The differential production of three forms of IL-1 receptor antagonist by human neutrophils and monocytes. J. Immunol. 1998;161:2004–2010. [PubMed] [Google Scholar]
- Malyak M., Guthridge J.M., Hance K.R., Dower S.K., Freed J.H., Arend W.P. Characterization of a low molecular weight isoform of IL-1 receptor antagonist. J. Immunol. 1998;161:1997–2003. [PubMed] [Google Scholar]
- Arend W.P., Malyak M., Guthridge C.J., Gabay C. Interleukin-1 receptor antagonistrole in biology. Annu. Rev. Immunol. 1998;16:27–55. doi: 10.1146/annurev.immunol.16.1.27. [DOI] [PubMed] [Google Scholar]
- Horai R., Asano M., Sudo K., Kanuka H., Suzuki M., Nishihara M., Takahashi M., Iwakura Y. Production of mice deficient in genes for interleukin (IL)-1α, IL-1β, IL-1α/β, and IL-1 receptor antagonist shows that IL-1β is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 1998;187:1463–1475. doi: 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C.A. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- Durum S.K., Oppenheim J.J. Proinflammatory cytokines and immunity. In: Paul W.E., editor. Fundamental Immunology. 3rd ed. Raven Press, Ltd; New York: 1993. pp. 801–835. [Google Scholar]
- Iwakura Y., Saijo S., Kioka Y., Nakayama-Yamada J., Itagaki K., Tosu M., Asano M., Kanai Y., Kakimoto K. Autoimmunity induction by human T cell leukemia virus type 1 in transgenic mice that develop chronic inflammatory arthropathy resembling rheumatoid arthritis in humans. J. Immunol. 1995;155:1588–1598. [PubMed] [Google Scholar]
- Ma Y., Thornton S., Boivin G.P., Hirsh D., Hirsch R., Hirsch E. Altered susceptibility to collagen-induced arthritis in transgenic mice with aberrant expression of interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:1798–1805. doi: 10.1002/1529-0131(199810)41:10<1798::AID-ART11>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Nicklin M.J.H., Hughes D.E., Barton J.L., Ure J.M., Duff G.W. Arterial inflammation in mice lacking the interleukin 1 receptor agonist gene. J. Exp. Med. 1999;191:303–311. doi: 10.1084/jem.191.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell T.L., Symons J.A., Ploski R., Forre O., Duff G.W. A genetic association between juvenile rheumatoid arthritis and a novel interleukin-1α polymorphism. Arthritis Rheum. 1995;38:221–228. doi: 10.1002/art.1780380210. [DOI] [PubMed] [Google Scholar]
- Mansfield J.C., Holden H., Tarlow J.K., Di Giovine F.S., McDowell T.L., Wilson A.G., Holdsworth C.D., Duff G.W. Novel genetic association between ulcerative colitis and the anti-inflammatory cytokine interleukin-1 receptor antagonist. Gastroenterology. 1994;106:637–642. doi: 10.1016/0016-5085(94)90696-3. [DOI] [PubMed] [Google Scholar]
- Blakemore A.I., Tarlow J.K., Cork M.J., Gordon C., Emery P., Duff G.W. Interleukin-1 receptor antagonist gene polymorphism as a disease severity factor in systemic lupus erythematosus. Arthritis Rheum. 1994;37:1380–1385. doi: 10.1002/art.1780370917. [DOI] [PubMed] [Google Scholar]
- Perrier S., Coussediere C., Dubost J.J., Albuisson E., Sauvezie B. IL-1 receptor antagonist (IL-1RA) gene polymorphism in Sjögren's syndrome and rheumatoid arthritis. Clin. Immunol. Immunopathol. 1998;87:309–313. doi: 10.1006/clin.1998.4520. [DOI] [PubMed] [Google Scholar]
- Tarlow J.K., Cork M.J., Clay F.E., Schmitt-Egenolf M., Crane A.M., Stierle C., Boehncke W.H., Eiermann T.H., Blakemore A.I., Bleehen S.S. Association between interleukin-1 receptor antagonist (IL-1ra) gene polymorphism and early and late-onset psoriasis. Br. J. Dermatol. 1997;136:147–148. doi: 10.1111/j.1365-2133.1997.tb08779.x. [DOI] [PubMed] [Google Scholar]
- Clay F.E., Cork M.J., Tarlow J.K., Blakemore A.I., Harrington C.I., Lewis F., Duff G.W. Interleukin 1 receptor antagonist gene polymorphism association with lichen sclerosus. Hum. Genet. 1994;94:407–410. doi: 10.1007/BF00201602. [DOI] [PubMed] [Google Scholar]
- Tarlow J.K., Clay F.E., Cork M.J., Blakemore A.I., McDonagh A.J., Messenger A.G., Duff G.W. Severity of alopecia areata is associated with a polymorphism in the interleukin-1 receptor antagonist gene. J. Invest. Dermatol. 1994;103:387–390. doi: 10.1111/1523-1747.ep12395398. [DOI] [PubMed] [Google Scholar]
- Firestein G.S., Boyle D.L., Yu C., Paine M.M., Whisenand T.D., Zvaifler N.J., Arend W.P. Synovial interleukin-1 receptor antagonist and interleukin-1 balance in rheumatoid arthritis. Arthritis Rheum. 1994;37:644–652. doi: 10.1002/art.1780370507. [DOI] [PubMed] [Google Scholar]
- Chomarat P., Vannier E., Dechanet J., Rissoan M.C., Banchereau J., Dinarello C.A., Miossec P. Balance of IL-1 receptor antagonist/IL-1β in rheumatoid synovium and its regulation by IL-4 and IL-10. J. Immunol. 1995;154:1432–1439. [PubMed] [Google Scholar]
- Chikanza I.C., Roux-Lombard P., Dayer J.M., Panayi G.S. Dysregulation of the in vivo production of interleukin-1 receptor antagonist in patients with rheumatoid arthritis. Pathogenetic implications. Arthritis Rheum. 1995;38:642–648. doi: 10.1002/art.1780380511. [DOI] [PubMed] [Google Scholar]
- Trentham D.E., Townes A.S., Kang A.H. Autoimmunity to type II collagenan experimental model of arthritis. J. Exp. Med. 1977;146:857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson C.M., Waksman B.H., Sharp J.T. Studies of arthritis and other lesions induced in rats by injection of mycobacterial adjuvant. J. Exp. Med. 1961;113:485–510. doi: 10.1084/jem.113.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromartie W.J., Craddock J.G., Schwab J.H., Anderle S.K., Yang C.H. Arthritis in rats after systemic injection of streptococcal cells or cell walls. J. Exp. Med. 1977;146:1585–1602. doi: 10.1084/jem.146.6.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang L., Theofilopoulos A.N., Dixon F.J. A spontaneous rheumatoid arthritis-like disease in MRL/l mice. J. Exp. Med. 1982;155:1690–1701. doi: 10.1084/jem.155.6.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakura Y., Tosu M., Yoshida E., Takiguchi M., Sato K., Kitajima I., Nishioka K., Yamamoto K., Takeda T., Hatanaka M. Induction of inflammatory arthropathy resembling rheumatoid arthritis in mice transgenic for HTLV-I. Science. 1991;253:1026–1028. doi: 10.1126/science.1887217. [DOI] [PubMed] [Google Scholar]