A Regulatory Role for Src Homology 2 Domain–Containing Inositol 5′-Phosphatase (Ship) in Phagocytosis Mediated by Fcγ Receptors and Complement Receptor 3 (αMβ2; Cd11b/Cd18) (original) (raw)
Abstract
The Src homology 2 domain–containing inositol 5′-phosphatase (SHIP) is recruited to immunoreceptor tyrosine-based inhibition motif (ITIM)–containing proteins, thereby suppressing phosphatidylinositol 3-kinase (PI 3-kinase)–dependent pathways. The role of SHIP in phagocytosis, a PI 3-kinase–dependent pathway, is unknown. Overexpression of SHIP in macrophages led to an inhibition of phagocytosis mediated by receptors for the Fc portion of IgG (FcγRs). In contrast, macrophages expressing catalytically inactive SHIP or lacking SHIP expression demonstrated enhanced phagocytosis. To determine whether SHIP regulates phagocytosis mediated by receptors that are not known to recruit ITIMs, we determined the effect of SHIP expression on complement receptor 3 (CR3; CD11b/CD18; αMβ2)–dependent phagocytosis. Macrophages overexpressing SHIP demonstrated impaired CR3-mediated phagocytosis, whereas macrophages expressing catalytically inactive SHIP demonstrated enhanced phagocytosis. CR3-mediated phagocytosis in macrophages derived from SHIP−/− mice was up to 2.5 times as efficient as that observed in macrophages derived from littermate controls. SHIP was localized to FcγR- and CR3-containing phagocytic cups and was recruited to the cytoskeleton upon clustering of CR3. In a transfected COS cell model of activation-independent CR3-mediated phagocytosis, catalytically active but not inactive SHIP also inhibited phagocytosis. We conclude that PI 3-kinase(s) and SHIP regulate multiple forms of phagocytosis and that endogenous SHIP plays a role in modulating β2 integrin outside-in signaling.
Keywords: macrophage, integrin, phosphatidylinositol 3-kinase, actin, leukocyte
Introduction
Phagocytosis, the process by which cells engulf microbial pathogens, apoptotic cells, and particulate debris, triggers the activation of multiple transmembrane signaling pathways that culminate in cytoskeletal assembly and particle ingestion. The requirement for specific signaling pathways in the various stages of phagocytosis is only partly understood. In the case of FcγR-mediated phagocytosis, particle ingestion requires the net assembly of actin in a phosphatidylinositol (PI) 3-kinase –independent fashion 1. PI 3-kinase activity is required for other aspects of phagocytic signaling, such as pseudopod extension 1 and phagosomal closure 2. The role of PI 3-kinase in promoting phagocytosis via other receptors is uncertain.
The Src homology 2 (SH2) domain–containing inositol 5′-phosphatase (SHIP) has been implicated in multiple signaling pathways in hematopoietic cells (for reviews, see references 3 and 4). SHIP is composed of multiple domains, including an SH2 domain, a catalytic domain, two NPXY motifs that confer binding to phosphotyrosine binding (PTB) domain–containing proteins, and several proline-rich regions. In B cells, coligation of the B cell antigen receptor with FcγRIIB results in the phosphorylation of the immunoreceptor tyrosine inhibitory motif (ITIM) of FcγRIIB. SHIP is subsequently recruited to the phosphorylated ITIM, which results in decreased phosphatidylinositol 3,4,5-trisphosphate (PtdIns[3,4,5]P3) content, decreased recruitment and activation of Bruton's tyrosine kinase and phospholipase Cγ, and impaired calcium signaling 5 6 7. Similarly, SHIP expression in mast cells is correlated with impaired degranulation after the addition of IgE-containing immune complexes 8 9. Although the role of SHIP in macrophage function is unknown, the phosphotyrosyl content of SHIP increases after ligation of FcγRs 10 11. Therefore, SHIP may subserve a negative regulatory role during engagement of FcγRs in macrophages, in part as a result of its interaction with the ITIM-containing FcγRIIB. In addition, SHIP has been shown to interact with phosphorylated immunoreceptor tyrosine activation motif (ITAM)-bearing peptides derived from the γ subunit of FcRs in vitro 10. Other SHIP-binding proteins that undergo enhanced tyrosine phosphorylation during engagement of FcγRs include Shc 12 and the p85 subunit of PI 3-kinase 13 14.
Ingestion of particles opsonized with the complement fragment C3bi is mediated predominantly by the β2 integrin, complement receptor 3 (CR3). Unlike FcγRs, which are constitutively active, CR3 requires prior activation for its phagocytic function (for a review, see reference 15). Although recent studies have emphasized differences in the mode of signaling via FcγRs and CR3 16 17, similarities do exist. For example, ligation of both FcγRs and CR3 leads to the enhanced production of PtdIns(3,4,5)P3 1 18 19, most likely as a result of the activation of one or more members of the PI 3-kinase family of enzymes. Although it is clear that PI 3-kinase activity is required for FcγR-mediated phagocytosis 1 2 18 20, a role for this enzymatic pathway in CR3-mediated phagocytosis has not been described. PI 3-kinase has been implicated in the adhesive function of β2 integrins, but its precise role in β2 integrin signaling is unknown. Some studies have emphasized the capacity of PI 3-kinase to upregulate the affinity or avidity of β2 integrins for ligand 21 22. In the case of LFA-1, this may occur by the recruitment of pleckstrin homology (PH) domain–containing proteins, such as cytohesins, to the cytosolic domain of the integrin 23. This suggests that PI 3-kinase regulates “inside-out” signaling of β2 integrins. It is unknown whether PI 3-kinase also influences “outside-in” integrin signaling.
To determine whether SHIP plays a role in phagocytosis via FcγRs and CR3, we modulated SHIP expression in a macrophage cell line and in COS cells transfected with the αM and β2 subunits of CR3. We also measured the phagocytic capacity of macrophages derived from SHIP−/− mice.
Materials and Methods
Cells and Reagents.
RAW LR5 cells were derived from RAW 264.7 cells 24. Primary macrophages were isolated from the peritoneal cavities of C57BL/6 mice by peritoneal lavage (resident peritoneal macrophages) or by peritoneal lavage 4 d after the intraperitoneal injection of thioglycollate (thio-macrophages) as described previously 25. COS-7 cells were obtained from American Type Culture Collection. Cells were maintained in RPMI medium containing 10% (vol/vol) FCS, 100 U/ml penicillin G, and 100 μg/ml streptomycin. Latex beads were from Polysciences, Inc. mAb 9E10 against the Myc epitope was from Boehringer. Rabbit antiserum against SHIP was a gift from Jeffrey Ravetch (The Rockefeller University, New York, NY). mAb CBRM1/5 against an activation-dependent epitope on CR3 26 was from Tim Springer (Harvard Medical School, Boston, MA). mAb U7.12 (mouse IgG2a) against DNP was a gift from Jay Unkeless (Mount Sinai School of Medicine, New York, NY). mAb M1/70 against CD11b and rat IgG2b were from Caltag. Goat IgG M19 against αM and rabbit IgG C-20 against c-fms were from Santa Cruz Biotechnology, Inc. mAb rmC5-3 against murine CD14 and mAb J11d against murine CD24 were from BD PharMingen. Goat IgG against human C3 was from Sigma-Aldrich. Rabbit anti–sheep erythrocyte IgG was from Cappel. Rhodamine-phalloidin, Alexa 568–conjugated goat anti–rabbit IgG, Alexa 488–conjugated goat anti–mouse IgG, Alexa 488–conjugated goat anti–rabbit IgG, and Alexa 488–streptavidin were from Molecular Probes. Horseradish peroxidase–conjugated donkey F(ab′)2 fragments against rabbit IgG, rat IgG, and goat IgG were from Jackson ImmunoResearch Laboratories. RC20H was from Transduction Laboratories. Superfect transfection reagent was from QIAGEN.
Construction of Plasmids and Transfection of Cells.
Murine SHIP (obtained from K. Mark Coggeshall, Ohio State University, Columbus, OH) was subcloned in pcDNA3.1/Myc-HisB (Invitrogen). The mutants used in this study, SHIPΔP′ase (D672A), a catalytically inactive mutant 27, SHIPR34G, an SH2 domain mutant 28 29, and SHIPY917,1020F, an NPXY motif mutant, were generated using the QuikChange Site-Directed Mutagenesis kit (Stratagene). All constructs were verified by DNA sequencing. CD18 and CD11b expression plasmids were gifts from Lloyd Klickstein (Harvard Medical School, Boston, MA) and Eric Brown (University of California at San Francisco, San Francisco, CA) respectively. Transfection of plasmids into either COS-7 or RAW LR5 cells was performed using Superfect according to the manufacturer's guidelines.
Particle Binding and Ingestion, and Immunofluorescence.
Sheep erythrocytes opsonized with rabbit IgG (EIgG) and carboxylate-modified latex beads ranging in size from 1 to 6 μm were opsonized with BSA/rabbit anti-BSA IgG as described 1. Sheep erythrocytes were opsonized with rabbit IgM and C3bi (EC3bi) as described 30. Thio-macrophages or RAW LR5 cells adherent to 13-mm round glass coverslips were incubated in buffer A (125 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 5 mM glucose, 10 mM NaHCO3, 1 mM MgCl2, 1 mM CaCl2, and 20 mM Hepes [pH 7.4]) and with either 5 × 106 EIgG or EC3bi, or with IgG-coated latex beads for the indicated times; association and phagocytosis assays were performed as described 1. For experiments in transfected RAW LR5 or COS cells, SHIP-expressing cells were identified by indirect immunofluorescence. After fixation in 3.7% formaldehyde, cells were permeabilized with 0.2% Triton X-100 and stained with mAb 9E10 and Alexa 488–conjugated anti–mouse IgG. For FcγR-mediated phagocytosis, 30 cells/coverslip were scored for association or phagocytosis. For CR3-mediated phagocytosis, 60–90 cells/coverslip were scored for association or phagocytosis. Nonexpressing cells on the same coverslips served as controls. Cells from a minimum of five separate microscopic fields per coverslip were analyzed.
For immunofluorescence experiments using rabbit IgG to detect SHIP, EIgG2a were obtained after derivitization of sheep erythrocytes with DNP using 0.3% picrylsulfonic acid and after incubation of the resultant dinitrophenylated erythrocytes with mouse IgG2a against DNP. For immunofluorescence localization of SHIP during phagocytosis of EC3bi, mouse erythrocytes were opsonized with purified rat IgM against CD24 followed by the addition of C5-deficient serum. Deposition verification of C3 was performed using indirect immunofluorescence. After incubation of adherent thio-macrophages with either EIgG2a or EC3bi, cells were fixed with 3.7% formaldehyde, permeabilized with 0.2% Triton X-100, and stained with rabbit antiserum against SHIP or rabbit IgG against c-fms followed by Alexa 568 anti–rabbit IgG and either Alexa 488 anti–mouse IgG to detect EIgG2a or goat anti-C3 followed by biotin-anti–goat IgG and Alexa 488–streptavidin to detect EC3bi. Cells were imaged using confocal laser scanning microscopy.
Isolation of Triton X-100–insoluble Cytoskeletons and Immunoblotting.
RAW LR5 cells were incubated with 2 μg/ml M1/70 or IgG2b in buffer A for 20 min at 4°C followed by 4 μg/ml F(ab′)2 goat anti–rat IgG for 5 min at 37°C. Cells were subjected to detergent lysis (1% Triton X-100, 150 mM NaCl, 50 mM Tris-HCl, 2 mM EDTA, 1 mM sodium orthovanadate, 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin; pH 7.4) at 4°C. Triton X-100–insoluble fractions were isolated after centrifugation (13,000 g for 5 min) and were solubilized in sample buffer after sonication and boiling. After SDS-PAGE and transfer onto nitrocellulose, immunoblotting was performed using the indicated antibodies. To determine the phosphotyrosyl content of SHIP, cells were incubated with M1/70 or IgG2b, as described above, and subjected to detergent lysis, immunoprecipitation using rabbit serum against SHIP, SDS-PAGE, and immunoblotting using rabbit antiserum against SHIP or RC20H, which recognizes phosphotyrosine. Quantitation of αM, SHIP, CD14, and phosphotyrosine content was performed using densitometry of immunoblots and NIH Image software.
Data Collection and Statistics.
Association and phagocytosis assays were performed in parallel. Experimental and control groups (e.g., SHIP −/− and SHIP+/+, or SHIP-expressing cells and nonexpressing controls) were analyzed in pairs. Statistical analysis of data was performed using two-tailed Student's t tests for paired samples.
Results
SHIP Is a Negative Regulator of FcγR-mediated Phagocytosis.
To test a role for SHIP in FcγR-mediated phagocytosis, we challenged thio-macrophages derived from SHIP−/− and SHIP+/+ mice with EIgG. SHIP−/− cells demonstrated a 45 ± 18% (mean ± SEM, n = 5) enhancement in the phagocytosis of EIgG. The increase in phagocytosis in SHIP−/− cells was not due to enhanced particle binding, as there was no significant difference in the binding of EIgG to these cells (Fig. 1). Also, there was no difference in surface FcγR expression between SHIP−/− and SHIP+/+ macrophages, as determined by flow cytometry using mAb 2.4G2 (data not shown). As PI 3-kinase inhibition resulted in decreased pseudopod extension 1, we tested whether sensitivity to SHIP modulation is dependent on the size of the phagocytic target; we reasoned that ingestion of large beads, which requires a greater extent of pseudopod extension, would be more sensitive to alteration in SHIP expression. There was no difference in phagocytosis efficiency of 2-μm beads between SHIP+/+ and SHIP−/− macrophages; however, larger particles (4.5- and 6-μm beads) were ingested more efficiently in SHIP−/− macrophages (Fig. 1 C). Both the rate (Fig. 1 D) and extent (Fig. 1B and Fig. C) of phagocytosis was enhanced in SHIP−/− macrophages. To confirm the role of endogenous SHIP in modulating FcγR-mediated phagocytosis, we transiently transfected Myc-tagged wild-type or catalytically inactive SHIP in RAW LR5 cells and challenged the transfected cells with EIgG. By microspectrofluorometry, both SHIP wild-type and SHIPΔP′ase were expressed at similar levels and overexpression of SHIP resulted in a 4.7 ± 0.4–fold (mean ± SEM, n = 57 cells) increase over endogenous SHIP. Overexpression of wild-type SHIP resulted in a 66 ± 4% (mean ± SEM, n = 5) inhibition of phagocytosis of EIgG. In contrast, expression of SHIPΔP′ase resulted in a 57 ± 17% (mean ± SEM, n = 5) enhancement of phagocytosis of EIgG, consistent with a dominant negative regulatory role for this construct in macrophage transfectants. Similar to SHIP−/− macrophages, macrophages expressing SHIPΔP′ase showed enhanced ingestion of large but not small IgG-coated particles (Fig. 2 A). Furthermore, SHIP was apparent in phagocytic cups in macrophages challenged with EIgG (Fig. 2 B). These data indicate that SHIP inhibits FcγR-mediated phagocytosis and that inhibition requires an intact catalytic domain.
Figure 1.

Macrophages lacking SHIP demonstrate enhanced FcγR-mediated phagocytosis. (A and B) Thio-macrophages derived from SHIP+/+ (black bars) and SHIP−/− (hatched bars) mice were challenged with EIgG for 15 min at 37°C as described in Materials and Methods. Association and phagocytic indices are shown. Data represent mean ± SEM, n = 5 (association) and 8 (phagocytosis). Differences between phagocytosis, but not binding, in SHIP+/+ and SHIP−/− macrophages were significant (P < 0.05). (C) Phagocytosis of SHIP−/− macrophages cells ingesting EIgG or IgG-coated latex beads expressed as a percentage of phagocytosis in SHIP+/+ macrophages. Phagocytic indices for 2-, 4.5-, and 6-μm beads, and EIgG, in SHIP+/+ macrophages were 540 ± 18, 1,284 ± 105, 140 ± 22, and 761 ± 55, respectively. Data represent mean ± SEM, n = 4–8. Differences between phagocytosis of 4.5- and 6-μm beads, and EIgG, but not 2-μm beads, in SHIP+/+ and SHIP−/− macrophages were significant (P < 0.05). (D) Time course of synchronized phagocytosis of EIgG in macrophages derived from SHIP+/+ (•) and SHIP_−_ /− (▪) mice. Macrophages were incubated with EIgG at 4°C to allow binding, but not ingestion, and further incubated for the indicated times at 37°C to allow for ingestion. Data represent mean ± SEM, n = 3.
Figure 2.

Endogenously expressed SHIP modulates FcγR-mediated phagocytosis. (A) Adherent RAW LR5 cells transfected with Myc-tagged wild-type SHIP (WT) or phosphatase-deficient SHIP (SHIPΔP′ase) were challenged with EIgG or IgG-coated beads for 30 min at 37°C as described in Materials and Methods. Phagocytosis of cells overexpressing SHIP (black bars) or expressing SHIPΔP′ase (hatched bars) is reported as a percentage of nonexpressing controls. Phagocytic indices for 2-, 4.5-, and 6-μm beads, and EIgG, in control macrophages were 652 ± 120, 423 ± 73, 261 ± 30, and 301 ± 34, respectively. Data represent mean ± SEM, n = 3–6. Differences between phagocytosis of macrophages expressing either wild-type SHIP or SHIPΔP′ase, and nonexpressing controls were significant (P values of 0.03 or less) for all but the 2-μm beads. (B) Adherent thio-macrophages obtained from SHIP+/+ mice were incubated with EIgG2a for 7 min at 37°C. After fixation, cells were stained for the presence of SHIP and EIgG2a as described in Materials and Methods. Bar, 10 μm.
SHIP Is a Negative Regulator of CR3-mediated Phagocytosis.
SHIP binds to phosphorylated ITIMs present in FcγRIIB, which is likely to be recruited by IgG ligand during phagocytosis. To determine whether SHIP plays a more general role in modulating phagocytosis (i.e., phagocytosis triggered by receptors that are not known to recruit ITIMs), we challenged thio-macrophages derived from SHIP−/− and SHIP+/+ mice with EC3bi, which selectively ligate CR3. Macrophages derived from SHIP−/− mice demonstrated enhanced ingestion of EC3bi compared with macrophages derived from SHIP+/+ mice (Fig. 3). The increased phagocytosis observed in SHIP−/− macrophages was due to enhanced phagocytic efficiency, as binding of EC3bi to these cells was not significantly increased (Fig. 3A and Fig. B) and there was no difference in the surface expression of the CR3 in SHIP−/− and SHIP+/+ macrophages (data not shown). CR3-mediated phagocytosis was twice as efficient in unstimulated SHIP−/− thio-macrophages; this effect was present, but less marked, in cells undergoing phagocytosis in the presence of PMA, which maximally stimulates CR3-mediated phagocytosis (Fig. 3 C).
Figure 3.

Lack of SHIP expression is associated with enhanced CR3-mediated phagocytosis. Adherent thio-macrophages obtained from SHIP+/+ (black bars) or SHIP−/− (hatched bars) mice were incubated with EC3bi for 30 min at 37°C. The association and phagocytosis indices were determined for cells incubated in the absence (A) and/or presence (B) of 100 nM PMA. Phagocytic indices in SHIP+/+ macrophages were 91 ± 17 in the absence of PMA and 479 ± 65 in the presence of PMA. (C) Phagocytosis of EC3bi expressed as a percentage, SHIP−/− versus SHIP+/+. Data represent mean ± SEM, n = 5–8. Differences between phagocytosis, but not binding, in SHIP+/+ and SHIP−/− macrophages were significant (P < 0.05).
PI 3-Kinase Activity Is Required for CR3-mediated Phagocytosis: A Role for PI 3-Kinase in Outside-In Signaling of CR3.
The preceding data indicate that SHIP regulates phagocytosis via CR3 and therefore raise the possibility that PI 3-kinase may also regulate CR3-mediated phagocytosis. Indeed, the PI 3-kinase inhibitor wortmannin (WM) caused a concentration-dependent inhibition of CR3-mediated phagocytosis in thio-macrophages. Similar results were obtained using thio-macrophages incubated with PMA, which maximally stimulates phagocytosis in these cells (Fig. 4 A). The IC50 of WM for CR3-mediated phagocytosis is consistent with the inhibition of PI 3-kinases, but not PI 4-kinases 31. These results implicate PI 3-kinase(s) in CR3-mediated phagocytosis.
Figure 4.

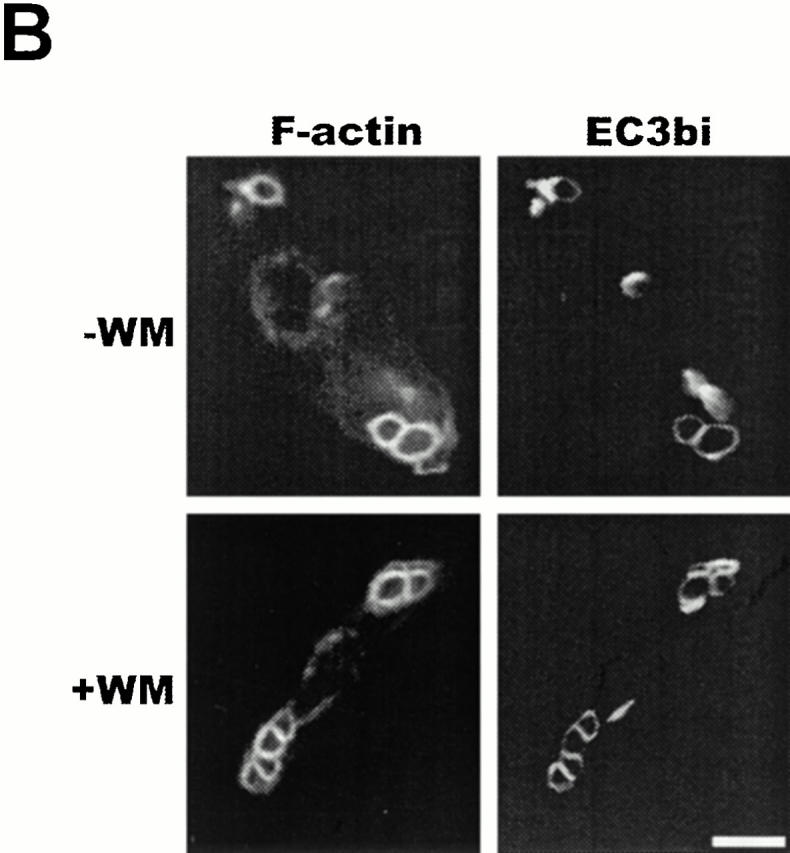
CR3-mediated phagocytosis, but not actin assembly, is dependent on PI 3-kinase. (A) Adherent thio-macrophages were preincubated for 15 min with the indicated concentrations of WM followed by the addition of EC3bi in the presence or absence of 100 nM PMA for 30 min at 37°C. Phagocytic indices were 84 ± 9 in the absence of PMA and 472 ± 125 in the presence of PMA. Phagocytosis of cells incubated with WM is expressed as the percentage of phagocytosis performed in the absence of WM. Data represent mean ± SEM, n = 3. (B) Adherent thio-macrophages were preincubated without (−WM) or with (+WM) 100 nM WM followed by further incubation with EC3bi for 12 min at 37°C. After fixation, cells were stained for F-actin with rhodamine-phalloidin and for EC3bi with anti-EIgG followed by FITC-anti–rabbit IgG. Bar, 10 μm.
Because integrin signaling encompasses pathways that lead to integrin activation (inside-out signaling) and pathways that ensue after integrin ligation (outside-in signaling), we examined the role of PI 3-kinase and SHIP in both components. For example, a prominent role for PI 3-kinase in CR3 activation would be suggested by a WM blockade of proximal signaling events after CR3 ligation. However, CR3 clustering induced the focal appearance of F-actin in cells incubated in the presence or absence of WM (Fig. 4 B), similar to the case of FcγR-mediated phagocytosis, and unlike that of growth factor receptors 32 33 34 35 36. The lack of inhibition of CR3-mediated F-actin mobilization by WM suggests that PI 3-kinase, and possibly SHIP, does not play a role in inside-out signaling by CR3 in macrophages. A lack of involvement of PI 3-kinase in inside-out signaling by CR3 in macrophages is also suggested by the similar extent of binding of EC3bi in SHIP−/− and SHIP+/+ macrophages (Fig. 3A and Fig. B). In contrast to human neutrophils, CR3-mediated phagocytosis in resident peritoneal macrophages derived from C57BL/6 mice is constitutive (i.e., does not require prior activation; reference 37 and data not shown). Phagocytosis of EC3bi in resident peritoneal macrophages derived from SHIP−/− mice was 252 ± 69% (mean ± SEM, n = 4; P < 0.05) of phagocytosis in macrophages derived from SHIP+/+ mice.
The above results are somewhat surprising in light of the data in human neutrophils and T cells, in which PI 3-kinase has been shown to be required for β2 integrin–mediated adhesion 21 22 38. To confirm a role for PI 3-kinase and SHIP in CR3-mediated outside-in rather than inside-out signaling, we transfected COS cells with the αM and β2 subunits of CR3, alone and together, and assessed the binding and ingestion of EC3bi. Only cells that expressed both subunits demonstrated binding of EC3bi (data not shown). CR3-expressing COS cells demonstrated phagocytosis of EC3bi in the absence of PMA, and the addition of PMA did not result in enhancement of phagocytosis (data not shown), indicating that CR3 was constitutively and maximally activated. In addition, CR3-expressing COS cells, but not nonexpressing controls, demonstrated marked staining with mAb CBRM1/5, an antibody that recognizes an activation-dependent epitope on CR3 (26, and data not shown). Addition of WM led to an 87% inhibition of phagocytosis of EC3bi in transfected COS (Fig. 5 A) but did not inhibit particle binding or staining with mAb CBRM1/5 (data not shown). Coexpression of wild-type SHIP, but not catalytically inactive SHIP, also led to a decrease in phagocytosis, further implicating the lipid product of PI 3-kinase, PIP3, in CR3-mediating outside-in signaling and phagocytosis. Similarly, in macrophages overexpressing SHIP, there was a significant decrease in phagocytosis of EC3bi (Fig. 5 B); however, in contrast to COS cells, which lack endogenous SHIP expression (data not shown), expression of a catalytically inactive allele of SHIP in macrophages led to enhanced phagocytosis of EC3bi (compare Fig. 5a and Fig. b), consistent with a suppressive role for endogenously expressed SHIP in these cells. Collectively, these data suggest that one or more isoforms of PI 3-kinase is required for CR3-mediated phagocytosis in macrophages and COS cells, that this requirement is not due to the enhancement of receptor affinity or avidity (i.e., inside-out signaling), and that endogenously expressed SHIP downregulates CR3-mediated phagocytosis in macrophages.
Figure 5.


Effect of SHIP or SHIPΔP′ase expression on CR3-mediated phagocytosis in COS cells and macrophages. (A) COS cells transfected with the αM and β2 subunits of CR3 and coexpressing the indicated constructs were challenged with EC3bi for 30 min at 37°C in the presence or absence of 100 nM WM. The phagocytosis index of COS cells expressing CR3 alone, in the absence of WM (controls) was 651 ± 108. Phagocytosis was determined as the percentage of control. Data represent mean ± SEM, n = 3. Differences in phagocytosis between cells either incubated in WM or expressing wild-type SHIP, and controls were significant (P < 0.05). (B) RAW LR5 cells expressing the indicated constructs were challenged with EC3bi and 100 nM PMA and incubated in the absence or presence of 100 nM WM for 30 min at 37°C. The phagocytosis index of nonexpressing cells in the absence of WM (controls) was 55 ± 10. Phagocytosis was determined as the percentage of controls. Data represent mean ± SEM, n = 5. Differences in phagocytosis between cells either incubated in WM, or expressing wild-type SHIP or SHIPΔP′ase, and controls were significant (P < 0.05).
Phagosomal and Cytoskeletal Localization of SHIP after Clustering of CR3.
To determine whether CR3 ligation results in an alteration in the distribution of SHIP, we performed indirect immunofluorescence in cells undergoing CR3-mediated phagocytosis. SHIP, but not the receptor for CSF-1, c-fms, localized to EC3bi-containing phagosomes (Fig. 6). The specificity of SHIP localization is evident from a lack of circumferential staining around phagosomes for another protein expressed in macrophages, c-fms (Fig. 6 E), and absence of SHIP staining in macrophages derived from SHIP−/− mice (data not shown). To confirm that CR3 ligation is sufficient to induce SHIP redistribution, we determined the cytoskeletal localization of SHIP and the αM subunit of CR3 after CR3 ligation. Both SHIP and αM, but not CD14, demonstrated enhanced cytoskeletal incorporation after mAb-induced clustering of CR3 (Fig. 7). In addition, there was a 2.2 ± 0.4–fold (mean ± SEM, n = 3) increase in the phosphotyrosyl content of SHIP after the clustering of CR3 using mAb M1/70 followed by a cross-linking antibody. In contrast, the phosphotyrosyl content of SHIP in macrophages incubated with an IgG2b control mAb followed by a cross-linking antibody was 0.8 ± 0.3–fold (mean ± SEM, n = 3) of unstimulated cells. Therefore, engagement of CR3 is sufficient to mediate an alteration in the localization of SHIP in macrophages.
Figure 6.

SHIP, but not c-fms, localizes to CR3-mediated phagocytic cups. Adherent thio-macrophages obtained from SHIP+/+ mice were incubated with EC3bi, as described in Materials and Methods, for 12 min at 37°C. After fixation, cells were stained for the presence of SHIP or c-fms (negative control), and C3bi as described in Materials and Methods. (A and D) Phase–contrast; (B) anti-SHIP; (E) anti–c-fms; (C and F) anti-C3. Bar, 10 μm.
Figure 7.
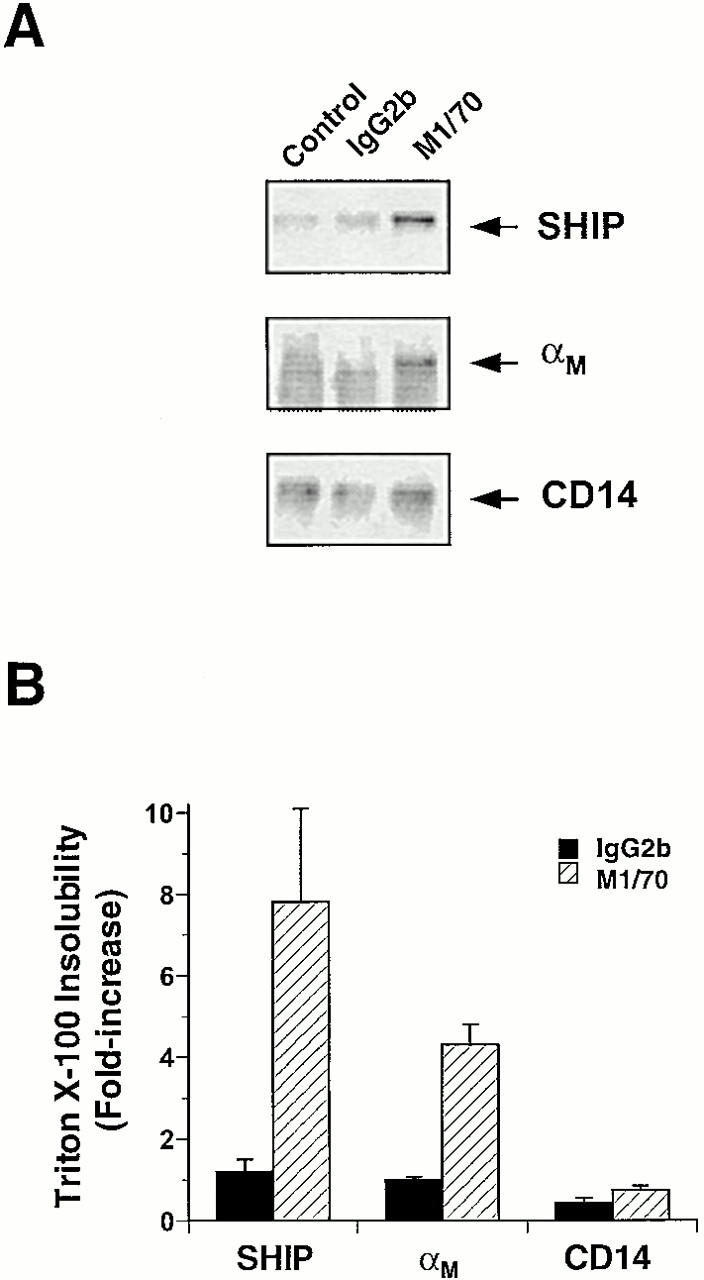
Incorporation of SHIP and αM into the Triton X-100–insoluble cytoskeleton after CR3 clustering. Triton X-100–insoluble fractions were isolated from control cells or from cells incubated with mAb M1/70 or an isotype-matched control mAb and F(ab′)2 fragments of goat anti–rat IgG as described in Materials and Methods. (A) Representative immunoblot of cytoskeletons probed with the indicated antibodies. (B) Fold increase in cytoskeletal incorporation of the indicated proteins. Data represent mean ± SEM, n = 3.
The NPXY Motifs of SHIP Are Required for Inhibition of CR3- but Not FcγR-mediated Phagocytosis.
Although SHIP has multiple domains that contribute to its association with other proteins, most studies have demonstrated an essential role for the SH2 domain in mediating interactions with other proteins 3 4. We determined the requirement of this domain in the modulation of both FcγR- and CR3-mediated phagocytosis. Expression of an allele of SHIP (SHIPR34G) that contains a mutation in a conserved residue required for SH2 domain–phosphotyrosine interactions 28 did not reproduce the phagocytic defect induced by the expression of wild-type SHIP (Fig. 8), indicating that the SH2 domain of SHIP is required for inhibition of both FcγR- and CR3-mediated phagocytosis. In contrast, expression of a SHIP allele lacking functional NPXY motifs (SHIPY917,1020F) did not abrogate SHIP-mediated inhibition of FcγR-mediated phagocytosis, but prevented the SHIP-mediated inhibition of CR3-mediated phagocytosis. Expression levels of both constructs in transfected cells were similar to those of SHIP and SHIPΔP′ase (i.e., all were within 12%, as determined by microspectrofluorometry). Taken together, these data indicate that both the SH2 domain and NPXY motifs of SHIP contribute to the antiphagocytic activity of SHIP for CR3-mediated phagocytosis; the NPXY motifs are dispensable for inhibition of FcγR-mediated phagocytosis.
Figure 8.

Differential requirements for the NPXY motifs of SHIP in inhibition of FcγR- and CR3-mediated phagocytosis. RAW LR5 cells transfected with the indicated constructs were challenged with either EIgG or EC3bi in the presence of 100 nM PMA, and incubated for 30 min at 37°C. Phagocytic indices were 530 ± 49 for EIgG and 104 ± 17 for EC3bi. Phagocytosis of cells expressing the indicated constructs is reported as the percentage of nonexpressing controls. Data represent mean ± SEM, n = 3. Difference in phagocytosis of EIgG by cells expressing SHIPY917,1020F and controls was significant (P < 0.05).
Discussion
A requirement for PI 3-kinase activity in FcγR-mediated phagocytosis is well established 1 2 18 20. Because SHIP has been shown to downmodulate the function of receptors that activate PI 3-kinase, and FcγRIIB contains a SHIP-binding site, it is not surprising that SHIP is capable of downmodulating FcγR-mediated phagocytosis. In contrast, little is known about the role of PI 3-kinase and SHIP in CR3-mediated phagocytosis. The results of this study are consistent with a role for both PI 3-kinase and SHIP in regulating CR3-mediated phagocytosis.
The involvement of PI 3-kinase in β2 integrin function was suggested by studies demonstrating a requirement for this family of enzymes in β2 integrin–dependent adhesion 21 22 38. Sensitivity to PI 3-kinase inhibitors was dependent on the nature of both the stimulus for adhesion and the adhesive substrate. Addition of immune complexes to human neutrophils resulted in a twofold increase in β2 integrin surface expression and a sixfold increase in the expression of a β2 integrin activation-dependent epitope, both of which were sensitive to WM 22. These results are consistent with a requirement for PI 3-kinase in immune complex–induced inside-out signaling of β2 integrins in neutrophils. Because β2 integrin function in most phagocytic leukocytes requires prior or concurrent activation, it would be difficult to discern an independent requirement for PI 3-kinase in β2 integrin outside-in signaling. The use of peritoneal macrophages from C57BL/6 mice and COS cells transfected with CR3 in this study circumvents this problem; in the case of the former, CR3 phagocytic function is apparent in the absence of additional stimuli. This capacity is sustained over several weeks while the cells are maintained in culture. The phagocytic capacity of COS cells transfected with CR3 is even greater, and essentially all of the CR3-expressing cells demonstrate high levels of surface expression of the activation-dependent epitope, CBRM1/5. The WM sensitivity of phagocytosis of EC3bi in these two cell types attests to a role for PI 3-kinase in outside-in signaling via CR3.
The finding that SHIP is capable of modulating FcγR-mediated phagocytosis is consistent with the recruitment of SHIP to the phosphorylated ITIM of FcγRIIB, which is likely to occur during binding of IgG. This model is supported by a recent study 11. However, it is unclear how SHIP becomes recruited to phagosomes during engagement of CR3. Mutational analysis indicates that both the SH2 domain and the NPXY motifs of SHIP are required for SHIP-mediated downmodulation of CR3-dependent phagocytosis, suggesting that the residues therein contribute to SHIP targeting. Several different proteins have been shown to interact with these motifs. The SH2 domain of SHIP has been shown to be required for the interaction of SHIP with FcγRIIB 8, Shc 29, Gab 1/2 39, SH2 domain–bearing protein tyrosine phosphatase 40, and Dok-3 41. The NPXY motifs of SHIP mediate interactions with Shc 42 and the p85 subunit of PI 3-kinase 43. For the most part, the significance of these interactions in vivo is unknown. Because SHIP translocates to the cytoskeleton after the clustering of CR3, it is anticipated that the identification of specific and direct SHIP–protein interactions that accompany CR3 ligation will be difficult. Further studies will be needed to address the mechanism of SHIP recruitment to the phagosome during CR3-mediated phagocytosis.
Ligation of either CR3 or FcγR leads to an increase in PtdIns(3,4,5)P3 accumulation 1 18 19 and PI 3-kinase activity is required for phagocytosis (1, 2, 18, 20, and this study). However, it is unknown which product of PI 3-kinase is essential for phagocytosis. PI 3-kinase catalyzes the addition of phosphate to the D-3 positions of phosphatidylinositol, phosphatidylinositol 4-phosphate, and phosphatidylinositol 4,5-bisphosphate to form phosphatidylinositol 3-phosphate, phosphatidylinositol 3,4-bisphosphate and PtdIns(3,4,5)P3, respectively. Of these, SHIP is known only to hydrolyze PtdIns(3,4,5)P3 44 45. Therefore, the reciprocal roles of PI 3-kinase and SHIP in phagocytosis suggests that PtdIns(3,4,5)P3 is the relevant target of these phosphoinositide-modulating enzymes. A similar role for PtdIns(3,4,5)P3 has been proposed for signaling via the B cell antigen receptor in B lymphocytes and FcεRI in mast cells 8 9. By binding the PH domain on members of the Tec family of tyrosine kinases, PtdIns(3,4,5)P3 contributes to the calcium signaling pathway in these cells 5 6 7. The identity of PH domain proteins that positively regulate phagocytosis is unknown, although several proteins that contain putative PtdIns(3,4,5)P3-binding PH domains 46 are expressed in macrophages (47 48; Greenberg, S., unpublished results).
It is possible that SHIP modulates phagocytosis by decreasing cellular d-_myo_-inositol 1,3,4,5-tetrakisphosphate (Ins[1,3,4,5]P4) content, although a role for Ins(1,3,4,5)P4 hydrolysis by SHIP in vivo is lacking. It is not known whether Ins(1,3,4,5)P4 is generated during the course of FcγR- or CR3-mediated phagocytosis. One study found a minimal and delayed increase in this phosphoinositide after the addition of serum-opsonized zymosan to human neutrophils 49. Ins(1,3,4,5)P4 has been shown to inhibit exocytosis in neurons, which has been attributed to the inhibition of synaptotagmin function by the binding of Ins(1,3,4,5)P4 50. Nonneuronal forms of synaptotagmin have also been implicated in exocytosis, and a recent study has shown that these isoforms of synaptotagmin bind Ins(1,3,4,5)P4 in vitro 51. Overexpression of SHIP might result in a decrease in basal or stimulated Ins(1,3,4,5)P4 levels, thereby enhancing exocytosis. Because phagocytosis requires the addition of new membrane during phagosome formation (i.e., exocytosis [1, 52]), it is unlikely that the mechanism of inhibition of phagocytosis by SHIP is via this mechanism. However, there are likely to be other cellular targets of Ins(1,3,4,5)P4, including PH domains that are capable of interacting with this water-soluble phosphoinositide in vitro 53 54. It is possible that PtdIns(3,4,5)P3 and Ins(1,3,4,5)P4 interact with a similar spectrum of PH domain–containing proteins in vivo.
Although this study did not address the precise mechanism by which products of PI 3-kinase and SHIP affect CR3-mediated phagocytosis, the lack of inhibition by WM of focal accumulations of F-actin beneath phagocytic cups strongly suggests that pseudopod extension and/or phagosomal closure requires PI 3-kinase activity. In this respect, CR3-mediated phagocytosis resembles FcγR-mediated phagocytosis 1. There are obvious differences in the morphology of phagosomes formed by clustered FcγRs and CR3: CR3-containing phagosomes appear to “sink” into the cell without demonstrating obvious pseudopod extension 16 55. Because the membrane surface area that is internalized during phagocytosis is a function only of the number and size of the particles engulfed, the requirement for new membrane in CR3- and FcγR-mediated phagocytosis must be similar. It is therefore likely that CR3-mediated phagocytosis requires exocytic insertion of new membrane 1 52, and that this process requires PI 3-kinase activity similar to FcγR-mediated phagocytosis 1.
Our findings are somewhat divergent from a recent study in which LFA-1–mediated adhesion to intracellular adhesion molecule (ICAM)-1 was enhanced in clones of the IL-3–dependent cell line, DA-ER, stably over-expressing SHIP 56. There are many possible explanations for this apparent disparity, including differences in the nature of the cell type used and differences in the integrins (CR3 versus LFA-1) and substrates (iC3b versus ICAM-1) that were studied. Although neither study addressed the in vivo consequences of modulation of SHIP activity on phagocytosis or leukocyte adhesion, it is interesting to speculate that the infiltration of leukocytes observed in the lungs of SHIP−/− mice 57 may reflect, in part, enhanced β2 integrin–dependent adhesive interactions with the endothelium.
Because SHIP plays a role in hematopoietic cell development and has been implicated in signaling via c-fms, the receptor for CSF-1 45, it is possible that enhanced phagocytosis in SHIP−/− cells is due to an alteration in their state of differentiation or activation. While this is difficult to test definitively, the observation that surface FcγR and CR3 expression and particle binding was unaltered in SHIP−/− cells suggests that this is not the case. In addition, the use of COS cells in some of the assays avoids the possible contribution of SHIP to CSF-1–dependent signaling.
In conclusion, we have presented evidence for a previously unsuspected role of SHIP in phagocytosis mediated by CR3, and for phagocytosis mediated by FcγRs. Together with data suggesting that PI 3-kinase is required for β2 integrin outside-in signaling, the relative expression and activities of PI 3-kinase(s) and SHIP in different macrophage populations are likely to determine the strength of the phagocytic signal. As CR3 has been implicated in other cellular functions besides phagocytosis, the expression of SHIP may influence other CR3-dependent activities, including leukocyte migration into inflammatory foci.
Acknowledgments
We thank Michael Cammer of the Analytic Imaging Facility of the Albert Einstein College of Medicine (Bronx, NY) for his help in imaging. We thank Rewa Grewal and Ann Clarke for genotyping the mice.
This work was supported by National Institutes of Health grants HL54164 and AR02158. C.D. Helgason is the recipient of a British Columbia Health Research Foundation Joint Research Scholarship. S. Greenberg is an Established Investigator of the American Heart Association.
Footnotes
Abbreviations used in this paper: CR3, complement receptor 3; ITAM, immunoreceptor tyrosine-based activation motif; ITIM, immunoreceptor tyrosine-based inhibition motif; PH, pleckstrin homology; SH2 , Src homology 2; SHIP, SH2 domain–containing inositol 5′-phosphatase; WM, wortmannin.
References
- Cox D., Tseng C.-C., Bjekic G., Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 1999;274:1240–1247. doi: 10.1074/jbc.274.3.1240. [DOI] [PubMed] [Google Scholar]
- Araki N., Johnson M.T., Swanson J.A. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M., Helgason C.D., Damen J.E., Scheid M., Duronio V., Liu L., Ware M.D., Humphries R.K., Krystal G. The role of SHIP in growth factor induced signalling. Prog. Biophys. Mol. Biol. 1999;71:423–434. doi: 10.1016/s0079-6107(98)00049-2. [DOI] [PubMed] [Google Scholar]
- Rohrschneider L.R., Fuller J.F., Wolf I., Liu Y., Lucas D.M. Structure, function, and biology of SHIP proteins. Genes Dev. 2000;14:505–520. [PubMed] [Google Scholar]
- Ono M., Okada H., Bolland S., Yanagi S., Kurosaki T., Ravetch J.V. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- Bolland S., Pearse R.N., Kurosaki T., Ravetch J.V. SHIP modulates immune receptor responses by regulating membrane association of BTK. Immunity. 1998;8:509–516. doi: 10.1016/s1074-7613(00)80555-5. [DOI] [PubMed] [Google Scholar]
- Scharenberg A.M., El-Hillal O., Fruman D.A., Beitz L.O., Li Z., Lin S., Gout I., Cantley L.C., Rawlings D.J., Kinet J.-P. Phosphatidylinositol-3,4,5-trisphosphate (PtdIns-3,4,5-P3)/Tec kinase-dependent calcium signaling pathwaya target for SHIP-mediated inhibitory signals. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1961–1972. doi: 10.1093/emboj/17.7.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M., Bolland S., Tempst P., Ravetch J.V. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fcγ RIIB. Nature. 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- Huber M., Helgason C.D., Damen J.E., Liu L., Humphries R.K., Krystal G. The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc. Natl. Acad. Sci. USA. 1998;95:11330–11335. doi: 10.1073/pnas.95.19.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresco D.L., Osborne J.M., Cooney D., Coggeshall K.M., Anderson C.L. The SH2-containing 5′-inositol phosphatase (SHIP) is tyrosine phosphorylated after Fcγ receptor clustering in monocytes. J. Immunol. 1999;162:6458–6465. [PubMed] [Google Scholar]
- Cameron A.J., Allen J.M. The human high-affinity immunoglobulin G receptor activates SH2-containing inositol phosphatase (SHIP) Immunology. 1999;97:641–647. doi: 10.1046/j.1365-2567.1999.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Lin C.T., Unkeless J.C. Correlations among tyrosine phosphorylation of Shc, p72syk, PLCγ1, and [Ca2+]i flux in Fcγ RIIA signaling. J. Immunol. 1994;152:3017–3023. [PubMed] [Google Scholar]
- Matsuo T., Hazeki K., Hazeki O., Katada T., Ui M. Specific association of phosphatidylinositol 3-kinase with the protooncogene product Cbl in Fcγ receptor signaling. FEBS Lett. 1996;382:11–14. doi: 10.1016/0014-5793(96)00122-6. [DOI] [PubMed] [Google Scholar]
- Barker S.A., Caldwell K.K., Hall A., Martinez A.M., Pfeiffer J.R., Oliver J.M., Wilson B.S. Wortmannin blocks lipid and protein kinase activities associated with PI 3-kinase and inhibits a subset of responses induced by FcεR1 cross-linking. Mol. Biol. Cell. 1995;6:1145–1158. doi: 10.1091/mbc.6.9.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E.J. Complement receptor, adhesion, and phagocytosis. Infect. Agents Dis. 1992;1:63–70. [PubMed] [Google Scholar]
- Allen L.A.H., Aderem A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor–mediated phagocytosis in macrophages. J. Exp. Med. 1996;184:627–637. doi: 10.1084/jem.184.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E., Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- Ninomiya N., Hazeki K., Fukui Y., Seya T., Okada T., Hazeki O., Ui M. Involvement of phosphatidylinositol 3-kinase in Fcγ receptor signaling. J. Biol. Chem. 1994;269:22732–22737. [PubMed] [Google Scholar]
- Lofgren R., Ng-Sikorski J., Sjolander A., Andersson T. β2 integrin engagement triggers actin polymerization and phosphatidylinositol trisphosphate formation in non-adherent human neutrophils. J. Cell Biol. 1993;123:1597–1605. doi: 10.1083/jcb.123.6.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley M.T., Costello P.S., Fitzer-Attas C.J., Turner M., Meng F., Lowell C., Tybulewicz V.L.J., DeFranco A.L. A critical role for Syk in signal transduction and phagocytosis mediated by Fcγ receptors on macrophages. J. Exp. Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner B., Heger M., Hofmann C., Czech W., Norgauer J. Evidence for the involvement of phosphatidylinositol 4, 5-bisphosphate 3-kinase in CD18-mediated adhesion of human neutrophils to fibrinogen. Biochem. Biophys. Res. Commun. 1997;232:719–723. doi: 10.1006/bbrc.1997.6350. [DOI] [PubMed] [Google Scholar]
- Jones S.L., Knaus U.G., Bokoch G.M., Brown E.J. Two signaling mechanisms for activation of αMβ2 avidity in polymorphonuclear neutrophils. J. Biol. Chem. 1998;273:10556–10566. doi: 10.1074/jbc.273.17.10556. [DOI] [PubMed] [Google Scholar]
- Kolanus W., Nagel W., Schiller B., Zeitlmann L., Godar S., Stockinger H., Seed B. αLβ2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell. 1996;86:233–242. doi: 10.1016/s0092-8674(00)80095-1. [DOI] [PubMed] [Google Scholar]
- Cox D., Chang P., Zhang Q., Reddy P.G., Bokoch G.M., Greenberg S. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 1997;186:1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F., Meyer B.C., Greenberg S., Silverstein S.C. Fc receptor–mediated phagocytosis occurs in macrophages at exceedingly low cytosolic Ca2+ levels. J. Cell Biol. 1988;106:657–666. doi: 10.1083/jcb.106.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Springer T.A. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 1993;120:545–556. doi: 10.1083/jcb.120.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuter-Reinhard M., Apell G., Pot D., Klippel A., Williams L.T., Kavanaugh W.M. SIP/SHIP inhibits Xenopus oocyte maturation induced by insulin and phosphatidylinositol 3-kinase. Mol. Cell. Biol. 1997;17:2559–2565. doi: 10.1128/mcb.17.5.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh L.E., Chen C.S., Cantley L.C. Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine-phosphorylated proteins. Cell. 1995;83:821–830. doi: 10.1016/0092-8674(95)90195-7. [DOI] [PubMed] [Google Scholar]
- Liu L., Damen J.E., Hughes M.R., Babic I., Jirik F.R., Krystal G. The Src homology 2 (SH2) domain of SH2-containing inositol phosphatase (SHIP) is essential for tyrosine phosphorylation of SHIP, its association with Shc, and its induction of apoptosis. J. Biol. Chem. 1997;272:8983–8988. doi: 10.1074/jbc.272.14.8983. [DOI] [PubMed] [Google Scholar]
- Wright S.D., Tobias P.S., Ulevitch R.J., Ramos R.A. Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for recognition by a novel receptor on macrophages. J. Exp. Med. 1989;170:1231–1241. doi: 10.1084/jem.170.4.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S., Catt K.J., Balla T. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl. Acad. Sci. USA. 1995;92:5317–5321. doi: 10.1073/pnas.92.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennstrom S., Hawkins P., Cooke F., Hara K., Yonezawa K., Kasuga M., Jackson T., Claesson-Welsh L., Stephens L. Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr. Biol. 1994;4:385–393. doi: 10.1016/s0960-9822(00)00087-7. [DOI] [PubMed] [Google Scholar]
- Wymann M., Arcaro A. Platelet-derived growth factor-induced phosphatidylinositol 3-kinase activation mediates actin rearrangements in fibroblasts. Biochem. J. 1994;298:517–520. doi: 10.1042/bj2980517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani K., Hara K., Kotani K., Yonezawa K., Kasuga M. Phosphoinositide 3-kinase as an upstream regulator of the small GTP-binding protein Rac in the insulin signaling of membrane ruffling. Biochem. Biophys. Res. Commun. 1995;208:985–990. doi: 10.1006/bbrc.1995.1431. [DOI] [PubMed] [Google Scholar]
- Jackson D.I., Verbi W., Lazarovits A.I., Crumpton M.J. T lymphocyte activationthe role of tyrosine phosphorylation. Cancer Surv. 1995;22:97–110. [PubMed] [Google Scholar]
- Martin S.S., Haruta T., Morris A.J., Klippel A., Williams L.T., Olefsky J.M. Activated phosphatidylinositol 3-kinase is sufficient to mediate actin rearrangement and GLUT4 translocation in 3T3-L1 adipocytes. J. Biol. Chem. 1996;271:17605–17608. doi: 10.1074/jbc.271.30.17605. [DOI] [PubMed] [Google Scholar]
- Bianco C., Griffin F.M., Jr., Silverstein S.C. Studies of the macrophage complement receptor. Alteration of receptor function upon macrophage activation. J. Exp. Med. 1975;141:1278–1290. doi: 10.1084/jem.141.6.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Svejgaard A., Skov S., Dobson P., Bendtzen K., Geisler C., Odum N. IL-2 induces β2-integrin adhesion via a wortmannin/LY294002-sensitive, rapamycin-resistant pathway. Phosphorylation of a 125-kilodalton protein correlates with induction of adhesion, but not mitogenesis. J. Immunol. 1996;157:5350–5358. [PubMed] [Google Scholar]
- Lecoq-Lafon C., Verdier F., Fichelson S., Chretien S., Gisselbrecht S., Lacombe C., Mayeux P. Erythropoietin induces the tyrosine phosphorylation of GAB1 and its association with SHC, SHP2, SHIP, and phosphatidylinositol 3-kinase. Blood. 1999;93:2578–2585. [PubMed] [Google Scholar]
- Liu L., Damen J.E., Ware M.D., Krystal G. Interleukin-3 induces the association of the inositol 5-phosphatase SHIP with SHP2. J. Biol. Chem. 1997;272:10998–11001. doi: 10.1074/jbc.272.17.10998. [DOI] [PubMed] [Google Scholar]
- Lemay S., Davidson D., Latour S., Veillette A. Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol. Cell. Biol. 2000;20:2743–2754. doi: 10.1128/mcb.20.8.2743-2754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkin T.D., Walk S.F., Liu L., Damen J.E., Krystal G., Ravichandran K.S. Shc interaction with Src homology 2 domain containing inositol phosphatase (SHIP) in vivo requires the Shc-phosphotyrosine binding domain and two specific phosphotyrosines on SHIP. J. Biol. Chem. 1997;272:10396–10401. doi: 10.1074/jbc.272.16.10396. [DOI] [PubMed] [Google Scholar]
- Gupta N., Scharenberg A.M., Fruman D.A., Cantley L.C., Kinet J.-P., Long E.O. The SH2 domain-containing inositol 5′-phosphatase (SHIP) recruits the p85 subunit of phosphoinositide 3-kinase during FcγRIIb1-mediated inhibition of B cell receptor signaling. J. Biol. Chem. 1999;274:7489–7494. doi: 10.1074/jbc.274.11.7489. [DOI] [PubMed] [Google Scholar]
- Damen J.E., Liu L., Rosten P., Humphries R.K., Jefferson A.B., Majerus P.W., Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioubin M.N., Algate P.A., Tsai S., Carlberg K., Aebersold A., Rohrschneider L.R. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev. 1996;10:1084–1095. doi: 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- Isakoff S.J., Cardozo T., Andreev J., Li Z., Ferguson K.M., Abagyan R., Lemmon M.A., Aronheim A., Skolnik E.Y. Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:5374–5387. doi: 10.1093/emboj/17.18.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Vik T.A. Growth factor stimulation of hematopoietic cells leads to membrane translocation of AKT1 protein kinase. Leuk. Res. 1997;21:849–856. doi: 10.1016/s0145-2126(97)00055-6. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S., Sahoo P.K., George A., Bal V., Rath S., Ravindran B. Delayed clearance of filarial infection and enhanced Th1 immunity due to modulation of macrophage APC functions in xid mice. J. Immunol. 1999;163:875–883. [PubMed] [Google Scholar]
- Leino L., Tuominen H., Lehtola K., Akerman K.E.O., Punnonen K. Biphasic formation of inositol phosphates in opsonized zymosan-stimulated human neutrophils. Cell. Signalling. 1995;7:397–402. doi: 10.1016/0898-6568(94)00094-r. [DOI] [PubMed] [Google Scholar]
- Fukuda M., Moreira J.E., Lewis F.M., Sugimori M., Niinobe M., Mikoshiba K., Llinas R. Role of the C2B domain of synaptotagmin in vesicular release and recycling as determined by specific antibody injection into the squid giant synapse preterminal. Proc. Natl. Acad. Sci. USA. 1995;92:10708–10712. doi: 10.1073/pnas.92.23.10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K., Fukuda M., Mikoshiba K. Inositol 1,3,4,5-tetrakisphosphate binding activities of neuronal and non-neuronal synaptotagmins. Identification of conserved amino acid substitutions that abolish inositol 1,3,4,5-tetrakisphosphate binding to synaptotagmins III, V, and X. J. Biol. Chem. 1998;273:12267–12273. doi: 10.1074/jbc.273.20.12267. [DOI] [PubMed] [Google Scholar]
- Hackam D.J., Rotstein O.D., Sjolin C., Schreiber A.D., Trimble W.S., Grinstein S. v-SNARE-dependent secretion is required for phagocytosis. Proc. Natl. Acad. Sci. USA. 1998;95:11691–11696. doi: 10.1073/pnas.95.20.11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M., Kojima T., Kabayama H., Mikoshiba K. Mutation of the pleckstrin homology domain of Bruton's tyrosine kinase in immunodeficiency impaired inositol 1,3,4,5-tetrakisphosphate binding capacity. J. Biol. Chem. 1996;271:30303–30306. doi: 10.1074/jbc.271.48.30303. [DOI] [PubMed] [Google Scholar]
- Fukuda M., Mikoshiba K. Structure-function relationships of the mouse Gap1m. Determination of the inositol 1,3,4,5-tetrakisphophate-binding domain. J. Biol. Chem. 1996;271:18838–18842. doi: 10.1074/jbc.271.31.18838. [DOI] [PubMed] [Google Scholar]
- Kaplan G. Differences in the mode of phagocytosis with Fc and C3 receptors in macrophages. Scand. J. Immunol. 1977;6:797–807. doi: 10.1111/j.1365-3083.1977.tb02153.x. [DOI] [PubMed] [Google Scholar]
- Rey-Ladino J.A., Huber M., Liu L., Damen J.E., Krystal G., Takei F. The SH2-containing inositol-5′-phosphatase enhances LFA-1-mediated cell adhesion and defines two signaling pathways for LFA-1 activation. J. Immunol. 1999;162:5792–5799. [PubMed] [Google Scholar]
- Helgason C.D., Damen J.E., Rosten P., Grewal R., Sorenson P., Chappel S.M., Borowski A., Jirik F., Krystal G., Humphries R.K. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]