Cell Contact–Dependent Immunosuppression by Cd4+Cd25+Regulatory T Cells Is Mediated by Cell Surface–Bound Transforming Growth Factor β (original) (raw)
Abstract
CD4+CD25+ T cells have been identified as a population of immunoregulatory T cells, which mediate suppression of CD4+CD25− T cells by cell–cell contact and not secretion of suppressor cytokines. In this study, we demonstrated that CD4+CD25+ T cells do produce high levels of transforming growth factor (TGF)-β1 and interleukin (IL)-10 compared with CD4+CD25− T cells when stimulated by plate-bound anti-CD3 and soluble anti-CD28 and/or IL-2, and secretion of TGF-β1 (but not other cytokines), is further enhanced by costimulation via cytotoxic T lymphocyte–associated antigen (CTLA)-4. As in prior studies, we found that CD4+CD25+ T cells suppress proliferation of CD4+CD25− T cells; however, we observed here that such suppression is abolished by the presence of anti–TGF-β. In addition, we found that CD4+CD25+ T cells suppress B cell immunoglobulin production and that anti–TGF-β again abolishes such suppression. Finally, we found that stimulated CD4+CD25+ T cells but not CD4+CD25− T cells express high and persistent levels of TGF-β1 on the cell surface. This, plus the fact that we could find no evidence that a soluble factor mediates suppression, strongly suggests that CD4+CD25+ T cells exert immunosuppression by a cell–cell interaction involving cell surface TGF-β1.
Keywords: T lymphocytes, suppressor-effector; CD4-positive T lymphocytes; receptors, interleukin 2; transforming growth factors; autoimmune diseases
Introduction
In recent years it has become evident that peripheral or postthymic tolerance is mediated, at least in part, by various types of regulatory T cells (suppressor T cells) 1. One such T cell is present in the 5–10% of unstimulated CD4+ T cells of adult mice that express CD25, the α-chain of IL-2R. This became evident from studies by Asano et al., who showed that thymectomy of certain mouse strains on day 3 of life results in elimination of the CD4+CD25+ T cell subset and the subsequent occurrence of various autoimmune diseases such as gastritis, orchitis, oophoritis, and thyroiditis 2. In addition, they showed that reconstitution of such neonatally thymectomized mice with CD4+CD25+ T cells prevents the development of these autoimmune diseases 2.
Another subset of CD4+ T cells, CD45RBlow T cells, has also been found to have immunoregulatory function. In particular, it was demonstrated that while transfer of CD4+CD45RBhigh T cells into SCID or recombination activating gene knockout recipient mice leads to colitis, transfer of these cells plus CD4+CD45RBlow T cells prevents colitis 3. Interestingly, the regulatory activity of CD4+CD45RBlow T cells has recently been shown to reside in the CD4+CD25+ T cell subpopulation 4, suggesting that T cells regulating gastritis and colitis are similar, if not the same.
One possible mechanism of the immunosuppression caused by both CD4+CD25+ and CD4+CD45RBlow T cells is that these cells either secret or cause the secretion of TGF-β and/or IL-10. This is suggested by cell transfer studies of colitis such as those mentioned above, in which it has been shown that Abs to TGF-β and/or IL-10R block suppressor activity of transferred cells 4 5 6. Also relevant here are studies showing that orally immunized mice in whom oral tolerance has been induced manifest T cells that produce TGF-β1, so-called Th3 T cells, and that T cell lines that produce IL-10, so-called Tr1 T cells, can prevent development of colitis in the above described SCID-transfer model 7 8 9. It should be noted, however, that these studies of CD4+CD25+ T cells and other types of suppressor T cells in the context of colitis do not correlate with other studies of CD4+CD25+ T cells. Thus, it has been shown repeatedly that when CD4+CD25+ T cells are cocultured with CD4+CD25− T cells in the presence of anti-CD3 and APCs, CD25− T cell proliferation is markedly suppressed, but this suppression depends on an as yet undefined cell–cell interaction and not on humoral factors such as TGF-β or IL-10 10 11.
In view of this discrepancy, we conducted studies to reexamine the mechanism of CD4+CD25+ T cell–induced suppression. Our main findings were that CD4+CD25+ T cells do produce TGF-β1 and IL-10 when stimulated in an appropriate fashion and, in addition, express high levels of TGF-β1 on their cell surfaces. Moreover, CD4+CD25+ T cells mediate suppression of both T cell and B cell function which is TGF-β dependent. Since such suppression requires cell–cell contact, as in prior studies, these data strongly suggest that CD4+CD25+ T cells mediate immunosuppression via cell surface presentation of TGF-β to TGF-βR on target cells.
Materials and Methods
Mice.
Specific pathogen-free, 8-wk-old female Balb/c mice were purchased from the National Cancer Institute (Frederick, MD). Animal use adhered to National Institutes of Health Laboratory Animal Care Guidelines.
Reagents.
Anti-CD3 mAb (145-2C11), anti-CD28 mAb (37.51), unconjugated and PE-conjugated anti-cytotoxic T lymphocyte–associated antigen (CTLA)-4 mAb (UC10-4F10-11), Cy-Chrome™–conjugated anti-CD4 mAb (H129.19), biotin-conjugated and FITC-conjugated anti-CD25 mAb (7D4), PE-conjugated anti-CD25 mAb (PC61), FITC-conjugated, PE-conjugated, and Cy-Chrome™–conjugated streptavidin, PE-conjugated anti-CD80 mAb (16-10A1), PE-conjugated anti-CD86 mAb (GL1), PE-conjugated anti-CD45RB mAb (23G2), PE-conjugated rat IgG2a, PE-conjugated rat IgG2b, unconjugated and PE-conjugated hamster IgG, anti-CD16/CD32 mAb (Fc Block™), PE-conjugated anti–mouse IL-4 mAb (BVD-1D11), PE-conjugated anti–mouse IL-10 mAb (JES5-16E3), and rat anti–mouse IL-10R mAb (1B1.3a) were purchased from BD PharMingen. Anti-FITC Microbeads, anti-PE Microbeads, and MS+ Separation Columns were purchased from Miltenyi Biotec. Mouse anti–TGF-β1, -β2, and -β3 mAb (clone 1D11) was purchased from Genzyme and R&D Systems. Unconjugated and biotin-conjugated chicken anti–TGF-β1 Ab, biotin-conjugated goat IgG, biotin-conjugated goat anti–human latency-associated protein (LAP) of TGF-β1 Ab, normal mouse IgG1, rat anti–mouse IL-10 mAb (clone JES052A5), and recombinant human latent TGF-β1were purchased from R&D Systems. Recombinant human active TGF-β1 was purchased from R&D Systems and PeproTech. Anti–human LAP of TGF-β1 mAb (clone 27232.11) was supplied by R&D Systems. Normal mouse IgG, normal rat IgG, unconjugated and biotin-conjugated chicken IgY (IgG), and goat anti–mouse IgG were purchased from Jackson ImmunoResearch Laboratories. Alkaline phosphatase–conjugated anti–mouse IgG was purchased from Pierce Chemical Co. _P_-nitro-phenyl phosphate was purchased from Sigma-Aldrich. Methyl-[3H]thymidine was purchased from NEN Life Science Products. Recombinant human IL-2 was purchased from Life Technologies. Biotin-conjugated rat anti–mouse IgG1 (H143.225.8) was purchased from Southern Biotechnology Associates, Inc. Horseradish peroxidase–conjugated streptavidin was purchased from Zymed Laboratories. Anti-actin Ab was purchased from Santa Cruz Biotechnology, Inc.
Cell Purification.
CD4+ T cells were purified using CD4+ T cell enrichment columns (R&D Systems) from total splenocytes. CD4+CD25+ T cells were purified by magnetic beads or FACS® sorting. Separation of CD4+CD25+ T cells with magnetic beads was reported elsewhere 11. In brief, CD4+ T cells were incubated with biotin-conjugated anti-CD25 for 20 min at 4°C, washed, incubated with FITC-conjugated streptavidin for 15 min at 4°C, and washed. The cells were then incubated with anti-FITC Microbeads for 15 min at 4°C and washed. CD25+ cells were isolated with MS+ positive selection column according to the manufacturer's protocol (Miltenyi Biotec). In some experiments, FITC-conjugated anti-CD25 was substituted for biotin-conjugated anti-CD25 and FITC-conjugated streptavidin. In some experiments, PE-conjugated anti-CD25 and anti-PE Microbeads were used. The magnetically retained cells were shown to be >90% CD4+CD25+ cells, and the flow-through were shown to be >95% CD4+CD25− cells by flow cytometric analysis.
For isolation of CD4+CD25+ and CD4+CD25− T cells by FACS® sorting, CD4+ T cells were stained with FITC-conjugated anti-CD25, or with PE-conjugated anti-CD25 and CD25-positive and -negative cells were sorted by FACS Vantage™ SE II (Becton Dickinson). To purify CD25+, CD25−CD45RBlow and CD25−CD45RBhigh CD4+ T cells, CD4+ cells were stained with FITC–anti-CD25 and PE–anti-CD45RB and sorted into three subpopulations. The purity of each population was >95%.
Cell Culture.
The culture medium used in all experiments was RPMI1640 (Life Technologies) with 10% FCS (Life Technologies), 100 U/ml penicillin, 100 μg/ml Streptomycin, 10 mM Hepes (pH 7.0), 1 mM sodium pyruvate, and 50 μM 2-ME unless mentioned. In some experiments, 1% Nutridoma SP (Roche Molecular Biochemicals) or 2.5% FCS was used instead of 10% FCS.
Proliferation Assays.
2.5 × 104 of CD4+CD25+ or CD4+ CD25− T cells were stimulated with plate-bound anti-CD3 mAb (10 μg/ml) with or without soluble anti-CD28 (2 μg/ml), and with or without IL-2 (20 U/ml) in flat-bottom 96-well plates (0.1 ml). In some experiments, plates were coated with anti-CD3 (10 μg/ml), plus either anti–CTLA-4 (15 μg/ml) or hamster IgG (15 μg/ml). For coculture of CD25+ and CD25− subsets of CD4+ T cells, 2.5 × 104 of CD4+CD25+ or CD4+CD25−, or both were stimulated with 10 μg/ml of soluble anti-CD3 and 5 × 104 irradiated (3,000 rad) T cell–depleted syngenic splenocytes (non–T cell) in flat-bottom 96-well plates (0.1 ml). Anti–TGF-β, anti–IL-10, anti–IL-10R, or control IgG was added to the culture. Cells were cultured at 37°C for 72 h and pulsed with 1 μCi of [3H]thymidine for the last 6 h of culture. Then, cells were harvested and assessed for thymidine incorporation in a liquid scintillation counter.
ELISA for Cytokine Production.
CD4+CD25+ or CD4+CD25− T cells were stimulated in a 106/ml concentration with plate-bound anti-CD3 mAb (10 μg/ml) with or without soluble anti-CD28 (2 μg/ml), and with or without IL-2 (20 U/ml) at 37°C. In some experiments, plates were coated with anti-CD3 (10 μg/ml), plus either anti–CTLA-4 (10 μg/ml) or hamster IgG (10 μg/ml). Culture supernatants were collected after 48 h or, in the case of TGF-β1, after 72 h. Cytokines secreted into culture fluid were assayed by commercial ELISA kits according to the manufacturer's protocol. IL-10, IL-4, and IFN-γ were measured by BD OptEIA™ ELISA Set (BD PharMingen). For TGF-β1 assay, samples were acidified by addition of HCl at 20 mM for 15 min, neutralized by NaOH, and then TGF-β1 content was measured by TGF-β1 Emax Immunoassay Kit (Promega) as described previously 12. Optical densities were measured at 450 nm using a microplate ELISA reader (MR5000; Dynatech).
Flow Cytometric FACS® Analysis.
The expression of CTLA-4 on CD4+CD25+ or CD4+CD25− T cells was analyzed as described previously 13. In brief, splenocytes were blocked with anti-CD16/CD32 at 4°C for 15 min, washed and stained with PE-conjugated anti–CTLA-4 or PE-conjugated control hamster IgG at 37°C for 2 h. Cells were then stained with FITC-conjugated anti-CD25 and Cy-Chrome™-conjugated anti-CD4 at 4°C for 20 min. Cells were washed and analyzed with a FACScan™ flow cytometer (Becton Dickinson). For analysis of the expression of CD80 and CD86, purified CD4+CD25+ and CD4+CD25− cells were stimulated separately with anti-CD3, irradiated non–T cells, and IL-2 (20 U/ml) for 72 h. Then, cells were blocked with anti-CD16/CD32 and stained with Cy-Chrome™-conjugated anti-CD4 and either of PE-conjugated anti-CD80 or PE-anti-CD86 at 4°C for 20 min. For staining of TGF-β1 on the cell surface, unstimulated CD4+ T cells, purified and stimulated CD4+CD25+ and CD4+CD25− T cells were stained with FITC-conjugated anti-CD25, biotin-conjugated anti-TGF-β1 (or biotin-conjugated chicken IgG for a negative control), and Cy-Chrome™-conjugated anti-CD4 at 4°C for 30 min, washed and stained with PE-conjugated streptavidin at 4°C for 15 min. When PE-conjugated Ab was used for cell purification, TGF-β was stained with a combination of biotin-conjugated anti–TGF-β1 and Cy-Chrome™-conjugated streptavidin. For the staining with anti-LAP mAb (27232.11), cells were incubated with 27232.11 Ab, washed, incubated with biotin-conjugated anti-mouse IgG1, washed, and incubated with PE-conjugated streptavidin and Cy-Chrome™–conjugated anti-CD4.
Ig Production.
We chose PWM as a stimulator of B cell Ig production as it is mitogenic for both B cell and T cell, was useful to measure suppressor activity of human regulatory T cell clones in our previous study 14 and is applicable also for mouse lymphocyte cultures 15. Non–T cells were purified from splenocytes by complement-mediated T cell depletion. 5 × 104 of CD4+CD25− cells or CD4+CD25+ cells, or both were cocultured with 5 × 104 non–T cells with 20 μg/ml of PWM and 20 U/ml of IL-2 and with or without neutralizing anti-cytokine (anti–TGF-β, anti–IL-10, or control IgG) at 37° C for 8 d. In some experiments, CD4+CD25+ and CD4+CD25− T cells were mixed at various ratios as indicated. Culture supernatants were collected and IgG concentration was determined by ELISA: 96-well ELISA plates (Immulon 1; Dynatech) were coated with 2.5 μg/ml of goat anti–mouse IgG, washed, blocked with 1% BSA/PBS, and then washed. Standards and samples were put in wells, incubated at room temperature for 2 h, and washed. Alkaline phosphatase–labeled anti–mouse IgG was added and incubated at room temperature for 1 h and washed. Finally, colorimetric substrate _p_-nitro-phenyl phosphate was added and OD410 was determined using a microplate ELISA reader.
Purification of Membrane Fractions of CD4+CD25+ T Cells and Immunoblot Analysis.
To obtain membrane preparation, CD4+ CD25+ and CD4+CD25− T cells were isolated from 30 Balb/c spleens and stimulated with plate-bound anti-CD3 (10 μg/ml) and anti–CTLA-4 (10 μg/ml), soluble anti-CD28 (2 μg/ml), and IL-2 (30 U/ml) for 3 d. To obtain total cell lysates, 3 × 106 cells were lysed in lysis buffer (1% NP-40, 150 mM NaCl, 20 mM Tris, pH 7.5, 2 mM EDTA) supplemented with protease inhibitor cocktails (Roche Molecular Biochemicals) for 30 min on ice, centrifuged at 12,000 g for 30 min at 4°C, and supernatants were collected. Membrane preparation was performed as described elsewhere 16. In brief, 2.5 × 107 cells were collected, washed in PBS, suspended in relaxation buffer (3 mM NaCl, 100 mM KCl, 3.5 mM MgCl2, 1.25 mM EGTA, 1 mM ATP, 1 mM PMSF, 10 mM Pipes, pH 7.4), sonicated for 10 s three times on ice, and then centrifuged at 1,000 g for 10 min at 4°C to remove nuclei. The supernatant was centrifuged over a 10% (wt/vol) sucrose cushion (100,000 g) for 30 min at 4°C. Cytoplasmic fraction (upper layer) was removed and saved, and the membrane pellets were washed in relaxation buffer, and then solubilized in lysis buffer.
The lysates were mixed with SDS-sample buffer, incubated at 95°C for 5 min and run in 12% SDS-PAGE at nonreducing condition, and transferred to a nitrocellulose membrane (Hybond™ ECL™; Amersham Pharmacia Biotech). The blotted membrane was blocked with 5% skim milk/TBS/0.1% Tween 20, washed, incubated with 0.2 μg/ml of biotin-conjugated chicken anti–TGF-β1 Ab, washed, and incubated with horseradish peroxidase–conjugated streptavidin. Then, the membrane was washed, developed by SuperSignal West Pico Chemiluminescent Substrate (Pierce Chemical Co.), and exposed to an X-ray film. After stripping, the membrane was reprobed with anti-actin Ab.
Results
Proliferation and Cytokine Secretion Profile of CD4+CD25+ T Cells.
In initial studies, we determined the conditions resulting in optimal stimulation of CD4+CD25+ regulatory T cells. As shown in Fig. 1 A, CD4+CD25+ T cells stimulated by plate-bound anti-CD3 Ab did not proliferate well whereas CD4+CD25− T cells stimulated with such Ab underwent vigorous proliferation. In contrast, addition of anti-CD28 Ab (2 μg/ml) and/or IL-2 (20 U/ml) to anti-CD3–stimulated cultures of CD4+CD25+ T cells resulted in proliferation comparable to that of CD4+CD25− T cells stimulated with anti-CD3 alone or in combination with anti-CD28 and/or IL-2 (the optimum concentration of anti-CD28 and IL-2 in these studies had been determined in preliminary experiments). These results indicate that consistent with previous findings, CD4+CD25+ T cell proliferation in vitro and, by inference, maintenance of these cells in vivo are more dependent on CD28 signaling and IL-2 than are CD4+CD25− T cells 10 17 18.
Figure 1.

Proliferation and cytokine production of CD4+CD25+ regulatory T cells are CD28 and IL-2 dependent. (A) 2.5 × 104 CD4+CD25+ or CD4+CD25− cells were stimulated with plate-bound anti-CD3 Ab (10 μg/ml) with or without soluble anti-CD28 Ab (2 μg/ml) and/or exogenous IL-2 (20 U/ml) in 96-well plates. Cell proliferation was measured by incorporation of [3H]thymidine after 72 h. The results shown represent the mean ± SEM of triplicate wells and representative of three independent experiments. (B–E) 105 CD4+CD25+ or CD4+CD25− cells were stimulated with plate-bound anti-CD3 Ab (10 μg/ml) with or without soluble anti-CD28 Ab (2 μg/ml) and/or exogenous IL-2 (20 U/ml) in 100 μl culture. The amount of TGF-β1 (B), IL-10 (C), IL-4 (D), and IFN-γ (E) in culture supernatant was measured by ELISA. In B, 1% Nutridoma/RPMI was used for culture media. The results shown represent the mean ± SEM of triplicate wells with each well measured in duplicate, and are representative of three independent experiments. n.d., not detected, *: P < 0.00007, **: P < 0.0002, ***: P < 0.0009, ****: P < 0.0008. (F) 105 CD4+CD25+ or CD4+CD25− cells were stimulated with soluble anti-CD3 Ab (10 μg/ml), 2 × 105 irradiated non–T cells and IL-2 (20 U/ml) in 100 μl culture. 2.5% FCS/RPMI was used for culture media and TGF-β1 content in media was subtracted as a background. The results shown represent the mean ± SEM of triplicate wells with each well measured in duplicate, and are representative of three independent experiments.
In further studies, we determined the ability of CD4+ CD25+ and CD4+CD25− T cells to produce cytokines under the above established condition of optimal proliferation. As shown in Fig. 1 B, we found that CD4+CD25+ T cells stimulated by surface-bound anti-CD3 produce low but detectable amounts of TGF-β1 and such production was considerably augmented by addition of anti-CD28 and/or IL-2 whereas CD4+CD25− T cells secreted only minimal amounts of TGF-β1 when stimulated under comparable conditions; this was most evident when cells were stimulated with anti-CD3, anti-CD28, and IL-2, in which case CD4+CD25+ T cells produced about 20 times more TGF-β1 than CD4+CD25− T cells. In addition, as shown in Fig. 1 C, we observed that CD4+CD25+ T cells also secrete high levels of IL-10 when stimulated with anti-CD3, anti-CD28, and/or IL-2 and again such secretion greatly exceeded that of CD4+CD25− T cells, in this instance by a factor of 10. Finally, as shown in Fig. 1D and Fig. E, we found that CD4+CD25+ T cells produce markedly less IL-4 and IFN-γ than CD4+CD25− T cells and were thus neither Th1 nor Th2 T cells. Taken together, these studies show for the first time that CD4+CD25+ T cells produce high levels of the regulatory cytokines TGF-β and IL-10 when appropriately stimulated. As such, they are consistent with previous reports showing a relative abundance of TGF-β and IL-10 mRNA in CD4+CD25+ T cells by reverse transcription (RT)-PCR 2 11, but contrast with previous reports that show that this cell population secrete low or undetectable amounts of TGF-β or IL-10 protein 10 11. It should be noted in this context that the high level production of TGF-β and IL-10 from a CD4+CD25+ T cell is not simply due to the fact that these cells are memory cells as CD4+CD25+ T cells produce less IL-4 and IFN-γ than CD4+CD25− T cells.
Costimulation through CTLA-4 Enhances Proliferation and TGF-β1 Production of CD4+CD25+ T Cells.
Recently, two groups of investigators reported that suppressor function of CD4+CD25+ T cells are mediated through CTLA-4 signaling both in vitro and in vivo 4 13. Furthermore, it has been shown that CTLA-4 is a negative regulator of T cell responses and crosslinking of CTLA-4 enhances TGF-β1 production by CD4+ T cells 14 19. These facts prompted us to investigate the involvement of the CTLA-4 signaling pathway in TGF-β1 production by CD4+CD25+ T cells. In preliminary studies we determined the expression of CTLA-4 in CD4+CD25+ T cells, both with respect to cell surface and intracellular CTLA-4, as the majority of CTLA-4 protein resides in the cytoplasm 20. For this purpose, cells were stained with anti–CTLA-4 at 37°C rather than 4°C as described in Materials and Methods. As shown in Fig. 2 A, 43.3% of CD4+CD25+ T cells were positive for CTLA-4 whereas only 2.8% of CD4+CD25− T cells were similarly positive. This result is consistent with previous data which demonstrated abundant expression of CTLA-4 in CD4+CD25+ T cells in comparison with CD4+CD25− T cells 4 13 17. Next, we determined the effect of anti–CTLA-4 stimulation on CD4+CD25+ T cells both with respect to proliferation and cytokine production. As shown in Fig. 3 A, addition of anti–CTLA-4 to cells stimulated by anti-CD3 plus anti-CD28 (in the presence or absence of IL-2) led to enhanced proliferation of CD4+CD25+ T cells, particularly in the presence of exogenous IL-2; in contrast, similar CTLA-4 engagement of CD4+CD25− T cells led to diminished proliferation in the presence and absence of IL-2. In previous studies it was shown that cross-linking of CTLA-4 inhibits T cell proliferation induced by the stimulation with anti-CD3 and anti-CD28 21. The present results show that while this may be true of mixed CD4+CD25+ and CD4+CD25− T cell subpopulations, or CD4+CD25− T cell populations, signaling of CD4+CD25+ T cells through CTLA-4 is a positive stimulus for these T cells. To our knowledge, this is the first demonstration that CTLA-4 signaling is a positive proliferation stimulus under some conditions. Interestingly, CD80 and CD86, ligands for CTLA-4 and CD28, are more strongly expressed on CD4+CD25+ T cells than on CD4+CD25− T cells after stimulation (Fig. 2B and Fig. C). These results suggest that CD80 and CD86 expressed on both APCs and on CD4+CD25+ T cells can provide costimulatory signals to CD4+CD25+ T cells and thus the latter cells are, in part, autostimulatory.
Figure 2.
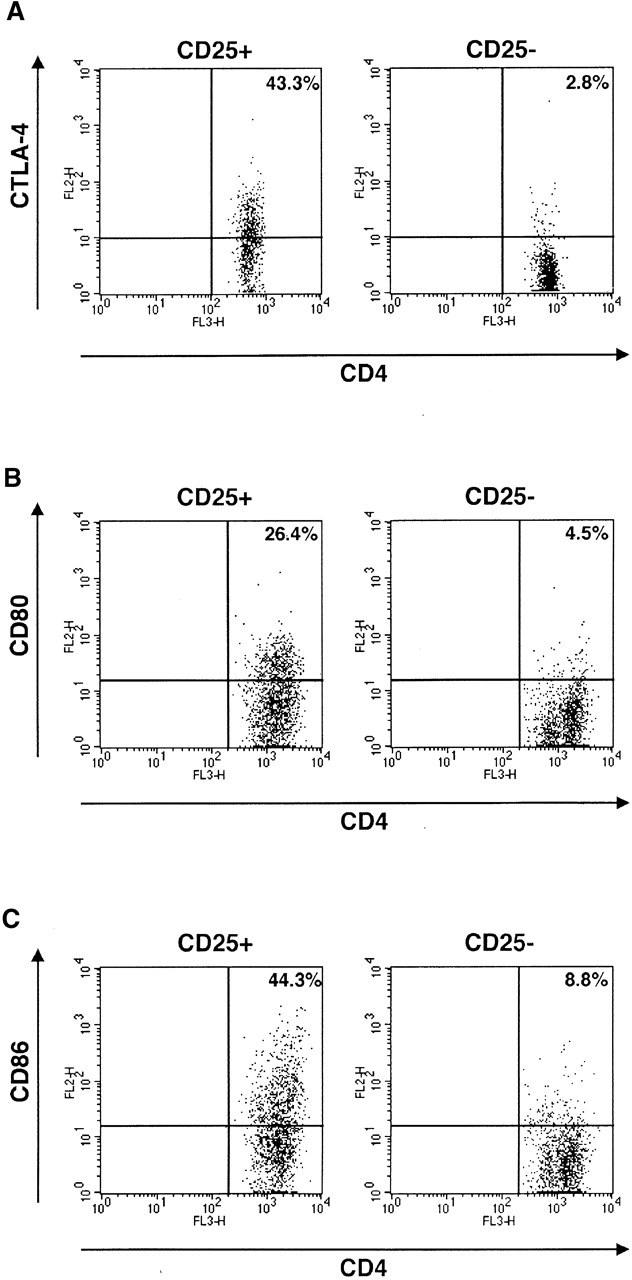
Expression of CTLA-4, CD80, and CD86 on CD4+CD25+ T cells. (A) CTLA-4 on/in splenocytes was stained as described in Materials and Methods. CD4+CD25+ and CD4+CD25− fractions were gated and expression of CTLA-4 on/in each population is shown. (B and C) Purified CD4+CD25+ and CD4+CD25− T cells were stimulated with anti-CD3 (10 μg/ml), irradiated non–T cells and IL-2 (20 U/ml) for 72 h and then stained with Cy-chrome™–conjugated anti-CD4 and either of PE-conjugated anti-CD80 or PE-conjugated anti-CD86. Expression of CD80 (B) and CD86 (C) in the CD4+ gate is shown.
Figure 3.
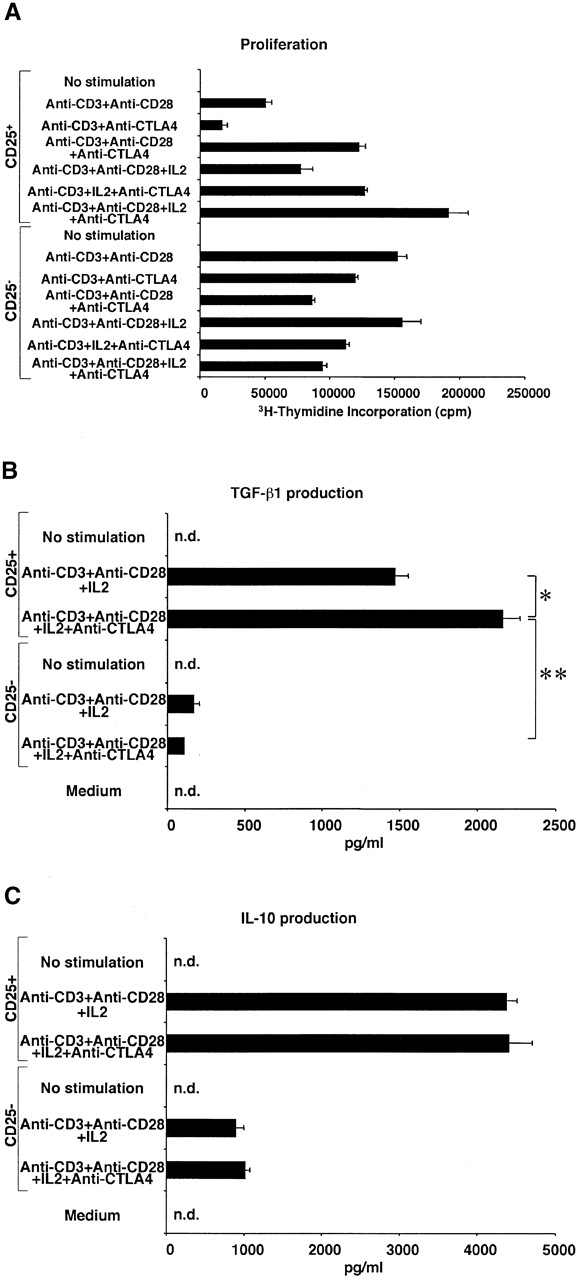
Signaling through CTLA-4 enhances proliferation and TGF-β1 secretion of CD4+CD25+ T cells. (A) 2.5 × 104 CD4+CD25+ or CD4+CD25− T cells were stimulated with plate-bound anti-CD3 with or without soluble anti-CD28 (2 μg/ml), plate-bound anti–CTLA-4, or exogenous IL-2 (20 U/ml) in 96-well plates. Cell proliferation was measured by incorporation of [3H]thymidine after 72 h. Plates were coated either with anti-CD3 (10 μg/ml) plus anti–CTLA-4 (15 μg/ml) or anti-CD3 plus hamster IgG (15 μg/ml). The results shown are the mean ± SEM of triplicate wells and representative of three independent experiments. (B and C) 105 CD4+CD25+ or CD4+CD25− T cells were stimulated with plate-bound anti-CD3 Ab, soluble anti-CD28 Ab (2 μg/ml), and exogenous IL-2 (20 U/ml) with or without plate-bound anti–CTLA-4 in 100 μl of culture. The amount of TGF-β1 (B) and IL-10 (C) was measured by ELISA. In B, 1% Nutridoma/RPMI was used for culture media. Plates were coated either with anti-CD3 (10 μg/ml) plus anti–CTLA-4 (10 μg/ml) or anti-CD3 plus hamster IgG (10 μg/ml). The results shown are the mean ± SEM of triplicate wells with each well measured in duplicate, and are representative of three independent experiments. n.d., not detected, *: P < 0.005, **: P < 0.00003.
In additional studies we examined the effect of anti–CTLA-4 on TGF-β production by CD4+CD25+ T cells. As shown in Fig. 3 B, we found that addition of anti–CTLA-4 to cultures of T cells stimulated with anti-CD3, anti-CD28, and IL-2 further enhanced the production of TGF-β1 by CD4+CD25+ T cells whereas such stimulation did not induce substantial TGF-β1 production by CD4+CD25− T cells. Furthermore, as shown in Fig. 3 C (and data not shown), this effect of anti–CTLA-4 Ab is specific for TGF-β production because anti–CTLA-4 Ab did not augment IL-10, IL-4, or IFN-γ production by either CD4+CD25+ or CD4+CD25− T cells. These data are consistent with the studies of Chen et al. who demonstrated that anti–CTLA-4 induces increased TGF-β1 production by naive mouse CD4+ T cells 19. However, in the latter case the amount of TGF-β1 produced was considerably lower than that in this study, presumably reflecting the fact that a mixed cell population containing both CD4+CD25+ and CD4+CD25− T cells was being stimulated in the earlier study.
Suppressor Function of CD4+CD25+ T Cells Is Mediated by TGF-β1.
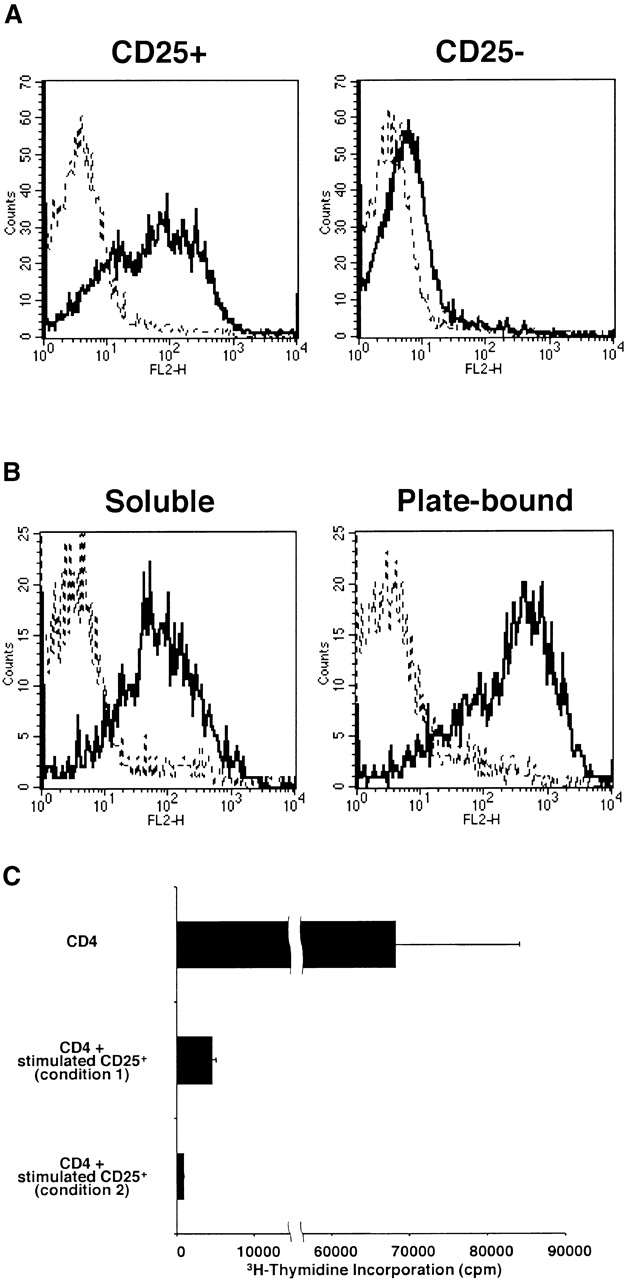
Having established that CD4+CD25+ T cells produce TGF-β1 when appropriately stimulated, we next investigated whether such production mediates immunosuppression. Our approach in these studies was to determine if CD4+CD25+ T cells mediate suppression in the absence and presence of anti-TGF-β. As shown in Fig. 4 A, we observed that in cultures containing control mouse IgG, CD4+CD25− T cells but not CD4+CD25+ T cells proliferated well in response to stimulation with soluble anti-CD3 Ab plus irradiated syngenic non–T cells; however, under this condition, if CD4+CD25+ T cells were cocultured with CD4+CD25− T cells, cell proliferation was profoundly suppressed. Thus, as previously reported 10 11, CD4+CD25+ T cells act as suppressor cells. In contrast, when the same cultures were carried out in cultures containing 50 μg/ml of anti–TGF-β (1D11) rather than mouse IgG, both CD4+CD25+ and CD4+CD25− T cells cultured alone exhibited increased proliferation, and, more importantly, CD4+CD25+ T cell–mediated suppression was completely abolished. The effect of anti–TGF-β was dose dependent because addition of the Ab at 100 μg/ml further increased the proliferation of CD4+CD25+ T cells, and consequently, of the coculture of CD4+CD25+ and CD4+CD25− T cells. 25 μg/ml of anti–TGF-β only partially restored cell proliferation (data not shown). Finally, as shown in Fig. 4 B, although CD4+CD25+ T cells produce large amounts of IL-10, addition of anti–IL-10 or anti–IL-10R at 100 μg/ml to the above cultures did not decrease the level of CD4+CD25+ T cell–mediated suppression. To further confirm that TGF-β is capable of suppressing T cell proliferation under these conditions, we added various amount of rTGF-β to the cell culture. As shown in Fig. 4 C, relatively small amount of recombinant active TGF-β1 (ED50: 31.3–62.5 pg/ml) significantly suppressed cell proliferation of the CD4+CD25− T cells. These results provide strong evidence that suppression of T cell proliferation by CD4+CD25+ T cells is mediated by TGF-β and, in addition, the unresponsiveness of CD4+CD25+ T cells to stimulation is due, at least in part, to autocrine suppression by TGF-β.
Figure 4.
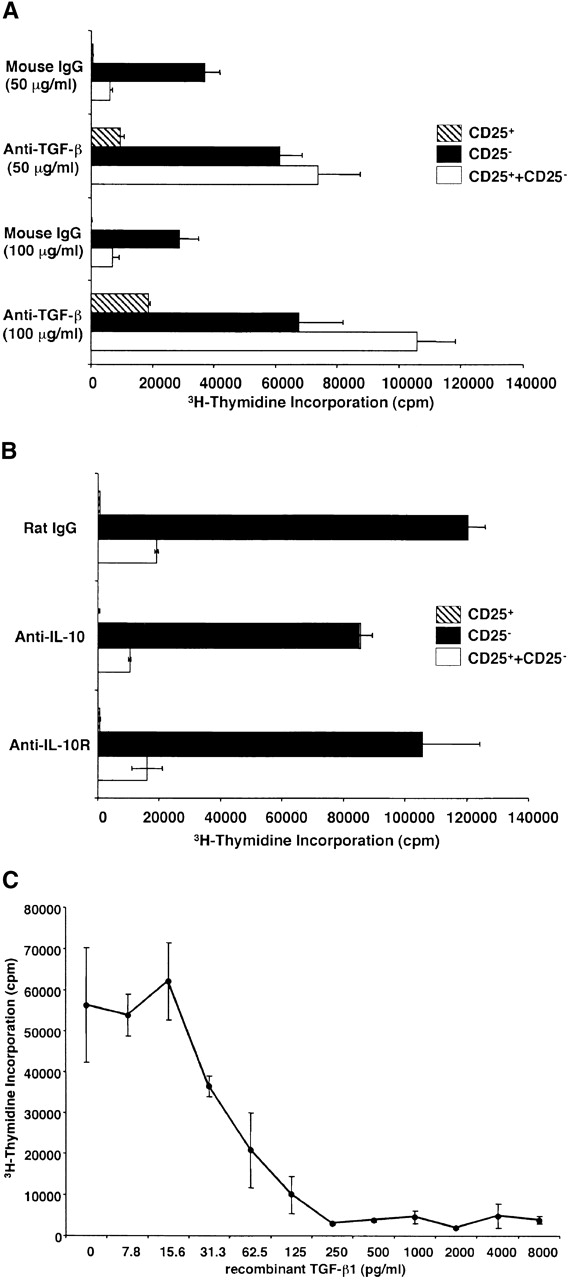
Suppression of T cell proliferation by CD4+CD25+ regulatory T cells is mediated by TGF-β. (A and B) 2.5 × 104 CD4+CD25− (black bars) or CD4+CD25+ T cells (hatched bars), or both (white bars) were stimulated with soluble anti-CD3 Ab (10 μg/ml) and 5 × 104 of irradiated syngenic non–T cells in 96-well plates in the presence of anti-cytokine or control IgG. (A) 50 μg/ml or 100 μg/ml of control mouse IgG or anti-TGF-β (1D11). (B) 100 μg/ml of control rat IgG, anti–IL-10, or anti–IL-10R. Cell proliferation was measured by incorporation of [3H]thymidine after 72 h. The results shown are the mean ± SEM of triplicate wells and are representative of four independent experiments. (C) 2.5 × 104 CD4+CD25− T cells were stimulated with soluble anti-CD3 Ab (10 μg/ml) and 5 × 104 of irradiated syngenic non–T cells in 96-well plates in the presence of various amounts of rTGF-β1 (active form). Cell proliferation was measured by incorporation of [3H]thymidine after 72 h. The results shown are the mean ± SEM of triplicate wells and are representative of three independent experiments.
In further studies we determined if CD4+CD25+ T cells inhibit T cell–dependent Ig synthesis by B cells as well as T cell proliferation via TGF-β. Here we set up cultures of CD4+CD25− T cells or CD4+CD25+ T cells with non–T cells stimulated with PWM and IL-2 (see Materials and Methods), and, as shown in Fig. 5 A, demonstrated first that the CD4+CD25− T cells help B cells produce IgG whereas CD4+CD25+ T cells do not help. We then showed that addition of CD4+CD25+ T cells to cocultures of CD4+CD25− T cell and non–T cells blocked IgG synthesis in a dose-dependent fashion. Then, as shown in Fig. 5 B, we showed that addition of anti–TGF-β Ab completely abolished the suppression of CD4+CD25+ T cells in the mixed cell cultures. For this experiment, we used a polyclonal chicken anti–TGF-β1 Ab because mouse anti–TGF-β mAb (1D11) used in the studies of T cell proliferation cross-reacts with mouse IgG in the usual ELISA system. According to the manufacturer, this polyclonal chicken Ab has a similar neutralizing capacity as the mouse monoclonal anti–TGF-β (1D11). Finally, addition of anti–IL-10 to cultures did not inhibit the suppression of CD4+CD25+ T cells, but instead decreased IgG production, presumably because IL-10 acts as a B cell growth factor under some conditions 22. From these results, we conclude that CD4+CD25+ T cells also suppress B cell IgG synthesis via TGF-β. These data, however, do not determine whether TGF-β acts on CD4+CD25− helper T cells or directly on B cells to mediate suppression.
Figure 5.


CD4+CD25+ regulatory T cells suppress B cell Ig synthesis through TGF-β. (A) 5 × 104 CD4+CD25− or CD4+CD25+ T cells, plus 5 × 104 non–T cells were stimulated with 20 μg/ml of PWM and 20 U/ml of IL-2 in 200 μl culture. In some wells, CD4+CD25+ cells were added to 5 × 104 CD4+CD25− cells at the ratio indicated in the figure. Cells were cultured at 37°C for 8 d and culture supernatant was collected. The concentration of mouse IgG in the supernatant was determined by ELISA. The results shown are the mean ± SEM of triplicate wells and are representative of three independent experiments. (B) 5 × 104 CD4+CD25− (black bars), or 5 × 104 of both CD4+CD25+ and CD4+CD25− T cells (white bars) were cocultured with 5 × 104 non–T cells, and stimulated with 20 μg/ml of PWM and 20 U/ml of IL-2 in 200 μl culture. 20 μg/ml of control chicken IgG or chicken anti–TGF-β1 Ab, or 100 μg/ml of rat anti–IL-10 mAb was added to the culture. Cells were cultured at 37°C for 8 d and culture supernatant was collected. Concentration of mouse IgG in the supernatant was determined by ELISA. The results shown are the mean ± SEM of triplicate wells and representative of three independent experiments.
In that the above experiments show that CD4+CD25+ T cells mediate suppression by TGF-β, they suggested that such suppression does not require cell contact between CD4+CD25+ T cells and their targets. To test this possibility, we conducted studies using a Transwell™ (Corning) culture system in which suppressor function was measured under conditions in which the putative CD4+CD25+ suppressor T cells and CD4+CD25− target T cells were separated by a membrane. In these studies, CD4+CD25+ or CD4+CD25− T cells were cultured in the inner well in the presence of soluble anti-CD3 Ab and APCs and CD4+CD25− “indicator” cells were cultured in the outer well again in the presence of soluble anti-CD3 and APCs; then, after 72 h, proliferation of the CD4+CD25− T cells in the outer well was assessed after transfer to 96-well plates and addition of [3H]thymidine. Control cultures, in which only non–T cells were cultivated with anti-CD3 in the inner well, were conducted in parallel. Contrary to expectation, we found that CD4+CD25+ T cells could not mediate suppression of CD4+CD25− T cell proliferation across a membrane (data not shown). In further studies to determine if CD4+CD25+ T cell–mediated suppression does not require cell contact we stimulated CD4+CD25− T cells in the presence of supernatants of culture of CD4+CD25+ T cells stimulated by soluble anti-CD3 and APCs. In parallel with the results of the Transwell™ studies, we found that the culture supernatant did not suppress CD4+CD25− T cell proliferation (data not shown). These results are consistent with the previous reports in which CD4+CD25+ T cells failed to suppress T cell proliferation through a membrane (Transwell™ system) or by transfer of their culture supernatants 10 11 and suggest that only low, subsuppressive amounts of TGF-β are secreted when CD4+ CD25+ T cells are stimulated by soluble anti-CD3 and APCs, the condition under which these cells mediate suppression. In fact, as shown in Fig. 1 F, we could detect only small amounts of (latent) TGF-β1 (100–200 pg/106 cells) in the supernatant of CD4+CD25+ T cells stimulated under this condition. This amount is not significantly different from that secreted by CD4+CD25− T cells and is not sufficient to cause suppression when added to T cell cultures. Note that the rTGF-β causing suppression in Fig. 4 C is active TGF-β.
CD4+CD25+ T Cells Express TGF-β1 on Their Cell Surface.
The above studies showing that cell contact is necessary for CD4+CD25+ T cell suppression, yet such suppression is mediated by TGF-β created a paradox that can conceivably be explained if we assume that suppression is mediated largely by cell-bound TGF-β. In initial studies to explore this possibility, we stained purified CD4+ T cells with FITC-conjugated anti-CD25 as well as with biotin-conjugated polyclonal anti–TGF-β1 and PE-conjugated streptavidin. As shown in Fig. 6 A, 15.4% of CD4+CD25+ T cells (1.4% of total CD4+ cells) were cell surface TGF-β1 positive, while virtually no CD4+CD25− T cells were cell surface TGF-β1 positive. Next, we stained purified CD4+CD25+ and CD4+CD25− T cells after stimulation with soluble anti-CD3, non–T cells, and IL-2. The biotin-conjugated anti-CD25 Ab was not used for isolation of CD4+CD25+ T cells in this experiment as biotin-conjugated anti–TGF-β was used for staining of cells. As shown in Fig. 6 B, 24 h after stimulation, the expression of TGF-β1 on CD4+CD25+ T cells was dramatically upregulated in that 64% of the cells were now surface TGF-β positive. While some CD4+CD25− T cells also expressed TGF-β1 on the cell surface after such stimulation (13%), in this case both the extent and intensity of staining was far below that of CD4+CD25+ T cells. Finally, as shown in Fig. 6 C, 6 d after stimulation CD4+CD25+ T cells still manifested a high level of surface TGF-β1, whereas the level of surface TGF-β1 on CD4+CD25− T cells had fallen to baseline. These results are summarized in the time course study in Fig. 6 D where it is shown that TGF-β1 expression on CD4+CD25+ and CD4+CD25− T cells after stimulation was quite different: the percentage of surface TGF-β1–positive cells in the CD4+CD25+ T cell subset increased from 15 to 64% in 24 h, reached a peak of 77% on day 3, and then maintained the level up to day 6. In contrast, the percentage of TGF-β1–bearing CD4+CD25− T cells attained a peak of 29% on day 2 and then rapidly decreased to baseline. We also isolated CD4+CD25+ T cells using another anti-CD25 mAb (PC61) and stained cell surface–bound TGF-β after stimulation, which resulted in virtually identical staining pattern as the experiments shown above in which cells were isolated using anti-CD25 (7D4) (data not shown). Cell surface TGF-β was also observed when CD4+CD25+ T cells were stimulated with anti-CD3 and APCs in the absence of exogenous IL-2 for 24 h but the expression level was lower than those stimulated with IL-2 (data not shown), suggesting that IL-2 is not necessary for the surface expression of TGF-β1 by these cells, but does enhances such expression. Finally, incubation of the polyclonal anti–TGF-β1 Ab with rTGF-β1 before the staining of CD4+CD25+ T cells significantly decreased the fluorescence intensity of the surface TGF-β staining (data not shown).
Figure 6.

CD4+CD25+ T cells express TGF-β1 on the cell surface. (A) Enriched CD4+ T cells were stained with FITC-conjugated anti-CD25, Cy-Chrome™–conjugated anti-CD4 and either biotin-conjugated chicken anti–TGF-β1 or biotin-conjugated normal chicken IgG, washed, and stained with PE-conjugated streptavidin. CD4+CD25+ and CD4+CD25− T cells were gated and expression of cell surface-bound TGF-β1 is shown. Thick lines: anti–TGF-β, thin lines: normal chicken IgG. The results shown are the representative of three independent experiments. (B and C) Purified CD4+CD25+ and CD4+CD25− T cells were stimulated with soluble anti-CD3 (10 μg/ml) and irradiated non–T cells in the presence of IL-2 (20 U/ml) for 24 h (B) or 6 d (C). Cells were then stained with Cy-Chrome™–conjugated anti-CD4 and either biotin-conjugated chicken anti–TGF-β1 or biotin-conjugated normal chicken IgG, washed and stained with PE-conjugated streptavidin. CD4+ cells were gated and expression of cell surface-bound TGF-β1 is shown. Thick lines: anti–TGF-β, thin lines: normal chicken IgG. The results shown are the representative of three independent experiments. (D) Graphic representation of the percentage of surface TGF-β1 positive cells in CD4+CD25+ (circle) and CD4+CD25− (triangle) T cells after stimulation.
Anti–TGF-β used in the above studies to detect cell surface TGF-β is a polyclonal (chicken) Ab raised against recombinant active (human) TGF-β1 and purified by TGF-β1 affinity chromatography; thus epitopes recognized by this Ab reside in active TGF-β1. To determine if latent TGF-β1 is in fact present on the cell surface, we also stained cells with Abs specific for LAP of TGF-β1. As shown in Fig. 7 A, we observed high levels of LAP expressed on the cell surface of stimulated CD4+CD25+ T cells using an anti-LAP mAb (clone 27232.11 [R&D Systems]) but only small amounts of LAP on the surface of stimulated CD4+CD25− T cells. Similar results were obtained with a polyclonal goat anti-LAP Ab (R&D Systems; data not shown). Finally, as shown in Fig. 7B and Fig. C, in contrast to the positive staining obtained with anti–TGF-β Ab or anti-LAP Abs, staining with anti–IL-10 or anti–IL-4 Abs for surface IL-10 or IL-4 was negative.
Figure 7.
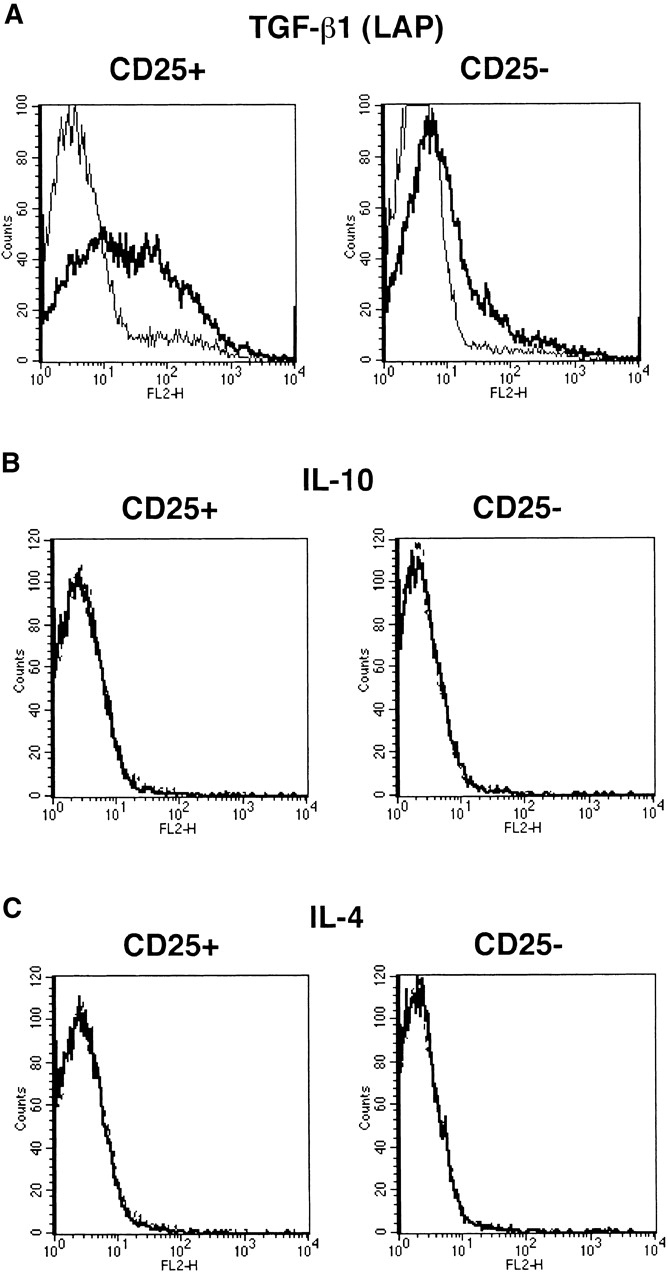
CD4+CD25+ T cells express LAP of TGF-β1 but not IL-10 or IL-4 on the cell surface. Purified CD4+CD25+ and CD4+CD25− T cells were stimulated with soluble anti-CD3 (10 μg/ml) and irradiated non–T cells in the presence of IL-2 (20 U/ml) for 24 h. Cells were incubated with Cy-Chrome™–conjugated anti-CD4 and either anti-LAP mAB (27232.11) (A), PE-conjugated anti–IL-10 (B), or PE-conjugated anti–IL-4 (C). Incubation with isotype-matched control IgG for each anti-cytokine was performed in parallel. In panel A, after incubation with anti-LAP, cells were washed, incubated with biotin-conjugated anti–mouse IgG1, washed, and incubated with PE-conjugated streptavidin. CD4+ cells were gated and expression of LAP (A), IL-10 (B), and IL-4 (C) on the cell surface was shown. Thick lines: anti-cytokine; thin lines: isotype-matched control IgG.
To further confirm that CD4+CD25+ T cells express high levels of TGF-β, we conducted immunoblot analyses. First, we purified total cell lysate from activated CD4+CD25−, CD4+CD25+ and whole CD4+ T cells and analyzed the lysates for TGF-β content. As shown in Fig. 8 A, TGF-β1 band was detected only in the CD4+CD25+ T cell lysate but not in CD4+CD25− or CD4+ lysates. Lysate from 3 × 106 CD4+CD25+ T cells and 2 ng rTGF-β1 gave almost identical density of TGF-β band in this assay (data not shown). Reprobing the same membrane with anti-actin Ab revealed bands of actin with almost identical intensity in each sample, showing that almost equal amount of protein had been loaded in each lane. Next, to confirm that TGF-β expressed by CD4+CD25+ T cells exists on the cell surface, we obtained purified membrane fraction of activated CD4+CD25+ and CD4+CD25− T cells (see Materials and Methods). As shown in Fig. 8 B, the TGF-β band was detected only in the membrane fraction of CD4+CD25+ T cells, but not in the membrane fraction of CD4+CD25− or cytoplasmic protein fraction of either cell population.
Figure 8.
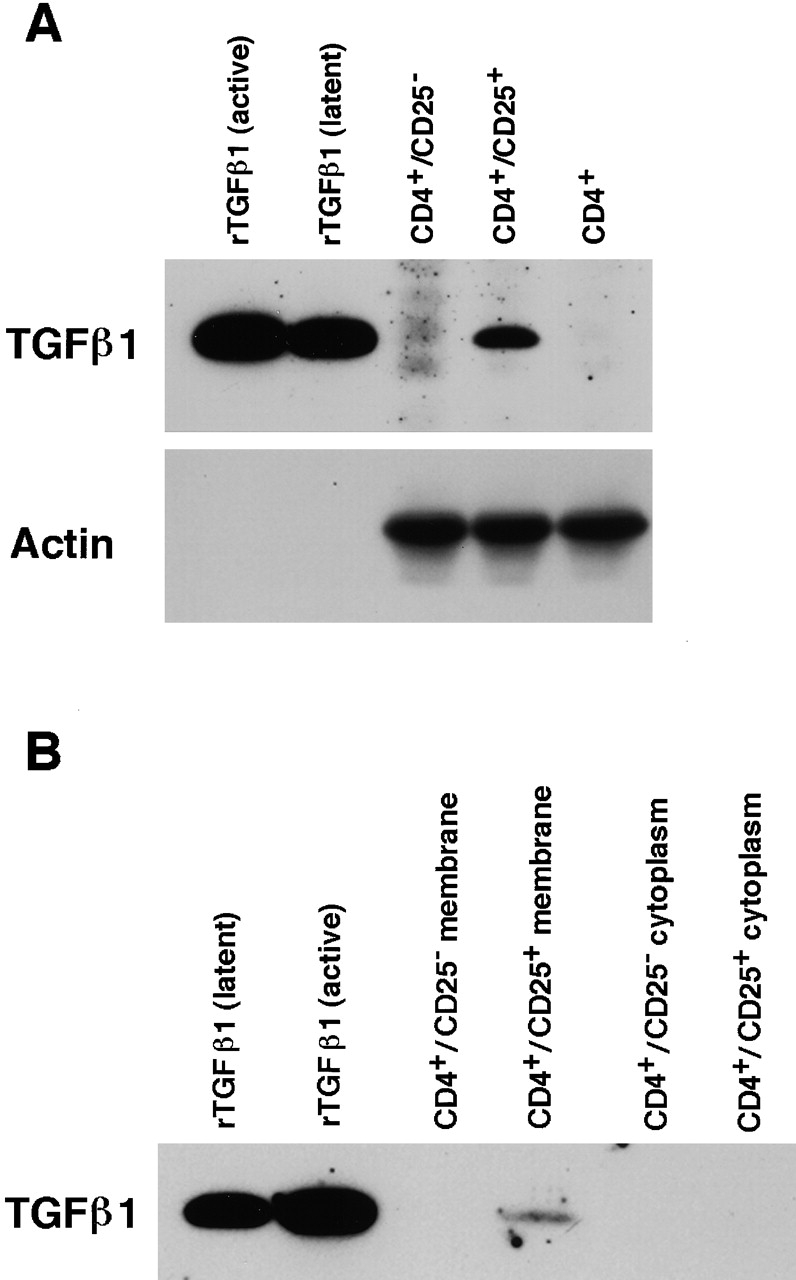
Immunoblot analyses for the expression of TGF-β1 in CD4+CD25+ T cells. (A) Total cell lysates purified from 3 × 106 of activated CD4+CD25−, CD4+CD25+ and CD4+ T cells were run in SDS/PAGE and blotted to the membrane. 5 ng of active and latent rTGF-β1 were loaded in parallel to serve as positive controls. Membrane was first probed using chicken anti–TGF-β1, stripped, and reprobed with anti-actin Ab. (B) Lysates from membrane (derived from 2.5 × 107 cells) and cytoplasmic (derived from 1.25 × 107 cells) preparations of activated CD4+CD25− and CD4+CD25+ T cells were subjected to SDS/PAGE and immunoblot analysis. Membrane was probed using chicken anti–TGF-β1.
In further studies we sought to verify that the surface TGF-β1 associated with CD4+CD25+ T cells arises from the cell itself and not from an exogenous source such as APCs in the culture or FCS in the culture medium. To this end, we stimulated purified CD4+CD25+ or CD4+CD25− T cells with plate-bound anti-CD3, soluble anti-CD28, plate-bound anti-CTLA-4, and IL-2 for 24 h in the presence of 1% Nutridoma/RPMI medium (rather than FCS/RPMI) without addition of APCs and then determined cell surface expression of TGF-β1 with the staining procedure described above. As shown in Fig. 9 A, we found that 73.4% of CD4+CD25+ T cells expressed TGF-β1 on the cell surface whereas only 8.5% of CD4+CD25− T cells expressed cell surface TGF-β1. As the only possible source of TGF-β1 in these cultures was the T cells themselves, we concluded that TGF-β1 detected on the surface of CD4+CD25+ T cells was produced by these cells and did not arise from an exogenous source.
Figure 9.
Expression of surface TGF-β1 after the stimulation with plate-bound anti-CD3 and anti–CTLA-4, soluble anti-CD28, and IL-2. (A) Purified CD4+CD25+ and CD4+CD25− T cells were stimulated with plate-bound anti-CD3 (10 μg/ml) and anti–CTLA-4 (10 μg/ml), soluble anti-CD28 (2 μg/ml), and IL-2 (20 U/ml). Cultures were carried out in 1% Nutridoma/RPMI. 24 h later, cells were incubated with Cy-Chrome™–conjugated anti-CD4 and either biotin-conjugated chicken anti–TGF-β1 or biotin-conjugated normal chicken IgG, washed, and stained with PE-conjugated streptavidin. CD4+ cells were gated and expression of cell surface-bound TGF-β1 is shown. Thick lines: anti–TGF-β; thin lines: normal chicken IgG. (B) Purified CD4+CD25+ T cells were stimulated with soluble anti-CD3 (10 μg/ml), non–T cells and IL-2 (20 U/ml) (left panel), or with plate-bound anti-CD3 (10 μg/ml) and anti–CTLA-4 (10 μg/ml), soluble anti-CD28 (2 μg/ml), and IL-2 (20 U/ml) (right panel). 60 h later, cells were incubated with Cy-Chrome™–conjugated anti-CD4 and either biotin-conjugated chicken anti–TGF-β1 or biotin-conjugated normal chicken IgG, washed, and stained with PE-conjugated streptavidin. CD4+ cells were gated and expression of cell surface-bound TGF-β1 is shown. Thick lines: anti–TGF-β; thin lines: normal chicken IgG. (C) Purified CD4+CD25+ T cells were stimulated with soluble anti-CD3 (10 μg/ml), non–T cells and IL-2 (20 U/ml) (condition 1) or with plate-bound anti-CD3 (10 μg/ml) and anti–CTLA-4 (10 μg/ml), soluble anti-CD28 (2 μg/ml), and IL-2 (20 U/ml) (condition 2) for 4 d and irradiated (1,000 rad). 2.5 × 104 CD4+ T cells were mixed with 2.5 × 104 CD4+CD25+ T cells stimulated in condition 1 or condition 2, and stimulated with soluble anti-CD3 Ab (10 μg/ml) and 5 × 104 of irradiated syngenic non–T cells in 96-well plates. Cell proliferation was measured by incorporation of [3H]thymidine after 72 h. The results shown are the mean ± SEM of triplicate wells and are representative of two independent experiments.
As shown in Fig. 3 B and Fig. 9 A, CD4+CD25+ T cells stimulated by plate-bound anti-CD3, soluble anti-CD28, plate-bound anti–CTLA-4, and IL-2 secret high levels of TGF-β1, indicating that these cells can produce both surface and secreted TGF-β1 when maximally stimulated. In addition, CD4+CD25+ T cells stimulated under these conditions express higher levels of surface-bound TGF-β1 than those stimulated with soluble anti-CD3, non–T cells, and IL-2 (Fig. 9 B); this allows us to determine if higher level of TGF-β1 expression is associated with enhanced suppressor activity. To this end, purified CD4+CD25+ T cells were stimulated with soluble anti-CD3, non–T cells and IL-2 (stimulation condition 1), or with plate-bound anti-CD3, soluble anti-CD28, plate-bound anti-CTLA-4, and IL-2 (stimulation condition 2) for 4 d, irradiated (1,000 rad) to inhibit cell proliferation, and then mixed with freshly isolated CD4+ T cells at 1:1 ratio to form cultures that were stimulated with anti-CD3 and non–T cells. We found that CD4+CD25+ T cells stimulated by stimulation condition 2 suppressed CD4+ T cell proliferation more profoundly than those stimulated by stimulation condition 1 when assessed by [3H]thymidine incorporation (Fig. 9 C). Mixtures of stimulated CD4+CD25+ T cells with fresh CD4+ T cells at 1:2 ratio gave almost the same results (data not shown).
CD4+CD25+ Population but Not CD4+CD25−CD45RBlow Population Expresses High Levels of TGF-β.
In the experiments shown above, we compared CD4+CD25+ T cells and CD4+CD25− T cells and showed that the former population is a high expresser of cell surface–bound and secreted TGF-β1. To exclude the possibility that this is simply because CD4+CD25+ T cells have been subject to prior stimulation through the TCR and that high level expression of TGF-β is simply a feature of previously stimulated T cells, we purified CD25+, CD25−CD45RBlow, and CD25−CD45RBhigh populations of CD4+ T cells. As reported previously 10 11 23, most of the CD4+CD25+ population was CD45RBlow (data not shown). As shown in Fig. 10 A, stimulated CD4+CD25+ T cells expressed abundant cell surface-bound TGF-β, whereas stimulated CD25− CD45RBlow and CD25−CD45RBhigh T cells expressed only small amount of cell surface–bound TGF-β1. In addition, as shown in Fig. 10 B, CD4+CD25+ T cells also secreted significantly higher amount of TGF-β1 into the culture supernatant after stimulation than CD25−CD45RBlow and CD25−CD45RBhigh populations. In contrast, as shown in Fig. 10C–E, CD25−CD45RBlow T cells secreted huge amounts of IL-10, IL-4, and IFN-γ which far exceeded that produced by CD25+ and CD25− CD45RBhigh T cells. Finally, while CD4+CD25+ T cells did not produce high levels of IL-4 or IFN-γ, they did produce quite high amount of IL-10, albeit at levels significantly lower than that of CD4+CD25−CD45RBlow T cells. Taken together, these data show that the expression of high levels of TGF-β in both membrane-bound and soluble forms is a unique feature of CD4+CD25+ regulatory T cells.
Figure 10.
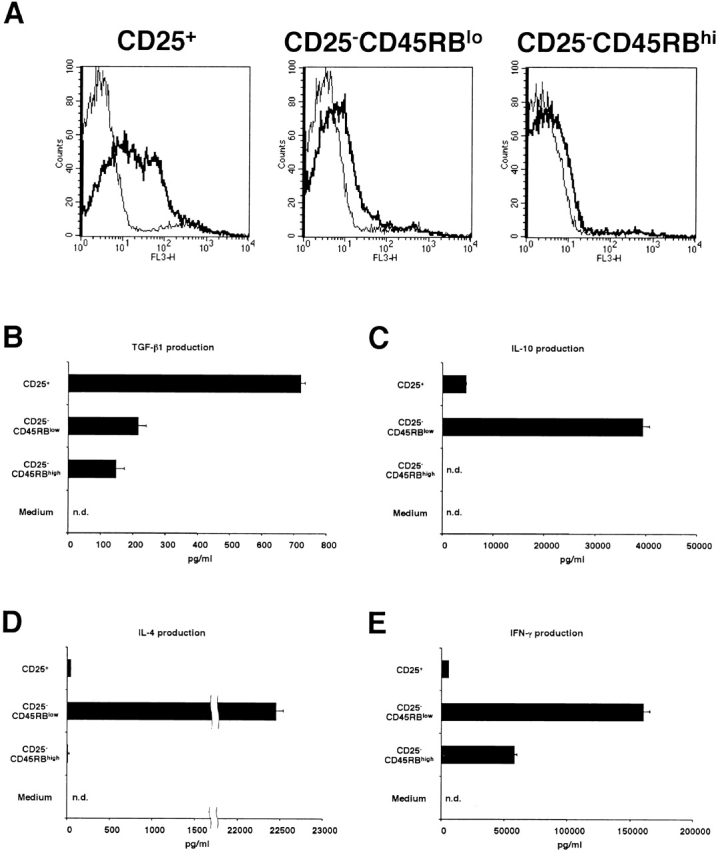
Comparison of cytokine expression among CD25+, CD25−CD45RBlow and CD25−CD45RBhigh populations of CD4+ T cells. (A) Purified CD4+CD25+, CD4+CD25− CD45RBlow and CD4+CD25−CD45RBhigh T cells were stimulated with plate-bound anti-CD3 (10 μg/ml) and anti–CTLA-4 (10 μg/ml), soluble anti-CD28 (2 μg/ml), and IL-2 (20 U/ml). 24 h later, cells were incubated with either biotin-conjugated chicken anti–TGF-β1 or biotin-conjugated normal chicken Ig G, washed, and stained with Cy-Chrome™–conjugated streptavidin. Expression of cell surface–bound TGF-β1 is shown. Thick lines: anti–TGF-β; thin lines: normal chicken IgG. (B–E) 105 CD4+ CD25+, CD4+ CD25−CD45RBlow and CD4+ CD25−CD45RBhigh T cells were stimulated with plate-bound anti-CD3 (10 μg/ml) and anti–CTLA-4 (10 μg/ml), soluble anti-CD28 (2 μg/ml) and IL-2 (20 U/ml) in 100 μl culture for 72 h. The amount of TGF-β1 (B), IL-10 (C), IL-4 (D), and IFN-γ (E) in culture supernatant was measured by ELISA. In B, 1% Nutridoma/RPMI was used for culture media. The results shown represent the mean ± SEM of triplicate wells with each well measured in duplicate. n.d., not detected.
From these results relating to surface TGF-β1 expression on CD4+CD25+ T cells, together with those relating to the twin observations that anti–TGF-β abolishes CD4+CD25+ T cell-mediated immunosuppression on T cell proliferation and B cell Ig synthesis and that such suppression requires cell–cell contact, we conclude that CD4+CD25+ T cells mediate immunosuppression via the expression of surface-bound TGF-β1.
Discussion
In this study we provide data showing that stimulated CD4+CD25+ T cells are a preferential source of TGF-β1, producing both surface-bound and secreted TGF-β1 under certain conditions, and mainly surface-bound TGF-β1 under others. The production of secreted TGF-β1 occurs when these cells are stimulated with plate-bound anti-CD3 and is enhanced by signaling via CD28, IL-2R, and CTLA-4, i.e., “maximal” stimulation conditions that may occur in vivo only in the presence of an intense immune response. In contrast, the production of surface-bound TGF-β (associated with the secretion of modest amounts of TGF-β1) occurs in the presence of “ordinary” levels of stimulation by APCs. Nevertheless, these latter levels of stimulation support the TGF-β1–mediated suppressor function of CD4+CD25+ T cells with respect to T cell proliferation and B cell Ig production in vitro, as in both cases suppression can be observed under these conditions which is inhibited by anti–TGF-β. These findings, plus the observation that suppression requires cell–cell contact, lead us to the conclusion that CD4+CD25+ T cell–mediated immunosuppression is mediated by cell surface–bound TGF-β1. Whether CD4+CD25+ T cells that both express cell surface–bound TGF-β1 and secrete TGF-β1 mediating suppression occur in vivo await further studies. In either case, it now becomes clear that autoimmune mouse models characterized by CD4+CD25+ T cell depletion such as mice thymectomized in the neonatal period and nude mouse recipients of syngenic CD25+ cell–depleted splenocytes develop autoantibodies because of the absence of suppressor T cells producing TGF-β1 2 24. In addition, mice with a conditioned knockout of TGF-βR type II gene, in which receptor signaling is inactivated in a B cell–specific manner, develop anti-double stranded DNA autoantibodies because of the lack of ability to respond to suppressor T cells producing TGF-β 25.
Thornton and Shevach reported that after stimulation, CD4+CD25+ T cells exhibit more potent suppressor function without an additional stimulation through TCR in an antigen nonspecific manner 26. Our time course study of cell surface TGF-β1 is consistent with their findings, as the expression of cell surface TGF-β1 on CD4+CD25+ T cells reaches a peak on day 3 poststimulation, and maintains a similar level of TGF-β1 expression for at least 6 d after stimulation; thus, as previously stimulated CD4+CD25+ T cells already express cell surface TGF-β1, they do not have to be re-stimulated to mediate suppression. Furthermore, inasmuch as CD4+CD25+ T cells maintain high levels of cell surface TGF-β1 for quite a long time, it is understandable that previously stimulated CD4+CD25+ T cells possess stronger suppressor function than unstimulated cells. This follows from the fact that previously stimulated CD4+ CD25+ T cells can immediately initiate suppression whereas resting CD4+CD25+ T cells require some time after stimulation through TCR to express surface TGF-β1 and mediate suppression.
As alluded above, previous studies of CD4+CD25+ T cell suppressor function have provided data contrary to those presented here in that they suggest that CD4+CD25+ T cells do not produce substantial amount of TGF-β and suppression mediated by such cells is not mediated by TGF-β 2 10 11. The question therefore arises as to why the present studies are in disagreement with these previous studies. We believe that the answer lies in the fact that CD4+CD25+ T cells only produce high, easily detectable amount of TGF-β when maximally stimulated (by plate-bound anti-CD3, soluble anti-CD28, or IL-2 plus anti–CTLA-4). In contrast, when they are stimulated by soluble anti-CD3 and APCs, i.e., conditions usually used to measure suppressor activity, they secrete relatively low amounts of TGF-β, yet express surface-bound TGF-β. Given the fact that soluble TGF-β1 is not readily detectable in cultures under these latter conditions, and moreover, the suppression requires cell–cell contact, it was understandably concluded that suppression was not mediated by TGF-β1. This conclusion, however, is unwarranted given the fact that CD4+CD25+ T cells express surface-bound TGF-β and that suppression is abolished by anti–TGF-β.
A second and related question that also needs explanation is why anti–TGF-β Ab did not reverse CD4+CD25+ T cell–mediated suppression in previous studies 10 11. One possibility (one that we favor) relates to the biology of TGF-β–mediated suppressor function. It is known that TGF-β is produced in a latent (inactive) form comprised of the active molecule encased in LAP and must be converted to an active form, TGF-β1 unassociated with LAP, to express biological activity. Although the mechanism of activation of latent TGF-β is not yet fully understood, recent evidence suggests that it requires binding to one or another protein on the cell surface. For instance, it may interact with thrombospondin-1, one of the major activators of TGF-β1 27 and the complex thus formed then interacts with CD36 on macrophages 28 or possibly with CD47 on T cells 29; to form a complex that allow plasmin to strip off LAP from latent TGF-β and convert the latter to active TGF-β 28. Another possible binding molecule on the cell surface is αvβ6 integrin which interacts with LAP to facilitate a conformational change of the latter protein which allows the exposure of active TGF-β 30. Our demonstration that LAP as well as TGF-β1 exists on the cell surface suggests that TGF-β1 bound to the cell surface is present as latent TGF-β1 which is activated upon cell–cell contact to mediate CD4+CD25+ T cell suppression. In this situation, it is difficult to inhibit TGF-β1–mediated suppression with anti-TGF-β, as the latter must interact with TGF-β1 in the short period between its conversion to an active form and its interaction with a relevant TGF-βR at a relatively protected site on the cell surface. In the present situation this roadblock to the identification of TGF-β production as the inhibiting mechanism was overcome by the fact that high concentrations of high affinity anti–TGF-β Ab were used in the inhibition studies that could presumably act even at the cell surface to inhibit TGF- β–mediated suppression.
A final question relates to whether CD4+CD25+ T cell suppression is mediated by TGF-β alone or whether other mechanisms may also play a role under some circumstances. First, with regard to possible effects of a second suppressor factor, IL-10, we have shown here that while addition of anti–TGF-β Ab to cultures results in reversal of suppression, addition of anti–IL-10 or anti–IL-10R Ab does not. Despite these results, it is still possible that IL-10 contributes to the suppressive effect. This view comes from recent studies showing that even though TGF-β and not IL-10 is the proximal cause of negative regulation resulting from feeding antigen (i.e., induction of oral tolerance), nevertheless the presence of IL-10 is necessary to downregulate IL-12/IFN-γ production which would otherwise inhibit expansion/proliferation of TGF-β–producing cells 31. Thus, in the in vitro system studied here where high levels of IL-12/IFN-γ are not present, IL-10 seems irrelevant, but in in vivo situations where high levels of the IL-12/IFN-γ are present, it may be highly relevant.
The mechanism by which TGF-β1 is retained on the surface of CD4+CD25+ T cells and becomes activated is presently unknown. TGF-β1 found on the surface of macrophages is bound to thrombospondin-1, and the latter, in turn, interacts with a cell surface molecule, CD36 28. Thus, it is likely that TGF-β1 is retained on the surface of CD4+CD25+ T cells by binding to certain as yet unidentified surface molecules. Inasmuch as the surface-bound TGF-β was detected by Abs whose target epitopes reside in both active TGF-β and LAP, it is likely that surface TGF-β1–positive CD4+CD25+ T cells bind functionally inactive TGF-β1 still associated with LAP. Then, upon cell–cell contact with a potential target cell, cell surface LAP is stripped away and functionally active TGF-β1 becomes available for suppression. This would imply that a proteolytic mechanism associated with the CD4+CD25+ T cell or its target becomes activated at this point. Another possibility not mutually exclusive with the idea that cell surface TGF-β1 is present associated with LAP, is that TGF-β on the cell surface binds to a TGF-βR. If this is so, however, it is likely that the receptor does not transduce a TGF-β signal (such as TGF-βR type III), as TGF-β is demonstrably retained on the cell surface of quite a long time without affecting cell function.
Although TGF-β could be stained with three different Abs, chicken anti-TGF-β1, mouse anti-LAP mAb, and goat anti-LAP polyclonal Ab, we could not observe positive staining using anti–TGF-β mAb, 1D11, which was used for neutralization of TGF-β. 1D11 anti–TGF-β mAb was raised against bovine TGF-β2 and reacts with active form of TGF-β1, β2, and β3, but not with latent TGF-β 27. Thus, if cell surface TGF-β exists in a latent form, the epitope recognized by 1D11 may be hidden by LAP. If surface TGF-β binds to some TGF-βR, again the epitope will be masked by the interaction between TGF-β and its receptor.
CTLA-4 signaling significantly enhanced the proliferation of CD4+CD25+ T cells induced by anti-CD3 and anti-CD28. At first glance this seems paradoxical as CTLA-4 engagement also enhances the production of TGF-β, a proliferation inhibitor. Indeed, CD4+CD25+ T cells are unresponsive to the stimulation with soluble anti-CD3 and APCs, which is at least in part due to the expression of TGF-β, as proliferation was partially restored by anti–TGF-β Ab. It should be noted, however, that when these cells are stimulated with plate-bound anti-CD3 and soluble anti-CD28, they proliferate to the same degree as CD4+CD25− T cells, and addition of active rTGF-β1 to such culture does not suppress T cell proliferation significantly (data not shown). Thus, the sensitivity of T cell proliferation to TGF-β–mediated suppression is dependent on the condition of T cell stimulation. Finally, CD4+CD25− T cell proliferation induced by plate-bound anti-CD3 and soluble anti-CD28 is also resistant to TGF-β–mediated suppression, whereas low amount of TGF-β leads to dramatic suppression of T cell proliferation stimulated with soluble anti-CD3 and APCs. Taken with the observation that CD4+CD25− T cell proliferation stimulated with plate-bound anti-CD3 is not suppressed by CD4+CD25+ T cells 11, these data relating to TGF-β–mediated suppression are in line with our conclusion that TGF-β mediates suppression of CD4+CD25+ T cells.
In future studies it will be important to demonstrate that production of TGF-β by CD4+CD25+ suppressor T cells also explain the regulatory activity of these cells in vivo as well as in vitro. In studies alluded to above, Powrie et al. have shown that CD4+CD25+ T cells mediate suppression of colitis in the SCID transfer models of colitis and, in addition, this suppression requires the presence of TGF-β 4 5. The present studies suggest that these cells are, in fact the source of the TGF-β, but further in vivo studies will be necessary to prove this point.
Acknowledgments
The authors would like to thank Drs. I. Fuss, T. Usui, and F. Scheiffele for helpful advice and discussion. We gratefully acknowledge Dr. H. Sato for his valuable advice and support with membrane preparation and Ms. S. Barbieri for excellent technical assistance with FACS® sorting. We wish to thank Dr. C. Prussin for his help in the acquisition of 27232.11 Ab.
Footnotes
Abbreviations used in this paper: CTLA, cytotoxic T lymphocyte–associated antigen; LAP, latency associated protein.
References
- Sakaguchi S. Regulatory T cellskey controllers of immunologic self-tolerance. Cell. 2000;101:455–458. doi: 10.1016/s0092-8674(00)80856-9. [DOI] [PubMed] [Google Scholar]
- Asano M., Toda M., Sakaguchi N., Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F., Leach M.W., Mauze S., Caddle L.B., Coffman R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- Read S., Malmstrom V., Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F., Carlino J., Leach M.W., Mauze S., Coffman R.L. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asseman C., Mauze S., Leach M.W., Coffman R.L., Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Kuchroo V.K., Inobe J., Hafler D.A., Weiner H.L. Regulatory T cell clones induced by oral tolerancesuppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- Neurath M.F., Fuss I., Kelsall B.L., Presky D.H., Waegell W., Strober W. Experimental granulomatous colitis in mice is abrogated by induction of TGF-beta–mediated oral tolerance. J. Exp. Med. 1996;183:2605–2616. doi: 10.1084/jem.183.6.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groux H., O'Garra A., Bigler M., Rouleau M., Antonenko S., de Vries J.E., Roncarolo M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Kuniyasu Y., Toda M., Sakaguchi N., Itoh M., Iwata M., Shimizu J., Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cellsinduction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- Thornton A.M., Shevach E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitani A., Fuss I.J., Nakamura K., Schwartz O.M., Usui T., Strober W. Treatment of experimental (trinitrobenzene sulfonic acid) colitis by intranasal administration of transforming growth factor (TGF)-beta1 plasmidTGF-beta1–mediated suppression of T helper cell type 1 response occurs by interleukin (IL)-10 induction and IL-12 receptor beta2 chain downregulation. J. Exp. Med. 2000;192:41–52. doi: 10.1084/jem.192.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Tagami T., Yamazaki S., Uede T., Shimizu J., Sakaguchi N., Mak T.W., Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J. Exp. Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitani A., Chua K., Nakamura K., Strober W. Activated self-MHC-reactive T cells have the cytokine phenotype of Th3/T regulatory cell 1 T cells. J. Immunol. 2000;165:691–702. doi: 10.4049/jimmunol.165.2.691. [DOI] [PubMed] [Google Scholar]
- Noro N., Adachi M., Yasuda K., Masuda T., Yodoi J. Murine IgA binding factors (IgA-BF) suppressing IgA productioncharacterization and target specificity of IgA-BF. J. Immunol. 1986;136:2910–2916. [PubMed] [Google Scholar]
- Cohen J.I., Seidel K.E. Varicella-Zoster virus open reading frame 1 encodes a membrane protein that is dispensable for growth of VZV in vitro . Virology. 1995;206:835–842. doi: 10.1006/viro.1995.1006. [DOI] [PubMed] [Google Scholar]
- Salomon B., Lenschow D.J., Rhee L., Ashourian N., Singh B., Sharpe A., Bluestone J.A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Papiernik M., de Moraes M.L., Pontoux C., Vasseur F., Penit C. Regulatory CD4 T cellsexpression of IL-2R alpha chain, resistance to clonal deletion and IL-2 dependency. Int. Immunol. 1998;10:371–378. doi: 10.1093/intimm/10.4.371. [DOI] [PubMed] [Google Scholar]
- Chen W., Jin W., Wahl S.M. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor beta (TGF-beta) production by murine CD4+ T cells. J. Exp. Med. 1998;188:1849–1857. doi: 10.1084/jem.188.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre M.L., Noel P.J., Eisfelder B.J., Chuang E., Clark M.R., Reiner S.L., Thompson C.B. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- Krummel M.F., Allison J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelletti J.F., Katz D.H. IL-10 stimulates murine antigen-driven antibody responses in vitro by regulating helper cell subset participation. Cell. Immunol. 1996;167:86–98. doi: 10.1006/cimm.1996.0011. [DOI] [PubMed] [Google Scholar]
- Annacker O., Pimenta-Araujo R., Burlen-Defranoux O., Barbosa T.C., Cumano A., Bandeira A. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J. Immunol. 2001;166:3008–3018. doi: 10.4049/jimmunol.166.5.3008. [DOI] [PubMed] [Google Scholar]
- Kuniyasu Y., Takahashi T., Itoh M., Shimizu J., Toda G., Sakaguchi S. Naturally anergic and suppressive CD25+CD4+ T cells as a functionally and phenotypically distinct immunoregulatory T cell subpopulation. Int. Immunol. 2000;12:1145–1155. doi: 10.1093/intimm/12.8.1145. [DOI] [PubMed] [Google Scholar]
- Cazac B.B., Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. 2000;13:443–451. doi: 10.1016/s1074-7613(00)00044-3. [DOI] [PubMed] [Google Scholar]
- Thornton A.M., Shevach E.M. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- Crawford S.E., Stellmach V., Murphy-Ullrich J.E., Ribeiro S.M., Lawler J., Hynes R.O., Boivin G.P., Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- Yehualaeshet T., O'Connor R., Green-Johnson J., Mai S., Silverstein R., Murphy-Ullrich J.E., Khalil N. Activation of rat alveolar macrophage-derived latent transforming growth factor beta-1 by plasmin requires interaction with thrombospondin-1 and its cell surface receptor, CD36. Am. J. Pathol. 1999;155:841–851. doi: 10.1016/s0002-9440(10)65183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo A.N., Mugge L.O., Klimiuk P.A., Weyand C.M., Goronzy J.J. Central role of thrombospondin-1 in the activation and clonal expansion of inflammatory T cells. J. Immunol. 2000;164:2947–2954. doi: 10.4049/jimmunol.164.6.2947. [DOI] [PubMed] [Google Scholar]
- Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A. The integrin alpha v beta 6 binds and activates latent TGF beta 1a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Fuss I.J., Boirivant M., Lacy B., Strober W. The role of regulatory cytokines (TGF-β and IL-10) in the suppression of experimental murine TNBS-colitis. Gastroenterology. 2000;118:A356. [Google Scholar]