Two Distinct Vps34 Phosphatidylinositol 3–Kinase Complexes Function in Autophagy and Carboxypeptidase Y Sorting inSaccharomyces cerevisiae (original) (raw)
Abstract
Vps30p/Apg6p is required for both autophagy and sorting of carboxypeptidase Y (CPY). Although Vps30p is known to interact with Apg14p, its precise role remains unclear. We found that two proteins copurify with Vps30p. They were identified by mass spectrometry to be Vps38p and Vps34p, a phosphatidylinositol (PtdIns) 3–kinase. Vps34p, Vps38p, Apg14p, and Vps15p, an activator of Vps34p, were coimmunoprecipitated with Vps30p. These results indicate that Vps30p functions as a subunit of a Vps34 PtdIns 3–kinase complex(es). Phenotypic analyses indicated that Apg14p and Vps38p are each required for autophagy and CPY sorting, respectively, whereas Vps30p, Vps34p, and Vps15p are required for both processes. Coimmunoprecipitation using anti-Apg14p and anti-Vps38p antibodies and pull-down experiments showed that two distinct Vps34 PtdIns 3–kinase complexes exist: one, containing Vps15p, Vps30p, and Apg14p, functions in autophagy and the other containing Vps15p, Vps30p, and Vps38p functions in CPY sorting. The vps34 and vps15 mutants displayed additional phenotypes such as defects in transport of proteinase A and proteinase B, implying the existence of another PtdIns 3–kinase complex(es). We propose that multiple Vps34p–Vps15p complexes associated with specific regulatory proteins might fulfill their membrane trafficking events at different sites.
Keywords: Phosphatidylinositol 3–kinase, autophagy, Vps34p, Vps30p/Apg6p, CPY sorting
Introduction
The yeast vacuole, equivalent to the lysosome of mammalian cells, is the major site of macromolecular turnover, which is carried out by various kinds of hydrolases. The transport pathways of hydrolases to the vacuole/lysosome are similar in both yeast and mammals. In yeast, newly synthesized vacuolar proteins translocate into the ER and subsequently traverse the Golgi complex. They are sorted away from secreted proteins in a late-Golgi compartment, which appears to be analogous to the mammalian TGN. Soluble vacuolar hydrolases such as carboxypeptidase Y (CPY) pass through the endosome/prevacuolar compartment (PVC) en route to the vacuole. Genetic screens that select for yeast strains that secrete CPY have identified more than 50 VPS (vacuolar protein-sorting) genes required for the correct targeting of CPY from the late-Golgi to the vacuole (for review see Horazdovsky et al. 1995).
The demonstration that one of the Vps proteins, Vps34p, is a phosphatidylinositol (PtdIns) 3–kinase (Schu et al. 1993) has focused attention on the involvement of lipid kinases in vesicular transport. Strains in which the VPS34 gene has been deleted are temperature-sensitive for growth at 37°C and have defects in the sorting of soluble vacuolar hydrolases (Robinson et al. 1988; Herman and Emr 1990; Herman et al. 1991a,Herman et al. 1991b). The vps15 mutants showed similar phenotypes to the vps34 mutants, suggesting that Vps15p acts at the same step of vacuolar protein transport. Subsequent biochemical analyses revealed that Vps15p is a serine/threonine kinase that interacts with Vps34p (Stack et al. 1993). Vps15p protein kinase activity is required for the Vps15p–Vps34p interaction and the PtdIns 3–kinase activity of Vps34p (Stack et al. 1993, Stack et al. 1995). Recently, it was demonstrated that the involvement of PtdIns 3–kinases in protein transport also extends to mammalian systems. The phosphoinositide 3–kinase inhibitors, wortmannin and LY294002, cause mammalian lysosomal proteins to be mistargeted (Brown et al. 1995; Davidson 1995), probably due to inhibition of a mammalian Vps34p homologue. The human homologue of Vps34p has been shown to associate with the Vps15p homologue, p150 (Volinia et al. 1995; Panaretou et al. 1997).
The requirement of PtdIns 3–kinases in membrane trafficking is not restricted to protein transport from the late-Golgi/TGN to the vacuole/lysosome. Recent data indicate that PtdIns 3–kinases are also required for autophagy in both yeast Hansenula polymorpha and human cells (Kiel et al. 1999; Petiot et al. 2000). Autophagy is a major eukaryotic process by which bulk cytoplasmic components are degraded in the vacuole/lysosome (for review see Dunn 1994). In response to starvation, double membrane structures known as autophagosomes nonselectively envelop a fraction of the cytoplasm, including its resident organelles, and target it to the vacuole/lysosome where the contents are degraded (Baba et al. 1994). In yeast, several autophagy-defective mutants (apg and aut) have been isolated (Tsukada and Ohsumi 1993; Thumm et al. 1994). Most of the responsible genes are novel and seem to function specifically in autophagy and the related cytoplasm-to-vacuole targeting (Cvt) pathway, which mediates transport of newly synthesized aminopeptidase I (API) to the vacuole (Harding et al. 1996). However, only APG6 is allelic to one of the VPS genes, VPS30, and thus is also required for CPY sorting (Seaman et al. 1997; Kametaka et al. 1998). Vps30p interacts with another Apg protein, Apg14p (Kametaka et al. 1998). However, curiously, disruption of APG14 does not affect CPY sorting (Kametaka et al. 1998). It has been proposed that Vps30p has two distinct functions in autophagy and CPY sorting and that only autophagy is Apg14p dependent.
In this study, we show that Vps34p forms at least two multisubunit PtdIns 3–kinase complexes: both contain Vps15p and Vps30p, whereas Apg14p and Vps38p are specific to each. Phenotypic analyses indicated that the complex containing Apg14p functions in autophagy and that containing Vps38p functions in CPY sorting. Moreover, phenotypic observation implied the existence of other PtdIns 3–kinase complexes. From these results, we propose that Vps34 PtdIns 3–kinase forms multiple complexes, each containing specific regulatory subunits that define the membrane trafficking pathway in which that complex is involved.
Materials and Methods
Yeast Strains and Media
Saccharomyces cerevisiae strains used are listed in Table . Yeast strains constructed in this study were derived from KA311A (Irie et al. 1993), YPH499 (Sikorski and Hieter 1989), or SEY6210 (Robinson et al. 1988). Construction of Δ_vps30::LEU2_, Δ_apg14::LEU2_, Δ_vps34::TRP1_, Δ_pep4::URA3_, and Δ_ypt7::LEU2_ was performed as described previously (Kametaka et al. 1998; Herman and Emr 1990; Kirisako et al. 1999; Noda et al. 2000). Δ_vps15::TRP1_ and Δ_vps15::HIS3_ cells were constructed to replace the 1.8-kb NcoI-StuI region in the VPS15 gene with the TRP1 marker and with the HIS3 marker, respectively. Δ_vps38::ADE2_ cells were constructed to replace the 20-base BamHI-PstI region in the VPS38 gene with the ADE2 marker. Cells were grown either in YPD medium (1% yeast extract, 2% peptone, and 2% glucose) or in synthetic complete (SC) medium containing nutritional supplements. For nitrogen starvation, SD(-N) medium (0.17% yeast nitrogen base with 2% glucose without amino acid and ammonium sulfate) was used.
Table 1.
Strains Used in This Study
| Strain | Genotype | Source |
|---|---|---|
| KA311A | MATa ura3 leu2 his3 trp1 | Irie et al. 1993 |
| AKY73 | KA311A; pep4_Δ::URA3_ | This study |
| AKY76 | KA311A; Δ_vps30::LEU2 pep4_Δ_::URA3_ | This study |
| YPH499 | _MATa ura3-52 lys2-801 ade2-101 trp1_Δ_63 his3-_Δ_200 leu2-Δ_1 | Sikorski and Hieter 1989 |
| TN125 | YPH499; Δ_pho8::PHO8_Δ_60_ | Noda and Ohsumi 1998 |
| AKY13 | TN125; Δ_apg14::LEU2_ | This study |
| AKY15 | TN125; Δ_vps30::LEU2_ | This study |
| AKY106 | YPH499; pep4_Δ::URA3_ | This study |
| AKY109 | N125; Δ_vps34::TRP1_ | This study |
| AKY110 | AKY106; Δ_vps34::TRP1_ | This study |
| AKY111 | AKY106; Δ_vps30::LEU2_ | This study |
| AKY112 | AKY106; Δ_apg14::LEU2_ | This study |
| AKY113 | AKY106; Δ_vps38::ADE2_ | This study |
| AKY114 | TN125; Δ_vps38::ADE2_ | This study |
| AKY115 | TN125; Δ_vps15::HIS3_ | This study |
| AKY116 | AKY106; Δ_vps15::HIS3_ | This study |
| AKY126 | AKY106; Δ_vps15::TRP1_ | This study |
| SEY6210 | _MAT_α _leu2-3,112 ura3-52 his3-_Δ_200 trp1_Δ_901 lys2-801 suc2-Δ_9 | Robinson et al. 1988 |
| KVY4 | SEY6210_;_ Δ_ypt7::LEU2_ | This study |
| AKY131 | SEY6210; Δ_vps34::TRP1_ | This study |
Plasmid Construction
pKHR1 is an Escherichia coli cloning vector constructed to produce fusion proteins containing an NH2-terminal His6, Myc epitope (EEQKLISEEDLLRKR) with a thrombin cleavage site (LVPRGS). The His6–Myc region was amplified from pTYE007 (Yoshihisa and Ito 1996) using the following primers: 5′-GGGAATTCATGAGAGGATCGCACCATCACCATCACCAC-3′ and 5′-GGGAATTCGGGGATCCACGCGGAACCAGACGTTTGCGCAGCAGGTCCTCTTCG-3′. The resulting fragments were digested with EcoRI and ligated into the EcoRI site of pKK223-3 (Amersham Pharmacia Biotech) to generate pKHR1.
To create the His6–Myc-tagged fusion proteins, the VPS30 and APG14 genes were cloned into pKHR1 to produce pKHR2 and pKHR3, respectively. To express His6–Myc–Vps30p in yeast, His6–Myc–VPS30 from pKHR2 was cloned into pRS424 (Christianson et al. 1992) to generate pKHR25. To express His6–Myc–Apg14p in yeast, His6–Myc–APG14 from pKHR3 was cloned into pAUR112 (TaKaRa) and pRS313 (Sikorski and Hieter 1989) to generate pKHR69 and pKHR75, respectively.
To create the COOH terminally triple hemagglutinin (HA)–tagged Vps38p expression construct, a vector plasmid that expresses the 3xHA fusion protein, was first constructed. The 3xHA epitope was amplified from pTK110 (Kirisako et al. 1999), using the following primers: 5′-TTTTCGACTAGTTACCCATACGATGTTCCTGAC-3′ and 5′-AAACGAGCTCTTAAGCGTAATCTGGAACGTCATATGG-3′. This PCR fragment was digested with SpeI and SacI and subcloned into pRS313 to generate pKHR45. Then, VPS38 was subcloned into pKHR45 to create Vps38p-3xHA construct pKHR65.
pKHR54 (VPS34) and pKHR55 (VPS15) contained a 2.9-kb Psp1406I-KpnI VPS34 genomic fragment and a 4.8-kb ScaI-SnaBI VPS15 genomic fragment in pRS316, respectively. pKHR60 (vps34-N736K) and pKHR59 (vps15-E200R) were constructed as described previously (Herman et al. 1991a; Schu et al. 1993).
Purification of the His6–Myc–Vps30p Complex(es)
A 4-liter culture of AKY76 cells harboring pKHR25 or pRS424 was converted to spheroplasts and lysed by osmotic shock and sonication in buffer A (50 mM Hepes-NaOH, pH 8.0, 200 mM sorbitol, 150 mM NaCl, 1 mM PMSF, 10 mM 2-mercaptoethanol [2-ME], protease inhibitor mixture [Complete, EDTA-free; Roche]). After removal of cell debris by centrifugation at 1,500 g for 5 min, the supernatant was solubilized by addition of five volumes of buffer B (50 mM Hepes-NaOH, pH 8.0, 1% Triton X-100, 150 mM NaCl, 1 mM PMSF, 10 mM 2-ME). Samples were centrifuged at 100,000 g for 30 min, and the supernatant was applied to a Ni-NTA agarose column (5 ml) equilibrated with buffer B. The column was washed with 20 ml buffer B and 60 ml buffer C (50 mM Hepes-NaOH, pH 8.0, 0.1% Triton X-100, 150 mM NaCl, 25 mM imidazole, 10 mM 2-ME) and eluted with 25 ml buffer D (50 mM Hepes-NaOH, pH 8.0, 0.1% Triton X-100, 150 mM NaCl, 10% glycerol, 250 mM imidazole, 10 mM 2-ME). The eluates were then incubated with immobilized affinity-purified antibodies to Vps30p. The column was prepared by coupling of Vps30p antibodies with protein A–Sepharose CL-4B beads, using dimethylpimelimidate as described previously (Ungermann et al. 1998). The resins were washed with 30 ml buffer E (10 mM Hepes-NaOH, pH 8.0, 0.5% Triton X-100, 150 mM NaCl, 10% glycerol, 0.1 mM EDTA, 10 mM 2-ME). Bound proteins were eluted with 10 ml 100 mM glycine-HCl, pH 2.5, precipitated by 5% TCA, washed with ice-cold acetone, and suspended in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 5% 2-ME, a trace of bromophenol blue). They were separated by SDS-PAGE, stained by Coomassie brilliant blue, and excised from the gel. The protein samples were digested with trypsin in the gel matrix. Extracted peptide mixtures were analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proteins were identified by comparing their peptide mass maps to the database.
Coimmunoprecipitation
Spheroplasts prepared from [35S]methionine/cysteine–labeled cells were lysed by extrusion through a polycarbonate filter with 3 μm pores as described previously (Vida and Gerhardt 1999) in buffer A and solubilized in buffer F (100 mM Hepes-NaOH, pH 8.0, 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 1 mM PMSF). Solubilized samples were then incubated with antibodies and protein A–Sepharose beads at 4°C for 2 h. The beads were washed with buffer F three times. Bound proteins were eluted with SDS sample buffer and separated by SDS-PAGE, followed by detection by autoradiography. In the case of unlabeled cells, spheroplasts were lysed by osmotic shock followed by sonication in buffer A. After removal of cell debris by centrifugation at 1,500 g for 5 min, the supernatant was solubilized with 1% Triton X-100. Samples were then incubated with the protein A–coupled antibodies and incubated at 4°C for 2 h. The beads were washed with buffer G (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% Triton X-100, 10 mM 2-ME). Bound material was eluted with 100 mM Glycine-HCl, pH 2.5, precipitated by 5% TCA, washed with ice-cold acetone, and suspended in SDS sample buffer. Proteins were separated by SDS-PAGE and transferred to nitrocellulose for immunoblotting. Anti-Vps34p antiserum was raised against His6–Myc–Vps34p fusion protein containing 345 amino acids (97–441) of Vps34p (His6–Myc–Vps34p [97–441]). Anti-Vps15p antiserum was raised against His6–Myc–Vps15p (222–768) or a mixture of His6–Myc–Vps15p (222–768) and His6–Myc–Vps15p (792–1189). Anti-Vps38p antiserum was raised against His6–Myc–Vps38p (53–293) or His6–Myc–Vps38p (53–439). Anti-Apg14p antiserum was raised against His6–Myc–Apg14p (2–62 and 271–344). Anti-Vps30p antiserum was raised against recombinant full-length Vps30p expressed as a glutathione _S_-transferase fusion protein.
Pulse–Chase, Immunoprecipitation, and Immunoblotting
Secretion of newly synthesized CPY was examined by pulse–chase and immunoprecipitation experiments as described previously (Cooper and Stevens 1996). Immunoblotting was carried out as described previously (Shintani et al. 1999). Antiserum against API was kindly provided by Dr. Daniel J. Klionsky (Michigan University, Ann Arbor, MI).
Protection Assay
Cells grown in YPD were transferred to SD(-N) for 4.5 h and converted to spheroplasts as previously described (Noda et al. 2000). The spheroplasts were suspended in a lysis buffer (25 mM Pipes-KOH, pH 6.8, 200 mM sorbitol). The lysate was centrifuged for 5 min at 500 g. The supernatant was spun at 13,000 g for 15 min, and the pellet was resuspended in lysis buffer. The samples were then treated with 10 mg/ml of proteinase K with or without 1% Triton X-100 on ice for 30 min.
Subcellular Fractionation
Yeast spheroplasts were lysed in buffer H (50 mM Hepes-NaOH, pH 7.5, 200 mM sorbitol, 150 mM NaCl, 1 mM PMSF, 10 mM 2-ME) by extrusion through a polycarbonate filter with 3-μm pores (Vida and Gerhardt 1999). The filter effluent was centrifuged at 500 g for 5 min to remove cell debris. The supernatant (total) was subsequently centrifuged at 13,000 g for 15 min to generate a low speed pellet (LSP) and low speed supernatant, which was further centrifuged at 100,000 g for 1 h to generate a high speed pellet (HSP) and high speed supernatant (HSS).
Pull-Down Assay
Yeast spheroplasts were lysed in buffer I (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM PMSF, 10 mM 2-ME, protease inhibitor mixture [Complete, EDTA-free]) by sonication. After removal of cell debris by centrifugation at 1,500 g for 5 min, the supernatant was solubilized with 1% Triton X-100. Unsolubilized material was removed by centrifugation (100,000 g, 30 min), and soluble samples were loaded on a Ni-NTA agarose column. The column was washed with buffer J (50 mM Tris-HCl, pH 8.0, 0.1% Triton X-100, 150 mM NaCl, 20 mM imidazole, 10 mM 2-ME) and eluted with buffer K (50 mM Tris-HCl, pH 8.0, 0.1% Triton X-100, 150 mM NaCl, 250 mM imidazole, 10 mM 2-ME).
Alkaline Phosphatase (ALP) Assays and PtdIns 3–kinase Assays
To measure autophagic activity, ALP assays were performed as described previously (Noda and Ohsumi 1998). PtdIns 3–kinase assays were performed as described previously (Stack et al. 1995).
Results
Vps30p Interacts with Vps15p, Vps34p, and Vps38p
Although Δ_vps30_ cells are defective in both autophagy and CPY sorting, Δ_apg14_ cells only have a defect in autophagy (Kametaka et al. 1998). Moreover, overproduction of Apg14p partially suppresses the autophagic defect caused by the apg6-1 mutation, which resulted in an ∼50% COOH-terminal truncation of Vps30p but not the CPY transport defect (Kametaka et al. 1998). These results prompted us to hypothesize that Vps30p/Apg6p may compose large protein complexes functioning in different processes. To test this, we first examined whether Vps30p interacts with proteins other than Apg14p. Wild-type or Δ_vps30_ cells were labeled with [35S]methionine/cysteine. Total lysates prepared from the wild-type or Δ_vps30_ cells were solubilized with Triton X-100 and subjected to immunoprecipitation using anti-Vps30p antibodies. In the immunoprecipitates from wild-type cells, many protein bands together with Vps30p were detected (Fig. 1 A, lane 1). Most of them were nonspecific backgrounds because they were also present in the immunoprecipitates from Δ_vps30_ cells (Fig. 1 A, lane 2). However, three bands, termed p160, p90, and p50, were present only in the immunoprecipitates from wild-type cells, indicating that these proteins specifically interact with Vps30p. To identify them, we constructed pKHR25, which carried His6_–_Myc_–_VPS30, a fusion gene encoding Vps30p with attached NH2-terminal His6 and Myc tag sequences. pKHR25 is a multicopy plasmid (2 μm) that expresses His6–Myc–Vps30p at levels 30-fold higher than chromosomally expressed Vps30p. Total lysates prepared from cells of AKY76 carrying pKHR25 were solubilized with Triton X-100, and the detergent extracts were loaded on a Ni-NTA agarose column (Fig. 1 B, lanes 1–4). The elutes were further purified by incubation with an immunoaffinity column prepared by covalently attaching anti-Vps30p antibodies to protein A–Sepharose beads (Fig. 1 B, lanes 5 and 6). Several proteins were eluted from the column along with His6–Myc–Vps30p (Fig. 1 B, lane 6). To discriminate bands that specifically interacted with His6–Myc–Vps30p from nonspecific background binding, the same purification procedure was repeated using the lysates of AKY76 carrying a plasmid that did not express His6–Myc–Vps30p. p90 and p50 but not p160 were specifically present in the purified fraction from His6–Myc–Vps30p-expressing cells (Fig. 1 C). Bands of p90 and p50 were excised from the gel, and the protein samples were digested with trypsin in the gel matrix. Extracted peptide mixtures were analyzed by matrix-assisted laser desorption/ionization mass spectrometry. The peptide mass maps were used to query a comprehensive sequence database for unambiguous protein identification. The results indicated that p90 and p50 were Vps34p and Vps38p, respectively. Vps34p is a PtdIns 3–kinase required for proper sorting of a subset of vacuolar proteins (Robinson et al. 1988; Herman and Emr 1990). Vps38p is involved in the targeting of CPY and mutant Pma1p to the vacuole (Luo and Chang 1997), although its precise role in these processes is unclear. To confirm that Vps34p and Vps38p indeed interact with Vps30p, coimmunoprecipitation experiments were performed using anti-Vps30p antibodies. Immunoblotting showed that Vps30p, Vps34p, and Vps38p were present in immunoprecipitates prepared from wild-type yeast, (Fig. 1 D, lane 1) but not in those from the Δ_vps30_ strain (Fig. 1 D, lane 2), indicating that Vps34p and Vps38p were precipitated specifically through the interaction with Vps30p. As reported previously, Apg14p also coimmunoprecipitated with Vps30p (Fig. 1 D, lane 1). Vps15p, a serine/threonine kinase, has been shown to interact with Vps34p (Stack et al. 1993). Therefore, Vps15p was the best candidate for p160. We next examined whether Vps15p existed in the immunoprecipitates obtained using anti-Vps30p antibodies. Immunoblotting using anti-Vps15p antibodies showed that Vps15p was also present (Fig. 1 D, lane 1). These results indicate that Vps30p form a complex(es) with Apg14p, Vps15p, Vps34p, and Vps38p. Until now, the role of Vps30p in autophagy and CPY sorting has remained unclear. We demonstrate here that Vps30p functions as a subunit of a Vps34 PtdIns 3–kinase complex(es). The reason why we failed to detect Apg14p in Fig. 1A and Fig. C, was probably due to its low abundance (see below). We could not detect Vps15p in Fig. 1 C. Vps15p might be concealed by nonspecific backgrounds because p160 was very close to nonspecific backgrounds in Fig. 1 A. Alternatively, Vps15p was degraded during the purification procedure due to its instability (see Discussion).
Figure 1.

Purification of Vps30p complexes. (A) AKY73 (wild type; lane 1) and AKY76 (Δ_vps30_; lane 2) cells grown in SC medium lacking methionine at 30°C were labeled with Express™ [35S]methionine/cysteine protein labeling mix (NEN Life Science Products) for 1 h. The labeled cells were converted to spheroplasts and lysed by extrusion through a polycarbonate filter with 3 μm pores. Thus, obtained lysates were solubilized with Triton X-100 and incubated with anti-Vps30p antibodies and protein A–Sepharose beads at 4°C for 2 h. Bound proteins were eluted, separated by SDS-PAGE, and detected by autoradiography using a PhosphorImager BAS2000 (Fuji Film). (B) Protein profiles during the purification of the Vps30p complexes. Total lysates prepared from AKY76 cells harboring pKHR25 (His6–Myc–VPS30; 2 μm) were solubilized with Triton X-100 and subjected to Ni-NTA agarose chromatography (load, lane 1; flow-through, lane 2). Proteins bound to Ni-NTA agarose were washed (lane 3) and eluted with 250 mM imidazole (lane 4). The eluates were then incubated with protein A–immobilized anti-Vps30p antibodies (flow-through, lane 5). The column was washed, and retained proteins were eluted with 100 mM glycine-HCl, pH 2.5 (lane 6; and C, lane 1). Proteins were separated by SDS-PAGE and visualized by Coomassie brilliant blue staining. (C) For control, a purification procedure as described in B was applied to lysates prepared from AKY76 cells harboring an empty pRS424 plasmid (lane 2). Proteins were analyzed by SDS-PAGE and visualized by silver staining. (D) Cells of AKY106 (wild type; lane 1) and AKY111 (Δ_vps30_; lane 2) were grown in YPD at 28°C. Total lysates were solubilized with Triton X-100 and incubated with protein A–immobilized anti-Vps30p antibodies. Retained proteins were eluted, separated by SDS-PAGE, and detected by immunoblotting with anti-Vps30p, anti-Vps34p, anti-Vps38p, anti-Apg14p, and anti-Vps15p antibodies.
Vps30p, Apg14p, and Vps38p Are Not Essential for Activation of Vps34p
Vps15 serine/threonine kinase is known to be essential for activation of Vps34p (Stack et al. 1993). One possible role of Vps30p, Apg14p, or Vps38p is a coactivator of Vps34p. To test this, total lysates prepared from each of the deletion strains were assayed for PtdIns 3–kinase activity by incubating with PtdIns and [γ-32P]ATP. Lipids were extracted and separated by TLC on Silica gel 60 plates (Walsh et al. 1991). Wild-type cells produced both PtdIns 4–phosphate, which was synthesized by PtdIns 4–kinases such as Stt4p and Pik1p, and PtdIns 3–phosphate [PtdIns(3)P] (Fig. 2, lane 1). As shown previously (Schu et al. 1993; Stack et al. 1993), Δ_vps34_ cells have no PtdIns 3–kinase activity at all (Fig. 2, lane 5), and Δ_vps15_ cells exhibit a very low but detectable level of PtdIns 3–kinase activity (Fig. 2, lane 6). Δ_vps30_ and Δ_vps38_ cells showed only a slight decrease in PtdIns 3–kinase activity (∼80% of wild-type cells) (Fig. 2, lanes 2 and 4). Δ_apg14_ cells exhibited an equivalent level of PtdIns 3–kinase activity to wild-type cells (Fig. 2, lane 3). These results indicate that Vps30p, Apg14p, and Vps38p are not essential for the PtdIns 3–kinase activity of Vps34p.
Figure 2.
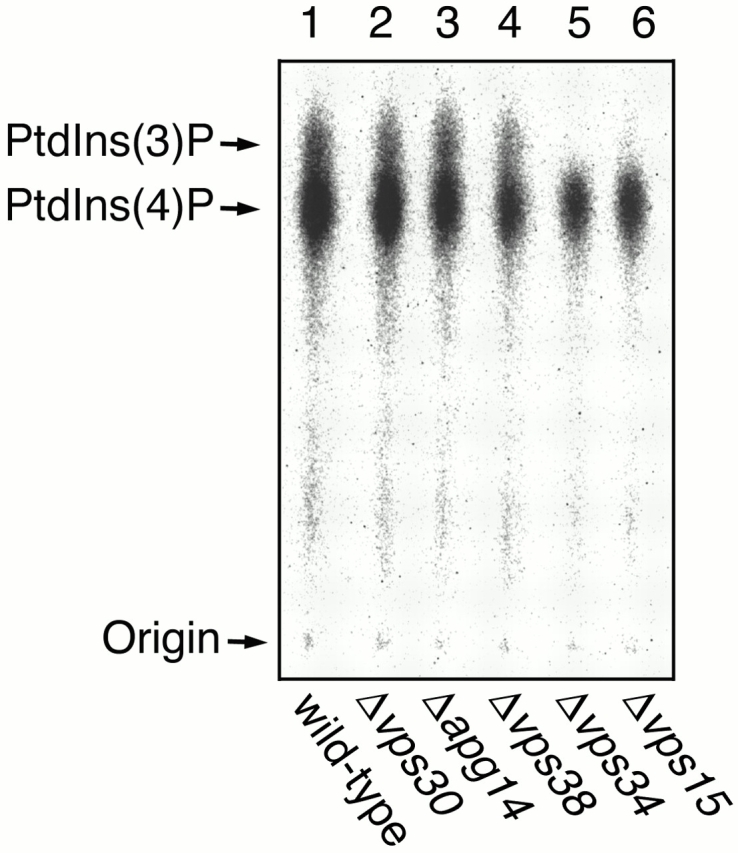
PtdIns 3–kinase activity in mutants lacking a component of Vps30p complexes. TN125 (wild type; lane 1), AKY15 (Δ_vps30_; lane 2), AKY13 (Δ_apg14_; lane 3), AKY114 (Δ_vps38_; lane 4), AKY109 (Δ_vps34_; lane 5), and AKY115 (Δ_vps15_; lane 6) cells were grown in YPD medium at 28°C. Total lysates were incubated with soybean PtdIns, [γ-32P]ATP, and 60 μM cold ATP in buffer L (20 mM Hepes-NaOH, pH 7.5, 10 mM MgCl2) for 5 min at 25°C. Lipids were extracted using chloroform–methanol, and samples were separated on Silica gel 60 TLC plates (Merck) with the borate system (Walsh et al. 1991) and detected by autoradiography using BAS2000.
Deletion Phenotypes Can Be Classified into Four Classes
We examined the effects of disruption of either VPS30, APG14, VPS38, VPS34, or VPS15 on several vacuolar trafficking pathways, including transport of CPY, proteinase A (PrA), proteinase B (PrB) and API, and autophagy. Transports of CPY, PrA, and PrB are initiated at the ER membrane, where they are translocated into lumen of the ER, and delivered by vesicular transport to the vacuole via the Golgi apparatus. The transport of API is completely different from that of CPY, PrA, and PrB. API is synthesized as a proform (pAPI) in the cytoplasm and targets directly to the vacuole via the Cvt pathway, where it is processed to the mature form (mAPI). The Cvt pathway uses a similar mechanism as the autophagic pathway, and most autophagy mutants are defective in the Cvt pathway (Harding et al. 1996; Scott et al. 1996; Baba et al. 1997). First, we examined transport of CPY by pulse–chase and immunoprecipitation experiments. Cells were pulse labeled with [35S]methionine/cysteine for 15 min and chased for 30 min at 28°C. The cells were then converted to spheroplasts and separated into intracellular (I) and extracellular (E) fractions, from which CPY was immunoprecipitated. In wild-type and Δ_apg14_ cells, >95% of the newly synthesized CPY was present as a mature form (mCPY) in an intracellular fraction (Fig. 3 A, lanes 1 and 3). Δ_vps30_, Δ_vps38_, Δ_vps34_, and Δ_vps15_ cells missorted and secreted virtually all CPY as the Golgi-modified p2 form (Fig. 3 A, lanes 6, 8, 10, and 12). These results indicate that all Vps proteins examined (Vps15p, Vps34p, Vps38p, and Vps30p/Apg6p), but not Apg14p, are required for proper sorting of CPY, as reported previously (Robinson et al. 1988; Herman and Emr 1990; Herman et al. 1991a; Luo and Chang 1997; Seaman et al. 1997; Kametaka et al. 1998). Transport of PrA, PrB, and API was examined by immunoblotting. Δ_vps15_ and Δ_vps34_ cells accumulated Golgi forms of PrA (pPrA) and PrB (pPrB) (Fig. 3B and Fig. C, lanes 4 and 5), as shown previously (Robinson et al. 1988), indicating that sorting of PrA and PrB is severely impaired in these cells. Alternatively, in Δ_vps38_ and Δ_vps30_ cells, most PrA and PrB were found as mature forms (mPrA and mPrB), although very low levels of precursor forms were detected as well (Fig. 3B and Fig. C, lanes 2 and 6). Δ_apg14_ cells exhibited normal sorting of PrA and PrB (Fig. 3B and Fig. C, lane 3). Transport of API was completely inhibited in Δ_vps30_, Δ_apg14_, Δ_vps34_, and Δ_vps15_ cells (Fig. 3 D, lanes 2–5), whereas Δ_vps38_ cells showed normal targeting of API (Fig. 3 D, lane 6). We next examined autophagy using an ALP assay (Noda et al. 1995), which monitors autophagy-dependent processing of ALP Pho8Δ60 (Noda et al. 1995). In wild-type cells, the ALP activity increased in response to starvation, whereas in Δ_vps30_, Δ_apg14_, Δ_vps34_, and Δ_vps15_ cells its elevation was severely inhibited (Fig. 3 E). The ALP activity of Δ_vps38_ cells was ∼70% of the activity of wild-type cells. These results indicate that Δ_vps30_, Δ_apg14_, Δ_vps34_, and Δ_vps15_ cells are defective both in the Cvt pathway and in the autophagy pathway, whereas in Δ_vps38_ cells both pathways are nearly intact. Table summarizes these transport activities in the mutant cells. We can thus classify the mutants into 4 classes. Class I, which includes Δ_vps15_ and Δ_vps34_, display the most severe phenotype: all transport of vacuolar proteins examined is inhibited, and their growth is slow at 28°C and arrested at 37°C. Δ_vps30_ (class II) is defective in both autophagy/Cvt and CPY sorting. However, Δ_apg14_ (class III) and Δ_vps38_ (class IV) are defective only in autophagy/Cvt or CPY sorting, respectively. These results suggest that Vps30p, Apg14p, and Vps38p may have regulatory roles, enabling the Vps34 PtdIns 3–kinase to perform specific functions.
Figure 3.
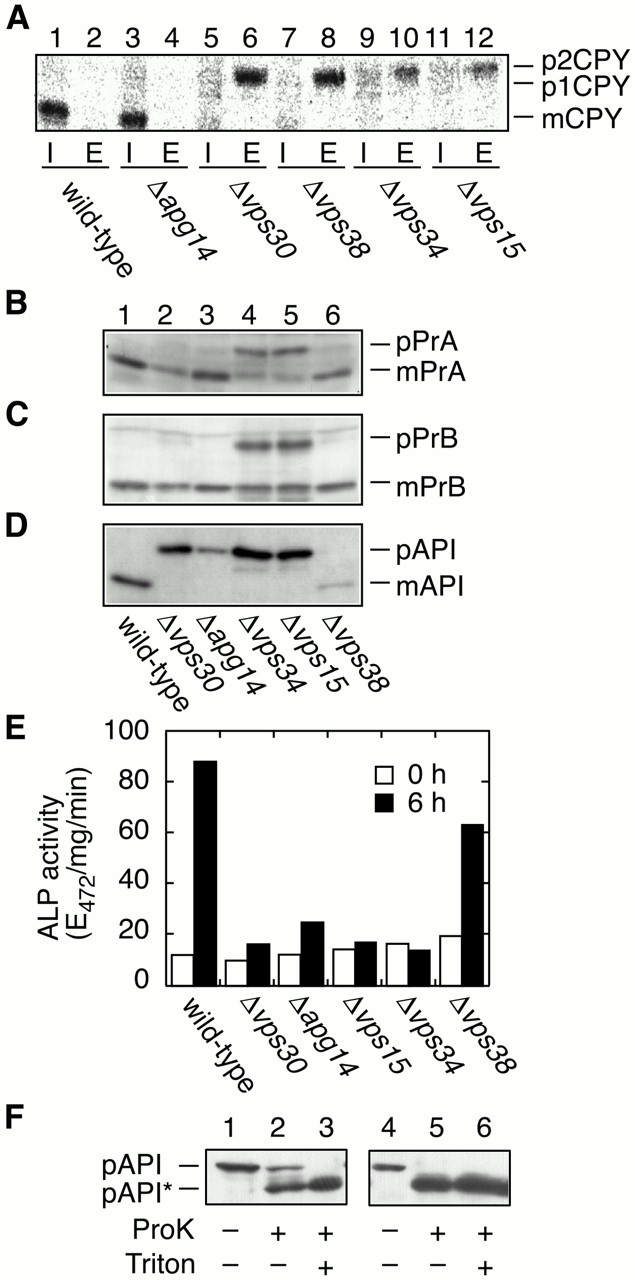
Transport of vacuolar proteins and autophagy. TN125 (wild type), AKY13 (Δ_apg14_), AKY15 (Δ_vps30_), AKY114 (Δ_vps38_), AKY109 (Δ_vps34_), and AKY115 (Δ_vps15_) cells were grown in SC medium lacking methionine (A) or in YPD (B–D) at 28°C. (A) Yeast cells were labeled with [35S]methionine/cysteine for 15 min and chased with unlabeled methionine and cysteine for 30 min. The labeled cells were converted to spheroplasts and separated into pellet (I, intracellular) and supernatant (E, extracellular) fractions. CPY was immunoprecipitated and visualized by autoradiography using BAS2000. (B–D) Total protein was separated by SDS-PAGE and detected by immunoblotting with anti-PrA (B), anti-Pr B (C), and anti-API (D). (E) Cells were grown in YPD (open bars) and shifted to SD(-N) medium for 6 h (filled bars) at 28°C. Lysates from each group of cells were subjected to the ALP assay (Noda and Ohsumi 1998) to measure autophagy activity. (F) KVY4 (Δ_ypt7_; lanes 1–3) and AKY131 (Δ_vps34_; lanes 4–6) cells grown in YPD to a log phase were transferred to SD(-N), and incubated for 4.5 h at 30°C. Total lysates were centrifuged at 13,000 g for 15 min. The pellets were treated with or without Triton X-100 and/or proteinase K as indicated on ice for 30 min. The samples were TCA-precipitated and subjected to immunoblotting with anti-API antibodies. pAPI*, digested pAPI fragment.
Table 2.
Summary of the Mutant Phenotypes
| WT | Δ_vps15_ | Δ_vps34_ | Δ_vps30_ | Δ_apg14_ | Δ_vps38_ | |
|---|---|---|---|---|---|---|
| Class I | Class I | Class II | Class III | Class IV | ||
| Autophagy | + | − | − | − | − | + |
| API | + | − | − | − | − | + |
| CPY | + | − | − | − | + | − |
| PrA | + | − | − | + | + | + |
| PrB | + | − | − | + | + | + |
| Growth at 37°C | + | − | − | + | + | + |
Next, we investigated whether the defect of autophagy in Δ_vps34_ cells is at the point of fusion or formation of the autophagosome using an API protection assay (Kim et al. 1999; Ishihara, N., T. Noda, Y. Kamada, T. Yoshimori, and Y. Ohsumi, unpublished material). API is transported into the vacuole via the Cvt pathway under nutrient-rich conditions (Klionsky and Ohsumi 1999). Additionally, API can be a marker of autophagosome cargo because it is selectively delivered to the vacuole via the autophagosome under starvation conditions. Ypt7 is a rab family GTPase whose mutant is defective in fusion between the autophagosome or Cvt vesicle and the vacuole and accumulates the autophagosomes and Cvt vesicles in the cytosol (Kim et al. 1999; Kirisako et al. 1999). We have shown that the autophagosome can be pelleted with a 13,000 g centrifugation in Δ_ypt7_ cells (Ishihara, N., T. Noda, Y. Kamada, T. Yoshimori, and Y. Ohsumi, unpublished material). Δ_ypt7_ and Δ_vps34_ cells grown in a starvation condition were converted to spheroplasts and lysed osmotically. The lysates were roughly precleared by low speed centrifugation, and the supernatants were spun at 13,000 g for 15 min. The loose pellets were dissolved in an osmotically stabilized buffer, treated with proteinase K in the presence or absence of detergent Triton X-100, and subjected to immunoblotting for API. As shown in Fig. 3 F, in Δ_ypt7_ cells, a population of protease K–resistant API existed and it was digested completely in the presence of Triton X-100, indicating the accumulation of autophagosomes in the cytosol. Conversely, in Δ_vps34_ cells, all API in the LSP was sensitive to proteinase K. Hence, Vps34p is required for formation of intact autophagosomes.
Apg14p and Vps38p Constitute Distinct Complexes
As shown above, Apg14p and Vps38p are required for different membrane trafficking pathways. To discriminate whether Apg14p and Vps38p compose a single complex that functions both in autophagy/Cvt and in CPY sorting, or Apg14p and Vps38p compose distinct complexes that function separately, detergent extracts prepared from wild-type cells were subjected to coimmunoprecipitation experiments using anti-Apg14p or anti-Vps38p antibodies. Immunoprecipitates were then visualized by immunoblotting using anti-Vps30p, anti-Vps34p, anti-Vps15p, anti-Apg14p, and anti-Vps38p antibodies (Fig. 4 A). Both Vps30p and Vps34p were coimmunoprecipitated with their respective specific antibodies (Fig. 4 A, lanes 1 and 2). A substantial level of Vps15p was detected in the immunoprecipitates of anti-Vps38p antibodies (Fig. 4 A, lane 2). Anti-Apg14p antibodies precipitated a very low but detectable amount of Vps15p (Fig. 4 A, lane 1). The low level of Vps15p was probably due to instability of Vps15p in cell lysates and low abundance of the Apg14p-containing complex (see below). However, Vps38p could not be detected in the immunoprecipitates obtained using anti-Apg14p antibodies at all (Fig. 4 A, lane 1). Immunoprecipitates obtained using anti-Vps38p antibodies did not contain Apg14p either (Fig. 4 A, lane 2). These results suggest that Vps38p and Apg14p are not present in the same complex. Both complexes possess PtdIns 3–kinase activity, although immunoprecipitates obtained using anti-Apg14p antibodies showed ∼10-fold less activity than anti-Vps38p antibody immunoprecipitates (Fig. 4 B).
Figure 4.

Apg14p and Vps38p exist in distinct complexes. (A and B) AKY73 cells were grown in YPD at 30°C. Total lysates were solubilized with Triton X-100 and incubated with protein A–immobilized anti-Apg14p antibodies (lane 1) or anti-Vps38p antibodies (lane 2). The beads were then separated into two fractions and were subjected to immunoblotting (A) or PtdIns 3–kinase assays (B). In A, bound proteins were eluted from the beads, separated by SDS-PAGE, and detected by immunoblotting with anti-Vps30p, anti-Vps34p, anti-Vps15p, anti-Apg14p, and anti-Vps38p antibodies. In B, labeled lipids were extracted, separated by TLC, and visualized by autoradiography using BAS2000. (C) AKY112/pAUR112 (vector; lane 1) and AKY112/pKHR69 (His6–Myc–APG14; lane 2) cell lysates were solubilized with Triton X-100 and loaded on a Ni-NTA agarose column. Bound proteins were eluted with 250 mM imidazole and were subjected to immunoblotting with anti-Vps30p, anti-Myc (9E10), anti-Vps34p, and anti-Vps38p antibodies.
The idea that Vps38p and Apg14p constitute different complexes was also supported by an independent method, a pull-down assay using a NH2 terminally fused His6–Myc–Apg14p protein. Lysates prepared from cells expressing His6–Myc–Apg14p were solubilized with Triton X-100 and incubated with Ni-NTA agarose. Retained proteins were eluted with imidazole and separated by SDS-PAGE followed by immunoblotting using anti-Myc, anti-Vps30p, anti-Vps34p, and anti-Vps38p antibodies (Fig. 4 C). His6–Myc–Apg14p bound to Ni-NTA agarose was eluted with imidazole (Fig. 4 C, lane 2). Vps30p and Vps34p were also found in the elution fraction (Fig. 4 C, lane 2). Binding of Vps30p and Vps34p to Ni-NTA agarose was dependent on His6–Myc–Apg14p; control experiments showed that they are not detected in the elution fraction in the absence of His6–Myc–Apg14p (Fig. 4 C, lane 1). Again, Vps38p was not detected in the elution fraction (Fig. 4 C, lane 2). These results confirm that Apg14p and Vps38p exist in distinct complexes. Taken along with the phenotypic analyses, these results lead to our conclusion that the Apg14p-containing complex (Vps34p–Vps15p–Vps30p–Apg14p) functions in autophagy, and the Vps38p-containing complex (Vps34p–Vps15p–Vps30p–Vps38p) functions in CPY sorting. Hereafter, we will refer the former complex as complex I and the latter as complex II.
Vps38p Mediates the Interaction between Vps30p and Vps34p–Vps15p
Previous studies suggested that Vps15p directly phosphorylates Vps34p (Stack et al. 1993). These proteins appear to form the cores of complexes I and II. To address the organization of complexes I and II, the coimmunoprecipitation experiments using anti-Vps30p antibodies, as shown in Fig. 1 D, were performed on Δ_apg14_, Δ_vps38_, Δ_vps34_, and Δ_vps15_ cells. Consistent with the results that Apg14p and Vps38p compose separate complexes, the interaction between Vps30p and Apg14p was not affected by deletion of VPS38 (Fig. 5 D, lane 2), and deletion of APG14 did not affect the Vps30p–Vps38p interaction (Fig. 5 E, lane 1). We also investigated their cellular amounts in the respective deletion strains by immunoblotting. The amounts of Apg14p and Vps38p were not changed compared with wild-type cells in Δ_vps38_ (Fig. 6 D, lane 4) or Δ_apg14_ (Fig. 6 E, lane 3) cells, respectively. The Vps30p–Vps38p interaction and the cellular amount of Vps38p were unchanged in Δ_vps34_ and Δ_vps15_ cells (Fig. 5 E, lanes 3 and 4; Fig. 6 E, lanes 5 and 6). However, Vps38p was not detected in Δ_vps30_ cells (Fig. 6 E, lane 2). These results suggest that Vps30p directly binds to Vps38p and protects Vps38p from degradation. In contrast, although both Vps15p and Vps34p were present in Δ_vps38_ cells (Fig. 6 B, lane 4; Fig. 6 C, lane 4), they could not interact with Vps30p in the absence of Vps38p (Fig. 5 B, lane 2; Fig. 5 C, lane 2). These results imply that the interaction between Vps30p and the Vps34p–Vps15p core complex is indirect and is mediated by Vps38p. Apg14p was barely detected in Δ_vps30_, Δ_vps34_, and Δ_vps15_ cells (Fig. 6 D, lanes 2, 5, and 6). Accordingly, a reduced amount of Apg14p was coimmunoprecipitated with Vps30p in Δvps34 cells (Fig. 5 D, lane 3), and Apg14p was not detected in the immunoprecipitates from Δ_vps15_ cells (Fig. 5 D, lane 4). These results suggest that direct binding of Vps30p and Vps34p–Vps15p to Apg14p may protect Apg14p from proteolysis by hindering recognition sites against proteases (proteolytic systems) or by inducing Apg14p to assume a tight conformation. Deletion of APG14 appears not to affect the interaction between Vps30p and Vps34p–Vps15p (Fig. 5 B, lane 1; Fig. 5 C, lane 1), although deletion of VPS38 leads to dramatic effects on it (Fig. 5 B, lane 2; Fig. 5 C, lane 2). These results suggest that complex I is much less abundant than complex II in yeast cells. In fact, the total amount of Apg14p is very low (data not shown).
Figure 5.

Coimmunoprecipitaion with Vps30p. AKY112 (Δ_apg14_; lane 1), AKY113 (Δ_vps38_; lane 2), AKY110 (Δ_vps34_; lane 3), and AKY116 (Δ_vps15_; lane 4) cells were grown in YPD at 28°C. Total lysates were solubilized with Triton X-100 and incubated with protein A–immobilized anti-Vps30p antibodies. Retained proteins were washed, eluted with 100 mM glycine-HCl, pH 2.5, precipitated with 5% TCA, and suspended in SDS sample buffer. Proteins were separated by SDS-PAGE and detected by immunoblotting with anti-Vps30p (A), anti-Apg14p (B), anti-Vps34p (C), anti-Vps15p (D), and anti-Vps38p (E) antibodies.
Figure 6.

Cellular amount of components of Vps30p complexes. AKY106 (wild type; lane 1), AKY111 (Δ_vps30_; lane 2), AKY112 (Δ_apg14_; lane 3), AKY113 (Δ_vps38_; lane 4), AKY110 (Δ_vps34_; lane 5), and AKY126 (Δ_vps15_; lane 6) cells were grown at 28°C. Total cellular protein was separated by SDS-PAGE for the detection of Vps30p (A), Vps34p (B), and Vps15p (C) by immunoblotting. For detection of Apg14p and Vps38p, pKHR75 expressing His6–Myc–Apg14p or pKHR65 expressing Vps38-3xHA was introduced into the above respective strains. Total protein prepared from them was separated by SDS-PAGE and detected by immunoblotting using anti-Myc (9E10) (D) or anti-HA (16B12) (E) antibodies.
Stack et al. 1995 reported that Vps15 kinase domain mutants are unable to interact with Vps34p, suggesting that Vps15p autophosphorylation or Vps15p-mediated phosphorylation of Vps34p may be involved in Vps15p–Vps34p complex formation. Alternatively, a Vps34p PtdIns 3–kinase domain mutant was able to associate with Vps15p in a manner indistinguishable from wild-type Vps34p (Stack et al. 1995). We examined the ability of the Vps15 kinase domain mutant and the Vps34 PtdIns 3–kinase domain mutant to form complexes I and II. The glutamic acid at position 200 of Vps15p is a highly conserved residue among protein kinases, and its mutation to arginine abolishes the in vivo phosphorylation of Vps15p, resulting in a temperature-sensitive growth defect and missorting of CPY (Herman et al. 1991a). The asparagine at position 736 of Vps34p is part of the catalytic loop region (DXHXXN) of proteins functioning in ATP binding and phosphate transfer (Knighton et al. 1991) and is conserved among protein kinases and lipid kinases. The N736K mutation results in a dramatic decrease in PtdIns 3–kinase activity and a severe defect in vacuolar protein sorting (Schu et al. 1993). Coimmunoprecipitation experiments with anti-Vps30p antibodies were performed using a Δ_vps15_ strain containing either wild-type or vps15-E200R allele on a low copy plasmid (Fig. 7, lanes 3 and 4). The effect of the kinase-negative vps15 mutant was similar to that of deletion of the VPS15 gene; the amount of Apg14p, Vps34p, and Vps15-E200R, but not Vps38p, precipitated with anti-Vps30p antibodies was severely reduced (Fig. 7, lane 4). Vps15p was not detected in the absence of Vps34p (Fig. 6 B, lane 5). We also found that Vps15-E200R, as well as Apg14p, was not detected in vps15-E200R cells (data not shown). Thus, Vps15p-mediated autophosphorylation of Vps15p or phosphorylation of Vps34p may be required for the interaction between Vps15p and Vps34p and for stabilization of Vps15p. In addition, Vps34p uncomplexed with Vps15p or unphosphorylated form of Vps34p does not complex with Vps30p, that is, does not bind to Vps38p for the reason described above. In contrast, all the interactions were normal in a Δ_vps34_ strain expressing the Vps34-N736K mutant protein (Fig. 7, lane 2). These results suggest that production of PtdIns(3)P is not required for these protein–protein interactions.
Figure 7.
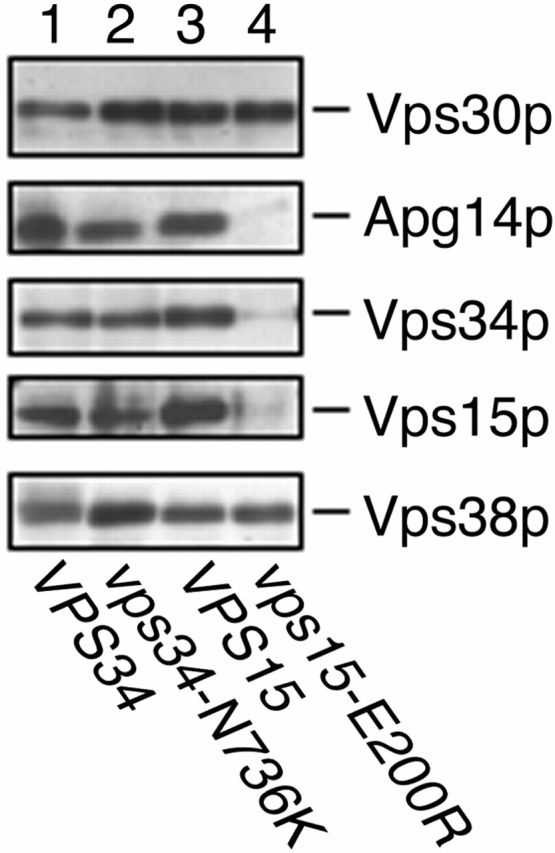
Kinase-defective Vps15p is unable to form Vps30p complexes. AKY109 (Δ_vps34_)/pKHR54 (VPS34) (lane 1), AKY109/pKHR60 (vps34-N736K) (lane 2), AKY115 (Δ_vps15_)/pKHR55 (VPS15) (lane 3), and AKY115/pKHR59 (vps15-E200R) (lane 4) cells were grown to mid-log phase in SC medium lacking uracil at 28°C. Total lysates were solubilized with Triton X-100 and incubated with protein A–immobilized anti-Vps30p antibodies. Bound proteins were washed and eluted with 100 mM glycine-HCl, pH 2.5. Proteins were separated by SDS-PAGE, followed by detection by immunoblotting with anti-Vps30p, anti-Apg14p, anti-Vps34p, anti-Vps15p, and anti-Vps38p antibodies.
Subcellular Distribution of Vps30p, Vps38p, and Vps34p
Although Vps30p has no apparent transmembrane domains or sites for lipid modification, Vps30p was found in the membrane fraction, in addition to the soluble fraction (Seaman et al. 1997). Membrane-associated Vps30p could be solubilized by salt or urea (Seaman et al. 1997; Kametaka et al. 1998). It is possible that the membrane association of Vps30p is mediated by a protein–protein interaction, with Vps15p, Vps34p, Vps38p, and Apg14p being candidates for a postulated membrane anchor. Therefore, we examined the subcellular location of Vps30p in these mutant cells. Total cell lysates were separated by sequential centrifugation at 13,000 and 100,000 g into LSP, HSP, and HSS fractions. In wild-type cells, most Vps30p was found in the LSP (60%) and HSS (34%), whereas only 6% of Vps30p was separated to the HSP (Fig. 8 A). A dramatic shift of Vps30p into the HSS fraction was observed in Δ_vps38_, Δ_vps34_, and Δ_vps15_ cells (Fig. 8 A). These results indicated that Vps15p, Vps34p, and Vps38p are required for recruitment of Vps30p to the membrane. Again, the absence of Apg14p had no effect on the subcellular location of Vps30p (Fig. 8 A), probably because complex I is in low abundance. Control experiments showed that ALP, a vacuolar membrane protein, was mainly localized to the LSP fraction (Fig. 8 D) and alcohol dehydrogenase was localized to the HSS fraction exclusively (data not shown) in all mutants tested. We also examined the subcellular distribution of Vps38p. The distribution pattern of Vps38p in wild-type cells was similar to that of Vps30p: 58, 8, and 34% of Vps38p was found in the LSP, HSP, and HSS, respectively (Fig. 8 B). We could not determine the subcellular location of Vps38p in Δ_vps30_ cells because Vps38p was scarcely detected in the absence of Vps30p (data not shown; Fig. 6 E, lane 2). The pattern was not changed in Δ_apg14_ cells (Fig. 8 B). Redistribution of Vps38p into the HSS fraction was observed both in Δ_vps34_ and Δ_vps15_ cells (Fig. 8 B). These results indicate that Vps38p is released to the cytoplasm together with Vps30p in Δ_vps34_ and Δ_vps15_ cells. A previous study indicated that Vps34p exists in both membrane and soluble fractions and that Vps15p is required for the membrane association of Vps34p (Stack et al. 1993). In our assay conditions, most Vps34p was found in the LSP (57%) and HSP (35%), and only 8% of Vps34p was present in the HSS fraction in wild-type cells (Fig. 8 C). In Δvps15 cells, 35% of Vps34p was released into the HSS fraction, whereas about half of Vps34p was detected in the HSP fraction (Fig. 8 C). Distribution of Vps34p was not changed in Δ_apg14_ cells (Fig. 8 C). In Δ_vps30_ and Δ_vps38_ cells, some shift of Vps34p to the HSP and HSS fractions was observed (Fig. 8 C). These results indicate that complex II resides on membranes in the LSP fraction, and disruption of the complex causes redistribution of Vps34p to the HSP membranes and the cytoplasm and a shift of Vps30p–Vps38p to the cytoplasm.
Figure 8.
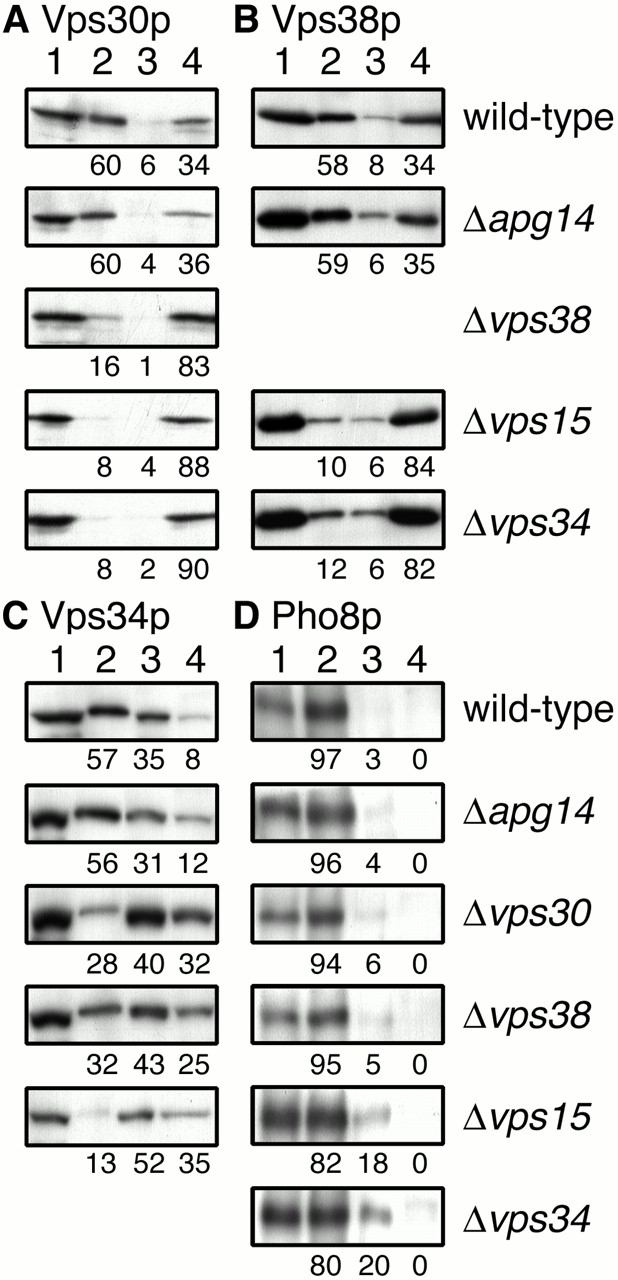
Subcellular fractionation of Vps30p, Vps38p, and Vps34p. AKY106 (wild type), AKY110 (Δ_vps34_), AKY111 (Δ_vps30_), AKY112 (Δ_apg14_), and AKY126 (Δ_vps15_) cells, each bearing pKHR65 (_VPS38-3xHA), and AKY113 (Δ_vps38) cells bearing pRS313 (empty vector) were grown in SC without histidine at 28°C. Cell lysates were subjected to subcellular fractionation by differential centrifugation as described in Materials and Methods. Total cell lysate (lane 1), LSP (lane 2), HSP (lane 3), and HSS (lane 4) fractions were subjected to SDS-PAGE, followed by immunoblotting with anti-Vps30p (A), anti-HA (16B12) (B), anti-Vps34p (C), or anti-Pho8p (D) antibodies. Relative amounts of each fraction were indicated. The values in A and C were averages of three independent experiments.
Discussion
Vps30p/Apg6p is required for both autophagy and CPY sorting (Kametaka et al. 1998). However, it was not yet known why and how Vps30p participates in these different membrane trafficking pathways. Recently, it was revealed that PtdIns 3–kinases also function both in autophagy (Kiel et al. 1999; Petiot et al. 2000) and protein transport to the vacuole/lysosome (Robinson et al. 1988; Herman and Emr 1990; Brown et al. 1995; Davidson 1995). Here, we provide evidence that Vps30p functions as a subunit of two distinct large PtdIns 3–kinase complexes: complexes I and II. Each complex contains a specific component, Apg14p (complex I) or Vps38p (complex II) together with three common proteins—Vps34p, Vps15p, and Vps30p. Gene disruption of one of the complex I components resulted in defects in autophagy, whereas gene disruption of one of the complex II components resulted in missorting of CPY. These results indicate that complexes I and II function in autophagy and CPY targeting, respectively. Vps10p is a late-Golgi transmembrane protein that acts as the sorting receptor for CPY (Marcusson et al. 1994). Mutation in VPS30 changes the subcellular distribution of Vps10p, resulting in a shift of Vps10p from the Golgi to the vacuolar membrane (Seaman et al. 1997). From these results, Seaman et al. 1997 proposed that Vps30p functions at the step essential for recycling of the Vps10p receptor from the endosome/PVC to the late-Golgi. However, it is still possible that Vps30p (complex II) functions in the anterograde transport of Vps10p–CPY-containing vesicle.
Vps38p could be coimmunoprecipitated with Vps30p in the absence of other factors (Fig. 5 E). Only Vps30p is required for stabilization of Vps38p (Fig. 6 E). These results suggest that Vps38p binds directly to Vps30p (Fig. 9). In contrast, although Vps15p and Vps34p were present in the Δ_vps38_ mutant (Fig. 6B and Fig. C), they could not be coprecipitated with Vps30p (Fig. 5B and Fig. C). Therefore, it seems that the interaction between Vps30p and the Vps34p–Vps15p core is not direct but mediated by Vps38p in complex II (Fig. 9). Apg14p is unstable in Δ_vps30_, Δ_vps15_, and Δ_vps34_ cells. These results suggest that both Vps30p and Vps15p–Vps34p may directly bind to Apg14p and conceal recognition sites for proteases (proteolytic systems) in Apg14p or induce Apg14p to adopt a protease-resistant conformation. Thus, Apg14p and Vps38p may act as connectors between Vps30p and Vps15p–Vps34p in complexes I and II, respectively (Fig. 9). However, deletion of APG14 appeared to have no effects on the interaction between Vps30p and Vps15p–Vps34p (Fig. 5B and Fig. C) and on the subcellular distribution of Vps30p (Fig. 8 A) and Vps34p (Fig. 8 C), whereas deletion of VPS38 had dramatic effects on them (Fig. 5B and Fig. C; Fig. 8A and Fig. C). These results suggest that complex I may represent only a minor population of PtdIns 3–kinase. In fact, the overall cellular amount of Apg14p is very low; we estimated that wild-type yeast cells contain ∼15-fold less Apg14p than Vps30p (data not shown). Moreover, the PtdIns 3–kinase activity of complex I was lower by ∼10-fold than that of complex II (Fig. 4 B). Therefore, the effects of the absence of Apg14p might be hidden by the abundant complex II. Although Apg14p and Vps38p have no significant sequence similarities, PairCoil (Berger et al. 1995) predicted that both proteins have potential coiled coil structures, which often mediate protein–protein interactions.
Figure 9.

Model for two distinct PtdIns 3–kinase complexes. Vps15p is anchored to membrane by myristic acid attached to the NH2 terminus of Vps15p (Herman et al. 1991b). Apg14p and Vps38p act as connectors between Vps30p and Vps34p. Phosphorylation of Vps34p by Vps15p is required for Vps34p–Vps15p and Vps34p–Apg14p/Vps38p interactions. White thick lines indicate sites of the interactions essential for the in vivo protein stabilization.
To obtain information about the molecular size of the Vps34 PtdIns 3–kinase complexes, gel filtration experiments were performed. When lysates were applied to a Superose 6 column, Vps30p, Apg14p, Vps34p, and Vps38p coeluted in a peak corresponding to ∼550 kD (data not shown). However, we could not detect Vps15p in any fractions because Vps15p was somewhat unstable in cell lysates and was gradually degraded by unknown proteases (data not shown). The 550-kD peak might be composed of a mixture of complexes I and II, both lacking Vps15p, that is, Vps30p–Apg14p–Vps34p and Vps30p–Vps38p–Vps34p. Although the artificial instability of Vps15p in vitro made it impossible to estimate the precise molecular weight of complex I and II, it provided two valuable insights into the complex formation. First, Apg14p and Vps38p connector molecules may directly bind to Vps34p. In the vps15 kinase-negative mutant, Vps34p was not coimmunoprecipitated with Vps30p (Fig. 7); that is, Vps34p could not bind to Vps38p, indicating that phosphorylation is required for the binding. From these results, together with the results of gel filtration, we derived the second conclusion: phosphorylation of Vps34p, but not the presence of Vps15p, may be required for the Apg14p–Vps34p and Vps38p–Vps34p interactions (Fig. 9). One attractive hypothesis is that Vps15p-mediated phosphorylation controls binding of Vps34p to Vps38p and Apg14p.
What is the function of PtdIns(3)P? One possibility is that PtdIns(3)P designates the vesicles that are not to be sorted away to the exocytic default pathway. PtdIns(3)P binding proteins may have important roles in presenting PtdIns(3)P as a marker molecule. The FYVE domain, a subfamily of the cysteine-rich RING motif, has been shown to bind directly to PtdIns(3)P (Burd and Emr 1998). In the yeast S. cerevisiae, five proteins are known to possess the FYVE domain. One of them, Vac1p, is involved in the fusion between the vesicle derived from the late-Golgi with the endosome/PVC (Peterson et al. 1999; Tall et al. 1999). Another FYVE domain–containing protein, Vps27p, is classified as a class E protein (Raymond et al. 1992). Mutations in class E VPS genes lead to an accumulation of vacuolar, endocytic, and late-Golgi markers in an exaggerated endosome/PVC, the class E compartment (Raymond et al. 1992; Piper et al. 1995). Vps27p may be required for delivery of proteins from the endosome/PVC: multivesicular body formation to the vacuole and for endosome/PVC-to-Golgi retrograde transport.
Alternatively, it is possible that PtdIns(3)P has a role in cargo selection at the vesicle budding step. In this model, binding of a cargo protein to the lumenal domain of the receptor transduces a signal through a conformational change that promotes receptor association with and/or activation of the Vps15p protein kinase. Activation of Vps15p leads to activation of the Vps34 PtdIns 3–kinase. Vps34p-mediated PtdIns(3)P production may recruit effector proteins that function in budding. In mammalian cells, PtdIns(3)P has been shown to play a role in adaptor (AP-2 and arrestin) incorporation into plasma membrane clathrin–coated pits (Gaidarov and Keen 1999; Gaidarov et al. 1999). Thus, only lipids surrounding the cargo receptor complex can bud, producing cargo-concentrated vesicles. It is also possible that PtdIns(3)P is required for vesicle formation by generating a driving force to curve the membrane into a bud by repulsive forces between the highly negative polar heads of PtdIns(3)P. However, wortmannin, a phosphoinositide 3–kinase inhibitor, did not inhibit the formation of TGN-derived vesicles but reduced the amount of receptor recruitment into those vesicles in mammalian cells (Gaffet et al. 1997). This observation is consistent with the idea that PtdIns(3)P is required for cargo selection but not for vesicle formation. In the case of autophagy and the Cvt pathways, complex I might act to load proteins essential for autophagosome/Cvt vesicle formation into vesicles, although it is not yet known if the constituent membranes and proteins of autophagosome/Cvt vesicles are supplied by vesicles.
Interestingly, the Δ_vps15_ and Δ_vps34_ mutants have additional phenotypes beyond that of the Δ_vps30_ mutant—impairment of PrA and PrB targeting and growth defects at 37°C. These results indicate that a fraction of the Vps34p–Vps15p complexes function independent of complexes I and II. Growth defects at high temperatures may be caused by defects in endocytosis because most of endocytosis mutations (end) confer a temperature-sensitive growth defect and VPS34 is allelic to END12 (Munn and Riezman 1994). It is attractive to speculate that Vps34p–Vps15p forms additional complexes with unknown factors to function in anterograde transport from the late-Golgi to the PVC/endosome for sorting of PrA and PrB and in endocytosis. PtdIns 3–kinase assays using cell lysates showed that deletion of either VPS30 or VPS38 caused only an ∼20% reduction of PtdIns 3–kinase activity and the APG14 disruptant showed equivalent PtdIns 3–kinase activity to wild-type cells (Fig. 2). Although it is possible that the postulated additional Vps34p–Vps15p complexes possess significant PtdIns 3–kinase activity, we propose instead that the roles of Vps30p, Apg14p, and Vps38p are not to activate Vps34p but to confer specificities on Vps34p for functions and/or locations where PtdIns(3)P should be produced. Consistent with this proposal, the absence of Vps30p or Vps38p caused some Vps34p to shift from the LSP membrane to the HSP membrane (Fig. 8 C). Further work is needed to determine the precise compartments where complexes I and II act.
Acknowledgments
Akio Kihara was supported by a Japan Society for the Promotion of Science (JSPS) Research Fellowship for Young Scientists.
Footnotes
N. Ishihara's present address is Department of Molecular Biology, Graduate School of Medical Science, Kyushu University, Maidashi Fukuoka, 812-8582, Japan.
Abbreviations used in this paper: 2-ME, 2-mercaptoethanol; ALP, alkaline phosphatase; API, aminopeptidase I; CPY, carboxypeptidase Y; Cvt, cytoplasm-to-vacuole targeting; HSP, high speed pellet; HSS, high speed supernatant; LSP, low speed pellet; PrA, proteinase A; PrB, proteinase B; PtdIns, phosphatidylinositol; PtdIns(3)P, PtdIns 3–phosphate; PVC, prevacuolar compartment; SC, synthetic complete.
References
- Baba M., Takeshige K., Baba N., Ohsumi Y. Ultrastructural analysis of the autophagic process in yeastdetection of autophagosomes and their characterization. J. Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M., Ohsumi M., Scott S.V., Klionsky D.J., Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B., Wilson D.B., Wolf E., Tonchev T., Milla M., Kim P.S. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. USA. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown W.J., DeWald D.B., Emr S.D., Plutner H., Balch W.E. Role for phosphatidylinositol 3–kinase in the sorting and transport of newly synthesized lysosomal enzymes in mammalian cells. J. Cell Biol. 1995;130:781–796. doi: 10.1083/jcb.130.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C.G., Emr S.D. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol. Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- Christianson T.W., Sikorski R.S., Dante M., Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Cooper A.A., Stevens T.H. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J. Cell Biol. 1996;133:529–541. doi: 10.1083/jcb.133.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson H.W. Wortmannin causes mistargeting of procathepsin D. Evidence for the involvement of a phosphatidylinositol 3–kinase in vesicular transport to lysosomes. J. Cell Biol. 1995;130:797–805. doi: 10.1083/jcb.130.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W.A., Jr. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994;4:139–143. doi: 10.1016/0962-8924(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Gaffet P., Jones A.T., Clague M.J. Inhibition of calcium-independent mannose 6-phosphate receptor incorporation into trans-Golgi network-derived clathrin-coated vesicles by wortmannin. J. Biol. Chem. 1997;272:24170–24175. doi: 10.1074/jbc.272.39.24170. [DOI] [PubMed] [Google Scholar]
- Gaidarov I., Keen J.H. Phosphoinositide–AP-2 interactions required for targeting to plasma membrane clathrin–coated pits. J. Cell Biol. 1999;146:755–764. doi: 10.1083/jcb.146.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov I., Krupnick J.G., Falck J.R., Benovic J.L., Keen J.H. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:871–881. doi: 10.1093/emboj/18.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding T.M., Hefner-Gravink A., Thumm M., Klionsky D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996;271:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- Herman P.K., Emr S.D. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae . Mol. Cell. Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman P.K., Stack J.H., DeModena J.A., Emr S.D. A novel protein kinase homologue essential for protein sorting to the yeast lysosome-like vacuole Cell 64 1991. 425 437a [DOI] [PubMed] [Google Scholar]
- Herman P.K., Stack J.H., Emr S.D. A genetic and structural analysis of the yeast Vps15 protein kinaseevidence for a direct role of Vps15p in vacuolar protein delivery EMBO (Eur. Mol. Biol. Organ.) J. 10 1991. 4049 4060b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horazdovsky B.F., DeWald D.B., Emr S.D. Protein transport to the yeast vacuole. Curr. Opin. Cell Biol. 1995;7:544–551. doi: 10.1016/0955-0674(95)80012-3. [DOI] [PubMed] [Google Scholar]
- Irie K., Takase M., Lee K.S., Levin D.E., Araki H., Matsumoto K., Ohima Y. MKK1 and MKK2, which encode Saccharomyces cerevisiae mitogen-activated protein kinase-kinase homologues, function in the pathway mediated by protein kinase C. Mol. Cell. Biol. 1993;13:3076–3083. doi: 10.1128/mcb.13.5.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kametaka S., Okano T., Ohsumi M., Ohsumi Y. Apg14p and Apg6p/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae . J. Biol. Chem. 1998;273:22284–22291. doi: 10.1074/jbc.273.35.22284. [DOI] [PubMed] [Google Scholar]
- Kiel J.A.K.W., Rechinger K.B., van der Klei I.J., Salomons F.A., Titorenko V.I., Veenhuis M. The Hansenula polymorpha PDD1 gene product, essential for the selective degradation of peroxisomes, is a homologue of Saccharomyces cerevisiae Vps34p. Yeast. 1999;15:741–754. doi: 10.1002/(SICI)1097-0061(19990630)15:9<741::AID-YEA416>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Kim J., Dalton V.M., Eggerton K.P., Scott S.V., Klinosky D.J. Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell. 1999;10:1337–1351. doi: 10.1091/mbc.10.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., Ohsumi Y. Formation process of autophagosome is traced with Apg8p/Aut7p in yeast. J. Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- Knighton D.R., Zheng J.H., Ten Eyck L.F., Ashford V.A., Xuong N.H., Taylor S.S., Sowadski J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Luo W., Chang A. Novel genes involved in endosomal traffic in yeast revealed by suppression of a targeting-defective plasma membrane ATPase mutant. J. Cell Biol. 1997;138:731–746. doi: 10.1083/jcb.138.4.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcusson E.G., Horazdovsky B.F., Cereghino J.L., Gharakhanian E., Emr S.D. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Munn A.L., Riezman H. Endocytosis is required for the growth of vacuolar H+-ATPase–defective yeastidentification of six new END genes. J. Cell Biol. 1994;127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T., Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Noda T., Matsuura A., Wada Y., Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae . Biochem. Biophys. Res. Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- Noda T., Kim J., Huang W.-P., Baba M., Tokunaga C., Ohsumi Y., Klionsky D.J. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol. 2000;148:465–479. doi: 10.1083/jcb.148.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretou C., Domin J., Cockcroft S., Waterfield M.D. Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3–kinase. J. Biol. Chem. 1997;272:2477–2485. doi: 10.1074/jbc.272.4.2477. [DOI] [PubMed] [Google Scholar]
- Peterson M.R., Burd C.G., Emr S.D. Vac1p coordinates Rab and phosphatidylinositol 3–kinase signaling in Vps45p-dependent vesicle docking/fusion at the endosome. Curr. Biol. 1999;9:159–162. doi: 10.1016/s0960-9822(99)80071-2. [DOI] [PubMed] [Google Scholar]
- Petiot A., Ogier-Denis E., Blommaart E.F., Meijer A.J., Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- Piper R.C., Cooper A.A., Yang H., Stevens T.H. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae . J. Cell Biol. 1995;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond C.K., Howald-Stevenson I., Vater C.A., Stevens T.H. Morphological classification of the yeast vacuolar protein sorting mutantsevidence for a prevacuolar compartment in class E vps mutants. Mol. Biol. Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.S., Klionsky D.J., Banta L.M., Emr S.D. Protein sorting in Saccharomyces cerevisiaeisolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schu P.V., Takegawa K., Fry M.J., Stack J.H., Waterfield M.D., Emr S.D. Phosphatidylinositol 3–kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- Scott S.V., Hefner-Gravink A., Morano K.A., Noda T., Ohsumi Y., Klionsky D.J. Cytoplasm-to-vacuole targeting and autophagy employ the same machinary to deliver proteins to the yeast vacuole. Proc. Natl. Acad. Sci. USA. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman M.N.J., Marcusson E.G., Cereghino J.L., Emr S.D. Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J. Cell Biol. 1997;137:79–92. doi: 10.1083/jcb.137.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T., Mizushima N., Ogawa Y., Matsuura A., Noda T., Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:5234–5241. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J.H., Herman P.K., Schu P.V., Emr S.D. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3–kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:2195–2204. doi: 10.1002/j.1460-2075.1993.tb05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J.H., DeWald D.B., Takegawa K., Emr S.D. Vesicle-mediated protein transportregulatory interactions between the Vps15 protein kinase and the Vps34 PtdIns 3–kinase essential for protein sorting to the vacuole in yeast. J. Cell Biol. 1995;129:321–334. doi: 10.1083/jcb.129.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall G.G., Hama H., DeWald D.B., Horazdovsky B.F. The phosphatidylinositol 3-phosphate binding protein Vac1p interacts with a Rab GTPase and a Sec1p homologue to facilitate vesicle-mediated vacuolar protein sorting. Mol. Biol. Cell. 1999;10:1873–1889. doi: 10.1091/mbc.10.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumm M., Egner R., Koch B., Schlumpberger M., Straub M., Veenhuis M., Wolf D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae . FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- Tsukada M., Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae . FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- Ungermann C., Nichols B.J., Pelham H.R., Wickner W. A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J. Cell Biol. 1998;140:61–69. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida T., Gerhardt B. A cell-free assay allows reconstitution of Vps33p-dependent transport to the yeast vacuole/lysosome. J. Cell Biol. 1999;146:85–97. doi: 10.1083/jcb.146.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S., Dhand R., Vanhaesebroeck B., MacDougall L.K., Stein R., Zvelebil M.J., Domin J., Panaretou C., Waterfield M.D. A human phosphatidylinositol 3–kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:3339–3348. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J.P., Caldwell K.K., Majerus P.W. Formation of phosphatidylinositol 3-phosphate by isomerization from phosphatidylinositol 4-phosphate. Proc. Natl. Acad. Sci. USA. 1991;88:9184–9187. doi: 10.1073/pnas.88.20.9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihisa T., Ito K. Pro-ompA derivatives with a His6 tag in their N-terminal “translocation initiation domains” are arrested by Ni2+ at an early post-targeting stage of translocation. J. Biol. Chem. 1996;271:9429–9436. doi: 10.1074/jbc.271.16.9429. [DOI] [PubMed] [Google Scholar]