Tryptophan-derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase (original) (raw)
Abstract
Macrophages exposed to macrophage colony-stimulating factor acquire the capacity to suppress T cell proliferation; this effect is associated with de novo expression of the tryptophan-catabolizing enzyme indoleamine 2,3-dioxygenase (IDO). We have purified IDO and tested its activity in in vitro models of T cell activation. IDO was able to inhibit proliferation of CD4+ T lymphocytes, CD8+ T lymphocytes, and natural killer (NK) cells; proliferation of B lymphocytes was not affected. The inhibitory role of tryptophan and of its catabolites was then tested. In the presence of tryptophan, only l-kynurenine and picolinic acid inhibit cell proliferation. In a tryptophan-free medium cell proliferation was not affected. In the absence of tryptophan inhibition induced by l-kynurenine and picolinic acid was observed at concentrations below the lowest concentration that was effective in the presence of tryptophan, and quinolinic acid acquired some inhibitory capacity. Inhibition of cell proliferation induced by the tryptophan catabolites resulting from IDO activity was selective, applying only to cells undergoing activation. Resting cells were not affected and could subsequently activate normally. We suggest that IDO exerts its effect on cell proliferation by (i) starting the cascade of biochemical reactions that produce the three catabolites and by (ii) enhancing their inhibitory potential by depriving the extracellular microenvironment of tryptophan.
Keywords: T cell proliferation; macrophages; indoleamine 2,3-dioxygenase; tryptophan; kynurenine
Introduction
Indoleamine 2,3-dioxygenase (IDO)* is an enzyme, ubiquitously distributed in mammalian tissues and cells, converting tryptophan to _N_-formylkynurenine. This substance is further catabolized to kynurenine and then to the terminal metabolites picolinic acid or quinolinic acid.
Evidences gathered in the last 10 y indicate that this enzyme functions as an antimicrobial defense mechanism by allowing cells to deplete tryptophan from intracellular pool or local microenvironment (1). Enhanced IDO activity was also found in vitro upon cell stimulation with Th1 type cytokines, and in vivo in diseases that are associated with chronic stimulation of Th1-mediated immunity (2).
Furthermore, a new role for IDO as immunosuppressive agent has been recently suggested (3). The authors provided evidence that expression of IDO during murine pregnancy could be instrumental in preventing rejection of the semiallogeneic fetus (4, 5). Further in vitro studies showed that monocyte-derived macrophages exposed to macrophage colony-stimulating factor (MCSF) acquire the capacity to suppress T cell proliferation by arresting T cells in the mid-G1 phase of the cell cycle (6). This effect is associated with de novo expression of IDO and with reduction of tryptophan concentration in the medium and can be prevented by IDO inhibitors (6). However, the demonstration that immunosuppression is achieved in this model solely by means of the IDO activity, and solely by means of the selective depletion of tryptophan is largely indirect. Furthermore, the analysis of the effects of tryptophan-derived catabolites on T cell proliferation is lacking (2).
Only the availability of purified IDO could allow to precisely identify the effects of the enzyme, and to elucidate the role played in this frame by the levels of tryptophan and of its catabolites in the extracellular environment.
Materials and Methods
IDO Purification.
The enzyme was purified following previously described techniques with major modifications (7).
Step 1: Crude Extract. Small intestine from 60 rabbits weighing ∼2 kg was provided by a breeding farm (A.R.C.E., Montemozzo di Temossi, Genoa, Italy). The distal 40-cm portion was cut longitudinally, luminal contents were accurately washed out with ice cold 0.9% NaCl solution, the intestine was cut in small pieces with a blade, and 200 g aliquots were then frozen at −80°C. When necessary, 300 ml of 50 mM phosphate buffer, pH 6.0, supplemented with 2 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, 10 μg/ml leupeptine and 10 μg/ml pepstatine (all from Sigma-Aldrich) were added to an aliquot. All subsequent procedures were performed at 4°C. The sample was then minced with a food blender, and lysis of tissue was completed using a Potter-Elvehjem homogenizer.
Step 2: Ammonium Sulfate Fractionation. After centrifugation at 20,000 g for 35 min, supernatant was collected and solid ammonium sulfate was added slowly with stirring till 70% of the saturating concentration was reached. The solution was stirred for 1 more hour, then the precipitate was recovered by centrifuging at 10,000 g for 10 min. Afterward, 30 ml ammonium sulfate at 50% of the saturating concentration were added to the precipitate, which was suspended with a Potter-Elvehjem homogenizer, and supernatant was collected. This procedure was repeated two more times, then the pooled supernatants were dialized against 50 mM phosphate buffer, pH 6.0.
Step 3: Hydroxyapatite Chromatography. 100 ml Bio-Gel hydroxyapatite (Bio-Rad Laboratories), rehydrated with 50 mM phosphate buffer, pH 6.0, were poured in a 2 × 40 cm column. After loading the sample, the column was washed with 50 mM phosphate buffer, pH 6.0, and then a linear gradient elution was performed between 200 ml each of 150 and 500 mM phosphate buffer, pH 6.0. Fractions between 240 and 340 mM phosphate concentration were pooled, and concentrated to 9 ml with Amicon YM-10 membrane (Amicon).
Step 4: Gel Filtration on Sephadex G-100. 4.5 ml of the sample were applied on a 3 × 50 cm column of Sephadex G-100 (Amersham Pharmacia Biotech) column previously equilibrated with 150 mM KCl in 20 mM phosphate buffer, pH 6.0. Blue Dextran and Xylene Cyanole were used as molecular weight markers: downward elution at a flow rate of 0.5 ml/min was performed using the same buffer, and fractions in between the two markers were collected. Fractions having a ratio of absorbance at 406 nm to absorbance at 280 above 0.4 and displaying IDO activity were pooled and concentrated to 3 ml with Amicon YM-10 membrane.
Step 5: Blue Sepharose Chromatography. 1 g of Blue Sepharose CL-6B powder (Amersham Pharmacia Biotech) was rehydrated with distilled water, loaded in a 20-cm column and equilibrated with 20 mM phosphate buffer, pH 6.0. The same buffer was used to dilute 1:10 the sample, then the solution was loaded on the column. After washing, a linear gradient elution was performed between 20 ml each of 20 mM phosphate buffer, pH 6.0, and 1 M KCl in 20 mM phosphate buffer, pH 6.0, at a flow rate of 0.4 ml/min. Ratio of absorbance at 406 nm to absorbance at 280 nm was evaluated for each fraction, and the fractions having a value >1.0 were pooled and concentrated to 140 μl with Amicon YM-10 membrane, followed by Centricon 10 (Amicon) ultrafiltration.
Purity of the sample was checked by SDS-PAGE, followed by Coomassie Blue staining, and by HPLC, using a TSK gel G3000SW (Toso Haas) gel filtration column.
The protein amount was evaluated by Bradford (8) method (Bradford Anal Biochem) using BSA as standard. The purified enzyme was stored in aliquots at −80°C at concentrations >3 mg/ml.
Assay of IDO.
The reaction was performed exactly following a technique described previously (9). The amount of L-kynurenine in the supernatant was measured by reversed phase HPLC (HP 1090; Hewlett-Packard), using a μ-Bondapack with C-18 column (10 × 30 mm, 5-μm particle size; Waters). 1 U of the enzyme activity was defined as the amount that produced 1 nmol of Kynurenine/h.
Cell Isolation and Culture.
Heparinized (heparin 10 U/ml) venous blood was obtained from healthy male volunteers after informed consent. PBMCs were isolated by centrifugation on a discontinuous density gradient of Fycoll-Hypaque (Biochrom KG). Afterward, adherent cells were removed by adherence, and PBLs were collected.
Isolation of CD4+ and CD8+ T lymphocytes, B lymphocytes, and NK cells was performed by means of immunomagnetic beads (IO beads; Immunotech-Coulter), exactly following manufacturer's instructions. Cells were separated by negative selection from PBLs, using the following murine mAbs: OKT 8 (anti-CD8; American Type Culture Collection), BU12 (anti-CD19, purified from the homonymous hybridoma, provided by I.C.M. MacLennan, University of Birmingham, Birmingham, UK), anti–human N-CAM (anti-CD56; BioSource), and RMO52 (anti-CD14; Beckman Coulter) for CD4+ T lymphocytes; OKT 4 (anti-CD4, American Type Culture Collection), BU 12, anti–human N-CAM, and RMO52 for CD8+ T lymphocytes; UCHT-1 (anti-CD3, Technogenetics, Cassina de' Pecchi, Italy), anti–human N-CAM, and RMO52 for B lymphocytes; and BU 12, UCHT-1, and RMO52 for NK cells. Monocytes to be used as feeder cells were separated by negative selection from adherent PBMCs by using BU 12, UCHT-1, and anti–human N-CAM.
Purity was checked by flow cytometry, measuring the membrane expression of CD4, CD8, CD19, CD56, and CD14, respectively.
Only cell preparations with purity ≥95% were taken into account. In some cases a second round of separation was required.
Cells were grown, unless otherwise stated, in RPMI 1640 supplemented with 10% FCS and 2 mM l-glutamine (all from Biochrom KG).
Cell Activation and Proliferation.
Cell proliferation studies were performed, unless otherwise indicated, on restimulated alloreactive T cells and on PHA-activated PBLs. Both models gave equivalent results; the data shown are from PHA-activated PBLs.
To produce alloreactive T cell lines 105 PBLs from responder were incubated with the same amount of irradiated (5,000 rad) PBLs from stimulator in round bottomed, 96-well plates. The day after, and every other day, 10 U/ml IL-2 (Chiron BV) were added. Each alloreactive CD3-positive cell line was maintained by adding, every 2 wk, 5 × 104 irradiated PBLs from stimulator. The addition of irradiated PBLs from stimulator marked the start of proliferation assays. After 24 and 48 h, 10 U/ml IL-2 were added.
To perform the MLRs indicated in Fig. 9 , 1.5 × 105 PBLs from responder were incubated with 105 irradiated (5,000 rad) PBLs from stimulator in round bottomed, 96-well plates, either in the absence or in the presence of 500 μM l-kynurenine. The day after, and every other day, 10 U/ml IL-2 were added. After 5 d supernatant was carefully removed, new medium was added, and cultures were restimulated with 105 irradiated PBLs from either the original stimulator or from a new stimulator. 6 d after 1 μCi 3[H]thymidine (Amersham Pharmacia Biotech) was added, and a standard thymidine incorporation assay was performed as indicated below.
Figure 9.

Effect of l-kynurenine on responsiveness of activated and resting cells. MLRs were initially performed either in the presence (white columns and black columns) or in the absence (gray columns) of l-kynurenine. 5 d after cells were washed and cultures were then restimulated with either cells from the initial stimulator (white columns) or from a different donor (black columns). After 7 more days cell proliferation was evaluated by measuring 3[H]thymidine incorporation. Three experiments, using different responders and stimulators were performed. Each column indicates the mean from three wells.
To obtain PHA blasts, 106 cells/ml PBLs were incubated for 1 h with PHA M form (Life Technologies) at a final concentration of 10 μl/ml. Afterward, cells were washed twice and seeded in round bottomed, 96-well plates at 5 × 104 cells per well to start proliferation assays. After 24 and 48 h, 10 U/ml IL-2 were added.
The protocol used to obtain PHA blasts was also used to activate purified CD4+ T lymphocytes, CD8+ T lymphocytes, and NK cells. However, for the latter subpopulation changes were made: NK cells were seeded on a feeder layer of 105 irradiated autologous monocytes, and IL-2 was used at a concentration of 20 U/ml.
B lymphocytes were seeded in round bottomed, 96-well plates at 5 × 104 cells per well and activated by coincubation with 1:64,000 dilution of Pansorbin cells (Calbiochem). After 24 and 48 h, 10 U/ml IL-2 were added.
When indicated, 0.1 μM methylene blue, 200 μM l-ascorbic acid (all from Sigma-Aldrich) or 1 mM 1-methyl-DL-tryptophan (Sigma-Aldrich) were added at the beginning of the test. These reagents, at the above mentioned concentrations, did not affect cell viability, as assessed by trypan blue dye exclusion technique and by flow cytometric evaluation of cell membrane permeability to propidium iodide (Sigma-Aldrich). IDO was added at time and concentrations indicated in the text, in a final volume of 200 μl.
To activate and grow the cells in the absence of tryptophan, a custom-made tryptophan-free RPMI 1640 medium (Biochrom KG) was supplemented with 2 mM l-Glutamine, 1× nonessential amino acids (GIBCO BRL, Life Technologies), and 20% dialyzed FCS. Dialysis was performed against 200 volumes of PBS with 10 changes. The resulting culture medium was devoid of tryptophan, as detected by the technique described below. Preliminary validation experiments confirmed that T cell proliferation in this medium was comparable to proliferation in the above mentioned normal medium. Preliminary tests also demonstrated that this medium did not affect cell viability.
In some cases l-tryptophan, l-kynurenine, picolinic acid (all from Sigma-Aldrich) and quinolinic acid (Sigma-Aldrich) were added to the cell cultures at the times and concentrations indicated.
Cocultures of MCSF-treated macrophages and activated PBLs were performed as described previously (10), with the exception that we used PHA-activated PBLs as model of T cell activation. MCSF-treated macrophages to PBLs ratios and monocytes to PBLs ratios are indicated in the text. MCSF (Vinci-Biochem, Florence, Italy) was used at a concentration of 200 U/ml. Supernatants were collected from cocultures 72 h after the addition of T cells, and levels of kynurenine were measured, as described below. After 24 more hours proliferation was measured.
Cell proliferation was evaluated mainly by standard thymidine incorporation assay. Unless otherwise indicated, 72 h after cell activation 1 μCi 3[H]thymidine (Amersham Pharmacia Biotech) was added, and incorporation was measured after 24 h in a β-Counter (Matrix 9600; Canberra Packard). In some tests the amount of cells in the different phases of the cell cycle was evaluated by exactly following the method of Nicoletti et al. (11). Flow cytometry analysis was performed on an EPICS XL flow cytometer (Beckman Coulter), cells were gated on the basis of physical properties (forward versus side light scatter) and at least 2,000 living cells were analyzed per sample.
Tryptophan and Kynurenine Assays.
Before the assay, tryptophan and kynurenine were extracted from the culture medium using a previously described technique, with minor changes (12). In brief, tricloroacetic acid (TCA) was added to the medium at the final concentration of 0.2 M and the vortexed mixture was centrifuged 5′ at 13,000 rpm. The pellet was trashed and the supernatant was injected into the HPLC system after its further centrifugation (5′ at 13,000 rpm).
The HPLC separation was performed at 40°C on a RP column (Beckman Ultrasphere C18 250 mm length, 2 mm internal diameter, Alltech Italia srl, Milan, Italy) using bidistilled and filtered water as unique eluent at the flow rate of 0.2 ml/min. The UV detector was used registering chromatograms at three different wavelengths (210, 280, and 360 nm) and collecting spectra in the 200–450 nm range. In any case, for standards or samples, the injected volume was of 100 μl and between two successive injections the delay time was of 25 min. The metabolites of interest were identified by the use of standards comparing retention times (9 min for l-kynurenine and 14 min for l-tryptophan) and spectra. The real nature of these compounds was confirmed by performing HPLC-mass spectrometric analysis in the negative ion mode, directly coupling the HPLC eluent to an electrospray ion source and identifying the analites by the expected m/z ratio. The quantitative analysis was performed performing two calibration curves injecting the l-tryptophan and l-kynurenine standard molecules at concentrations between 0.1 and 100 μM.
The sensitivity threshold of the assay was 0.1 μM for both kynurenine and tryptophan.
RPMI 1640 was found to contain 25 μM tryptophan; addition of 10% FCS increased tryptophan concentration to 26 μM. Tryptophan concentration in the above mentioned tryptophan-free medium was below the sensitivity of the assay.
Results
Effect of Purified IDO on PBL Proliferation.
IDO was purified according to the protocol described in Materials and Methods section. The final preparation was homogeneous by SDS-PAGE and HPLC. The overall purification was 480-folds and the specific activity was of 156,000 U/mg of protein.
The capacity of the purified enzyme to inhibit PBL proliferation was tested by adding the enzyme at increasing concentrations at the beginning of the test, together with methylene blue and l-ascorbic acid. These cofactors were added to optimize the efficiency of the enzyme, possibly by increasing production of O2 − (13). Under these conditions a dose-dependent inhibition of cell proliferation was detected, both by evaluating 3[H]thymidine incorporation (Fig. 1 A) and by counting the cells entering the cell cycle (Fig. 1 B).
Figure 1.


Dose–response relationship to IDO for T cell proliferation. Increasing doses of IDO, together with 0.1 μM methylene blue and 200 μM l-ascorbic acid, were added at the beginning of the test to PHA-activated PBLs. Untreated cells were used as control. Cell culture was stopped at 96 h and proliferation was evaluated by measuring 3[H]thymidine incorporation (A) or by counting the cells in the cell cycle, as indicated in the Materials and Methods section (B). m ± 1 SD, n = 3.
The role of cofactors is elucidated in Fig. 2 , showing the effect of various combinations of IDO and cofactors on the proliferation of PHA-stimulated PBLs, evaluated both by measuring 3[H]thymidine incorporation (Fig. 2 A) and by counting the cells entering the cell cycle (Fig. 2 B). When the sole IDO, at the final concentration of 4,000 U/ml, was added at the beginning of the test, half of proliferation was retained. The same result was obtained by adding to IDO either methylene blue or l-ascorbic acid. Instead, coincubation of IDO with both cofactors reduced proliferation to one fourth of the control. The same result was obtained when 800 U IDO/well were added daily to the culture, in the absence of cofactors.
Figure 2.


Role of cofactors on IDO activity. PHA-activated PBLs were incubated with the indicated combinations of 4,000 U/ml IDO, 0.1 μM methylene blue, and 200 μM l-ascorbic acid. In a set of experiments 800 U IDO were added daily to the cell culture, in the absence of cofactors. Control is represented by cells incubated with medium alone. Cell culture was stopped after 96 h and proliferation was evaluated by measuring 3[H]thymidine incorporation (A) or by counting the cells in the cell cycle (B). m ± 1 SD, n = 3.
To reach the confirmation that the inhibition of PBL proliferation was due solely to the enzymatic activity of IDO, inhibition tests were performed in the presence of 1 mM 1-methyl-DL-tryptophan, a compound reported to be a potent inhibitor of IDO activity (14). As shown in Fig. 3, A and B, 1 -methyl-DL-tryptophan was able to almost completely prevent the inhibition of proliferation induced by IDO and cofactors.
Figure 3.


Effect of the IDO competitive inhibitor 1-methyl-DL-tryptophan. PHA-activated PBLs were incubated with the indicated combinations of 4,000 U/ml IDO, 0.1 μM methylene blue, 200 μM l-ascorbic acid, and 1 mM 1-methyl-DL-tryptophan. Control is represented by cells incubated with medium alone. Cell culture was stopped after 96 h and proliferation was evaluated by measuring 3[H]thymidine incorporation (A) or by counting the cells in the cell cycle (B). m ± 1 SD, n = 3.
On the basis of previous studies, indicating that macrophage-dependent inhibition of T cell proliferation could be related to a sustained arrest of the cell cycle in the mid-G1 phase (6), we checked whether PBLs activated with PHA in the presence of IDO showed an arrest in cell cycle progression. Indeed, by counting the cells in the different phases of the cell cycle with the method of Nicoletti et al. (11), we found that PHA-activated PBLs in the presence of IDO showed a marked reduction in the number of cells in the phases from S to M (Fig. 1 B).
Effect of Tryptophan and of Its Catabolites on PBL Proliferation.
Tryptophan is converted by IDO to _N_-formylkynurenine; this substance is further catabolized to kynurenine and then to the terminal metabolites picolinic acid or quinolinic acid. To understand the mechanisms of the inhibitory effect of the enzyme on PBL proliferation, it is mandatory to elucidate the effects that reduction of tryptophan and/or increase of downstream catabolites in the medium have on cell proliferation. Therefore, we started to investigate the effects exerted on PBL proliferation by three tryptophan-derived catabolites: l-kynurenine, picolinic acid, and quinolinic acid. The test was performed in complete culture medium, that contains 26 μM tryptophan, and the substances were added at the beginning of the test (Fig. 4 A). At concentrations above 1 mM a toxic effect was detected for all the three substances. Below this threshold, a dose-dependent inhibition of proliferation was observed with l-kynurenine and, to a lesser extent, with picolinic acid; instead, quinolinic acid was ineffective. Upon coincubation, l-kynurenine and picolinic acid showed a clear synergistic effect (Fig. 4 B). The two substances exerted their effect by blocking cell cycle in the mid-G1 phase (Fig. 4 C).
Figure 4.



Effect of tryptophan catabolites on T cell proliferation. (A) Increasing concentrations of l-kynurenine (▪––▪), picolinic acid (•- - -•), or quinolinic acid (▴…▴) were added to PHA-activated PBLs. Cell culture was stopped after 96 h and proliferation was evaluated by measuring 3[H]thymidine incorporation. (B) PHA-activated PBLs were incubated with the indicated combinations of 250 μM l-kynurenine, picolinic acid, or quinolinic acid. Control is represented by cells incubated with medium alone. Cell culture was stopped after 96 h and proliferation was evaluated by measuring 3[H]thymidine incorporation. (C) PHA-activated PBLs were incubated with 250 μM of l-kynurenine, picolinic acid, or quinolinic acid, respectively. Control is represented by untreated PHA-blasts. Cell culture was stopped after 96 h and the cells in the cell cycle were counted. m ± 1 SD, n = 3.
To further investigate the characteristics of this inhibitory effect, we focused our attention on l-kynurenine that was the most effective, among the tryptophan-derived catabolites examined, in inhibiting cell proliferation. The results of this investigation are summarized in Fig. 5 . As it can be seen, both 0.5 and 1 mM l-kynurenine abrogated proliferation when administered till 36 h after activation. Inhibition with 1 mM l-kynurenine could not be reverted by washing out the substance or by restimulating the cells after wash-out. Instead, when 0.5 mM l-kynurenine was used, cell proliferation was restored either partially or completely by washing out the substance, the amount of recovery being inversely related to the length of incubation with l-kynurenine; the amount of recovery was not significantly increased by restimulating the cells with PHA after wash-out.
Figure 5.
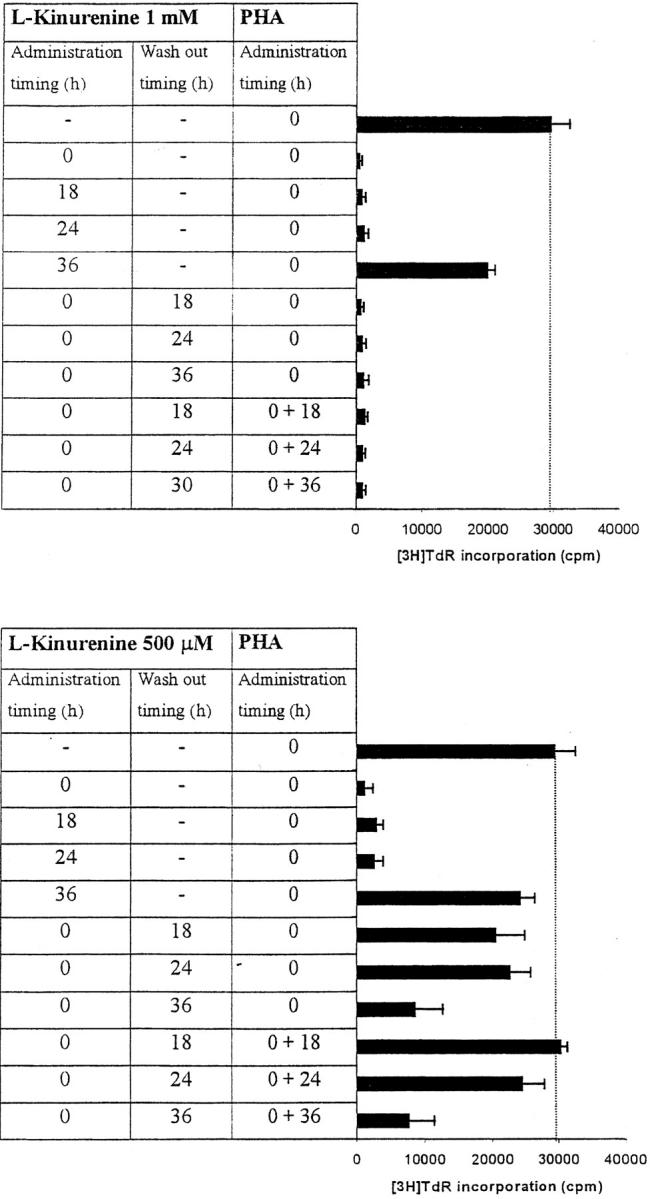
Timing of l-kynurenine–dependent inhibition of T cell proliferation. l-kynurenine at 1 mM concentration (top) or at 500 μM (bottom) was added to PHA-activated PBLs at the beginning of the test, or 18, 24 or 36 h after PHA activation, timing of l-kynurenine administration being indicated in the left column. In a subset of experiments l-kynurenine, administered at the beginning of the test, was removed from the samples at the times indicated in the central column: samples were centrifuged, supernatant was discarded, and cells were resuspended in medium. In a further subset of experiments PHA was administered again to the cells immediately after l-kynurenine wash-out (right column). Control is represented by untreated PHA-activated PBLs. Cell culture was stopped after 96 h from PHA activation, and proliferation was evaluated by measuring 3[H]thymidine incorporation. The degree of proliferation is indicated by the bars in the right part of each panel. The dotted line shows thymidine incorporation of control. m ± 1 SD, n = 3.
It has to be noted that exogenously added l-kynurenine is degraded during incubation with PBLs. In the case of 100 μM l-kynurenine added at the beginnning of the test to PHA-activated PBLs, a residual concentration of 25 μM l-kynurenine could be detected 72 h later.
To evaluate the relevance of the tryptophan levels in the extracellular environment, we used a culture medium devoid of tryptophan. As shown in Fig. 6 A, PHA-activated PBLs retained the capacity to proliferate in this medium. Furthermore, proliferation was not increased by adding increasing concentrations of l-tryptophan at the beginning of the test. In particular, no increase in proliferation was observed after adding 30 μM l-tryptophan, that is above the concentration of tryptophan in the normal culture medium. Afterward, the degree of inhibition of PBL proliferation was evaluated for each of the three catabolites both in tryptophan-free medium and in tryptophan-free medium supplemented with 26 μM l-tryptophan, that is the concentration of tryptophan in normal complete medium (Fig. 6, B–D). In the absence of tryptophan, l-kynurenine and, to a lesser extent, picolinic acid were endowed with inhibitory activity at concentrations that were below the concentrations that inhibited PBL proliferation in the presence of the amino acid (Fig. 6, B and C). Quinolinic acid, that was ineffective in the presence of l-tryptophan, in the absence of tryptophan showed a dose–response relationship for inhibition of proliferation (Fig. 6 D).
Figure 6.




Effect of tryptophan concentration on tryptophan catabolites-dependent inhibition of T cell proliferation. (A) Increasing concentrations of l-tryptophan were added to PBLs activated and cultured in a medium devoid of tryptophan. Cell culture was stopped after 96 h from PHA activation, and proliferation was evaluated by measuring 3[H]thymidine incorporation. (B–D) Increasing concentrations of l-kynurenine (B), picolinic acid (C), and quinolinic acid (D) were added to PBLs activated and cultured in tryptophan-free medium (▪——▪), or in the tryptophan-free medium supplemented with 26 μM l-tryptophan (▴——▴). Controls were represented by untreated PHA-activated PBLs grown in tryptophan-free medium and in the same medium supplemented with l-tryptophan, respectively. Cell culture was stopped after 96 h from PHA activation, and proliferation was evaluated by measuring 3[H]thymidine incorporation. To permit comparison, proliferation has been normalized to the control cultures (PHA-activated PBLs grown in tryptophan-free medium: 19,134 ± 1,109 cpm; PHA-activated PBLs grown in tryptophan-free medium supplemented with l-tryptophan: 20,021 ± 1,776 cpm). m ± 1 SD, n = 3.
The next step was aimed at comparing the effect of exogenously added l-kynurenine with the effect displayed by kynurenine produced as consequence of IDO expression in MCSF-treated macrophages upon incubation with activated PBLs. Cocultures of the two cell populations were performed, and kynurenine was measured in supernatants. Accordingly to previous data (10), different degrees of inhibition of PBL proliferation could be observed, depending on the macrophages to PBLs ratio (Fig. 7 A). Instead, untreated monocytes did not affect PBL proliferation. Afterward, the amount of kynurenine in the supernatant of cocultures at different macrophages to PBLs ratios was measured, and related to the degree of cell proliferation. A clear inverse relationship was found (Fig. 7 B). In this model substantial inhibition of proliferation was achieved with concentrations of kynurenine at 72 h above 10 μM.
Figure 7.


Relationship between degree of proliferation and concentration of kynurenine in coculture supernatants. (A) PHA-activated PBLs (PHA blasts) were cultured alone, in the presence of monocytes at the ratios indicated, in the presence of MCSF-treated macrophages at the ratios indicated, or in the presence of the MCSF solution (200 U/ml) used with macrophages. Cell culture was stopped after 96 h from PHA activation, and proliferation was evaluated by measuring 3[H] thymidine incorporation. m ± 1CD, n = 3. (B) PHA-activated PBLs were cultured in the presence of MCSF-treated macrophages at ratios of 4:1 (♦), 16:1 (▴), 64:1(▪), each ratio being performed in triplicate. Supernatants were collected from cocultures 72 h after the addition of PBLs, and levels of kynurenine were measured, as described in Materials and Methods section. Cell culture was stopped after 24 more hours, and proliferation was evaluated by measuring 3[H]thymidine incorporation. The amount of kynurenine in each supernatant was related to the level of proliferation in the well. The regression line is indicated. Star indicates the mean of thymidine incorporation of three wells with PHA-activated PBLs alone.
T Lymphocytes and NK Cells Are the Targets of IDO-dependent Inhibition.
Since PBLs are made up of different subpopulations, we wanted to get some insight into the sensitivity of each subpopulation to IDO activity. For this reason IDO effect was tested on CD4+ and on CD8+ T lymphocytes, on B lymphocytes, and on NK cells purified from peripheral blood (Fig. 8 A). We found that IDO-dependent inhibition of proliferation only applies to T lymphocytes and NK cells responding to a proliferative stimulus, proliferation of B lymphocytes not being affected. As in the case of PBLs, proliferation of each subpopulation in tryptophan-free medium did not differ from proliferation in the same medium supplemented with 26 μM tryptophan (proliferation in tryptophan-supplemented medium being 83.4 ± 9.7% of proliferation in tryptophan-free medium for CD4+ T lymphocytes, 114.1 ± 11.5% for CD8+ T lymphocytes, 108.7 ± 10.3 for B lymphocytes, and 81.3 ± 14.8 for NK cells; m ± 1 SD, n = 3). Instead, tryptophan-derived catabolites were effective, in the IDO-sensitive subpopulations, in inhibiting cell proliferation (Fig. 8, B–D), Consistent with the findings from PBLs, the inhibitory activity was enhanced in the absence of tryptophan.
Figure 8.




Effect of IDO and tryptophan-derived catabolites on different PBL subpopulations. (A) The different cell subpopulations were activated as indicated in the Materials and Methods section, and cultured in the presence of 4,000 U/ml IDO, 0.1 μM methylene blue, and 200 μM l-ascorbic acid. Cell culture was stopped after 96 h from activation, and proliferation was evaluated by measuring 3[H]thymidine incorporation. For each subpopulation untreated activated cells were used as control. m ± 1 SD, n = 3. (B–D) The effect of 1 mM l-kynurenine (B), 1 mM picolinic acid (C), and 1 mM quinolinic acid (D) was tested on each PBL subpopulation. The different cell subpopulations were activated as indicated in the Materials and Methods section, and cultured either in tryptophan-free medium (white columns), or in tryptophan-free medium supplemented with 26 μM l-tryptophan (black columns). Cell culture was stopped after 96 h from activation, and proliferation was evaluated by measuring 3[H]thymidine incorporation. For each subpopulation untreated cells, activated and cultured in the same medium, were used as control. m ± 1 SD, n = 3.
Inhibition of Cell Proliferation only Applies to Cells Responding to a Stimulus.
We noted in the experiments showed in Fig. 5 that cell proliferation could be irreversibly affected by l-kynurenine. This finding allowed us to address the question on whether IDO-dependent inhibition only applies to cells responding to a stimulus, or to all the cells in the culture, both activated and resting. MLRs were initially performed either in the presence or in the absence of l-kynurenine. 5 d after cells were washed to remove the catabolite and cultures were then restimulated with either cells from the initial stimulator or from a different donor. After 7 more days cell proliferation was measured. Results are shown in Fig. 9. Responsiveness to the original donor was dramatically reduced in cells pretreated with l-kynurenine, as compared with untreated cells. Instead, responsiveness of the same cells to the new donor was not affected.
This finding indicates that l-kynurenine-dependent block is specific to cells responding to an activating stimulus; resting cells remain viable, and can subsequently activate normally.
Discussion
In this study, we have investigated the mechanisms of IDO-dependent inhibition of PBL proliferation. For this purpose we have set up a new protocol for IDO purification. Our approach allows the same recovery, but is both easier and faster than the ones described previously (7, 9).
Afterward, we tested the capacity of purified IDO to inhibit PBL proliferation in in vitro models. We have used PHA as mitogen, instead of anti-CD3 (6, 10), to avoid any possible interference via Fc receptors, that are highly expressed on macrophage surface (15). The other model we used was based on alloreactive T cell lines. The two models gave equivalent results, but for brevity only results with PHA-activated PBLs are shown here.
In detail, we found that purified IDO at a concentration of 4,000 U/ml was able to reduce proliferation of PBLs to the half, when administered at the beginning of the test. However, because of the short half-life of the enzyme at 37°C in culture medium (unpublished data and reference 16), purified IDO was more effective when administered repeatedly than in a single dose. Better results were obtained by coincubating the enzyme with methylene blue and l-ascorbic acid: these cofactors optimize the efficiency of the enzyme, possibly by increasing production of O2 − (13). It is possible that cofactors exist in macrophage cytosol that perform the same function of the two above mentioned substances. We got a confirmation that the inhibition of PBL proliferation was due to the enzymatic activity of IDO by demonstrating that 1-methyl-DL-tryptophan, a competitive inhibitor of IDO activity (14), was able to almost completely prevent the inhibition of proliferation induced by the enzyme.
In agreement with previous studies (6), indicating that macrophage-dependent inhibition of T cell proliferation was related to a sustained arrest of the cell cycle, we found that also IDO treatment blocks cells in the mid-G1 phase.
These data, taken together, suggest that macrophage-dependent inhibition of PBL proliferation can result from the sole IDO enzymatic activity, and other antiproliferative factors released by macrophages might be not required.
We also found that, among the different subpopulations making up PBLs, only CD4+ and CD8+ T lymphocytes and NK cells were sensitive to IDO, proliferation of B lymphocytes not being affected. This finding, together with previous data indicating that proliferation of fibroblasts and of vascular endothelial cells is not affected by the presence of macrophages (10), rules out the possibility that IDO might act as a nonspecific inhibitor for proliferating cells.
Instead, inhibition of cell proliferation induced by the tryptophan catabolites resulting from IDO activity is selective, applying to specific leukocyte subpopulations and only to cells undergoing activation. Resting cells are not affected and can subsequently activate normally.
Previous data show that both macrophages (6) and dendritic cells (17) can inhibit T cell proliferation via IDO-dependent mechanisms. Our results confirm the immunological role played by the enzyme.
In this regard, it has been hypothesized that IDO expression by APCs helps maintain peripheral tolerance by regulating autoreactive T cells that have escaped intrathymic deletion (6, 10). It has also been suggested that IDO production by APCs could be part of a mechanism self-limiting immune response (17). As a matter of fact, IDO expression is induced in APCs by signals, namely IFN-γ and CD40 ligand, produced by T cells undergoing activation. Therefore, IDO might be part of a negative feedback loop.
To gain insights into the mechanisms of IDO-dependent inhibition of T and NK cell proliferation, we analyzed the effects of changes in the concentration of tryptophan and its catabolites in the extracellular microenvironment. We found that in the absence of tryptophan substantial levels of proliferation were retained. It is conceivable that cells could get the tryptophan that is required for proliferation by catabolizing proteins contained in the medium. The slight reduction in cell proliferation that was observed under these conditions could not be related to the lack of tryptophan, since addition of l-tryptophan did not increase proliferation. Instead, we think that part of factors sustaining proliferation, that are normally contained in FCS, were probably lost during the dialysis performed to remove unbound tryptophan.
The three tryptophan-derived catabolites we tested were found to be able, at various degree and under different conditions, to inhibit cell proliferation. In the presence of tryptophan, inhibition of proliferation was observed with l-kynurenine and, to a lesser extent, with picolinic acid; instead, quinolinic acid was ineffective. When the same test was performed in the absence of thryptophan, all the three catabolites were effective, at various degrees, in inducing a dose-dependent inhibition of proliferation. In the case of l-kynurenine and, to a lesser extent, of picolinic acid, inhibition was observed at concentrations below the lowest concentration that was effective in the presence of tryptophan, while in the case of quinolinic acid the inhibitory capacity was acquired de novo.
Confirming their role as major cause of IDO effect, tryptophan-derived catabolites were found to exert their inhibitory effect by blocking the cell cycle progression.
It has to be noted that at concentrations of l-kynurenine up to 500 μM the inhibition of PBL proliferation occurs only when the substance is added within 36 h from cell activation and inhibition can be overcome by removing the substance from the medium within the same period. This indicates that the target mechanisms of l-kynurenine-dependent inhibition are operative only at the early stages of cell activation. This finding is in agreement with data from (10), indicating that macrophage-dependent suppression of T cell proliferation specifically affects the initial transition from quiescence to proliferation.
Some considerations can also be done on the results of the attempt to compare the effect of exogenously added l-kynurenine with the effect displayed by kynurenine resulting from IDO expression by MCSF-treated macrophages in the presence of activated PBLs. Cocultures were performed, and kynurenine levels in the supernatants were related to the degree of inhibition observed. Confirming the role of kynurenine in IDO-dependent inhibition of cell proliferation, the amount of kynurenine in the supernatant was inversely related to the degree of cell proliferation (Fig. 7 B). We are well aware of the fact that we do not know the kinetics of release and consumption of kynurenine in cocultures of MCSF-treated macrophages and activated PBLs, therefore a precise comparison of the activity of kynurenine in the two systems cannot be done. However, it is noteworthy that the range of concentration of kynurenine that in this system is related with reduced cell proliferation is close to the range of concentration of exogenous l-kynurenine that is active in tryptophan-free medium in the absence of MCSF-treated macrophages. As a matter of fact, we found that in cocultures of MCSF-treated macrophages and activated PBLs, proliferation >50% was achieved with concentrations of l-kynurenine at 72 h below 10 μM. A degree of proliferation in the range of 50% of control was achieved in tryptophan-free medium in the absence of MCSF-treated macrophages by exogenously adding l-kynurenine at concentrations between 200 and 40 μM at the beginning of the test. Since l-kynurenine is quickly degraded in the supernatant of PBL cultures, it is conceivable that the concentration that is effective during the test is well below the concentration at the beginning of the test.
We have to take into account also the probable occurrence of synergic effect of different tryptophan-derived catabolites, as in the case of l-kynurenine and picolinic acid.
These considerations prompt us to suggest that l-kynurenine and, possibly, also other tryptophan-derived catabolites are the substances responsible for the inhibitory effect displayed by the enzyme on proliferation of IDO-sensitive cells. Our data indicate in tryptophan-derived catabolites a group of substances endowed with immunosuppressive activity.
These conclusions are in contrast with previous findings from D.H. Munn and coworkers, indicating that the inhibitory effect exerted by IDO on T cell proliferation can be due to the sole tryptophan starvation (6). The authors reach their conclusions on the basis of the fact that conditioned medium from cocultures of MCSF-treated macrophages and activated T cells was unable to support T cell proliferation unless tryptophan was added. However, in the light of our results, the data displayed by Munn and coworkers do not rule out the possibility of tryptophan-derived immunosuppressive catabolites. As a matter of fact, we have found kynurenine in the supernatant of cocultures performed as described by Munn and coworkers. Therefore, we suggest that this substance, and presumably other tryptophan-derived catabolites in the conditioned medium, was responsible of the inhibitory effect on T cell proliferation displayed by conditioned medium in (6) Also, the capacity of the same conditioned medium to support T cell proliferation after the addition of tryptophan could be explained on the basis of our results. To exert the same inhibitory effect, l-kynurenine has to be one order of magnitude more concentrate in the presence of tryptophan than in the absence of the amino acid. As it regards the experiment described in (6), we suggest that after tryptophan was added the concentration of kynurenine in the conditioned medium was not able anymore to inhibit T cell proliferation.
Instead, we speculate that IDO expressed by macrophages upon interaction with activated T cells exerts its effect on T and NK cell proliferation via two different mechanisms. On one side it starts the cascade of biochemical reactions that leads to the production of tryptophan-derived catabolites, themselves endowed with the capacity to inhibit cell proliferation. On the other side it depletes the medium of tryptophan, allowing at least three catabolites to put in action their inhibitory potential at concentrations that probably can be achieved in the extracellular microenvironment.
Acknowledgments
This work was supported by funds from PF CNR Target Project Biotechnology, MURST-CNR 2000, and Ministero della Sanità RF 1999, RF 2000 and RF2001. M. Tonetti was in part supported by CNR Target Project Biotechnology. R. Rotondo was recipient of a Villa Rusconi fellowship.
Preliminary data was presented to the XVIII International Congress of the Transplantation Society, August 27–September 1, 2000, Rome, Italy.
Footnotes
*
Abbreviation used in this paper: IDO, indoleamine 2,3-dioxygenase.
References
- 1.Taylor, M.W., and G. Feng. 1991. Relationship between interferon-γ, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 5:2516–2522. [PubMed] [Google Scholar]
- 2.Widner, B., G. Weiss, and D. Fuchs. 2000. Tryptophan degradation to control T-cell responsiveness. Immunol. Today. 21:250. [DOI] [PubMed] [Google Scholar]
- 3.Mellor, A.L., and D.H. Munn. 1999. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol. Today. 20:469–473. [DOI] [PubMed] [Google Scholar]
- 4.Munn, D.H., M. Zhou, J.T. Attwood, I. Bondarev, S.J. Conway, B. Marshall, C. Brown, and L.A. Mellor. 1998. Prevention of allogenic fetal rejection by tryptophan catabolism. Science. 281:1191–1193. [DOI] [PubMed] [Google Scholar]
- 5.Mellor, A.L., J. Sivakumar, P. Chandler, K. Smith, H. Molina, D. Mao, and D.H. Mellor. 2001. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat. Immunol. 2:64–68. [DOI] [PubMed] [Google Scholar]
- 6.Munn, D.H., E. Shafizadeh, J.T. Attwood, I. Bondarev, A. Pashine, and A.L. Mellor. 1999. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 189:1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimizu, T., S. Nomiyama, F. Hirata, and O. Hayaishi. 1978. Indoleamine 2,3-dioxygenase. Purification and some properties. J. Biol. Chem. 253:4700–4706. [PubMed] [Google Scholar]
- 8.Bradford, M. 1976. A rapid and sensitive method for the quantitation of microgram of protein utilizing the principles of protein-dye binding. Anal. Biochem. 72:248–254. [DOI] [PubMed] [Google Scholar]
- 9.Takikawa, O., T. Kuroiwa, F. Yamazaki, and R. Kido. 1988. Mechanism of interferon-γ action. Characterization of indoleamine 2,3-Dioxygenase in cultured human cells induced by interferon-γ and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J. Biol. Chem. 263:2041–2048. [PubMed] [Google Scholar]
- 10.Munn, D.H., J. Pressey, A.C. Beall, R. Hudes, and M.R. Alderson. 1996. Selective activation-induced apoptosis of peripheral T cells imposed by macrophages. A potential mechanism of antigen-specific peripheral lymphocyte deletion. J. Immunol. 156:523–532. [PubMed] [Google Scholar]
- 11.Nicoletti, I., G. Migliorati, M.C. Pagliacci, F. Grignani, and C. Riccardi. 1991. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139:271–279. [DOI] [PubMed] [Google Scholar]
- 12.Damonte, G., A. Sdraffa, E. Zocchi, L. Guida, C. Polvani, M. Tonetti, U. Benatti, P. Boquet, and A. De Flora. 1990. Multiple small molecular weight guanine nucleotide-binding proteins in human erythrocyte membranes. Biochem. Biophys. Res. Commun. 166:1398–1405. [DOI] [PubMed] [Google Scholar]
- 13.Hirata, F., and O. Hayaishi. 1975. Studies on indoleamine 2,3-dyoxigenase. I. Superoxid anion as substrate. J. Biol. Chem. 250:5960–5966. [PubMed] [Google Scholar]
- 14.Cady, S.G., and M. Sono. 1991. 1-methyl-DL-tryptophan, β-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and β-[3-Benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch. Biochem. Biophys. 291:326–333. [DOI] [PubMed] [Google Scholar]
- 15.Fanger, M.W., L. Shen, R.F. Graziano, and P.M. Guyre. 1989. Cytotoxicity mediated by human Fc receptors for IgG. Immunol. Today. 10:92–99. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto, S., and O. Hayaishi. 1967. Tryptophan pyrrolase of rabbit intestine. D- and L-tryptophan-cleaving enzyme or enzymes. J. Biol. Chem. 242:5260–5266. [PubMed] [Google Scholar]
- 17.Hwu, P., M.X. Du, R. Lapointe, M. Do, M.W. Taylor, and H.A. Young. 2000. Indoleamine 2,3-dioxigenase production by human dendritic cells results in the inhibition of T cell proliferation. J. Immunol. 164:3596–3599. [DOI] [PubMed] [Google Scholar]