Crucial Role of Interferon Consensus Sequence Binding Protein, but neither of Interferon Regulatory Factor 1 nor of Nitric Oxide Synthesis for Protection Against Murine Listeriosis (original) (raw)
Abstract
Listeria monocytogenes is widely used as a model to study immune responses against intracellular bacteria. It has been shown that neutrophils and macrophages play an important role to restrict bacterial replication in the early phase of primary infection in mice, and that the cytokines interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) are essential for protection. However, the involved signaling pathways and effector mechanisms are still poorly understood. This study investigated mouse strains deficient for the IFN-dependent transcription factors interferon consensus sequence binding protein (ICSBP), interferon regulatory factor (IRF)1 or 2 for their capacity to eliminate Listeria in vivo and in vitro and for production of inducible reactive nitrogen intermediates (RNI) or reactive oxygen intermediates (ROI) in macrophages. ICSBP−/− and to a lesser degree also IRF2−/− mice were highly susceptible to Listeria infection. This correlated with impaired elimination of Listeria from infected peritoneal macrophage (PEM) cultures stimulated with IFN-γ in vitro; in addition these cultures showed reduced and delayed oxidative burst upon IFN-γ stimulation, whereas nitric oxide production was normal. In contrast, mice deficient for IRF1 were not able to produce nitric oxide, but they efficiently controlled Listeria in vivo and in vitro. These results indicate that (a) the ICSBP/IRF2 complex is essential for IFN-γ–mediated protection against Listeria and that (b) ROI together with additional still unknown effector mechanisms may be responsible for the anti-Listeria activity of macrophages, whereas IRF1-induced RNI are not limiting.
L isteria monocytogenes, a grampositive facultative intracellular bacterium, infects macrophages and hepatocytes in mice and has been used as a classic model to study immune responses against intracellular bacteria (1). Neutrophile granulocytes (2), γδ T cells (3), and above all macrophages (4) are important during the early phase of the immune response. In SCID mice lacking mature B and T lymphocytes, NK cells activated by macrophage-derived TNF-α have been shown to activate the listericidal effector mechanisms of macrophages via secretion of IFN-γ (5). These cells are able to restrict initial replication of Listeria in murine liver and spleen, since IFN-γ inhibits evasion of Listeria from phagosomes into the cytoplasm (6). Specific T cells are needed for final elimination of the pathogen (7) and also for protection against secondary infection (8–10). Studies of Listeria infection in mice deficient for IFN-γ (11), IFN-γ receptor (12) or TNF receptor 1 (13) have shown that the two cytokines IFN-γ and TNF-α are crucial for survival. However, the involved signaling pathways are not known, and the effector mechanisms used by macrophages for killing of Listeria are still debated. The role of reactive oxygen intermediates (ROI)1 (14–19) as well as reactive nitrogen intermediates (RNI) (20–23) has been analyzed repeatedly; these experiments revealed variations between different experimental setups and analyzed species.
Much information has accumulated about molecular and in vivo biological function of IFNs over the past 15 years (for review: see 24, 25). Two different pathways can be distinguished: IFN-α and -β are binding to the type I IFN receptor, whereas IFN-γ binds to the type II IFN receptor. Analysis of gene-targeted mice deficient for only one (12, 26) or both of these receptors (27) have revealed that depending upon the type of the pathogen these two systems are either redundant or complementary in their antimicrobial activity (for review see 28).
A variety of IFN-induced transcription factors have now been described, most of them belonging to the structurally related family of the interferon regulatory factors (IRFs) and some being identical with signal transducers and activators of transcription (STATs; Table 1). It has been revealed that there is an overlap between the two IFN systems at the level of transcription. Whereas some components of the interferon-stimulated gene factor (ISGF) 3α are only induced by type I IFN (29), IRF1 (30, 31) and STAT1 (32, 33) can be upregulated via both IFN receptors or by viruses directly (31), and interferon consensus sequence binding protein (ICSBP) is the prototype of a type II IFN-induced factor (34, 35). IRF2 is omitted from Table 1, because the way of its induction has not been clearly elucidated so far. The fact that IRF2 is lacking in ICSBP −/− mice (36) suggests induction via IFN-γ pathway. In vitro transfection systems with reporter genes have revealed that IRF1 (37) and ISGF3 (29) are activating transcription of genes containing the interferon-stimulated response element (ISRE) in their promotor sequence, whereas ICSBP (38) and IRF2 (37) have repressor activity for ISRE-containing genes.
Table 1.
Interferon Signal Transduction
| Interferons | ||
|---|---|---|
| IFN type I (α/β) | IFN type II (γ) | |
| Receptors | ||
| Type I IFN-R | Type II IFN-R | |
| Protein kinases | ||
| Only Type I | Type I/II | Only Type II |
| Tyk-2 | Jak-1, PKC | Jak-2 |
| Transcription factors | ||
| Only Type I | Type I/II | Only Type II |
| ISGF-3a (+) | STAT 1 (+) | ISGF-3γ (+) |
| (= STAT1b/STAT2) | (= p 91 monomer) | (= p48) |
| IRF-1 (+) | GAF (+) | |
| (= p91 dimer) | ||
| ICSBP (−) | ||
| IFN-stimulated genes | ||
| Only type I | Type I/II | Only Type II |
| Mx-1 | MHC class I | MHC class II |
| IFN type I | ||
| iNOS | ||
| antimicrobicidal activity |
The generation of gene-targeted mice for the transcription factors IRF1 (39, 40), IRF2 (39) and ICSBP (36) allows to test for their biological role and their induction in different infectious disease models, especially for activation of macrophages. This study therefore evaluated the susceptibility of these mouse strains to Listeria infection in vivo and compared it to some macrophage effector functions upon IFN-γ stimulation in vitro.
Materials and Methods
Mice.
Mice deficient for ICSBP (background C57BL6× 129Sv), IRF1(129Sv), IRF2 (C57BL/6), IFN I and II receptor (both 129Sv) were generated by gene targeting in embryonic stem cells as described (12, 26, 36, 39). IRF1-deficient mice were kindly provided by Prof. Charles Weissmann (Institute for Molecular Biology I, University of Zürich, Switzerland). IFN type I receptor−/− (A129) and IFN type II receptor−/− (G129) mice were obtained from the breeding colony of Prof. M. Aguet (Institute for Molecular Biology I, University of Zürich, Switzerland). Control C57BL/6 or 129Sv mice as well as RAG2−/− mice were obtained from the Institute for Laboratory Animals (Veterinary Hospital, Zürich, Switzerland). Mice were used at 6–10 wk of age. The different breedings (except A129 and G129) and all the experiments were performed under conventional (non-SPF) conditions.
Listeria Culture and Infection.
Listeria monocytogenes was originally obtained from B. Blanden (Canberra, Australia). It was cultured in trypticase soy broth (BBL Microbiology Systems, Cockeysville, MD), and overnight cultures were titrated on tryptose blood agar plates (Difco Laboratories, Detroit, MI). For injection, the original culture was diluted in BSS to inject the indicated dose in 200 μl for i.v. or 30 μl for injection into the footpad (i.f.).
Determination of Bacterial Titers.
On the indicated days after infection the whole spleen and one lobe of the liver were taken out and homogenized. Bacterial titers were determined by plating out four serial 10-fold dilutions of organ suspensions on tryptose blood agar plates.
Adoptive Transfer of Spleen Cells.
On day 0, spleen single cell suspensions were let to adhere to plastic to deplete them from macrophages. After 2 h 3 × 107 splenocytes were transferred into nonirradiated RAG2−/− recipients. On day 1 the recipients were infected with 2 × 105 CFU of Listeria, and on day 10 liver and spleen were taken out to determine bacterial titers.
Peritoneal Macrophage Cultures.
Peritoneal macrophages (PEM) of different strains were elicited by injection of 2 ml of a starch solution (2%; Merck, Darmstadt, Germany) intraperitoneally on day −5 and harvested on day 0 by rinsing the peritoneal cavity with 10 ml of cold BSS. The macrophages were washed three times with BSS supplemented with albumin to prevent clumping and then plated on cover slips in 24-well plates. Cells were cultured in IMDM (Gibco, Basel, Switzerland) supplemented with 10% FCS, glutamine, and 50 μg/ml gentamicin, an only extracellularly effective antibiotic. After 2 h of adherence the cover slips were washed twice and put in 1 ml IMDM. The cultures were stimulated with 200 ng/ml LPS, with 200 U/ml recombinant murine IFN-γ (Genzyme, Cambridge, MA) or a combination of both for 42 h and then used for determination of nitric oxide (NO) production, of respiratory burst or of Listeria killing in vitro. In those cultures used for killing assays, the medium was changed to antibiotic-free after 24 h.
Determination of NO and Respiratory Burst.
NO production was measured by determination of nitrite accumulation in PEM cultures with Griess reagent (0.05% _N_-1-naphthyl-ethylene-diamine-dihydrochloride/0.5% sulfanilamide/2.5% phosphoric acid; all from Fluka, Buchs, Switzerland) as described (41). In brief, 50 μl cell culture supernatant was added to 150 μl Griess reagent in 96well plates and incubated at room temperature for 10 min. Absorption was read with an ELISA reader at 570 and 630 nm.
Respiratory burst was measured as H2O2 production by cultured PEM upon PMA (Sigma, Buchs, Switzerland) stimulation as described (18). In brief, H2O2 secretion of macrophages was quantified by chemiluminescence under presence of horseradish peroxidase type I (Sigma) and 5-amino-2,3-dihydro-1,4-phthalazinedione (luminol; Sigma) after triggering with 50 ng/ml PMA. Light emission was discontinuously measured over 15 min in a LKB 1251 luminometer (LKB, Bromma, Sweden). Values in mV were converted into pmol H2O2 after calibration by the scopoletin method (42). The cells on the cover slips were counted, and values for NO and H2O2 calculated as nmol/105 cells. In Fig. 4 B stimulation index of stimulated versus unstimulated cultures is shown, because absolute values of respiratory burst varied between the experiments. PMA was used to trigger respiratory burst because, as a chemically defined substance, it is the most reliable burst trigger. Also opsonized Listeria, BCG or zymosan could be used with similar capacities to trigger burst (23, 43), but more variability. Because we investigated mouse strains deficient for various IFNdependent transcription factors, the induction phase of NADPH oxidase during 2 d under IFN-γ stimulation is important in our experiment, whereas the effector phase of the burst trigger is only used as read-out to measure the enzyme activity by providing the best stimulator (PMA) and eccess of substrate (luminol).
Figure 4.


NO production and respiratory burst in PEM cultures. PEMs of mice deficient for different IFN-related transcription factors were cultured as described in Fig. 3. (A) After 42 h NO production was measured by determination of nitrite accumulation in the culture supernatants by using Griess reagent. Values are calculated as nmol nitrite per 105 cells. One of three independent and comparable experiments is shown. (B) Respiratory burst capacity of the same PEMs was determined by measuring H2O2 production by chemiluminescence after stimulation with PMA. Values were first calculated as nmol H2O2 per 105 cells, and then a stimulation index of stimulated versus unstimulated cultures was determined (stimulation index of unstimulated culture = 1). Error bars indicate standard deviation.
In Vitro Killing Assay.
PEM cultures in antibiotic-free medium as described above were infected with 107 CFU of Listeria from an overnight culture, washed three times and opsonized with normal human serum. After 15 min of phagocytosis the infected cultures were washed thoroughly, and gentamicin-containing medium and the respective stimulators were added. To determine the infection rate at time point t 0, three cover slips were taken out. The remaining ones were further cultured for 7 h to allow digestion of Listeria by macrophages. After 7 h the cover slips were taken out, dried, and then stained according to May-Grünwald-Giemsa. For each mouse strain and each stimulation a total of 600 macrophages were counted under the microscope to determine the number of _Listeria_-infected cells. The change of infected macrophages was calculated in percentage of the infection rate at t 0. For details, see reference 18.
Immunohistochemistry.
Mice infected with 5 × 103 CFU of Listeria i.v. were sacrificed on day 5 or 6. Organs were immersed in Hank's BSS and frozen in liquid nitrogen. 5-μm cryosections were fixed with acetone for 10 min, immunostained for Listeria with a polyclonal rabbit anti-Listeria serum (diluted 1/2,000; kindly provided by Professor J. Bille, Institute of Microbiology, University Hospital of Lausanne, Switzerland) and for iNOS with a polyclonal rabbit anti-iNOS serum (diluted 1/1,500; Biomol, Plymouth, PA). Bound primary antibodies were detected using a sandwich staining procedure. Sections were incubated with alkaline phosphatase-labeled goat anti–rabbit Ig (diluted 1/80; Jackson Laboratories, Bar Harbor, Maine) followed by alkaline phosphataselabeled donkey anti–goat Ig (diluted 1/80; Tago). Dilutions of secondary reagents were made in TBS containing 5% normal mouse serum. All incubation steps were done for 30 min at room temperature. Alkaline phosphatase was visualized using naphthol ASBI phosphate and New Fuchsin (Sigma) as substrate, which yields a red color reaction product. Endogenous alkaline phosphatase was blocked by levamisole. Sections were counterstained with hemalum, and cover slips were mounted with glycerol/gelatin.
Results
Enhanced Bacterial Replication and Increased Lethality after Listeria Monocytogenes Infection in ICSBP-deficient Mice.
Gene targeted mice deficient for ICSBP, IRF1, or IRF2 were infected with various doses of Listeria intravenously or peripherally i.f., and survival was monitored daily (Table 2). All ICSBP−/− mice died after injection of a dose as low as 50 CFU of Listeria, whereas five of six IRF2−/− mice succumbed to a dose of 5 × 103 CFU within 12 d. In contrast, IRF1−/− and wild-type mice resisted to a dose of 5 × 103 CFU injected intravenously. However, IRF1−/− mice on C57BL/6 background and held under strict SPF conditions also showed enhanced susceptibility to Listeria, when injected with a 5–10-times higher dose intraperitoneally (Ferrick, D., and H.W. Mittrücker, personal communication). Listeria titers in liver and spleen were determined 24 h after a high dose (2 × 105 CFU) and 5 d after an intermediate dose (5 × 103 CFU) of Listeria injected intravenously. In the first 24 h, when neutrophils seem to play an important role (2), there was almost no titer difference between the three strains and only a 10-fold difference compared to control mice (data not shown). However, after 5 d when activated macrophages are essential for control of Listeria infection, ICSBP−/− and IRF2−/− showed between 102- and 106-fold higher titers in liver and spleen, whereas IRF1−/− mice controlled Listeria replication comparable to controls (Fig. 1 A). In vitro gene regulation studies have revealed that ICSBP and IRF2 form complexes which then have a markedly enhanced DNA binding capacity to ISRE compared to the single factors (44). In contrast to IRF1, they are both negative regulators of classical IFN-induced genes. However, both transcription factors are obviously of major importance for early anti-Listeria immune responses. Since it has been shown that ICSBP−/− mice do not express IRF2 (although the gene is intact [36]), this can explain the even more drastic phenotype of ICSBP−/− compared to IRF2−/− mice, because they represent functionally a double knock-out phenotype.
Table 2.
Resistance of Different Mouse Strains against Listeria Infection
| Mouse strain | Listeria dose | Route of infection | Surviving/ infected | |
|---|---|---|---|---|
| d6 | d12 | |||
| CFU | ||||
| ICSBP−/− | 5 × 103 | i.v. | 0/7 | 0/7 |
| 5 × 102 | i.f. | 0/7 | 0/7 | |
| 5 × 101 | i.f. | 3/5 | 0/5 | |
| IRF1−/− | 5 × 103 | i.v. | 11/12* | 5/6 |
| IRF2−/− | 5 × 103 | i.v. | 12/13* | 1/6 |
| A129 | 5 × 103 | i.v. | 7/7 | 7/7 |
| G129 | 5 × 103 | i.v. | 0/9 | 0/9 |
| 5 × 102 | i.f. | 5/6 | 0/6 | |
| 5 × 101 | i.f. | 6/6 | 1/6 | |
| wt C57BL/6 | 5 × 103 | i.v. | 8/8 | 7/8 |
| wt 129/Sv | 5 × 103 | i.v. | 9/9 | 9/9 |
Figure 1.


Listeria titers in organs of different mouse strains deficient for molecules in the IFN signaling pathway. (A) Mice deficient for IFN- related transcription factors (ICSBP, IRF1, IRF2; filled diamonds) or (B) mice lacking functional IFN type I (A129; filled diamonds) or type II (G129; filled circles) receptors and control littermates (open diamonds) were infected with 5 × 103 CFU of Listeria intravenously. After 5 d bacterial titers were determined in liver and spleen. Groups of three to four mice were analyzed. Each symbol represents one mouse. One representative experiment of two is shown for each strain.
Competition of different transcription factors of the IRF family at the DNA binding level has been demonstrated in in vitro studies (45). It was therefore possible that lack of IFN type I–induced transcription factors would lead to increased activity of IFN type II–induced factors. To test this in vivo, we infected mice deficient for the type II (G129) or the type I (A129) IFN receptor and control mice (wt129) with 5 × 103 CFU of Listeria and determined bacterial titers in liver and spleen on day 5 (Fig. 1 B). As demonstrated earlier (12), G129 mice showed drastically enhanced bacterial replication and lethality (Table 2), whereas A129 eliminated the pathogen even more efficiently than wt129 mice. This result suggests that competition between the two signaling pathways at the transcription factor level occurs. IFN type II–induced transcription factors (and among them especially ICSBP) may compensate for the lack of IFN type I–induced factors in the A129 mouse, thereby conferring even higher resistance to Listeria infection than in control mice. Because early Listeria clearance in nude mice (46) has been shown to be more efficient than in immunocompetent controls because their macrophages are preactivated (probably by LPS derived from normal intestinal bacteria leaking into circulation), this may be an additional factor explaining the results in A129 mice and also the difference between IRF1−/− mice held under conventional versus SPF conditions.
Capacity of Adoptively Transferred ICSBP− /− Spleen Cells to Correct Immunodeficiency of RAG2− /− Mice.
Our results of anti-Listeria immune response in ICSBP−/− mice suggested a major defect of IFN-γ-induced macrophage function, because lymphocytes, especially cytotoxic T cells, but also B cells, had been shown to function almost normally after viral infections (36, 39). Therefore macrophage functions were tested in vivo by adoptive transfer experiments and in vitro by PEM cultures.
RAG2−/− mice are devoid of functional T and B cells, but have normal macrophages and natural killer cells (47). When infected with an intermediate dose of Listeria, they are able to control bacterial replication comparable to nude mice (46), but cannot eliminate the pathogen. To test whether ICSBP-deficient T cells could develop normal specific anti- Listeria immunity, we transferred on day 0 macrophage- depleted ICSBP−/− spleen cells into RAG2−/− mice, challenged them with a high dose of Listeria (2 × 105 CFU i.v.) on day 1 and evaluated Listeria titers in liver and spleen on day 10 to look for efficiency of the specific immune response. As a positive control normal spleen cells and as a negative control no spleen cells were transferred. The result (Fig. 2) revealed no difference of Listeria counts between recipients of ICSBP−/− and ICSBP+/+ spleen cells; in contrast RAG2−/− mice that did not receive spleen cells exhibited 100- (spleen) to 1,000-fold (liver) higher bacterial counts. This result indicates that ICSBP−/− splenocytes (especially the mutant T cells) were able to promote elimination of Listeria as successfully as normal lymphocytes in cooperation with the intact macrophage compartment of the RAG2−/− mouse.
Figure 2.

Capacity of ICSBP−/− spleen cells to transfer Listeria resistance to RAG2−/− mice. Non adherent spleen cells of ICSBP+/+ (open diamonds) or ICSBP−/− (filled diamonds) mice were transferred to RAG2−/− mice. After 24 h the recipients as well as untreated RAG2−/− mice (open circles) were infected with 2 × 105 CFU of Listeria, and bacterial titers were determined on day 9 after infection to assess overall protection. Groups of four to five recipient mice were used. Each symbol represents one mouse. One of three comparable experiments is shown.
Analysis of Listeria Killing in an In Vitro PEM Culture.
To evaluate listericidal activity of macrophages of the different mutant mouse strains, we tested PEM in an in vitro killing assay. ICSBP−/−, IRF1−/−, and IRF2−/− PEM were elicited by starch injection intraperitoneally, plated onto cover slips and cultured as described in Materials and Methods. After 42 h the cultures were infected with Listeria in vitro, and the number of infected cells determined at time point t 0 and after 7 h of infection. The results (Fig. 3) show that PEM of ICSBP−/− and, to a lesser degree, IRF2−/− mice allowed enhanced replication of Listeria, whereas PEM of IRF1−/− and normal mice were able to reduce the bacterial load in these macrophage cultures. Also the number of bacteria per macrophage was higher in ICSBP−/− mice (mostly more than 10 bacteria/cell) compared to their controls (0–4 bacteria/cell), revealing some macrophages with plenty of Listeria and typical comet tails (18). This finding confirms the defect in macrophage effector function, which correlates with the in vivo susceptibility of these mouse strains to Listeria (ICSBP−/− >IRF2−/− >IRF1−/−).
Figure 3.

Elimination of Listeria from a PEM culture stimulated with LPS, IFN-γ or both. PEM of mice deficient for different IFN-related transcription factors were elicited with starch solution, harvested after 5 d and put into culture on cover slips under presence of the indicated stimulators. After 42 h (at time point t 0) they were infected with Listeria and allowed to digest the bacteria during 7 h. Then, cover slips were taken out, dried and May-Grünwald-Giemsa stained. The number of infected macrophages was counted under the microscope, compared to the infection rate at t 0 and expressed as difference in percentage of infected cells. Error bars represent standard deviation. Examples for absolute numbers: the number of infected macrophages at t 0 was between 80 and 106/200 for all strains; the number of infected cells in IFN-γ-stimulated cultures after 7 h of digestion was 19/200 for wild-type and 168/200 for ICSBP−/− mice.
Evaluation of NO Synthesis and Respiratory Burst as AntiListeria Effector Functions of Macrophages.
The effector mechansim responsible for listericidal properties of macrophages is widely studied and still not clearly defined. ROI (14–19) as well as RNI (20–23) have been proposed to be of major importance. Therefore NO production (measured as nitrite accumulation in culture medium) and respiratory burst upon PMA stimulation (H2O2 production measured by chemiluminescence) were tested in PEM cultures stimulated with LPS and/or IFN-γ as described in Materials and Methods. NO production (Fig. 4 A) was absent in IRF1−/− mice confirming earlier results that iNOS cannot be induced by IFN type I or II combined with LPS and/or TNF in the absence of IRF1 (48, 49). In contrast, iNOS activity was normal in ICSBP−/− and in IRF2−/− mice as well as in G129 mice (50). This finding in IRF2−/− mice differs from recently published results (51). The high susceptibility of the ICSBP−/− and G129 strains to Listeria infection in vivo and in vitro indicates that the NO effector mechanism does not play a limiting role in Listeria clearance. The phenotype of IRF1−/− mice found here is compatible to the published results of Listeria infection in iNOS-deficient mice (23). These mice showed also a slightly enhanced bacterial replication (10–100-fold) and a higher lethality to Listeria infection, but only after injection of 6 × 104 CFU i.v. The LD50 of iNOS-deficient mice was only a factor 10 lower compared to their normal littermates, whereas in the case of ICSBP−/− mice this difference is more than 10,000fold (Table 1; LD50 for C57BL/6 mice is ∼3 × 105 CFU [52]). The fact that iNOS induction in the susceptible strains (ICSBP−/−, IRF2−/−) was higher than in controls (Figs. 4 A, and 5) could even indicate that NO may have a toxic effect on infected cells during murine listeriosis.
Figure 5.
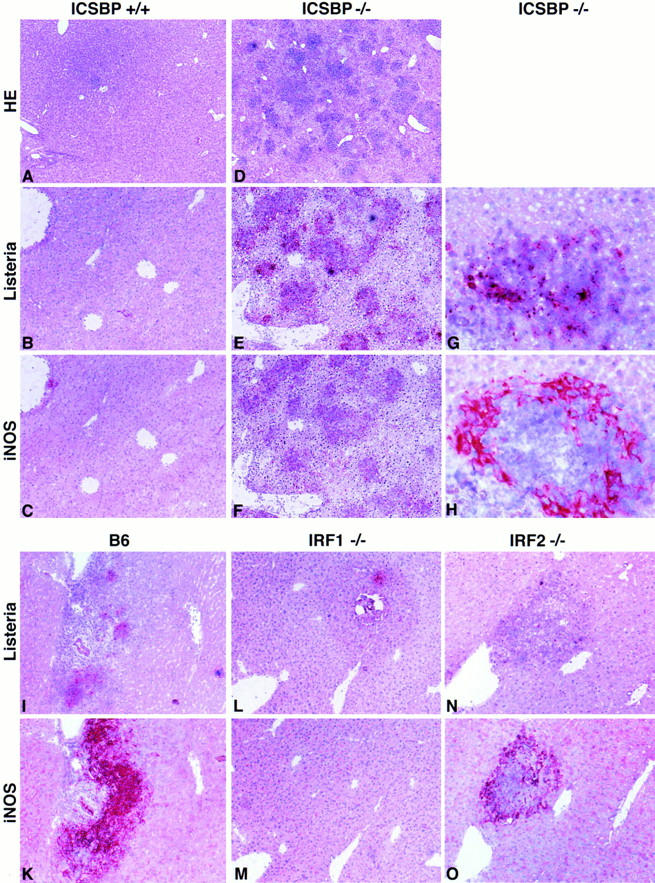
Listeria replication and iNOS expression in the liver after infection with Listeria. ICSBP−/− (D–H), IRF1−/− (L, M), IRF2−/− (N, O), and control mice (A–C, I, K) were infected with 5 × 103 CFU of Listeria. After 5 or 6 d liver and spleen were taken out. Conventional HE staining (A, D) and immunohistology for Listeria (B, E, G, I, L, N) and iNOS (C, F, H, K, M, O) was performed using polyclonal primary antibodies. Magnifications: (A, D), ×35, (B, C, E, F, I–O), ×60 (G, H), ×220.
With respect to respiratory burst upon PMA challenge, all mouse strains showed comparable basal activity of unstimulated cultures; however, in ICSBP−/− and IRF2−/− mice ROI production could not be stimulated by IFN-γ (Fig. 4 B) and was 3–5 min delayed compared to control mice (data not shown). This finding suggests that deficient ROI production might be partially responsible for the high susceptibility of ICSBP−/− mice to Listeria, but it cannot fully explain the drastic phenotype of these mice. At least a third effector pathway not yet known may have to be evoked to explain this phenotype (see Discussion). In addition, the fact that LPS-induced respiratory burst was enhanced in IRF2−/− mice correlates inversely with a recently published finding of high IRF2 levels in LPS-hyporesponsive mouse strains (53). Thus, IRF2 may mediate the macrophage deactivating effect of LPS (42).
Expression of iNOS in Liver and Spleen after Listeria Infection of ICSBP− /− and IRF2− /− Mice, but not of IRF1− /− Mice.
Apart from macrophages hepatocytes are a major target cell in murine listeriosis. They are infected by direct cell-to-cell spread of the pathogen that is able to associate with actin filaments of the cytoskeleton (54). Hepatocytes can produce NO. Therefore, to see whether the phenotype of ICSBP−/− and IRF2−/− mice is due to a localized inability of iNOS expression in the liver, immunohistological analysis of iNOS expression in liver (Fig. 5) and spleen (not shown) after Listeria infection was performed. Mice of the three gene-targeted and control strains were infected with Listeria (5 × 103 CFU i.v.). On day 5 or 6 liver and spleen were taken, cryosectioned, and then immunostained for Listeria and for iNOS with an appropriate polyclonal rabbit antiserum. Induction of iNOS comparable to wild-type mice could be demonstrated in all strains except IRF1−/− (Fig. 5, C, F, K, M, O). It was abundant in regions where Listeria and abcesses were found (detail shown in Fig. 5, G and H). In addition fulminant Listeria proliferation in liver (Fig. 5 E) and spleen of ICSBP−/− mice was found with accompanying tissue destruction (Fig. 5 D) correlating with the high bacterial titers (Fig 1). This analysis also shows that the susceptibility of ICSBP−/− and IRF2−/− mice to Listeria is not due to inefficient NO production in the liver. In contrast, IRF1−/− mice were well protected and did not express iNOS in the liver. This cannot be explained by earlier decline of the bacterial load because these mice had equal Listeria titers in the liver as wild-type mice 5 d after infection (Fig. 1 A). The same analysis was performed on spleen sections with comparable results (not shown).
Discussion
The type II IFN system has been shown to be of crucial importance for immunity against Listeria, because mice deficient for IFN-γ (11) or the type II IFN receptor (12) are highly susceptible to this bacterium. However, the intracellular signalling pathway and the final effector mechanisms involved in Listeria clearance are only incompletely understood. The presented analysis of three gene-targeted mouse strains deficient of ICSBP, IRF1, or IRF2 with respect to their capacity to survive and eliminate Listeria in vivo and in vitro and to the ability of their macrophages to respond with ROI or RNI production upon IFN-γ stimulation suggests three major conclusions: (a) ICSBP/IRF2 complex (but not IRF1) is of crucial importance for murine innate immunity to Listeria in vivo and in vitro, (b) iNOS induction and NO synthesis play no limiting role for anti- Listeria activity of macrophages upon IFN-γ stimulation, (c) stimulation of ROI production by IFN-γ together with a postulated third yet unknown effector pathway in macrophages may be responsible for protection in the early phase of primary Listeria infection. The role of NO for antimicrobial activity of macrophages has been tested in other infectious model systems. It seems to play an important role in leishmaniasis (55, 56) and tuberculosis (48), but has no limiting effect in toxoplasmosis (57) and listeriosis (this study, references 58, 59).
Our results of the analysis of mice deficient for IFN type I or II receptors revealed surprisingly that the type I IFN receptor-deficient mice (A129) were better protected than their normal littermates. This finding may reveal in vivo competition of transcription factors of both signaling pathways at the DNA binding level (45) suggesting that absence of the type I system enhances function of the type II system and concurrently Listeria protection. A potentiating effect of LPS leaking through from intestinal bacteria leading to macrophage preactivation may be involved.
From the analysis of TNF receptor 1-deficient (13) mice it is known that TNF-α is a second important cytokine for protection against Listeria. It is produced by macrophages upon infection with Listeria and may act via the following two pathways: (a) the SCID model revealed that TNF-α is necessary for activation of NK cells that then produce IFN-γ to further induce TNF receptor 1 and TNF-α expression (60, 61) and macrophage effector functions (4); (b) macrophage- or γδ T cell–derived TNF-α may act in an autocrine or paracrine fashion directly on macrophages to activate anti-Listeria effector molecules. Involvement of the ICSBP/ IRF2 complex in the signaling cascade of the TNF receptor could theoretically explain the described in vivo findings, but this has not been formally demonstrated so far. In addition, another transcription factor, NF-IL6, which can be upregulated by LPS/CD14 and also by TNF-α (62), is important for clearance of Listeria as demonstrated in the NF-IL6–deficient mice (63).
From our findings in three different mouse strains and from the published literature, the following model of signaling events in activation of anti-Listeria immunity may be proposed (Fig. 6): after activation of IFN receptors various tyrosine kinases are induced and STAT proteins phosphorylated (Table 1); they regulate the induction and activation of transcription factors of the IRF family among which the exclusively IFN-γ-dependent ICSBP mediates protection against Listeria. Two major questions remain open: (a) What molecules are involved in the signalling of the TNF receptor that could explain its importance for anti-Listeria immunity (ICSBP, NF-IL6, other transcription factors, indirect effect via NK cell activation)? (b) How do macrophages kill Listeria? Our results, but also the published ones on IFN type II receptor- and iNOS-deficient mice, rather argue against RNI production being a limiting factor. ROI may be involved since ICSBP- and NF-IL6–deficient mice had reduced respiratory burst, and this correlated with high susceptibility to Listeria infection. But still there may be a potential third mechanism involved to explain the drastic phenotype of ICSBP−/− mice. Studies on iron metabolism of peritoneal macrophages (64) and murine β-thalassemia (65) suggested that iron scavengers lead to enhanced, and iron overload to reduced, resistance to Listeria by direct interference with the essential bacterial iron metabolism. Whether IFN-γ– and/or TNF-α–mediated enhancement of iron-binding proteins can explain resistance to murine listeriosis remains to be investigated.
Figure 6.

Proposed possible signaling events in macrophages after Listeria infection. IFN-γ–induced ICSBP is of crucial importance for protection in murine listeriosis, probably partly via ROI production. G-CSF (66) plays a minor and IRF1-induced RNI (23) no limiting role for bacterial resistance. A potentiating effect or an additional factor is postulated. How ICSBP, NF-IL6 or other transcription factors are involved in TNF-mediated protection against Listeria remains to be determined.
Acknowledgments
We would like to thank A. Schaffner and H. Hengartner for expert advice and helpful discussion; C. Weissmann and M. Aguet for mutant mouse strains; J. Bille for anti-Listeria serum; A. Althage, L. Vlk, and H. Haber for excellent technical support; H. Neff and N. Wey for photographs; and E. Hörhager, and S. Kläusli for secretarial help.
This work was supported by the Swiss National Science Foundation grant no. 31-32195.91 to R.M. Zinkernagel, and grants no. 31-4577.95 and 32-42536.94 to G. Schoedon), the Kanton of Zürich and the German Science Foundation (DFG).
Footnotes
1 Abbreviations used in this paper: ICSBP, interferon consensus sequence binding protein; i.f., into the footpad; iNOS, inducible nitric oxide synthase; IRF1/2, interferon regulatory factor 1/2; ISGF, interferon-stimulated gene factor; ISRE, interferon-stimulated response element; NO, nitric oxide; PEM, peritoneal macrophage; RNI, reactive nitrogen intermediates; ROI, reactive oxygen intermediates; STAT, signal transducers and activators of transcription.
References
- 1.Kaufmann SHE. Immunity to intracellular bacteria. Annu Rev Immunol. 1993;11:129–163. doi: 10.1146/annurev.iy.11.040193.001021. [DOI] [PubMed] [Google Scholar]
- 2.Conlan JW, North RJ. Neutrophil-mediated dissolution of infected host cells as a defense strategy against a facultative intracellular bacterium. J Exp Med. 1991;174:741–744. doi: 10.1084/jem.174.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hiromatsu K, Yoshikai Y, Matsuzaki G, Ohga S, Muramori K, Matsumoto K, Bluestone JA, Nomoto K. A protective role of gamma/delta T cells in primary infection with Listeria monocytogenes in mice. J Exp Med. 1992;175:49–56. doi: 10.1084/jem.175.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bancroft GJ, Schreiber RD, Unanue ER. Natural immunity: a T-cell-independent pathway of macrophage activation, defined in the scid mouse. Immunol Rev. 1991;124:5–24. doi: 10.1111/j.1600-065x.1991.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 5.Wherry JC, Schreiber RD, Unanue ER. Regulation of gamma interferon production by natural killer cells in scid mice: roles of tumor necrosis factor and bacterial stimuli. Infect Immun. 1991;59:1709–1715. doi: 10.1128/iai.59.5.1709-1715.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Portnoy DA, Schreiber RD, Connelly P, Tilney LG. Gamma interferon limits access of Listeria monocytogenesto the macrophage cytoplasm. J Exp Med. 1989;170:2141–2146. doi: 10.1084/jem.170.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bancroft GJ, Schreiber RD, Bosma GC, Bosma MJ, Unanue ER. A T cell-independent mechanism of macrophage activation by interferon-gamma. J Immunol. 1987;139:1104–1107. [PubMed] [Google Scholar]
- 8.Mielke MEA, Ehlers S, Hahn H. T-cell subsets in delayed-type hypersensitivity, protection, and granuloma formation in primary and secondary Listeria infection in mice: superior role of Lyt-2+ cells in acquired immunity. Infect Immun. 1988;56:1920–1925. doi: 10.1128/iai.56.8.1920-1925.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harty JT, Schreiber RD, Bevan MJ. CD8 T cells can protect against an intracellular bacterium in an interferon gamma-independent fashion. Proc Natl Acad Sci USA. 1992;89:11612–11616. doi: 10.1073/pnas.89.23.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kägi D, Ledermann B, Bürki K, Hengartner H, Zinkernagel RM. CD8+ T cell-mediated protection against an intracellular bacterium by perforin-dependent cytotoxicity. Eur J Immunol. 1994;24:3068–3072. doi: 10.1002/eji.1830241223. [DOI] [PubMed] [Google Scholar]
- 11.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science (Wash DC) 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 12.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor [see comments] Science (Wash DC) 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 13.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature (Lond) 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 14.Lepay DA, Steinman RM, Nathan CF, Murray HW, Cohn ZA. Liver macrophages in murine listeriosis. J Exp Med. 1985;161:1503–1512. doi: 10.1084/jem.161.6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peck R. Gamma interferon induces monocyte killing of Listeria monocytogenesby an oxygen-dependent pathway; alpha- or beta-interferons by oxygen-independent pathways. J Leukoc Biol. 1989;46:434–440. doi: 10.1002/jlb.46.5.434. [DOI] [PubMed] [Google Scholar]
- 16.van Dissel JT, Stikkelbroek JJM, van Furth R. Differences in the rate of intracellular killing of catalase-negative and catalase-positive Listeria moncytogenes by normal and interferon-gamma-activated macrophages. Scand J Immunol. 1993;37:443–446. doi: 10.1111/j.1365-3083.1993.tb03316.x. [DOI] [PubMed] [Google Scholar]
- 17.Leenen PJM, Canono BP, Drevets DA, Voerman JSA, Campbell PA. TNF-α and IFN-γ stimulate a macrophage precursor cell line to kill Listeria moncytogenes in a nitric oxide-independent manner. J Immunol. 1994;154:5141–5147. [PubMed] [Google Scholar]
- 18.Bläuer F, Groscurth P, Schneemann M, Schoedon G, Schaffner A. Modulation of the antilisterial activity of human blood-derived macrophages by activating and deactivating cytokines. J Interferon Cytokine Res. 1995;15:105–114. doi: 10.1089/jir.1995.15.105. [DOI] [PubMed] [Google Scholar]
- 19.Inoue S, Itagaki S-I, Amano F. Intracellular killing of Listeria monocytogenes in the J774.1 macrophagelike cell line and the lipopolysaccharide (LPS)-resistant mutant LPS1916 cell line defective in the generation of reactive oxygen intermediates after LPS treatment. Infect Immun. 1995;63:1876–1886. doi: 10.1128/iai.63.5.1876-1886.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beckerman KP, Rogers HW, Corbett JA, Schreiber RD, McDaniel ML, Unanue ER. Release of nitric oxide during the T cell-independent pathway of macrophage activation. J Immunol. 1993;150:888–895. [PubMed] [Google Scholar]
- 21.Flesch IEA, Hess JH, Kaufmann SHE. NADPH diaphorase staining suggests a transient and localized contribution of nitric oxide to host defence against an intracellular pathogen in situ. Int Immunol. 1994;6:1751–1757. doi: 10.1093/intimm/6.11.1751. [DOI] [PubMed] [Google Scholar]
- 22.Boockvar KS, Granger DL, Poston RM, Maybodi M, Washington MK, Hibbs JB, Jr, Kurlander RL. Nitric oxide produced during murine listeriosis is protective. Infect Immun. 1994;62:1089–1100. doi: 10.1128/iai.62.3.1089-1100.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie Q-w, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 24.Williams BRG. Transcriptional regulation of interferon-stimulated genes. Eur J Biochem. 1991;200:1–11. doi: 10.1111/j.1432-1033.1991.tb21041.x. [DOI] [PubMed] [Google Scholar]
- 25.Sen GC, Lengyel P. The interferon system; a bird's eye view of its biochemistry. J Biol Chem. 1992;267:5017–5020. [PubMed] [Google Scholar]
- 26.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science (Wash DC) 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 27.van den Broek MF, Müller U, Huang S, Aguet M, Zinkernagel RM. Antiviral defence in mice lacking both α/β and γ interferon receptors. J Virol. 1995;69:4792–4796. doi: 10.1128/jvi.69.8.4792-4796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van den Broek MF, Müller U, Huang S, Zinkernagel RM, Aguet M. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol Rev. 1995;148:5–18. doi: 10.1111/j.1600-065x.1995.tb00090.x. [DOI] [PubMed] [Google Scholar]
- 29.Levy DE, Lew DJ, Decker T, Kessler DS, Darnell JE., Jr Synergistc interaction between interferon-alpha and interferon-gamma through induced synthesis of one subunit of the transcription factor ISGF3. EMBO (Eur Mol Biol Organ) J. 1990;9:1105–1111. doi: 10.1002/j.1460-2075.1990.tb08216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coccia EM, Marziali G, Stellacci E, Perrotti E, Ilari R, Orsatti R, Battistini A. Cells resistant to interferon-beta respond to interferon-gamma via the Stat1-IRF-1 pathway. Virology. 1995;211:113–122. doi: 10.1006/viro.1995.1384. [DOI] [PubMed] [Google Scholar]
- 31.Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, Miyata T, Taniguchi T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell. 1988;54:903–913. doi: 10.1016/s0092-8674(88)91307-4. [DOI] [PubMed] [Google Scholar]
- 32.Meraz MA, White JM, Sheehan KCF, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 33.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 34.Driggers PH, Ennist DL, Gleason SL, Mak W-H, Marks MS, Levi B-Z, Flanagan JR, Appella E, Ozato K. An interferon gamma-regulated protein that binds the interferon-inducible enhancer element of major histocompatibility complex class I genes. Proc Natl Acad Sci USA. 1990;87:3743–3747. doi: 10.1073/pnas.87.10.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Politis AD, Sivo J, Driggers PH, Ozato K, Vogel SN. Modulation of interferon consensus sequence binding protein mRNA in murine peritoneal macrophages. J Immunol. 1992;148:801–807. [PubMed] [Google Scholar]
- 36.Holtschke T, Löhler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, Lou J, Knobeloch K-P, Gabriele L, Waring JF, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 37.Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, Taniguchi T. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell. 1989;58:729–739. doi: 10.1016/0092-8674(89)90107-4. [DOI] [PubMed] [Google Scholar]
- 38.Nelson N, Marks MS, Driggers PH, Ozato K. Interferon consensus sequence-binding protein, a member of the interferon regulatory factor family, suppresses interferoninduced gene transcription. Mol Cell Biol. 1993;13:588–599. doi: 10.1128/mcb.13.1.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kündig TM, Amakawa R, Kishihara K, Wakeham A, et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- 40.Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW, Taniguchi T. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science (Wash DC) 1994;264:1921–1923. doi: 10.1126/science.8009222. [DOI] [PubMed] [Google Scholar]
- 41.Schoedon G, Schneemann M, Hofer S, Guerrero L, Blau N, Schaffner A. Regulation of the L-argininedependent and tetrahydrobiopterin-dependent biosynthesis of nitric oxide in murine macrophages. Eur J Biochem. 1993;213:833–839. doi: 10.1111/j.1432-1033.1993.tb17826.x. [DOI] [PubMed] [Google Scholar]
- 42.Rellstab P, Schaffner A. Endotoxin suppresses the generation of O2- and H2O2by “resting” and lymphokine-activated human blood-derived macrophages. J Immunol. 1989;142:2813–2820. [PubMed] [Google Scholar]
- 43.Schaffner A, Rellstab P. Gamma-interferon restores listericidal activity and concurrently enhances release of reactive oxygen metabolites in dexamethasone-treated human monocytes. J Clin Invest. 1988;82:913–919. doi: 10.1172/JCI113698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bovolenta C, Driggers PH, Marks MS, Medin JA, Politis AD, Vogel SN, Levy DE, Sakaguchi K, Appella E, Coligan JE, et al. Molecular interactions between interferon consensus sequence binding protein and members of the interferon regulatory factor family. Proc Natl Acad Sci USA. 1994;91:5046–5050. doi: 10.1073/pnas.91.11.5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weisz A, Kirchhoff S, Levi B-Z. IFN consensus sequence binding protein (ICSBP) is a conditional repressor of IFN inducible promoters. Int Immunol. 1994;6:1125–1131. doi: 10.1093/intimm/6.8.1125. [DOI] [PubMed] [Google Scholar]
- 46.Emmerling P, Finger H, Hof H. Cell-mediated resistance to infection with Listeria monocytogenes in nude mice. Infect Immun. 1977;15:382–385. doi: 10.1128/iai.15.2.382-385.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 48.Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, Le J, Koh SI, Kimura T, Green SJ, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science (Wash DC) 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 49.Martin E, Nathan C, Xie Q-w. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med. 1994;180:977–984. doi: 10.1084/jem.180.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamijo R, Shapiro D, Le J, Huang S, Aguet M, Vilcek J. Generation of nitric oxide and induction of major histocompatibility complex class II antigen in macrophages from mice lacking the interferon gamma receptor. Proc Natl Acad Sci USA. 1993;90:6626–6630. doi: 10.1073/pnas.90.14.6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salkowski CA, Barber SA, Detore GR, Vogel SN. Differential dysregulation of nitric oxide production in macrophages with targeted disruptions in IFN regulatory factor-1 and -2 genes. J Immunol. 1996;157:3107–3110. [PubMed] [Google Scholar]
- 52.Berche PA. Resistance to listeriosis in two lines of mice genetically selected for high and low antibody production. Immunology. 1985;56:707–715. [PMC free article] [PubMed] [Google Scholar]
- 53.Barber SA, Fultz MJ, Salkowski CA, Vogel SN. Differential expression of interferon regulatory factor 1 (IRF-1), IRF-2, and interferon consensus sequence binding protein genes in lipopolysaccharide (LPS)-responsive and LPS-hyporesponsive macrophages. Infect Immun. 1995;63:601–608. doi: 10.1128/iai.63.2.601-608.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dabiri GA, Sanger JM, Portnoy DA, Southwick FS. Listeria monocytogenesmoves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci USA. 1990;87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stenger S, Thüring H, Röllinghoff M, Bogdan C. Tissue expression of inducible nitric oxide synthase is closely associated with resistance to Leishmania major. . J Exp Med. 1994;180:783–793. doi: 10.1084/jem.180.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature (Lond) 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 57.Khan IA, Matsuura T, Fonseka S, Kasper LH. Production of nitric oxide (NO) is not essential for protection against acute Toxoplasma gondiiinfection in IRF-1−/− mice. J Immunol. 1996;156:636–643. [PubMed] [Google Scholar]
- 58.Samsom JN, Langermans JA, Groeneveld PH, van Furth R. Acquired resistance against a secondary infection with Listeriamonocytogenes in mice is not dependent on reactive nitrogen intermediates. Infect Immun. 1996;64:1197–1202. doi: 10.1128/iai.64.4.1197-1202.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gregory SH, Wing EJ, Hoffman RA, Simmons RL. Reactive nitrogen intermediates suppress the primary immunologic response to Listeria. . J Immunol. 1993;150:2901–2909. [PubMed] [Google Scholar]
- 60.Aggarwal BB, Eessalu TE, Hass PE. Characterization of receptors for human tumor necrosis factor and their regulation by gamma-interferon. Nature (Lond) 1985;318:665–667. doi: 10.1038/318665a0. [DOI] [PubMed] [Google Scholar]
- 61.Collart MA, Belin D, Vassalli J-D, de Kossodo S, Vassalli P. Gamma-interferon enhances macrophage transcription of the tumor necrosis factor/cachectin, interleukin 1, and urokinase genes, which are controlled by short-lived repressors. J Exp Med. 1986;164:2113–2118. doi: 10.1084/jem.164.6.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akira S, Yoshida K, Tanaka T, Taga T, Kishimoto T. Targeted disruption of the IL-6 related genes: gp130 and NF-IL-6. Immunol Rev. 1995;148:221–253. doi: 10.1111/j.1600-065x.1995.tb00100.x. [DOI] [PubMed] [Google Scholar]
- 63.Tanaka T, Akira S, Yoshida K, Umemoto M, Yoneda Y, Shirafuji N, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell. 1995;80:353–361. doi: 10.1016/0092-8674(95)90418-2. [DOI] [PubMed] [Google Scholar]
- 64.Alford CE, King TE, Jr, Campbell PA. Role of transferrin, transferrin receptors, and iron in macrophage listericidal activity. J Exp Med. 1991;174:459–466. doi: 10.1084/jem.174.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ampel NM, Van Wyck DB, Aguirre ML, Willis DG, Popp RA. Resistance to infection in murine beta-Thalassemia. Infect Immun. 1989;57:1011–1017. doi: 10.1128/iai.57.4.1011-1017.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- 67.Fujita T, Reis LF, Watanabe N, Kimura Y, Taniguchi T, Vilcek J. Induction of the transcription factor IRF-1 and interferon-beta mRNAs by cytokines and activators of second-messenger pathways. Proc Natl Acad Sci USA. 1989;86:9936–9940. doi: 10.1073/pnas.86.24.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]