CCR6, a CC Chemokine Receptor that Interacts with Macrophage Inflammatory Protein 3α and Is Highly Expressed in Human Dendritic Cells (original) (raw)
Abstract
Dendritic cells initiate immune responses by ferrying antigen from the tissues to the lymphoid organs for presentation to lymphocytes. Little is known about the molecular mechanisms underlying this migratory behavior. We have identified a chemokine receptor which appears to be selectively expressed in human dendritic cells derived from CD34+ cord blood precursors, but not in dendritic cells derived from peripheral blood monocytes. When stably expressed as a recombinant protein in a variety of host cell backgrounds, the receptor shows a strong interaction with only one chemokine among 25 tested: the recently reported CC chemokine macrophage inflammatory protein 3α. Thus, we have designated this receptor as the CC chemokine receptor 6. The cloning and characterization of a dendritic cell CC chemokine receptor suggests a role for chemokines in the control of the migration of dendritic cells and the regulation of dendritic cell function in immunity and infection.
Dendritic cells (DCs)1 are central to antigen-specific immunity in vertebrates. They act in vivo as immune potentiating cells, sampling the antigenic environment and presenting those antigens to effector lymphocytes (for reviews see references 1 and 2). DCs are found at many sites in the body, particularly at or near surface areas where antigen contact is likely, and in the lymphoid organs where they form close associations with T cells. Migration is an integral part of DC function; precursor cells must migrate from the bone marrow to resident sites in the tissue. After exposure to antigen or inflammatory stimuli, many DCs migrate from their resident sites to the secondary lymphoid organs (1–4). Much of how DCs achieve their migratory functions remains a mystery. Recent attention has focused on the chemokine system, a multipartite superfamily of chemoattractant cytokines that induce the directed migration of leukocytes and other cells, and on the superfamily of G protein–coupled, seven transmembrane receptor proteins through which chemokines act (for reviews see references 5–8). Chemokines are involved not only in immune cell trafficking and inflammation, but are also central to infectious disease processes. For example, certain chemokines have been shown to be endogenous inhibitors of HIV-1 entry (9), and some chemokine receptors are ‘cofactors' or ‘second receptors' for HIV binding and fusion with target cells (10–15).
The understanding of how chemokines might regulate the migration of DCs is still rather sparse, though a few reports clearly demonstrate that certain CC chemokines are indeed chemoattractants for some types of DC in vitro (16, 17). It has been also demonstrated that HIV-1 can infect DCs in vitro through interactions with CC chemokine receptor (CCR)5, CXC chemokine receptor (CXCR)4, and probably another chemokine receptor (18). To address some of the questions surrounding the control of DC function at the molecular level, we sought to identify new elements of the chemokine system in these cells. Here, we report on the isolation of a new chemokine receptor, CCR6, which is highly expressed in CD34+ cell-derived DCs, but not in monocyte-derived DCs, and its interaction with a recently described CC chemokine, macrophage inflammatory protein (MIP)-3α (19).
Materials and Methods
PCR Screening of cDNA Libraries.
cDNA plasmid libraries from DCs, eosinophils, and T cells were used as templates in PCR reactions as follows: 50 μl total volume in 0.2-ml tubes with 5 μM degenerate primers second intracellular loop (IC2) sense (5′ GATCGNTAGCTNGCNATNGTNCA T/C GC) and transmembrane (TM)7 antisense (5′ GCATANATNANNGG G/A T C/T NAN G/A CAGCAGTG) in buffer containing 20 mM NH4S04, 75 mM Tris HCl, pH 9.0, at 25°C, 0.01% Tween 20, 2 mM MgCl2, 200 mM deoxynucleotide triphosphates and 2.5 units Taq DNA polymerase (ABT, London, UK). “Touchdown” PCR was performed as follows: 15 cycles of 94°C for 1 min, 65– 50°C annealing for 1 min decreasing by 1° per cycle, and at 72°C for 2 min followed by 20 cycles of 94°C for 30 s, 50°C for 1 min, and 72°C for 2 min. PCR products (550 bp) were purified after agarose gel electrophoresis by absorption to glass beads (Qiaex II; Qiagen GmbH, Hilden, Germany) and cloned via Taq DNA polymerase A overhangs into the T-tailed vector pTAg1 (R&D Sys. Inc., Abingdon, UK). Recombinant plasmids were identified by colony PCR and restriction mapping, and double-strand sequencing was performed using flanking M13 and T7 primers. Full-length BN-1 cDNA sequence was obtained by PCR amplification using BN-1–specific oligonucleotides and M13 forward and reverse primers to PCR amplify BN-1 sequences from human DC cDNA libraries constructed in the cloning vector pSPORT (GIBCO BRL, Gaithersburg, MD). In addition, radiolabeled probes generated from the PCR products were used to isolate full-length BN-1 clones by colony hybridization.
Analysis of Gene Expression Distribution.
Northern blot analysis was performed using a multiple tissue Northern blot purchased from Clontech (Palo Alto, CA). Where limited amounts of messenger RNA (mRNA) precluded direct Northern analysis, expression was assessed on Southern blots of cDNA libraries. Libraries were constructed from polyA+ RNA selected with oligotex beads (Qiagen, Chatsworth, CA). 2 μg of mRNA was primed by oligo dT (Superscript cDNA synthesis kit; GIBCO BRL), and the quality of the resulting cDNA was evaluated by three criteria: (a) a size range of cDNA from the first strand synthesis ranging from >0.5 to 5 kb on alkaline gel analysis, (b) independent clones numbering greater than 106/100 ng of unamplified cDNA, and (c) a high proportion of full-length clones with low levels of genomic or ribosomal RNA contamination (<5%), as assessed by random sequence analysis of each library. For Southern analysis, large scale plasmid DNA preparations of amplified libraries were done using a Giga prep (Qiagen). 5 μg from the primary amplified cDNA library was digested with NotI and SalI (Boehringer Mannheim, Indianapolis, IN) to release the inserts, run on a 1% agarose gel, and transferred to a nylon membrane (Schleicher & Schuell, Keene, NH). Blots were probed with a 32P-labeled BN-1 cDNA fragment under high stringency hybridization and wash conditions. The resultant hybridization patterns reflect the different sized cDNA fragments in the libraries and their relative abundance. To confirm that the restricted distribution was reflective of unamplified mRNA, semiquantitiative PCR was performed on total RNA isolated directly from monocytes, monocyte-derived DCs, and CD34+ cell-derived DCs. The amplification pattern mirrored the hybridization patterns seen in the library Southern blots.
Chromosomal Localization.
Genomic DNA (400 ng) from a panel of 20 human–rodent somatic cell hybrids cell lines (Biosmap; BIOS Labs., New Haven, CT) was used as a template in a 50-μl PCR reaction. The primer pairs were: BN1, 5′ GACCGGTACATCGCCATTGTACAGGC; BN2, 5′ CTGAACTTC- TGCCCAATAAAAGCGTAG; BN3, 5′ GTACAAGTCCTCAGGCTTCTCCTG; and BN4, 5′ TGCATAACATCTATGAGTATGTTTCAC. Human TNF-α promoter primers TNF1: 5′ CAAACACAGGCCTCAGGACTC and TNF2: 5′ AGGGAGCGTCTGCTGGCTG were a gift of Dominic Kwiatkowski. Genomic DNA from a panel of 16 somatic cell hybrids containing deletions and translocations of human chromosome 6 were a gift of Dr. J.M. Boyle (Paterson Institute, Manchester, UK). Genomic DNAs of the radiation hybrid mapping panel Genebridge 4 were obtained from the Medical Research Council HGMP Resource Centre (Cambridge, UK).
Receptor–Ligand Binding and Signaling Analyses.
Full-length BN-1 expression constructs were first made in pcDNA3 (Invitrogen, San Diego, CA) and used to generated stable transfectants in human embryonic kidney 293 (HEK293) cells as described (20). To control for levels of cell-surface expression of the receptor, subsequent constructs were made in which the M2 “Flag” epitope was attached to the NH2-terminal portion of the receptor. Anti-M2 antibody was then used to detect cell-surface BN-1–encoded polypeptide and to sort the highest expressing cells for stable propagation. Intracellular calcium signaling analyses were performed as described (21). Equlibrium binding was done using a standard filtration protocol using 106 cells incubated with 0.1 nM [125I]MIP-3α (custom labeled by Amersham, Arlington Heights, IL) in the presence increasing amounts of unlabeled MIP-3α competitor. Reactions were incubated for 2 h at 22°C before being aspirated onto GF/C filters, washed, and measured by scintillation counting. Data were analyzed using IgorPro software (WaveMetrics, Lake Oswego, Oswego, OR).
Results
Cloning of Novel Chemokine Receptor Candidate cDNAs.
Degenerate reverse transcription PCR was used to identify novel chemokine receptor-like sequences from several cell types: monocytes, cultured T cells, eosinophils, and DCs. A combination of primer pairs was used representing homologous regions within the family of known chemokine receptors and other seven transmembrane–spanning receptors associated with cell motility. Primer pairs from regions encoding TM2 and TM7, as well as the IC2, were used on substrate mRNA or cDNA from the cells listed above. Of these, primer pair IC2 sense and TM7 antisense generated from DCs, and to a lesser extent eosinophils, a 550-bp PCR product whose sequence was at that time not present in any of the available databases. The 550-bp cDNA representing the IC2–TM7 segment were then used to isolate from a DC library a larger cDNA clone, designated ‘Barney' or BN-1, encoding the entire presumptive open reading frame (ORF) of a novel chemokine receptor-like protein.
The predicted protein sequence encoded by the BN-1 ORF is shown in Fig. 1, aligned with human CXCR2 (formerly known as IL-8RB) and human CCR4. During further characterization of the function of the BN-1 protein, the same sequence was subsequently deposited as an “orphan” TM7 receptor in the EMBL/GenBank/DDBJ database as accession No. U68030, and was reported as a potential chemokine-like orphan receptor sequence of unknown function (22, 23). Multiple sequence alignment of the protein encoded by BN-1 with other human chemokine receptor sequences showed amino acid identity with CXCR1 and CXCR2 of ∼38%, but this is only slightly more identity than to the closest human CC chemokine receptor (CCR4, ∼36%). The sequence alignment of Fig. 1 shows that parts of the BN-1–encoded ORF share extensive homology with CXC receptors, but not CC receptors (e.g., within TM3), whereas other parts of the BN-1 ORF show closer homology to CCR4 (e.g., the intracellular TM3–TM4 loop). Phylogenetic depictions of the evolutionary relatedness of the various chemokine receptors show BN-1 to be on a branch containing CXCR1, CXCR2, and CXCR4, and distinct from the branch containing the previously described human CC chemokine receptors CCR1–CCR5 (not shown).
Figure 1.

Sequence homology of the BN-1–coding region with other chemokine receptors. The deduced 374–amino acid sequence encoded by the BN-1 cDNA was compared to other human and viral chemokine receptors using the multiple sequence alignment program Pileup of the Wisconsin EGCG DNA analysis software package. Shown here is the BN-1 amino acid sequence aligned with the human CXCR2 and CCR4. The positions of the hydrophobic membrane spanning regions TM1–TM7 are indicated by bars above the sequence. Amino acids identical between BN-1 and either CXCR2 or CCR4 are boxed.
Chromosomal Localization of the BN-1 Gene.
We analyzed a panel of human–rodent somatic cell hybrids to ascertain the location of the human BN-1 gene. PCR using two pairs of BN-1–specific oligonucleotides consistently yielded a PCR product in only those hybrids containing human chromosome 6 (Fig. 2 A). Positive controls using PCR primers against a known human chromosome 6 gene, human TNF, confirmed the hybrid cell line karyotyping (Fig. 2 B), and localization of BN-1 on human chromosome 6 was confirmed by PCR typing of chromosome 6 deletion and translocation hybrids (not shown). We next used a radiation hybrid panel (Fig. 2 legend) to unambiguously localize the BN-1 gene to 6q26-27, consistent with the fluorescent in situ hybridization analysis of Liao, et al. (23). This constitutes a new locus for chemokine receptors; the CC chemokine receptors CCR1–CCR5 are closely linked on chromosome 3p21, the CXC receptors CXCR1 and CXCR2 map to chromosome 2q35, and CXCR4 maps to chromosome 2q21.
Figure 2.

The BN-1 gene maps to chromosome 6. (A) PCR analysis of hamster, mouse, human and rodent–human hybrid cell line genomic DNAs (samples 1-20 Biosmap; BIOS Labs., New Haven, CT) using BN-1–specific primers BN1 and BN2. The lane marked Blank is the result of PCR in the absence of template, lanes M1 and M2 contain DNA molecular weight markers. The same genomic DNA samples gave positive PCR signals of the expected size with a second pair of BN-1–specific PCR primers, BN3 and BN4 (see Materials and Methods). (B) The result of PCR analysis of the same genomic DNA samples with human TNF-α promoter primers. The assignment of the BN-1 gene to chromosome 6 was confirmed by PCR analysis of a panel of chromosome 6 deletion and translocation hybrids (data not shown). PCR analysis of the Genebridge 4 radiation hybrid panel with BN1 and BN2 primers gave the following data vector: 12202021002210000101200020000000000000000112010000100 0010200011000020210100001020010100000001. The BN-1 data vector was compared to the WICGR human genome radiation hybrid map using the Whitehead Institute/Massachusetts Institute of Technology Center for Genome Research automapper version 1.0 (http//:www-genome.wi.mit.edu/). The BN-1 STS was placed on chromosome 6, 3.36 centiRay from the framework STS D6S1008 with a lod score >3.0.
Distribution of BN-1 Message.
Various direct and indirect approaches were used to assess the distribution of BN-1 mRNA. A Northern blot containing mRNA from a variety of organs and tissues showed an ∼3.5-kb message predominantly in spleen, and to a lesser extent in thymus, testis, small intestine, and peripheral blood (Fig. 3 A, top left). In addition, the spleen showed two smaller transcripts of ∼2.7 and 1.7 kb. mRNAs from various lymphoid and hematopoietic cell lines (TF-1, Jurkat, MRC5, JY, and U937; Fig 3 A, top right) were negative for BN-1 expression. For cell lines and tissues where limited amounts of mRNA precluded direct Northern analysis, expression was determined by the extent of hybridization among the gel-fractionated population of cDNA inserts from libraries made from those cells. BN-1 expression was examined first in 19 cDNA libraries made from various cells of lymphoid lineage (Fig. 3 A, bottom). BN-1 cDNA was present in one library made from resting PBMCs, consistent with the observation of Zaballos et al. for expression of the CKR-L3 orphan cDNA in CD4 and CD8 cells (22), and of the STRL22 orphan in PBLs (23). Interestingly, however, a matched PBMC library made after the cells were activated with anti-CD3 antibody and PMA showed no BN-1 cDNAs, which was further notable by their absence from virtually every other library made from T cell lines and clones (in various states of activation and anergy), pooled B cells, and NK cells (Fig. 3 A, bottom). Resting human splenocytes contain BN-1 cDNA (Fig. 3 A, bottom, Splen), but a matched library made after activation of splenocytes with anti-CD40 and IL-4 (Splen Act) showed diminished levels of BN-1 cDNA. Thus it appears that BN-1 may not be abundantly expressed in the lymphoid lineage, or that its expression is downregulated with cellular activation or growth in lymphocyte cultures.
Figure 3.

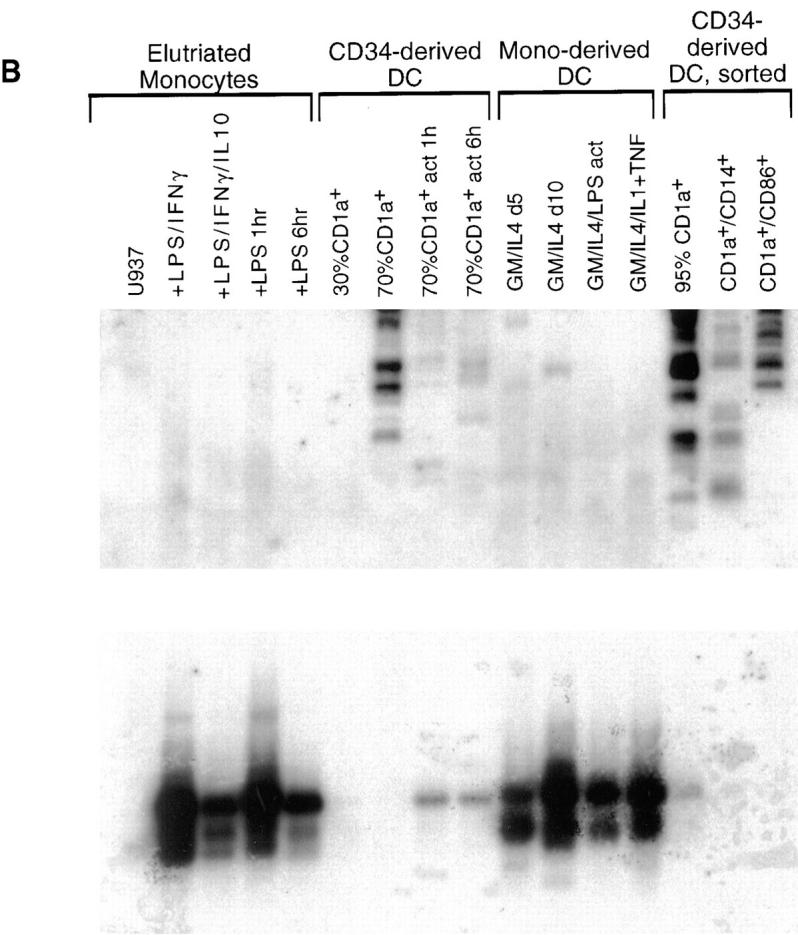

Analysis of BN-1 gene expression. (A) Northern blot analysis of BN-1 expression in RNA prepared from the various human tissues and cell lines (top); sizes of RNA markers (in kilobases) are indicated in the left margin. (Bottom) A Southern blot analysis of BN-1 in cDNA libraries prepared from the human PBL and primary cell T, B, and NK cell cultures. PBMC, human peripheral blood mononuclear cells; PBMC Act, the same PBMC stimulated with anti-CD3 and PMA for 2, 6, and 12 h and pooled; Mot 72 and Mot 81, human Th0 T cell clones. Act, Stimulation with anti-CD3 and anti-CD28 for 2, 6, and 12 h and pooled; and Anergic, stimulation with a specific peptide rendering the cells nonresponsive to antigen stimulation. HY06 and HY93 are human Th1 and Th2 T cell clones, respectively, with activation and anergy treatments as above except for the peptide specificity; B cell pool, a collection of EBV cell lines; Splen, total human splenocytes, resting; Splen Act, the same population stimulated with anti-CD40 and IL-4 for 2, 6, and 16 h and pooled; NK pool, a pool of primary NK cell clones; NK Act 6h, the same pool stimulated 6 h with PMA and ionomycin; NK B1, a single primary human NK cell clone; NK Act 6h, that clone activated as above. Probing replicate blots with human β actin cDNA gave readily detectable ∼2.0-kb species in all lanes (data not shown). References upon request. (B) Southern blot of BN-1 distribution (top) in cDNA libraries made from monocytes and DCs. U937, human monocyte cell line. Human elutriated monocytes have been stimulated as follows: LPS/IFNγ, cultured in the presence of these activators and blocking antibodies for IL-10 for 1, 2, 6, 12, and 24 h and pooled; LPS/IFNγ/IL10, the same pool without blocking antibodies to IL-10; LPS 1 h and LPS 6 h, monocytes stimulated for 1 and 6 h with LPS, respectively; CD34-derived DC, 30 and 70% DCs are CD1a+ DCs derived from CD34+ human cord blood stem cells by growth in GM-CSF and TNF-α, with the percent CD1a+ cells determined by FACs® over time in culture. The cultures were 70% CD1a+ after 12 d. 70% CD1a + act 1 h and act 6 h, the same cultured DC stimulated with PMA and ionomycin for 1 and 6 h, respectively. Mono-derived DC, DCs derived from human elutriated monocytes by growth in GM-CSF and IL-4 for 5 and 10 d as listed; GM/IL4/LPS act, 10-d cultures stimulated for 6 h with LPS; GM/IL4/IL1+TNF, are 10-d monocyte-derived DCs stimulated with IL-1α and TNF-α for 4 and 16 h and pooled. CD34-derived DC, sorted, DCs derived from CD34 cells as described that were then sorted on the basis on the cell surface expression of the markers listed: 95% CD1a+, DC derived from CD1a-sorted cells (Langerhans-like); CD1a+/CD14+, DC derived from CD14-sorted cells (dermal/interstitial). (Bottom) The same blot stripped and reprobed with the human CCF-18/MIP-1γ-chemokine. In all cases, the ladder effect represents different sizes of cDNA inserts in the library ranging from ∼0.6 to 3.5 kbp for BN-1. The two predominant CCF-18/MIP-1γ cDNAS are ∼0.7 and 1.3 kbp. (C) PCR analysis of unamplified mRNA from DC to confirm BN-1 distribution. Lane 1, CD34+ cord blood cells cultured 12 d in GM-CSF and TNF-α; lane 2, CD34-derived DCs stimulated with PMA and ionomycin; lane 3, CD34-derived DC purified by FACS® sorting for 98% CD1a+ expression; lanes 4–8, various cultures of monocyte-derived DCs (cultured in GM-CSF + IL-4 for 8 d) under conditions of stimulation as shown; + Ctl, positive control amplification using 1 pg BN-1 plasmid as starting substrate; − Ctl, same reaction with identical reagents in the absence of the plasmid substrate. Elutriated monocytes (not shown) were also negative. Each lane is representative of at least three independent experiments.
Since the original BN-1 PCR product was generated from a library derived from human DCs, we assessed the distribution of the receptor among these cells. There are two principle methods by which DCs can be generated in vitro: (a) stimulation of peripheral monocyte progenitors by prolonged exposure of these cells to GM-CSF and IL-4 (24, 25), and (b) by culture of CD34+ precursor cells from bone marrow or cord blood with GM-CSF and TNF. Resultant DCs can be tracked via expression of differentiation markers such as CD1a and CD86, and purified by sorting for one or more of these markers. DCs generated by both procedures take on the typical veiled morphology and achieve antigen presentation capability (26, 27).
A striking distribution of BN-1 cDNA was seen in a panel of libraries made from monocytes and two types of in vitro–derived dendritic cells, suggesting marked differences between monocyte- and CD34-derived DCs. BN-1 was not present in any of the libraries derived from purified elutriated monocytes, and there were few if any BN-1 cDNAs in the libraries prepared from monocyte-derived DCs (Fig. 3 B, top). By contrast, there is clear expression of BN-1 as the CD34+ cells develop into differentiated DCs (Fig. 3 B, top). Although little signal is seen when these cultures are 30% CD1a+ (at about day 6), abundant message is present by the time the cells are 70% CD1a+ (about day 12). Interestingly, when 70% CD1a+ cultures are stimulated for 1–6 h with PMA and ionomycin (70% CD1a+ act 6 h), much less BN-1 cDNA is found in the libraries subsequently made from those cells (Fig. 3 B, top). BN-1 is most abundantly represented in libraries made from DC cultures derived from cells which were sorted on the basis of CD1a expression (95% CD1a; Langerhans-like), and less well in those DCs that were derived from CD14+-sorted cells (dermal/interstitial-like) (Fig. 3 B, top; reference 27). To control for the quality of monocyte and monocyte-derived DC libraries, we probed with other markers, including the CC chemokine human CCF-18/MIP-1γ (28). Strikingly, the cDNA distribution pattern for this chemokine is almost directly inverse of that for BN-1 (Fig. 3 B, bottom). Finally, PCR performed directly on primary, unamplified poly A+ mRNA harvested from monocyte- and CD34-derived DCs yielded amplification products whose pattern mirrored the hybridizations seen in the library southern blots (Fig. 3 C). Taken together, the distribution data suggest that BN-1 seems to be expressed most highly in a mature phenotype of DCs derived from the differentiation of CD34+ cells, and is not well expressed in DCs generated from monocytes.
Chemokine Specificity of the Receptor Encoded by BN-1.
The chemokine literature is replete with cDNA sequences encoding orphan chemokine receptors of unknown ligand specificity and unknown function. To assess the ligand-binding specificity of the putative chemokine receptor encoded by BN-1, we constructed expression plasmids (pFlagBN-1) encoding a receptor with an added NH2-terminal Flag epitope. This allowed for detection and selection of the most highly expressing stable transfectants using an anti-Flag monoclonal antibody. To decrease the chance of a species- or lineage-specific G protein–dependent response, we also tested BN-1 expression constructs in the following different types of cells from different species: human embryonic kidney 293 (HEK293), murine 3T3 fibroblasts, and chinese hamster ovary (CHO) cells. Stable transfectants were generated from each of these cell lines; for some, cells were sorted by anti-Flag antibody for high expression and further propagation, an example of which is shown in Fig. 4 A.
Figure 4.

Chemokine specificity of the receptor encoded by BN-1. (A) A sorted population of transfected CHO cells stably expressing BN-1 protein containing an NH2-terminal Flag epitope (CHO-FlagBN-1) showing intensity of anti-Flag mAb staining relative to wild-type CHO cells. (B) Panel of purified recombinant or synthetic chemokines assayed for intracellular calcium mobilizing activity in various BN-1 transfectants. At 100 nM final concentration − indicates no detectable intracellular calcium mobilization in any of the transfectants (n >3), whereas ++ denotes a robust response in all transfectants (CHO, 3T3, HEK293) bearing BN-1. (C) Dose response of MIP-3α in the induction of intracellular calcium in CHO-FlagBN-1 cells. CHO wild-type cells are shown as a control. (D) Binding and homologous competition of radiolabeled MIP-3α to BN-1–bearing cells. (Inset) Scatchard transformation of the binding data to reveal a _K_d of ∼0.1 nM.
A panel of purified chemokine ligands was then used to challenge the various stable transfectants expressing the BN-1 gene product. Calcium mobilization in the cytoplasm of BN-1 transfectants was used to assess receptor engagement and functional coupling to G proteins, as has been established for other chemokine receptors (21). We tested a panel of 25 purified recombinant or synthetic chemokines (listed in Fig. 4 B) including all of the standard CXC, C, and traditional CC chemokines, as well as the CX3C chemokine Fractalkine (20). With one exception, all chemokines were negative for calcium mobilization in recombinant BN-1–bearing cells of all backgrounds. One chemokine, however, the recently described CC chemokine MIP-3α, showed a robust calcium signal, whereas the wild-type and mock-transfected cells were unresponsive. MIP-3α exhibited a dose-dependent response, reaching a plateau at ∼10 nM (Fig. 4 C). MIP-3α was positive in every test on the stable transfectants in all three cellular backgrounds and also in transfectants expressing the native form of the receptor (without the NH2-terminal flag), as well as in a variety of transiently transfected cells of different types (not shown). We did not detect MIP-3α activity on other chemokine receptors tested (CCR1–CCR5, and CXCR1 and 2 [not shown]). Competition binding and dissociation experiments were also performed. Homologous competition of radioloableled MIP-3α with unlabeled ligand showed clear dissociation; Scatchard transformation revealed a dissociation constant (_K_d) of ∼0.1 nM (Fig. 4 D). From the signaling and binding data, we therefore concluded that the BN-1 gene product is a receptor for the CC chemokine MIP-3α, hence its designation as CCR6.
Discussion
We report the identification of a cDNA encoding a chemokine receptor abundantly expressed in certain types of cultured human DC populations, and the characterization of the protein that it encodes. This receptor, designated here as CCR6, is notable in that of the over two dozen purified chemokines tested, it appears to interact only with the newly reported CC chemokine MIP-3α.
CCR6 is expressed most highly in DCs derived from CD34+ cells, but not in monocyte-derived DCs. We have also noted CCR6 expression in resting PBMCs, consistent with reports of the presence of transcripts for the orphan clones CKR-L3 and STRL22 (22, 23). Those reports, however, did not examine DC populations, nor did they identify any ligands which bound the orphan cDNA gene product. The robust but differential patterns of CCR6 suggest that it plays a role in the function of certain types of DCs. At least three populations of cells with dendritic morphology have been reported in peripheral blood (29, 30). Each may represent discrete maturational and functional stages; the possibility has been discussed that the CD34-derived DCs in vitro are enriched for Langerhans-like cells. It will be therefore be of interest to assess whether CCR6 will prove to be DC lineage marker in vivo. It is known that of the different DC populations identified in the periphery, only one appears to efficiently support HIV-1 infection (30). DCs have been proposed to act in a variety of sites as a reservoir for HIV-1 (31), although most efficient infection seems to occur in cocultures of DC and T cells. The virus can clearly infect DCs alone, and can use the chemokine receptors CXCR4 and CCR5 as infection cofactors (18). Additionally, some infection of DC from CCR5-deficient individuals by M-tropic HIV has been observed (18), suggesting an additional coreceptor on these cells. Although we do not yet know how CCR6 expression relates to the different types of DCs in vivo, it may be a candidate for an infection cofactor for strains of HIV that infect DC (32).
It has been shown that monocyte-derived DCs and CD34+ umbilical cord blood–derived DCs are chemotactically responsive to the CC chemokines RANTES, MIP-1α, and monocyte chemotactic protein 3 (16, 17). The chemotactic response of DCs to MIP-3α, however, has not been reported. Indeed, much remains to be determined as to the properties of MIP-3α. Recently identified as an orphan chemokine of unknown function (19), it has also been described as LARC (liver activation–related chemokine) by Hieshima et al. (33). Expressed sequence tags containing parts of the MIP-3α–coding region were generated from sequence analysis of fetal lung, liver, and pancreatic islet cDNA libraries (19, 33). Although a CC chemokine, MIP-3α is widely diverged from other CC chemokines, showing only 20–28% identity with other members of the CC family and being encoded on chromosome 2 rather than chromosome 17 (33). The chemotactic properties of MIP-3α, especially as mediated through CCR6, are currently being studied. LARC protein was shown to induce chemotaxis of T cells, albeit less potently and efficiently than RANTES (33). This would be consistent with CCR6 expression in T cells as noted here and for CKR-L3 (22), but the existence of other MIP-3α receptors on lymphocytes cannot be excluded. Our experiments suggest that levels of CCR6 expression in other cells are low relative to the levels seen in DCs, and it is notable that CCR6 so far exhibits specificity only for MIP-3α among the extensive panel of chemokines tested. The specificity of CCR6 binding is supported by the evidence of Baba et al., whose study appeared while the present manuscript was under revision, reporting a receptor of identical sequence, GPR-CY4, that binds specifically to LARC/MIP-3α (34). Again, that study did not examine dendritic cell distribution or function.
In summary, the identification of CCR6 as a provisionally specific receptor for the CC chemokine MIP-3α defines another piece in the puzzle of chemokine–chemokine receptor interactions. Detailed analysis of CCR6 and its CC chemokine ligand may shed new light on the control of DC migration and function, providing a potential therapeutic avenue for treatment of a wide range of pathologies including HIV infection, autoimmunity, and transplant rejection.
Acknowledgments
We thank Dr. K. Bacon for help and advice, and Drs. J.M. Boyle and M.J. Greaves for gifts of human chromosome 6 hybrid DNAs and advice on human gene mapping studies. We are grateful to Jasmine and Shona for stimulating discussions regarding cell motility in the early stages of this project.
Footnotes
DNAX is supported by Schering-Plough; work in the laboratory of S. Gordon is supported by the United Kingdom Medical Research Council; D.R. Greaves is supported by Glaxo Wellcome.
1
Abbreviations used in this paper: CCR, a receptor for CC chemokines; CHO, Chinese hamster ovary; CXCR, a receptor for CXC chemokines; DC, dendritic cell; HEK293, human embryonic kidney 293; IC2, second intracellular loop; LARC, liver activation–related chemokine; MIP, macrophage inflammatory protein; mRNA, messenger RNA; ORF, open reading frame; TM, transmembrane.
D.R. Greaves and W. Wang contributed equally to this study.
References
- 1.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 2.Caux C, Liu Y-J, Banchereau J. Recent advances in the study of dendritic cells and follicular dendritic cells. Immunol Today. 1995;16:2–4. doi: 10.1016/0167-5699(95)80061-1. [DOI] [PubMed] [Google Scholar]
- 3.Austyn, J.M., M.I. Liddington, and G.G. MacPherson. 1997. Dendritic cells: migration in vivo. In Handbook of Experimental Immunology. 5th edition. D.M. Weir, C. Blackwell, and L.A. Herzenberg, editors. Blackwell, Oxford.
- 4.Austyn JM. New insights into the mobilization and phagocytic activity of dendritic cells. J Exp Med. 1996;183:1287–1292. doi: 10.1084/jem.183.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schall TJ, Bacon KB. Chemokines, leucocyte trafficking, and inflammation. Curr Opin Immunol. 1994;6:865–873. doi: 10.1016/0952-7915(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 6.Baggiolini M, DeWold B, Moser B. Interleukin-8 and related chemotactic cytokines–CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 7.Premack BP, Schall TJ. Chemokines receptors: gateways to inflammation and infection. Nat Med. 1996;2:1174–1178. doi: 10.1038/nm1196-1174. [DOI] [PubMed] [Google Scholar]
- 8.Adams DH, Lloyd AR. Chemokines: leucocyte recruitment and activation cytokines. Lancet. 1997;349:490–495. doi: 10.1016/s0140-6736(96)07524-1. [DOI] [PubMed] [Google Scholar]
- 9.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+T cells. Science (Wash DC) 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 10.Deng D, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature (Lond) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 11.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane G protein–coupled receptor. Science (Wash DC) 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 12.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, et al. The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 13.Dragic T, Litwin V, Allway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature (Lond) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 14.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 15.Endres MJ, Clapham PR, Marsh M, Ahuja M, Turner JD, MacKnight A, Thomas JF, Stoebenau-Haggarty B, Choe S, Vance PJ, et al. CD4-independent infection by HIV-2 is mediated by Fusin/CXCR4. Cell. 1996;87:745–756. doi: 10.1016/s0092-8674(00)81393-8. [DOI] [PubMed] [Google Scholar]
- 16.Sozzani S, Sallusto F, Luni W, Zhou D, Piemonti L, Allavena P, Van Damme J, Valitutti S, Lanzavecchia A, Mantovani A. Migration of dendritic cells in response to formyl peptides, C5a and a distinct set of chemokines. J Immunol. 1995;155:3292–3295. [PubMed] [Google Scholar]
- 17.Xu LL, Warren MK, Rose WL, Gong WH, Wang JM. Human recombinant monocyte chemotactic protein and other C–C chemokines bind and induce directional migration of dendritic cells in vitro. J Leukocyte Biol. 1996;60:365–371. doi: 10.1002/jlb.60.3.365. [DOI] [PubMed] [Google Scholar]
- 18.Granelli-Piperno A, Moser B, Pope M, Chen D, Wei Y, Isdell F, O'Doherty U, Paxton W, Koup R, Mojsov S, et al. Efficient interaction of HIV-1 with purified dendritic cells via multiple chemokine receptors. J Exp Med. 1996;184:2433–2438. doi: 10.1084/jem.184.6.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi DL, Cicari AP, Franz-Bacon K, McClanahan TK, Zlotnik A. Identification through bioinformatics of two new proinflammatory human chemokines MIP3α and MIP3β. J Immunol. 1997;158:1033–1036. [PubMed] [Google Scholar]
- 20.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature (Lond) 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 21.Bacon KB, Premack BA, Gardner P, Schall TJ. Activation of dual T-cell signaling pathways by the chemokine RANTES. Science (Wash DC) 1995;269:1727–1730. doi: 10.1126/science.7569902. [DOI] [PubMed] [Google Scholar]
- 22.Zaballos A, Varona R, Gutierrez J, Lind P, Marquez G. Molecular cloning and RNA expression of two new human chemokine receptor-like genes. BBRC (Biochem Biophys Res Commun) 1996;227:846–853. doi: 10.1006/bbrc.1996.1595. [DOI] [PubMed] [Google Scholar]
- 23.Liao F, Lee H-H, Farber J. Cloning of STRL22, a new human gene encoding a G-protein–coupled receptor related to chemokine receptors and located on chromosome 6q27. Genomics. 1997;40:175–180. doi: 10.1006/geno.1996.4544. [DOI] [PubMed] [Google Scholar]
- 24.Bender A, Sapp M, Schuler G, Steinman RM, Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J Immunol Methods. 1996;196:121–135. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- 25.Reid CDL, Stackpoole A, Meager A, Tikerpae J. Interactions of tumor necrosis factor with granulocyte-macrophage colony-stimulating factor and other cytokines in the regulation of dendritic cell growth in vitro from early bipotent CD34+ progenitors in human bone marrow. J Immunol. 1992;149:2681–2689. [PubMed] [Google Scholar]
- 26.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor α. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caux C, Vanbervliet B, Massacrier C, Dezutter-Dambuyant C, de Saint-Vis B, Jaquet C, Yoneda K, Imamura S, Schmitt D, Banchereau J. CD34+hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNFα. J Exp Med. 1996;184:695–706. doi: 10.1084/jem.184.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hara T, Bacon KB, Cho LC, Yoshimura A, Morikawa Y, Copeland NG, Gilbert DJ, Jenkins NA, Schall TJ, Miyajima A. Molecular-cloning and functional-characterization of a novel member of the C–C chemokine family. J Immunol. 1995;155:5352–5358. [PubMed] [Google Scholar]
- 29.O'Doherty U, Peng M, Gezelter S, Swiggard WJ, Betjes M, Bhardwaj N, Steinman RM. Human blood contains two subsets of dendritic cells, one immunologically mature and the other immature. Immunology. 1994;82:487–493. [PMC free article] [PubMed] [Google Scholar]
- 30.Weissman D, Li Y, Ananworanich J, Zhou L-J, Adelsberger J, Tedder TF, Baseler M, Fauci A. Three populations of cells with dendritic morphology exist in peripheral blood, only one of which is infectable with human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1995;92:826–830. doi: 10.1073/pnas.92.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livingstone WJ, Moore M, Innes D, Bell JE, Simmonds P the Edinburgh Heterosexual Transmission Study Group. Frequent infection of peripheral blood CD8-positive T-lymphocytes with HIV-1. Lancet. 1996;348:649–654. doi: 10.1016/s0140-6736(96)02091-0. [DOI] [PubMed] [Google Scholar]
- 32.Soto-Ramirez L, Renjifo B, McLane MF, Marlink R, O'Hara C, Sutthent R, Wasi C, Vithayasai P, Vithayasai V, Apichartpiyakul C, et al. HIV-1 Langerhans' cell tropism associated with heterosexual transmission of HIV. Science (Wash DC) 1996;271:1291–1295. doi: 10.1126/science.271.5253.1291. [DOI] [PubMed] [Google Scholar]
- 33.Hieshima K, Imai T, Opdendakker G, Van Damme J, Kusud J, Tei H, Sakkai Y, Takatsuki K, Miura R, Yoshie O, Nomiyama H. Molecular cloning of a novel human CC chemokine, liver and activation related chemokine (LARC) expressed in liver. J Biol Chem. 1997;272:5846–5849. doi: 10.1074/jbc.272.9.5846. [DOI] [PubMed] [Google Scholar]
- 34.Baba M, Imai T, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Hieshima K, Yoshie O. Identification of CCR6, the specific receptor for a novel lymphocyte-directed CC chemokine LARC. J Biol Chem. 1997;272:14893–14898. doi: 10.1074/jbc.272.23.14893. [DOI] [PubMed] [Google Scholar]