Dendritic Cells Genetically Modified with an Adenovirus Vector Encoding the cDNA for a Model Antigen Induce Protective and Therapeutic Antitumor Immunity (original) (raw)
Abstract
Dendritic cells (DCs) are potent antigen-presenting cells that play a critical role in the initiation of antitumor immune responses. In this study, we show that genetic modifications of a murine epidermis-derived DC line and primary bone marrow–derived DCs to express a model antigen β-galactosidase (βgal) can be achieved through the use of a replication-deficient, recombinant adenovirus vector, and that the modified DCs are capable of eliciting antigen-specific, MHC-restricted CTL responses. Importantly, using a murine metastatic lung tumor model with syngeneic colon carcinoma cells expressing βgal, we show that immunization of mice with the genetically modified DC line or bone marrow DCs confers potent protection against a lethal tumor challenge, as well as suppression of preestablished tumors, resulting in a significant survival advantage. We conclude that genetic modification of DCs to express antigens that are also expressed in tumors can lead to antigen-specific, antitumor killer cells, with a concomitant resistance to tumor challenge and a decrease in the size of existing tumors.
Many tumor cells express epitopes on their surface that provide a potential target for therapeutic vaccine strategies that boost natural immune-mediated tumor defense mechanisms (1–3). One of the challenges to developing antitumor vaccines is to develop strategies that focus the immune response to the relevant tumor antigen. The present study approaches this challenge by using replication-deficient, recombinant adenovirus (Ad)1 vectors to transfer a gene coding for a model antigen (β-galactosidase; βgal) to dendritic cells (DCs) ex vivo, and administering the genetically modified DCs to a syngeneic host. We choose this approach to capitalize on the biologic properties of Ad vectors and DCs that are relevant to inducing antigen-specific CTLs directed against specific antigens.
In this context, there is emerging evidence that, in some circumstances, Ad vectors can act as adjuvants to boost CTL response against heterologous transgenes (4–9). This evidence, together with the knowledge that genes expressed by Ad vectors are expressed through the class I pathway (10, 11), and that DCs are the most potent members of the general class of antigen presenting cells (12–15), led us to the hypothesis that genetically modifying DCs with an Ad vector coding for a tumor antigen might help focus the immune response to generating antigen-specific CTLs.
With this background, the objectives of this study are to assess the ability of Ad vectors to genetically modify DCs ex vivo, and to assess the ability of DCs modified with model antigens transferred to a syngeneic host to (a) induce antigen-specific CTLs, (b) prevent tumors from developing in animals challenged with tumor cells expressing the model antigen, and (c) suppress the growth of preexisting tumors composed of the same tumor cells. To accomplish this, we used the XS52 BALB/c DC line (16, 17), as well as freshly isolated DCs from bone marrow of BALB/c mice. By using an Ad vector expressing βgal and the BALB/c syngeneic colon carcinoma cell line CT26.CL25 expressing βgal (18), the data dramatically demonstrate that administration of DCs modified to express βgal induces βgal-specific CTLs, prevents tumor growth, and suppresses the growth of preexisting tumors.
Materials and Methods
Mice and Cell Lines.
6–8-wk-old male BALB/c (H-2d) mice were obtained from the Jackson Lab. (Bar Harbor, ME). Animals were housed under specific pathogen-free conditions and treated according to National Institutes of Health guidelines.
XS52 (H-2d) clone 4D, a DC line established from newborn BALB/c mouse epidermis, was grown in complete RPMI media (10% fetal bovine serum, 2 mM l-glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin) supplemented with recombinant murine GM-CSF (2 ng/ml; Sigma Chemical Co., St. Louis, MO) and supernatants collected from the stromal NS47 cell line (both cell lines were provided by A. Takashima, University of Texas Southwestern Medical Center, Dallas, TX; references 16, 17). CT26.WT (H-2d) is a clone of the _N_-nitroso-_N_-methylurethane–induced BALB/c undifferentiated colon adenocarcinoma (18). CT26.CL25 is a clone of CT26.WT that has been transduced with the Escherichia coli βgal gene (both cell lines provided by N.P. Restifo, National Cancer Institute, Bethesda, MD; reference 18). E22 cells, a clone of mouse thymoma EL4 (H-2b) cell line transduced with the βgal gene, was used as a negative control in the CTL assay (E22 was provided by Y. Paterson, University of Pennsylvania, Philadelphia, PA; reference 18). The SVBalb (H-2d) fibroblast cell line was used as in vitro stimulator cells in the CTL assay; this cell line is syngeneic to BALB/c mice (provided by L. Gooding, Emory University, Atlanta, GA; reference 19). CT26.WT and SVBalb were grown in complete RPMI media. CT26.CL25 and E22 were grown in complete RPMI containing 400 μg/ml G418 (GIBCO BRL, Gaithersburg, MD).
Generation of Dendritic Cells In Vitro from Bone Marrow.
Primary bone marrow DCs were obtained from mouse bone marrow precursors as described by Inaba et al. (20). In brief, lymphocyte- and erythrocyte-depleted murine bone marrow cells harvested from femurs and tibias were plated in completed RPMI media supplemented with recombinant murine GM-CSF (100 U/ml) and recombinant murine interleukin 4 (20 ng/ml; Genzyme, Farmington, MA). On days 2 and 4, nonadherent granulocytes were gently removed and fresh media was added. On day 6, loosely adherent proliferating DC aggregates were dislodged and replated. On day 8 of culture, released, mature, nonadherent cells with the typical morphological features of DCs were used for in vitro phenotypic and functional analysis as well as for the immunization of mice. For phenotypic analysis, the nonadherent cells from the bone marrow culture were stained with a panel of antibodies characteristic of DCs, including CD11b (No. 01714D, PharMingen, San Diego, CA), CD44 (No. 01225A, PharMingen), and IAd (No. 06274D, PharMingen) and then quantified by flow cytometry. Isotype-matched monoclonal antibodies were used as controls. Bone marrow DCs were also tested for their ability to activate naive allogeneic T cells in vitro using a mixed lymphocyte reaction assay (20).
Ad Vector–mediated Gene Transfer and Expression in DCs In Vitro.
The replication-deficient Ad vectors used in this study are E1a−, partially E1b−, and partially E3− vectors based on human Ad5 (21, 22). The construction of these Ad vectors have been described previously, including vectors expressing no transgene (AdNull) and the E. coli βgal gene (Adβgal; reference 23). All Ad vectors were propagated in 293 cells, purified by two rounds of CsCl density centrifugation, dialyzed, and stored at −70°C as previously described (21, 22). The titer of viral stock was determined by plaque-forming assay using 293 cells. All vector preparations were demonstrated to be free of replication competent adenovirus (24).
To assess the ability of Ad vectors to transfer and express genes in DCs in vitro, the XS52 DC line and primary bone marrow DCs were infected with Adβgal or AdNull for 2 h at various multiplicities of infection (moi). 24 h later, βgal expression was quantified by flow cytometry using fluorescein di-β-galactoside (Molecular Probes, Inc., Eugene, OR; reference 25). Labeling with propidium iodide (1 μg/ml) was used to facilitate live/dead cell discrimination.
Induction of Antigen-specific CTLs.
To determine if the administration of DCs by in vitro infection with Adβgal would evoke βgal-specific CTLs, the XS52 DC line or primary bone marrow DCs were infected with Adβgal or AdNull (moi 100) as described above. BALB/c mice were immunized once by subcutaneous injection of 3 × 105 modified DCs suspended in 100 μl HBSS. Spleens were removed 14 d after DC inoculation, the in vivo primed splenocytes were pooled and cocultured (2 × 106/ ml) with irradiated (5,000 rad) syngeneic SVBalb cells (2 × 105/ ml) pulsed with the synthetic βgal peptide (1 μM) in a 6-well plate (5 ml/well). After 5 d of coculture, the in vitro restimulated viable splenocytes were assayed in a 51Cr–release assay for specific cell lysis against a variety of target cells, including syngeneic cell lines CT26.WT, βgal-expressing CT26.CL25, and βgal peptide-pulsed CT26.WT, as well as a MHC-mismatched, βgal-expressing E22 (H-2b) cell line. The βgal peptide used in the CTL studies is the nanomer TPHPARIGL (residues 876–885) that is naturally presented by the H-2 Ld molecule (26). The peptide was synthesized (Biosynthesis, Lewisville, TX) to a purity >99% as determined by high pressure liquid chromatography and amino acid analysis. The 51Cr–release assay was performed as previously described (9, 27).
Protection against Lethal Tumor Challenge after XS52 DC Immunization.
To evaluate the ability of the XS52 DC line infected with Adβgal to protect against a tumor challenge, 6–8-wk-old BALB/c mice were immunized with 3 × 105 Adβgal-modified (moi 100, 24 h, 37°C) XS52 (referred to as XS52-Adβgal) in 100 μl HBSS injected subcutaneously in the left lower quadrant of the ventral abdominal wall (n = 4). Control animals received either no XS52 (n = 5), XS52 infected with AdNull (XS52-AdNull; n = 2), or XS52-Adβgal that were frozen and thawed four times before injection to lyse the cells (n = 5). 2 wk later, naive and XS52-immunized mice were challenged with syngeneic 105 CT26.CL25 murine colon carcinoma cells injected intravenously through the jugular vein. This model generated multiple diffuse pulmonary metastases that were highly lethal. 12–16 d after tumor challenge, the mice were killed, their lungs harvested, fixed in 4% paraformaldehyde, and stained for βgal expression with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). To quantify the tumor burden, the number of blue staining lung metastases was carefully counted in a blinded fashion under a dissecting microscope. Only metastatic deposits ⩽250 could be reliably enumerated; lungs with >250 deposits were assigned an empirical number of 250.
To evaluate if an effective antitumor protection would translate into significant prolongation of survival, an identical, parallel experiment was performed in which the mice (n = 9 in XS52 Adβgal group; n = 10 in XS52-AdNull group) were not killed after XS52 immunization and tumor challenge, but were followed for survival. In addition, to evaluate if the antitumor immunity was βgal specific, another group of mice (n = 10) immunized with XS52-Adβgal was challenged with CT26.WT tumor cells that did not express βgal and followed for survival.
Suppression of Preestablished Lung Metastases by XS52 DC Line Immunization.
To mimic the clinical scenario more closely, we evaluated whether preexisting lung metastases could be suppressed after immunization with XS52 cell line modified with Adβgal (n = 4) or AdNull (n = 4). BALB/c mice were first injected with 3 × 104 CT26.CL25 tumor cells intravenously to generate multiple lung metastases. 3 d later, tumor-bearing mice were either left untreated, or treated with subcutaneous injections of XS52-AdNull (n = 4), XS52-Adβgal (n = 4), or XS52-Adβgal that were frozen and thawed four times to lyse the cells (n = 3). 20 d after tumor implantation, the animals were killed and the lungs evaluated for the presence of metastases in a similar fashion as described in the prevention experiment.
To evaluate if treatment of preestablished lung metastases would result in a prolongation of survival in tumor-bearing animals, a separate but similar experiment was performed in which the mice were not sacrificed following treatment with XS52 cells modified with Adβgal (n = 7) or AdNull (n = 5), but were followed for survival. To evaluate if the treatment effect was βgal specific, a group of mice (n = 6) with preestablished CT26.WT tumors that did not express βgal was treated with XS52 cells modified with Adβgal and followed for survival.
Protection against Lethal Tumor Challenge after Bone Marrow DC Immunization.
To evaluate the ability of bone marrow DCs infected with Adβgal to protect against a tumor challenge, 6–8-wk-old BALB/c mice (n = 8/group) were immunized with 3 × 105 bone marrow DC–Adβgal in 100 μl HBSS injected subcutaneously in the left lower quadrant of the ventral abdominal wall. Control animals received either no bone marrow DCs, bone marrow DCs infected with AdNull (bone marrow DC–AdNull), or bone marrow DC–Adβgal that were frozen and thawed four times before injection to lyse the cells. 2 wk later, naive and immunized mice were challenged with syngeneic 105 CT26.CL25 murine colon carcinoma cells injected intravenously through the jugular vein. The mice were then followed for survival. In addition, to evaluate if the antitumor immunity was βgal specific, another group of mice (n = 8) immunized with bone marrow DC– Adβgal was challenged with CT26.WT tumor cells that did not express βgal and followed for survival.
Suppression of Preestablished Lung Metastases by Bone Marrow DC Immunization.
To mimic the clinical scenario more closely, we evaluated whether preexisting lung metastases could be suppressed after immunization with βgal-expressing bone marrow DCs. BALB/c mice were first injected with 3 × 104 CT26.CL25 tumor cells intravenously to generate multiple lung metastases. 3 d later, tumor-bearing mice (n = 8/group) were either left untreated, or treated with subcutaneous injections of bone marrow DC–AdNull, bone marrow DC–Adβgal, or bone marrow DC– Adβgal that were frozen and thawed four times to lyse the cells. The mice were then followed for survival. To evaluate if the treatment effect was βgal specific, a group of mice (n = 8) with preestablished CT26.WT tumors that did not express βgal was treated with bone marrow DC–Adβgal and followed for survival.
Statistical Analysis.
All animal experiments used a minimum of four animals except for the XS52 prevention experiment in which the XS52-AdNull group had only two animals (inadvertently, two animals in this group were left out of the study; in a repeat study, this group had five animals). All studies were repeated at least twice with similar results. Quantitative results are expressed as mean ± standard error of the mean. Statistical analysis was performed using the unpaired two-tailed Student's t test, with the exception of the survival data which was analyzed using the log-rank test (28).
Results
Adenoviral Transduction of XS52 Cells In Vitro.
To evaluate the ability of Ad vectors to transfer and express genes in XS52 DCs, XS52 DCs were infected with Adβgal, and the cells were quantified for βgal activity at 24 h after infection by flow cytometry. Although naive XS52 (Fig. 1, A) and AdNull-infected XS52 (B) showed only background levels of βgal activity, expression of βgal was readily detected in the majority (73%) of XS52 infected with Adβgal at moi of 100 (C), demonstrating that successful gene transfer and expression can be achieved in DCs using an Ad vector. Parallel studies carried out using other moi demonstrated 14% XS52 cells expressing βgal with a moi of 30 and 62% at a moi of 300 (not shown). On the basis of these studies, all subsequent studies were carried out with a moi of 100.
Figure 1.

Ad vector–mediated gene transfer and expression of βgal to the murine XS52 DC line in vitro. The XS52 cells were infected in vitro with an Ad vector expressing βgal (Adβgal) or the AdNull control vector at moi of 100 for 2 h. 24 h later, βgal expression was quantified by flow cytometry using fluorescein di-β-galactoside. Cells were stained with propidum iodide to facilitate discrimination among live and dead cells. For all panels, the y-axis reflects DC number and the x-axis reflects log fluorescein intensity. The K gate has been set according to the negative control in A. The percentage of βgal-expressing DCs was determined by the right shift of the curve along the K gate. (A) Uninfected XS52 cells. (B) XS52 infected with AdNull. (C) XS52 infected with Adβgal.
Induction of CTLs Using Ad Vector–transduced XS52 Cells.
To determine if Ad-mediated gene expression in DCs would induce an antigen-specific CTL response in vivo, BALB/c mice were first immunized with 3 × 105 XS52 cells that had been modified in vitro with AdNull- or Adβgal-infection. Splenocytes from immunized mice were then harvested and stimulated in vitro with βgal peptide and were then assayed for specific cell lysis against a panel of target cells in a standared 51Cr–release assay. Although immunization of naive XS52 or XS52-AdNull infected cells did not result in a measurable βgal-specific CTL response (Fig. 2, A), significant lysis of βgal-expressing CT26.CL25 target cells or βgal peptide-pulsed CT26.WT target cells were observed in mice immunized with XS52-Adβgal (B). Only negligible lysis of non–βgal-expressing CT26.WT target cells was detected in mice immunized with XS52-Adβgal, indicating that the cellular immune response induced by XS52-Adβgal was βgal specific. This specific cytotoxicity was mediated predominantly by MHC-restricted CTLs rather than by natural killer cells since E22, an MHC-mismatched (H-2b), βgal-expressing murine tumor cell line, was not significantly lysed in the same assay.
Figure 2.

Induction of CTL response in BALB/c mice after in vivo administration of Adβgal-infected XS52 DCs. The XS52 DCs were transduced in vitro with AdNull or Adβgal at moi of 100 for 2 h. 24 h later, 3 × 105 cells were administered subcutaneously to syngeneic BALB/ c mice. 14 d later, splenocytes harvested from immunized mice were stimulated for 5 d in vitro with syngeneic fibroblasts (SVBalb) pulsed with a βgal peptide, and then assayed for specific cell lysis against three syngeneic colon carcinoma target cells (parental CT26.WT cells, βgal-expressing CT26.CL25 cells, or CT26.WT pulsed with βgal peptide in vitro [CT26.WT-βgal peptide]) as well as the E22 βgal-expressing allogeneic tumor cell line. (A) CTL response in mice immunized with XS52 DCs infected with AdNull. (B) CTL response in mice immunized with XS52 DCs infected with Adβgal.
Protection against Lethal Tumor Challenge after Immunization with the Adβgal-modified XS52 DC Line.
Between 12 and 16 d after an intravenous tumor challenge, tumor-bearing mice which were not treated showed signs of respiratory distress and developed hemorrhagic pleural effusion and diffuse lung metastases (Fig. 3, A). Similarly, mice which were previously immunized with XS52-AdNull developed numerous lung metastases (B). In marked contrast, mice which were previously immunized with XS52-Adβgal showed very few visible lung metastases under the dissecting microscope (C and D), indicating that immunization of mice with DCs genetically modified to express the βgal model antigen could effectively protect against a lethal tumor challenge. Quantitative analysis of the number of lung metastases confirmed the significant difference between XS52-Adβgal–immunized mice and untreated mice (P <0.0001) or XS52-AdNull–immunized mice (_E_). Further, immunization with XS52-Adβgal that had been frozen and thawed did not confer protection when compared to untreated and XS52-AdNull–immunized groups (_P_ >0.1; not shown), i.e., live DCs were a prerequisite to generate an effective antitumor immunity.
Figure 3.
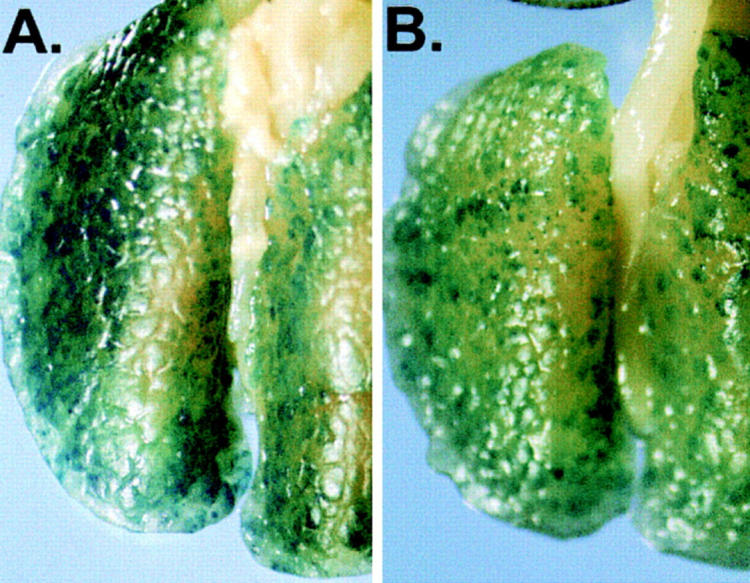


Protection against lethal tumor challenge using modified XS52 DCs. Shown are examples and quantitative data of protection against lung metastases after XS52 immunization, using the βgal as a model antigen, BALB/c mice, and syngeneic CT26.CL25 colon carcinoma cells (expressing βgal). XS52 cells were genetically modified by in vitro infection with AdNull or Adβgal (moi 100, 2 h). 24 h after infection, 3 × 105 XS52-AdNull or XS52-Adβgal were injected subcutaneously to syngeneic BALB/c mice. 14 d later, the mice were challenged with intravenous administration of 3 × 105 CT26.CL25 cells. 12–16 d after tumor challenge, the mice were killed, their lungs harvested, fixed, and stained for βgal expression with X-Gal. (A) Example of lungs from a nonimmunized mouse. (B) Example of lungs from a mouse immunized with XS52-AdNull. (C and D) Examples of lungs from mice immunized with XS52-Adβgal. (E) Quantification of the number of lung metastases in nonimmunized, XS52-AdNull–immunized and XS52-Adβgal–immunized mice after tumor challenge. Using a dissecting microscope, surface blue-staining (βgal+) lung metastases were enumerated. Each data point represents an individual animal. Only metastatic deposits ⩽250 could be reliably enumerated; lungs with >250 metastases were assigned an empirical number of 250.
The metastatic lung tumor model in this study was highly lethal, such that mice immunized with the negative control DCs (XS52-AdNull) succumbed to advanced pulmonary malignancy an average of two wk after tumor implantation (Fig. 4). In contrast, mice which had been preimmunized with XS52-Adβgal exhibited a markedly prolonged survival (P <0.0001, Fig. 4), consistent with the findings that XS52-βgal–immunized mice had far fewer lung metastases than the control treatment groups after a tumor challenge. Importantly, no protection was conferred by XS52-Adβgal immunization against CT26.WT tumor cells which did not express βgal (_P_ >0.06; not shown), suggesting that the antitumor immunity generated by administration of the Adβgal-modified DCs was antigen (βgal)- specific.
Figure 4.

Survival advantage in mice immunized with modified XS52 cells after a tumor challenge. Animals were immunized with XS52 cells modified with Adβgal or AdNull. The experiment is similar to that depicted in Fig. 3, except that the animals were not killed but were followed for survival. The data is expressed as percent survival as a function of time. Survival for the mice immunized with XS52-Adβgal was significantly prolonged over the XS52-AdNull control, as determined by log-rank analysis of the Kaplan-Meier survival curves (P <0.0001).
Suppression of Preestablished Lung Metastases after XS52-Adβgal Treatment.
To evaluate if tumor protection after DC immunization could be extended to a more stringent and clinically relevant model, Adβgal-modified XS52 were used to treat mice with preexisting tumors. In this experiment, diffuse lung metastases were first generated in BALB/c mice by intravenous administration of 3 × 104 CT26.CL25 tumor cells. 3 d after tumor implantation, the mice were either left untreated or were treated with subcutaneous administration of 3 × 105 XS52–AdNull modified cells or XS52-Adβgal–modified cells. Tumor-bearing mice that were not immunized (Fig. 5, A) or those that were immunized with the control DCs (XS52-AdNull; B) exhibited multiple lung metastases 20 d after tumor implantation. In contrast, tumor-bearing mice which received XS52-Adβgal treatment demonstrated a striking reduction in the number of metastases (C and D), demonstrating that DCs genetically modified to express a tumor antigen could effectively suppress preexisting tumors. Quantitative analysis of the number of metastatic nodules confirmed the significant treatment differences between XS52-Adβgal–treated group and the control groups (P <0.009; _E_). In contrast, XS52-Adβgal cells that had been lysed by repeated freezing and thawing did not confer any therapeutic effect when compared to untreated or XS52-AdNull–treated animals (_P_ >0.4; not shown).
Figure 5.



Suppression of preestablished lung metastases by administration of modified XS52 DCs. Shown are examples and quantitative data of treatment effect on preestablished lung metastases induced by XS52-Adβgal immunization. 3 d after the establishment of diffuse lung metastases in BALB/c mice with intravenous administration of 3 × 104 CT26.CL25 cells, 3 × 105 XS52-AdNull or XS52-Adβgal were administered subcutaneously to the tumor-bearing mice. 20 d after tumor implantation, the mice were killed, and their lungs were harvested, fixed, and stained for βgal expression with X-Gal. (A) Example of lungs from a nonimmunized mouse. (B) Example of lungs from a mouse receiving XS52-AdNull treatment. (C and D) Examples of lungs from mice receiving XS52-Adβgal treatment. (E) Quantification of the number of lung metastases in untreated, XS52-AdNull– and XS52-Adβgal–treated mice with preestablished lung metastases. Using a dissecting microscope, surface blue-staining (βgal+) lung metastases were enumerated. Each data point represents an individual animal. Only metastatic deposits ⩽250 could be reliably enumerated; lungs with >250 metastases were assigned an empirical number of 250.
In cancer treatment, any observed treatment response becomes even more meaningful if it translates into a survival advantage. To evaluate if the reduction in the number of lung metastases in mice treated with XS52-Adβgal conferred a survival advantage, a group of animals were followed for survival. Tumor-bearing mice that were treated with XS52-Adβgal lived significantly longer than the animals in the negative control groups (P <0.002, Fig. 6). Consistent with the observation in the prevention experiment, XS52-Adβgal treatment did not suppress preestablished non–βgal-expressing CT26.WT tumors (not shown), again suggesting that the therapeutic effect is antigen (βgal)-specific (_P_ >0.2). Further, lysed XS52-Adβgal cells did not confer a survival advantage as compared to untreated mice or mice treated with XS52-AdNull (P >0.8; not shown).
Figure 6.

Survival advantage in tumor-bearing mice treated with modified XS52 cells. Animals were immunized with XS52 cells modified with Adβgal or AdNull. The experiment is similar to that depicted in Fig. 5, except that the animals were not killed but were followed for survival. The data is expressed as percentage of survival as a function of time. Survival for mice which were treated with XS52-Adβgal was significantly prolonged over the XS52-AdNull control, as determined by log-rank analysis of the Kaplan-Meier survival curves (P <0.002).
Adenovirus Vector Modification of Primary Bone Marrow DCs.
To evaluate if the observations made using XS52 cell line could also be reproduced using primary DCs, DC modification, tumor prevention, and tumor treatment experiments were carried out similar to that with the XS52 DC line. Bone marrow DC from BALB/c mice were isolated and expanded ex vivo for 7 d, followed by their characterization using composite criteria of typical morphology, cell surface markers, and functional mixed lymphocyte reaction assay. Cell surface phenotypic analysis by flow cytometry revealed high frequency of expression of CD11b (two separate preparations, 72, 73%), CD44 (98, 99%), and MHC class II antigen IAd (75, 80%; not shown). Consistent with the known function of DCs, as few as 100 DCs induced an allogenic T lymphocyte proliferation in a mixed lymphocyte reaction with stimulation indices of >20 (not shown).
To evaluate the ability of Ad vectors to transfer and express genes in primary bone marrow DCs, Adβgal was used to infect bone marrow DCs an moi of 100, and the cells were quantified for βgal activity at 24 h after infection by flow cytometry. Similar to the findings with XS52-Adβgal (Fig. 1, C), expression of βgal was readily detectable (95%) in bone marrow DCs infected with Adβgal (Fig. 7, C), but not in naive bone marrow DCs (A) or bone marrow DCs infected with AdNull (B), demonstrating that Ad vectors can transfer and express genes in primary DCs. Parallel studies carried out using other moi demonstrated 17% bone marrow DC cells expressing βgal with an moi of 30 and 96% at an moi of 300 (not shown). Based on these studies, all subsequent studies were carried out at an moi 100.
Figure 7.

Ad vector–mediated gene transfer and expression of βgal in bone marrow DC (BMDC) in vitro. The primary murine DCs were infected in vitro with Adβgal or AdNull control vector at moi of 100 for 2 h. 24 h later, βgal expression was quantified by flow cytometry using fluorescein di-β-galactoside. For all panels, the y-axis reflects DC number and the x-axis reflects log fluorescein intensity. The percentage of βgal-expressing DCs was determined by the right shift of the curve along the K gate. (A) Uninfected bone marrow DCs. (B) Bone marrow DCs infected with AdNull. (C) Bone marrow DCs infected with Adβgal.
Induction of CTL Response In Vivo Using Ad Vector –transduced Primary Bone Marrow DCs.
To evaluate if genetically modified primary bone marrow DCs would induce an antigen-specific CTL response in vivo, splenocytes from mice immunized with subcutaneous administration of bone marrow DCs transduced with Adβgal were stimulated in vitro with βgal peptide and were then evaluated for specific cell lysis against a panel of target cells in a standard 51Cr–release assay. Immunization with bone marrow DC–Adβgal cells induced a βgal-specific CTL response (Fig. 8, B), whereas the negative control bone marrow DC–AdNull cells did not (A). Only negligible lysis of non–βgal-expressing CT26.WT target cells were detected in mice immunized with bone marrow DC–Adβgal cells, indicating that the cellular immune response induced was βgal specific. This specific cytotoxicity was mediated predominately by MHC-restricted CTLs rather than by natural killer cells since E22, an MHC-mismatched (H-2b) βgal-expressing murine tumor cell line, was not significantly lysed in the same assay.
Figure 8.

Induction of CTL response in BALB/c mice after in vivo administration of bone marrow DCs infected with Adβgal. Bone marrow DCs were transduced in vitro with AdNull or Adβgal at moi of 100 for 2 h. 24 h later, 3 × 105 cells were administered subcutaneously into syngeneic BALB/c mice. 14 d later, splenocytes harvested from immunized mice were stimulated for 5 d in vitro with syngeneic fibroblasts (SVBalb) pulsed with βgal peptide and then assayed for specific cell lysis against three syngeneic colon carcinoma target cells (parental CT26.WT cells, βgal-expressing CT26.CL25 cells, or CT26.WT pulsed with βgal peptide in vitro [CT26.WT-βgal peptide]) as well as the E22 βgal-expressing allogeneic tumor cell line. (A) CTL response in mice immunized with bone marrow DCs infected with AdNull. (B) CTL response in mice immunized with bone marrow DCs infected with Adβgal.
Prevention against Lethal Tumor Challenge after Bone Marrow DC Immunization.
Consistent with the studies using XS52-Adβgal modified DCs, BALB/c mice that were immunized with bone marrow DC–Adβgal before challenge with tumor cells were effectively protected against the tumor challenge, as evidenced by a prolonged survival in the bone marrow DC–Adβgal group compared to the bone marrow DC–AdNull group (P <0.0001, Fig. 9). In contrast, mice immunized with bone marrow DC–Adβgal were not protected against a challenge by CT26.WT cells (_P_ >0.9; not shown), indicating that the immune response was βgal specific.
Figure 9.

Survival advantage in mice immunized with modified bone marrow DCs after a tumor challenge. Animals were immunized with bone marrow DCs modified with Adβgal or AdNull, followed 14 d later by intravenous challenge of 105 CT26.CL25 tumor cells. The animals were not killed, but were followed for survival. The data is expressed as percentage of survival as a function of time. Survival for the mice immunized with bone marrow DC–Adβgal was significantly prolonged over the bone marrow DC–AdNull control, as determined by log-rank analysis of the Kaplan-Meier survival curves (P <0.0001).
Suppression of Preestablished Lung Metastases by Bone Marrow DC Immunization.
Mice with preexisting lung metastases that received bone marrow DC–Adβgal treatment lived significantly longer than the bone marrow DC–AdNull control group (P <0.002, Fig. 10), demonstrating that primary DCs that are genetically modified to express the tumor antigen could suppress preestablished tumors. As in the prevention experiment, bone marrow DC–Adβgal immunization had no therapeutic effect on CT26.WT tumors that did not express βgal (_P_ >0.5; not shown), indicating that the antitumor effect was tumor antigen specific. Similarly, lysed bone marrow DCs were ineffective in prolonging survival compared to control groups (P >0.1; not shown).
Figure 10.

Survival advantage in tumor-bearing mice treated with modified bone marrow DCs. 3 d after the establishment of lung metastases with intravenous administration of 3 × 104 CT26.CL25 tumor cells, BALB/c mice were immunized with bone marrow DCs modified with Adβgal or AdNull. The animals were not killed, but were followed for survival. The data is expressed as percent survival as a function of time. Survival for mice that were treated with bone marrow DC–Adβgal was significantly prolonged over the bone marrow DC–AdNull control, as determined by log-rank analysis of the Kaplan-Meier survival curves (P <0.002).
Discussion
This study evaluates the use of an adenovirus gene transfer vector to deliver a model antigen into DCs in vitro, and the ability of subsequent in vivo administration of these genetically modified DCs to induce tumor antigen–specific antitumor immunity. The data demonstrate that an Ad vector encoding βgal as a model antigen can effectively transfer and express the transgene in a DC line and primary bone marrow DCs. In vivo administration of these modified DCs elicits an MHC-restricted, antigen-specific CTL response, protects syngeneic mice against tumor challenge, and suppresses preexisting lethal tumors. Importantly, a reduction in tumor burden in all studies was translated into significant survival advantage. These observations underscore the ability of genetically modified DCs to effectively present tumor antigens and induce immunity against tumors. With the discovery and definition of more tumor antigens, this strategy of genetic manipulation of DCs to express specific tumor antigens may provide an effective treatment for an increasing number of malignancies.
Genetic Manipulation of DCs.
DCs are the most potent professional antigen presenting cells that are capable of initiating T cell–dependent immune responses (12–15). Strategies that have been used to adapt this biologic potential to tumor therapy includes manipulating DCs by coculturing (pulsing) DCs with whole tumor antigens, defined tumor antigenic peptides, or total RNA from tumor cells (29–38). Using such approaches, it has been shown that effective antigen-specific immunity and antitumor responses can be elicited (29–38). Genetically modifying DCs by genes coding for specific tumor antigens to DCs has potential advantages. First, the genetically modified DCs may present previously unknown epitopes in association with different MHC molecules. Second, gene transfer to DCs ensures that the gene product is endogenously processed, leading to the generation of MHC class I–restricted CTLs which is the effector arm of cell-mediated immune response against tumor cells. Third, by using viral vectors to modify DCs in vitro, there is far less administration of viral proteins, in contrast to direct in vivo vaccination with viral vectors, thus minimizing the generation of neutralizing antibodies that prevent repeat treatment (27, 39).
Theoretically, direct gene transfer to DCs has advantages in generating CTLs toward the protein coded by the transgene, in that the gene will be transferred to the DC nucleus, expressed, and presented in the context of class I MHC for the immune system to recognize as self or nonself (10, 11). In this regard, there is emerging evidence that when Ad vectors expressing heterologous transgenes are administered in vivo the Ad functions as an “adjuvant” to enhance cellular immune responses against the heterologous transgene product, including the generation of transgene product–specific CTLs (4–9). It is not known whether or not this is secondary to in vivo transfer of the transgene to DCs in the local milieu, or is by other mechanisms. However, independent of the process by which Ad vectors induce CTLs against heterologous transgenes, the data in the present study clearly demonstrate that when an Ad vector expressing a heterologous transgene is used to transfer the transgene to DCs in vitro, subsequent transfer of the genetically modified DCs to a syngeneic host will result in the induction of CTLs against the heterologous transgene product.
Different gene transfer approaches have been explored to use genetically modified DCs in antitumor therapy (40– 44). Several groups have reported the successful transduction of human DCs in vitro by retrovirus vectors encoding a model antigen (βgal) and tumor antigens including the melanoma MART-1 antigen and the human epithelial tumor antigen mucin (40, 41, 43). Brossart et al. (44) has recently shown that adenovirus and vaccinia virus vectors could transfer a model antigen (ovalbumin) into murine immortalized and primary DCs, and that the virus–infected DCs elicited stronger antigen-specific CTL response in vivo than immunization with DCs which were pulsed with ovalbumin peptide or protein. Importantly, immunization of mice twice with the virus-infected immortalized DCs protected the mice from a lethal challenge with tumor cells expressing ovalbumin. Consistent with these observations, the present study demonstrated that a single immunization of genetically modified DC line and primary DCs elicited antigen-specific CTL responses in vivo that were highly effective in protecting the mice from a lethal tumor challenge. Importantly, our study also shows that treatment of tumor-bearing mice with a single immunization of genetically modified DCs led to a marked suppression of the growth of established tumors. To further improve the therapeutic efficacy of this strategy, the codelivery of cytokine gene and tumor antigen gene into DCs and/or repeat immunization of DCs may enhance the antigen-specific antitumor immunity.
Clinical Relevance.
In addition to demonstrating that Ad vector–transduced DCs could generate an antigen-specific CTL response that protected immunized animals against a tumor challenge, this study also showed that the genetically modified DCs effectively suppress preestablished, rapidly growing metastatic tumors. The latter observation is clinically relevant since patients have preexisting tumors at the time of presentation and the Achilles heel of conventional cancer treatment today remains the failure to effectively control metastatic disease. In this regard, one clinical scenario to which this study is particularly relevant is one of micrometastases which are often present at the time of primary detection of many malignancies such as breast, colon, and lung cancer (45, 46). A possible clinical strategy to approach this problem would be to modify autologous DCs from such patients using an Ad vector encoding the relevant tumor antigen, and then administering the genetically modified DCs as adjuvant treatment after primary therapy.
Acknowledgments
We would like to thank Susan Cho and Philip L. Leopold for helpful advice and assistance in carrying out these studies, and N. Mohamed for help in preparation of the manuscript.
Footnotes
These studies were supported, in part, by GenVec, Inc. (Rockville, MD) and the Will Rogers Memorial Fund (White Plains, NY). M.A.S. Moore is supported by the Gar Reichman Fund of the Cancer Research Institute (New York), S. Rafii is supported by a grant from the National Institutes of Health (K08-HL 02926), and Dorothy Rodbell Foundation for Sarcoma Research, (New York), R. Granstein is supported by a grant from the National Institutes of Health (R01-AR 40667) and a grant from the Edith C. Blum Foundation (New York), H.-L. Kong is supported, in part, by the Ministry of Health of Singapore, the Singapore National Medical Research Council, and the National University Hospital (Singapore, Republic of Singapore).
W. Song and H.-L. Kong participated equally in this study.
1
Abbreviations used in this paper: Ad, adenovirus; βgal, β galactosidase; DC, dendritic cell; moi, multiplicity of infection.
References
- 1.Pardoll DM. Cancer vaccines. Immunol Today. 1993;14:310–316. doi: 10.1016/0167-5699(93)90051-L. [DOI] [PubMed] [Google Scholar]
- 2.Houghton AN. Cancer antigens: immune recognition of self and altered self. J Exp Med. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juillard V, Villefroy P, Godfrin D, Pavirani A, Venet A, Guillet JG. Long-term humoral and cellular immunity induced by a single immunization with replication-defective adenovirus recombinant vector. Eur J Immunol. 1995;25:3467–3473. doi: 10.1002/eji.1830251239. [DOI] [PubMed] [Google Scholar]
- 5.Chen PW, Wang M, Bronte V, Zhai Y, Rosenberg SA, Restifo NP. Therapeutic antitumor response after immunization with a recombinant adenovirus encoding a model tumor-associated antigen. J Immunol. 1996;156:224–231. [PMC free article] [PubMed] [Google Scholar]
- 6.Xiang ZQ, Yang Y, Wilson JM, Ertl HC. A replication-defective human adenovirus recombinant serves as a highly efficacious vaccine carrier. Virology. 1996;219:220–227. doi: 10.1006/viro.1996.0239. [DOI] [PubMed] [Google Scholar]
- 7.Flanagan B, Pringle CR, Leppard KN. A recombinant adenovirus expressing the simian immunodeficiency virus Gag antigen can induce long-lived immune responses in mice. J Gen Virol. 1997;78:991–997. doi: 10.1099/0022-1317-78-5-991. [DOI] [PubMed] [Google Scholar]
- 8.Rodriques EG, Zavala F, Eichinger D, Wilson JM, Tsuji M. Single immunizing dose of recombinant adenovirus efficiently induces CD8+ T cell–mediated protective immunity against malaria. J Immunol. 1997;158:1268–1274. [PubMed] [Google Scholar]
- 9.Song W, Kong H, Traktman P, Crystal RG. Cytotoxic T lymphocyte responses to proteins encoded by heterologous transgenes transferred in vivo by adenoviral vectors. Hum Gene Ther. 1997;8:1207–1217. doi: 10.1089/hum.1997.8.10-1207. [DOI] [PubMed] [Google Scholar]
- 10.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 11.Bevan MJ. Antigen presentation to cytotoxic T lymphocytes in vivo. J Exp Med. 1995;182:639–641. doi: 10.1084/jem.182.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 13.Grabbe S, Beissert S, Schwarz T, Granstein RD. Dendritic cells as initiators of tumor immune responses: a possible strategy for tumor immunotherapy? . Immunol Today. 1995;16:117–121. doi: 10.1016/0167-5699(95)80125-1. [DOI] [PubMed] [Google Scholar]
- 14.Young JW, Inaba K. Dendritic cells as adjuvants for class I major histocompatibility complex–restricted antitumor immunity. J Exp Med. 1996;183:7–11. doi: 10.1084/jem.183.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinman RM. Dendritic cells and immune-based therapies. Exp Hematol (NY) 1997;24:859–862. [PubMed] [Google Scholar]
- 16.Xu S, Bergstresser PR, Takashima A. Phenotypic and functional heterogeneity among murine epidermal-derived dendritic cell clones. J Invest Dermatol. 1995;105:831–836. doi: 10.1111/1523-1747.ep12326625. [DOI] [PubMed] [Google Scholar]
- 17.Xu S, Ariizumi K, Caceres-Dittmar G, Edelbaum D, Hashimoto K, Bergstresser PR, Takashima A. Successive generation of antigen-presenting, dendritic cell lines from murine epidermis. J Immunol. 1995;154:2697–2705. [PubMed] [Google Scholar]
- 18.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, Restifo NP. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685–4692. [PMC free article] [PubMed] [Google Scholar]
- 19.Rawle FC, Knowles BB, Ricciardi RP, Brahmacheri V, Duerksen-Hughes P, Wold WS, Gooding LR. Specificity of the mouse cytotoxic T lymphocyte response to adenovirus 5. E1a is immunodominant in H-2b, but not in H-2d or H-2k mice. J Immunol. 1991;146:3977–3984. [PubMed] [Google Scholar]
- 20.Inaba K, Inaba M, Deguchi M, Hagi K, Yasumizu R, Ikehara S, Muramatsu S, Steinman RM. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II–negative progenitor in mouse bone marrow. Proc Natl Acad Sci USA. 1993;90:3038–3042. doi: 10.1073/pnas.90.7.3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenfeld MA, Siegfried W, Yoshimura K, Yoneyama K, Fukayama M, Stier LE, Paakko PK, Gilardi P, Stratford-Perricaudet LD, Perricaudet M, et al. Adenovirus-mediated transfer of a recombinant alpha 1-antitrypsin gene to the lung epithelium in vivo. Science (Wash DC) 1991;252:431–434. doi: 10.1126/science.2017680. [DOI] [PubMed] [Google Scholar]
- 22.Rosenfeld MA, Yoshimura K, Trapnell BC, Yoneyama K, Rosenthal ER, Dalemans W, Fukayama M, Bargon J, Stier LE, Stratford-Perricaudet L, et al. In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the airway epithelium. Cell. 1992;68:143–155. doi: 10.1016/0092-8674(92)90213-v. [DOI] [PubMed] [Google Scholar]
- 23.Hersh J, Crystal RG, Bewig B. Modulation of gene expression after replication-deficient, recombinant adenovirus–mediated gene transfer by the product of a second adenovirus vector. Gene Ther. 1995;2:124–131. [PubMed] [Google Scholar]
- 24.Crystal RG, McElvaney NG, Rosenfeld MA, Chu CS, Mastrangeli A, Hay JG, Brody SL, Jaffe HA, Eissa NT, Danel C. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat Genet. 1994;8:42–51. doi: 10.1038/ng0994-42. [DOI] [PubMed] [Google Scholar]
- 25.MacGregor, G.R., G.P. Nolan, S. Fiering, M. Roederer, and L.A. Herzenberg. 1991. Use of E. coli lacZ (β-galactosidase) as a reporter gene. In Methods in Molecular Biology. E.J. Murray, editor. The Humana Press Inc., Clifton, NJ. 217– 235. [DOI] [PubMed]
- 26.Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J Immunol. 1993;151:3971–3980. [PubMed] [Google Scholar]
- 27.Mack CA, Song WR, Carpenter H, Wickham TJ, Kovesdi I, Harvey BG, Magovern CJ, Isom OW, Rosengart T, Falck-Pedersen E, et al. Circumvention of anti-adenovirus neutralizing immunity by administration of an adenoviral vector of an alternate serotype. Hum Gene Ther. 1997;8:99–109. doi: 10.1089/hum.1997.8.1-99. [DOI] [PubMed] [Google Scholar]
- 28.Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, Mantel N, McPherson K, Peto J, Smith PG. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. Analysis and examples. Br J Cancer. 1977;35:1–39. doi: 10.1038/bjc.1977.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grabbe S, Bruvers S, Gallo RL, Knisely TL, Nazareno R, Granstein RD. Tumor antigen presentation by murine epidermal cells. J Immunol. 1991;146:3656–3661. [PubMed] [Google Scholar]
- 30.Mayordomo JI, Zorina T, Storkus WJ, Zitvogel L, Celluzzi C, Falo LD, Melief CJ, Ildstad ST, Kast WM, DeLeo AB, et al. Bone marrow–derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med. 1995;1:1297–1302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- 31.Ossevoort MA, Feltkamp MC, van Veen KJ, Melief CJ, Kast WM. Dendritic cells as carriers for a cytotoxic T-lymphocyte epitope-based peptide vaccine in protection against a human papillomavirus type 16–induced tumor. J Immunother Emphasis Tumor Immunol. 1995;18:86–94. doi: 10.1097/00002371-199508000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184:465–472. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD. Peptide-pulsed dendritic cells induce antigen-specific, CTL-mediated protective tumor immunity. J Exp Med. 1996;183:283–287. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 35.Mayordomo JI, Loftus DJ, Sakamoto H, De Cesare CM, Appasamy PM, Lotze MT, Storkus WJ, Appella E, De Leo AB. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med. 1996;183:1357–1365. doi: 10.1084/jem.183.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J Exp Med. 1996;183:317–322. doi: 10.1084/jem.183.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porgador A, Snyder D, Gilboa E. Induction of antitumor immunity using bone marrow–generated dendritic cells. J Immunol. 1996;156:2918–2926. [PubMed] [Google Scholar]
- 38.Zitvogel L, Mayordomo JI, Tjandrawan T, DeLeo AB, Clarke MR, Lotze MT, Storkus WJ. Therapy of murine tumors with tumor peptide-pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell 1–associated cytokines. J Exp Med. 1996;183:87–97. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y, Li Q, Ertl HCJ, Wilson JM. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J Virol. 1995;69:2004–2015. doi: 10.1128/jvi.69.4.2004-2015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henderson RA, Nimgaonkar MT, Watkins SC, Robbins PD, Ball ED, Finn OJ. Human dendritic cells genetically engineered to express high levels of the human epithelial tumor antigen mucin (MUC-1) Cancer Res. 1996;56:3763–3770. [PubMed] [Google Scholar]
- 41.Reeves ME, Royal RE, Lam JS, Rosenberg SA, Hwu P. Retroviral transduction of human dendritic cells with a tumor-associated antigen gene. Cancer Res. 1996;56:5672–5677. [PubMed] [Google Scholar]
- 42.Wan, Y., J. Bramson, R. Cater, F. Graham, and J. Gauldie. 1996. Dendritic cells transfected with adenovirus vectors encoding a model tumor-associated antigen for tumor immunotherapy. Cold Spring Harbor–Gene Therapy Meeting, Cold Spring Harbor, NY. 316. (Abstr.).
- 43.Aicher A, Westermann J, Cayeux S, Willimsky G, Daemen K, Blankenstein T, Uckert W, Dorken B, Pezzutto A. Successful retroviral mediated transduction of a reporter gene in human dendritic cells: feasibility of therapy with gene-modified antigen presenting cells. Exp Hematol (NY) 1997;25:39–44. [PubMed] [Google Scholar]
- 44.Brossart P, Goldrath AW, Butz EA, Martin S, Bevan MJ. Virus-mediated delivery of antigenic epitopes into dendritic cells as a means to induce CTL. J Immunol. 1997;158:3270–3276. [PubMed] [Google Scholar]
- 45.Fisher B. The evolution of paradigms for the management of breast cancer: a personal perspective. Cancer Res. 1992;52:2371–2383. [PubMed] [Google Scholar]
- 46.Fidler, I.J. 1995. Invasion and metastasis. In Clinical Oncology. M.D. Abeloff, J.O. Armitage, A.S. Lichter, and J.E. Niederhuber, editors. Churchill Livingstone, New York. 55–76.