Induction of Apoptosis of Metastatic Mammary Carcinoma Cells In Vivo by Disruption of Tumor Cell Surface CD44 Function (original) (raw)
Abstract
To understand how the hyaluronan receptor CD44 regulates tumor metastasis, the murine mammary carcinoma TA3/St, which constitutively expresses cell surface CD44, was transfected with cDNAs encoding soluble isoforms of CD44 and the transfectants (TA3sCD44) were compared with parental cells (transfected with expression vector only) for growth in vivo and in vitro. Local release of soluble CD44 by the transfectants inhibited the ability of endogenous cell surface CD44 to bind and internalize hyaluronan and to mediate TA3 cell invasion of hyaluronan-producing cell monolayers. Mice intravenously injected with parental TA3/St cells developed massive pulmonary metastases within 21–28 d, whereas animals injected with TA3sCD44 cells developed few or no tumors. Tracing of labeled parental and transfectant tumor cells revealed that both cell types initially adhered to pulmonary endothelium and penetrated the interstitial stroma. However, although parental cells were dividing and forming clusters within lung tissue 48 h following injection, >80% of TA3sCD44 cells underwent apoptosis. Although sCD44 transfectants displayed a marked reduction in their ability to internalize and degrade hyaluronan, they elicited abundant local hyaluronan production within invaded lung tissue, comparable to that induced by parental cells. These observations provide direct evidence that cell surface CD44 function promotes tumor cell survival in invaded tissue and that its suppression can induce apoptosis of the invading tumor cells, possibly as a result of impairing their ability to penetrate the host tissue hyaluronan barrier.
Obligatory stages in tumor metastasis include tumor cell adhesion to the endothelium of organs distant to the site of primary tumor growth, transmigration of the endothelium, and formation of new cellular colonies within the invaded tissue. Formation of new colonies depends, at least in part, on the ability of tumor cells to communicate with their microenvironment, which includes the extracellular matrix (ECM)1 and growth factors and cytokines sequestered by the ECM and stromal cells. Adhesion to the ECM provides normal epithelial cells with signals that promote their survival and proliferation in vivo (1). Similarly, despite the anchorage-independent nature of malignant cell growth, appropriate interactions with the ECM are likely to foster tumor cell survival and growth in newly invaded tissues. Thus, some members of the integrin family of adhesion receptors, which play a major role in tumor cell attachment to ECM molecules such as collagens, laminin, and fibronectin (2), can promote tumor cell metastasis, and the integrin repertoire of any given tumor cell is likely to help determine whether and where the cell can metastasize (3). In addition to integrins, several unrelated cell surface structures are likely to influence tumor growth and dissemination. Recent evidence has shown that cell surface expression of CD44, a receptor for the ECM glycosaminoglycan hyaluronan (HA), may have a highly significant effect on the regulation of tumor growth and metastasis (4–6).
CD44 is a polymorphic glycoprotein whose diversity is determined by differential splicing of at least 10 variable exons encoding a segment of the extracellular domain, termed exons v1–v10, and cell type–specific glycosylation (7, 8). Using a variety of experimental models, CD44 has been shown to mediate cell–cell and cell–ECM interactions (9–11), augment tumor cell motility on HA-coated substrates (12), costimulate lymphocyte activation and tissue infiltration (13, 14), and promote growth and metastasis of some tumor types (4, 5). Although glycosaminoglycan side chains associated with some CD44 isoforms can also bind a subset of heparin-binding growth factors (15), cytokines (16), and ECM proteins such as fibronectin (17), most of the functions ascribed to CD44 thus far can be attributed to its ability to bind and internalize HA (18, 19). HA is a large polysaccharide, composed of multiple repeats of the β1,3 _N_-acetylglucosaminyl-β1,4 glucuronide disaccharide unit, principally produced by stromal cells and deposited in the pericellular matrices and the ECM of most tissues. In the ECM, HA is bound to several proteins, helping to create a lattice that may regulate cell adhesion and migration (20, 21). HA production is increased at sites of cell proliferation, including limb bud formation, inflammation, tissue remodeling, and tumor cell invasion, and is thought to modulate cell behavior by binding to specific cell surface receptors, most notably CD44 (22). Consistent with this notion, CD44–HA interaction has been observed to enhance growth of certain tumors in vivo (23). However, neither the mechanism by which CD44–HA interaction enhances tumor growth and metastasis nor the stage of the metastatic process at which the interaction is functionally relevant have been elucidated.
To obtain insight into the stage(s) of tumor metastasis that may be facilitated by CD44 expression and the in vivo fate of tumor cells in which endogenous CD44 function has been impaired, we transfected cDNAs encoding soluble CD44 receptors into the TA3/St murine mammary carcinoma, which constitutively expresses cell surface CD44, and studied the effect of soluble CD44 expression on tumor growth and dissemination in vivo. Our results show that expression of soluble CD44 reduces the HA binding and internalizing ability of TA3/St cells and abrogates their ability to form lung metastases after tail vein injection. Moreover, we show that TA3 cells with impaired endogenous CD44 function are unable to invade HA-producing cell monolayers and undergo apoptosis after penetration of lung tissue in vivo. We propose that one function of CD44 in tumor cells may be to facilitate penetration of stromal cell–derived HA, which, at least for some tumor cell types, may be a critical step toward establishing metastatic colonies.
Materials and Methods
Cell Culture and Antibodies.
G8 mouse fetal myoblasts were obtained from American Type Culture Collection (ATCC, Rockville, MD), and cultured in DMEM with 10% fetal bovine serum (FBS, both from Irvine Scientific, Santa Ana, CA) and 10% horse serum (GIBCO BRL, Gaithersburg, MD). TA3/St cells were maintained in DMEM supplemented with 10% FBS. All the transfected TA3/St cells were cultured in DMEM with 10% FBS and 0.5 mg/ml of G418 (GIBCO BRL). The KM201 and HB-233 hybridomas (ATCC), which produce mAbs against mouse CD44 and intracellular adhesion molecule (ICAM)-1, respectively, were both cultured in DMEM with 10% FBS. Partially purified mAbs were obtained by 50% (NH4)2SO4 precipitation of the hybridoma culture media.
Truncated Soluble CD44 and ICAM-1 Constructs and PCR Analysis of CD44 Isoform Expression.
Total RNA was isolated from G8 myoblasts, TA3/St cells, and transfected TA3/St cells using TRIzol reagent (GIBCO BRL) according to the manufacturer's instructions. cDNA was synthesized from 5 μg of total RNA using Superscript II RNase H− reverse transcriptase (GIBCO BRL) according to the vendor's recommendations. Three sets of PCR were performed using cDNA generated from G8 myoblasts, TA3, and transfected TA3 cells, respectively. The first set of PCRs was performed to generate soluble CD44 isoforms from the cDNA derived from G8 myoblasts using the sense exon 1f primer and the antisense new v10r primer (24), and the products were inserted into pCR 3-Uni eukaryotic expression vectors (Invitrogen Corp., San Diego, CA). The authenticity and orientation of the nucleotide sequences of the inserts were confirmed by DNA sequencing using the dideoxy chain termination method. The second set of PCRs was performed to assess TA3/St cell expression of CD44 isoforms at the mRNA level, using the antisense exon 16r primer along with either the sense exon 5f primer, to identify the standard CD44H isoform, or the sense variant exon v1f to v10f primers. The third set of PCRs was performed to identify the G418-resistant colonies that express soluble CD44 at the mRNA level after transfection. In this case, cDNA was made from G418-resistant colony-derived RNA, and PCR was performed using the sense exon 1f primer and the antisense M13r primer, which complements the sequence of the expression vector immediately 3′ to the insert. Taq DNA polymerase (Perkin Elmer, Norwalk, CT) was employed for 30 (in the first set) or 35 (in the second and third sets) cycles at 94°C for 40 s, 55°C for 40 s, and 72°C for 90 s, followed by a final 7 min at 72°C. 25-μl aliquots of the PCR reaction products were analyzed by 1% agarose gel electrophoresis. The remaining portion of the products were used for subcloning if desired. The primers used were as follows: Exon 1f, 5′-CGCCATGGACAAGTTTTGGT-3′; Exon 5f, 5′-GCCTACTGGAGATCAGGATG-3′; Exon 16r, 5′-GATCCATGAGTCACAGTGCG-3′; v1f, 5′-TTGCCTCAACTGTGCACTCA-3′; v2f, 5′-TGATGACCACCCCTGAAACA-3′; v3f, 5′-GTACGGAGTCAAATACCAAC-3′; v4f, 5′-TTGCAAGTACTCCACGGGTT-3′; v5f, 5′-ATAGACAGAATCAGCACCAG-3′; v6f, 5′-CTCCTAATAGTACAGCAGAA-3′; v7f, 5′-CTTCGGCCCACAACAACCAT-3′; v8f, 5′-ATACAGACTCCAGTCATAGT-3′; v9f, 5′-CACAGAGTCATTCTCAGAAC-3′; v10f, 5′-CTAAGAGCGGCGCTAAAGAT-3′; v20r, 5′-CACCCCAATCTTCATGTCCA-3′; New v10r, 5′-CTATAAAGCAGAAAAATCAGCAACC-3′; and M13r, 5′-TAGAAGGCACAGTCGAGGCT-3′; f and r indicate forward and reverse orientations, respectively.
Truncated soluble CD44v6-10 containing the R43A mutation was generated using oligonucleotide primers designed to contain an internal NarI site that allowed convenient substitution of R by A without having to introduce silent mutations. The dipeptide G–R at positions 42–43 was therefore mutated to G–A. The oligonucleotide primers used were: m44R43A r, 5′-CACGCGGGCGCCATTTTTCTCCACATGGAATACTGG-3′; and m44R43A f, 5′-CACGCGGGCGCCTACAGTATCTCCCGGACTGAGGCA-3′. The primer pairs Exon1f and m44R43A r and m44R43A f and New v10r were used to amplify 5′ and 3′ segments of the truncated CD44 cDNA, respectively, and the amplified fragments were subjected to NarI endonuclease hydrolysis and inserted into the pCR 3-Uni vector in a three-way ligation. The orientation of the construct and the appropriate mutation were verified by sequencing.
Soluble truncated ICAM-1 was PCR-amplified from mouse spleen cDNA and ligated to the pCR 3-Uni vector, as described above. The primers used were: mICAM-1 f, 5′-GCAATGGCTTCAACCCGTGCCAAGCCCACGCTACCT-3′; and mICAM-1 r, 5′-TTAGTGGTACAGTACTGTCAGGTACACATTCCT-3′.
Transfection.
TA3/St cells were seeded at 2 × 105/well into a 6-well plate, and cultured overnight in DMEM supplemented with 10% FBS. On the next day, the cells were transfected with mixtures of Lipofectamine (15 μl/ml of serum-free DMEM, GIBCO BRL) and pCR 3-Uni eukaryotic expression vector alone (5 μg/ml) or pCR 3-Uni vector containing truncated CD44 cDNAs encoding soluble CD44 isoforms with variant exons v6–v10 or v8–v10, with or without the R43A mutation, or truncated ICAM-1 cDNA encoding soluble ICAM-1, for 5 h. The transfection medium was then replaced with fresh 10% FBS DMEM medium for an additional 48 h, whereupon the cells were subjected to G418 drug selection (1.5 mg/ml G418 in DMEM/10% FBS). G418 resistant colonies were picked 2–3 wk after application of the selection medium. Expression of the transfected cDNAs was assessed by both reverse transcriptase (RT)–PCR and Western blot analysis.
Fluorescein-labeled Binding Assay and FACS® Analysis of Cell Surface CD44 Expression.
Cells were detached from culture dishes with EDTA (Irvine Scientific), washed once with 10% FBS DMEM, and once with PBS, preincubated for 1 h on ice with mAb against mouse CD44 or mouse ICAM-1 or with PBS alone, and then incubated with 20 μg/ml fluorescein-labeled hyaluronan (FL-HA; Anika Research, Inc., Woburn, MA) for an additional 2 h on ice. After three washes with PBS, the cells were suspended in 1 ml PBS, and analyzed on a FACScan® (Becton Dickinson, Mountain View, CA). For assessment of cell surface CD44 expression, cells were detached from plates as described above, washed, and incubated with mAb IM7.8 (∼10 μg/ml) on ice for 1 h, washed five times with PBS, and incubated with fluorescein-conjugated goat anti–rat secondary antibody for 30 min on ice. The cells were then washed extensively and analyzed on a FACScan® immediately. The cells were incubated with an unrelated isotype-matched antibody and fluorescein-labeled secondary antibody as a negative control.
Cell Fluorescence Labeling and Tumor Metastasis Assays.
The transfected TA3/St cells (TA3neo and TA3sCD44, 1 × 106 in 0.2 ml Hank's balanced salt solution per mouse) were injected into the tail vein of male A/jax syngeneic mice, (The Jackson Laboratory, Bar Harbor, ME). The animals were observed daily, and were killed between 3 and 4 wk after injection. Tissues were excised, the extent of metastasis was documented, and then the tissues were fixed and sectioned for histologic and immunohistochemical analysis.
For fluorescence labeling, TA3neo and TA3sCD44 cells were cultured in 10% FBS DMEM at 70–80% confluence. Cells were then incubated at 37°C for 30 min in fresh, prewarmed culture medium containing 10 μM of green 5-chloromethyl-fluorescein diacetate (CMFDA, Molecular Probes, Inc., Eugene, OR), followed by fresh 10% FBS DMEM for an additional 30 min at 37°C. Next, cells were detached from culture dishes with EDTA (Irvine Scientific), and washed once with 10% FBS DMEM, and once with PBS. 5 × 106 labeled cells, resuspended in 0.2 ml Hank's balanced salt solution, were injected into the tail vein of each A/jax mouse.
Immunofluorescence Microscopy and Histology.
The lungs of injected mice were removed 1, 24, and 48 h after injection, fixed, and 5-μm-thick paraffin sections were cut. After deparaffinization, rehydration, and mounting, the sections were observed under a fluorescence microscope. TUNEL assays were performed on the paraffin sections of the lungs from mice injected with Green CMFDA–labeled tumor cells using an ApopTag Plus kit (Oncor, Inc., Gaithersburg, MD) in which rhodamine-conjugated anti-digoxigenin Fab fragments (Boehringer Mannheim Biochemicals, Indianapolis, IN) were substituted for fluorescein-conjugated antidigoxigenin antibody.
Invasion Assays.
5 × 104 G8 myoblasts were seeded onto each well of 6-well plates and cultured until they reached confluence. The monolayers were then fixed in DMSO for 1 h at room temperature, and washed extensively with PBS. All of the TA3/St transfectants were maintained in DMEM supplemented with 10% FBS and 0.5 mg/ml of G418 (GIBCO BRL). For the invasion assay, the TA3neo and TA3sCD44 cells were detached from culture dishes with EDTA solution (Irvine Scientific), washed once with 10% FBS DMEM and once with PBS, and seeded onto the fixed G8 monolayers at 5 × 103 cells/well of a 6-well plate. The invasiveness of TA3 cells was monitored daily under an inverted microscope.
Western Blot Analysis.
Supernatants of cultured TA3 cells were collected and concentrated through centricon-30 filters (Amicon, Danvers, MA). Lysates from attached cells were extracted with 50 mM Tri-HCl buffer, pH 7.4, containing 500 mM NaCl, 5 mM EDTA, 1% Triton X-100, 2 mM phenylmethylsulfonyl fluoride, 2 mg/ml leupeptin, and 0.5-ml aprotinin. 30 μg of protein from cell extracts and proteins from 2 ml of the culture medium, which were concentrated into 30 μl, were loaded per lane, subjected to 10% SDS-PAGE, and transferred onto Hybond-ECL (Amersham Corp., Arlington Heights, IL) membranes. The anti-CD44 mAb KM201 or IM7.8 was used to detect CD44 on the membranes.
HA-affinity Histochemistry.
Biotinylated proteoglycan (bPG) was used as a probe to detect HA (provided by Dr. Charles B. Underhill, Georgetown University, Washington, DC) according to previously described methodology (25). In brief, after inactivation of endogenous peroxidase with 10% H2O2 in methanol, tissue sections were incubated with 2 μg/ml bPG in PBS containing 10% calf serum (GIBCO BRL) for 30 min at room temperature. To determine background staining, 2 μg/ml bPG was first mixed with 100 μg/ml HA before use. After extensive washing with PBS, bound bPG was detected using Vector A and B reagents (Vector Laboratories, Burlingame, CA) and diaminobenzidine tetrahydrochloride dihydrate (Aldrich Chemical Co., Milwaukee, WI) according to the manufacturer's instructions.
Cell Adhesion Assays.
Adhesion of the transfected TA3/St cells to immobilized HA was performed as previously described (23). In brief, 2 × 105 51Cr-labeled cells were seeded onto 24-well plates coated with HA (5 mg/ml; Sigma Chemical Co., St. Louis, MO), 20 μg/ml laminin, 20 μg/ml collagen type IV, or fibronectin (20 μg/ml; Collaborative Biomedical Products, Bedford, MA) and incubated at room temperature for 1 h. Unattached cells were washed away by PBS, and bound cells were solubilized with 1 N NaOH, and an incorporated radiolabel was quantified in a γ counter. (Beckman Instruments, Inc., Fullerton, CA).
Cell Proliferation Assays.
Transfected TA3/St cells were seeded into 24-well plates at 2 × 105 cells/ml in 10% FBS DMEM, and cultured overnight. 0.5 μCi/ml [3H]thymidine were then added to the culture medium and the cells were incubated for 12–16 h. The cells were then washed twice with PBS, fixed with 10% cold trichloroacetic acid (TCA, Fisher Scientific Co., Santa Clara, CA), and washed twice with 7% TCA and once with 70% ethanol. The radioactive material on the plates was dissolved in 0.5 N NaOH, and quantified in a β counter.
HA Uptake and Degradation.
[3H]-HA was provided by Dr. Charles Underhill. The assay was performed as previously described (18). In brief, TA3neo and TA3sCD44 transfectants were seeded into 24-well plates at 2 × 105 cells/ml, and cultured in 10% FBS DMEM overnight. Fresh medium containing 2 μg/ml [3H]-HA (specific activity 7.6 × 104 cpm/μg) was then applied to the cultured cells, and after 40 h of incubation the medium and the cells were subjected to protease E (Sigma Chemical Co.) digestion overnight at 55°C. The supernatants of the digested mixtures were centrifuged in Centricon 30 tubes (Amicon), the solutions containing degraded [3H]-HA were collected from bottom chambers of the centricon tubes, and the corresponding radiolabel was quantified in a β counter (Beckman Instruments Inc.).
Results
Development of Tumor Cell Transfectants Expressing Soluble CD44.
To investigate how tumor cell surface CD44 expression influences the metastatatic process, we expressed soluble, truncated CD44 isoforms, composed of the extracellular domain containing different combinations of variant exons but lacking the transmembrane and intracellular domains, in the mouse mammary carcinoma TA3/St. TA3/St cells constitutively express several cell surface CD44 isoforms ranging from 80 to 220 kD (Fig. 1, B and C), display CD44-mediated binding of HA (Fig. 1 A), and rapidly form tumors in the lung after intravenous injection. We reasoned that the soluble CD44 isoforms should compete with and suppress or abrogate the normal function of endogenous cell surface CD44, thereby providing insight into how CD44 expression regulates TA3/St cell behavior in vivo. Naturally occurring soluble truncated CD44 receptors, which bear a stop codon in exon v10, have recently been identified in G8 mouse fetal myoblasts (24). Two soluble truncated CD44 isoforms containing variable exons v6–v10 and v8–v10 were isolated from G8 cell RNA by RT-PCR, introduced into the pCR 3-Uni eukaryotic expression vector (Invitrogen Corp.) and stably expressed in TA3/St cells. Four independent TA3/St transfectants expressing two different soluble CD44 isoforms (termed sCD44v6-10 No. 12 and No. 17 and sCD44v8-10 No. 13 and No. 19) and two independent clones transfected with the expression vector alone (neo No. 1 and No. 8) were selected for further study. All six transfectants expressed comparable levels of cell surface CD44, as assessed by FACS® analysis, and displayed a similar baseline proliferation rate as measured by [3H]thymidine incorporation in vitro (Table 1). Western blot analysis of transfectant supernatants using the mAb KM201, which recognizes an epitope common to all CD44 isoforms in the NH2 domain, revealed that sCD44v6-10 and sCD44v8-10 transfectants, but not neotransfectants, secreted soluble CD44 into their culture medium (Fig. 1 C, b).
Figure 1.
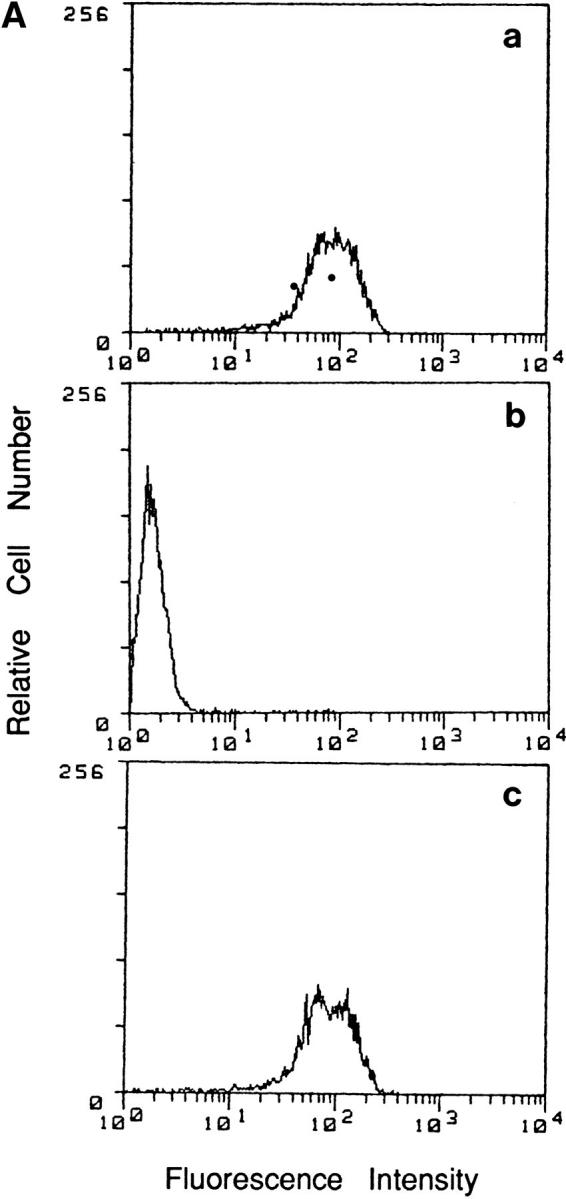


Binding of HA and CD44 expression by TA3/St cells. (A) Binding of FL-HA by TA3/ST cells as assessed by FACS® analysis (a). The ability of TA3/St cells to bind FL-HA completely abrogated by the blocking anti-CD44 monoclonal antibody KM201 (b), whereas control anti–ICAM-1 mAb has no effect (c). (B) Expression of multiple CD44 isoforms by TA3/St cells: RT-PCR was performed using the oligonucleotide primers listed in Materials and Methods and total RNA from TA3/St cells, and the products were analyzed on 1% agarose gels. Lane 1, 100-bp DNA reference ladder (GIBCO BRL); lane 2, PCR products using a forward primer corresponding to exon 5 (5f) and a reverse primer corresponding to exon 16 (16r). A 120-bp product representing the expression of the standard CD44 isoform containing no variant exons (CD44H) is indicated (white arrow); the presence of larger products indicates expression of multiple CD44 variants. Lanes 3–12, the reverse 16r primer was used together with forward primers v1f through v10f. The products in these lanes demonstrate that TA3/St cells express a range of CD44 variants containing variant exons v1 through v10. (C) Western blot analysis of CD44 expression in lysates (a) and supernatants (b) of parental and soluble CD44-transfected TA3/St cells. Lysates in a and supernatants in b were from: lane 1, TA3neo No. 1; lane 2, TA3neo No. 8; lane 3, TA3sCD44 v8-v10 No. 13; lane 4, TA3sCD44v8-10 No. 19; lane 5, TA3sCD44v6-v10 No. 12; lane 6, TA3sCD44v6-10 No. 17 cells. TA3neo No. 1 and No. 8 cells express CD44H (∼80 kD), and several larger CD44 isoforms; lysates from TA3 cells expressing soluble CD44 show a similar pattern of CD44 isoform expression, except that the abundance of CD44 proteins of ∼80 Kd, which corresponds to the Mr of soluble CD44 isoforms, is increased as expected. Western blot analysis of TA3/St cell culture supernatants (b) reveals that TA3neo No. 1 and No. 8 cells (lanes 1 and 2) do not produce soluble CD44, whereas TA3sCD44 transfectants produce variable amounts of soluble CD44 (lanes 3–6). Arrows indicate molecular weight markers, which are, from top to bottom, 203, 118, 86, and 52 kD.
Table 1.
Characterization of the Transfected TA3/St Cells
| Cell line | Endogenous CD44 (MFI) | Soluble CD44 | sICAM-1 | Adhesion to HA | [3H]Thymidine incorporation (CPM) | |
|---|---|---|---|---|---|---|
| RT-PCR | Western blot | RT-PCR/ Western blot | ||||
| TA3neo No. 1 | 273.38 | − | − | − | 100% ± 19.8% | 148923.33 ± 10517.66 |
| TA3neo No. 8 | 251.71 | − | − | − | 140% ± 11.4% | 128886.67 ± 4010.27 |
| TA3sCD44 v8-v10 No. 13 | 269.99 | ++ | ++ | − | 35.5% ± 8.5% | 139566.67 ± 8826.41 |
| TA3sCD44 v8-v10 No. 19 | 285.75 | + | + | − | 64.4% ± 10.1% | 129980 ± 504.78 |
| TA3sCD44 v6-v10 No. 12 | 259.39 | ++ | ++ | − | 55.35 ± 10.6% | 130726 ± 14282.39 |
| TA3sCD44 v6-v10 No. 17 | 304.24 | +++ | +++ | − | 29.6% ± 3.64% | 144903.33 ± 8025.62 |
| TA3sICAM-1 No. 13 | 300.00 | − | − | +++ | 105.68% ± 4.33% | 141993.33 + 7503.13 |
| TA3sICAM No. 24 | 271.61 | − | − | ++ | 81.46% ± 8.53% | 128663.33 ± 8637.74 |
| TA3sCD44 v6-v10R43A No. 43 | 299.48 | +++ | +++ | − | 81.97% ± 9.65% | 140363.33 ± 3871.39 |
| TA3sCD44 v6-v10R43A No. 55 | 316.00 | ++ | ++ | − | 155.88% ± 9.65% | 130490 ± 14086.41 |
Soluble CD44 Expression Abrogates TA3 Cell Metastatic Proclivity.
Each of the six transfectants was injected into the tail vein of nude (nu/nu) or A/jax mice in three independent experiments. In the first and second experiments, three nude and three A/jax mice were injected with 1 × 106 cells of each transfectant. In the third experiment, a minimum of six A/jax mice were injected with each transfectant (summarized in Table 2). 3 wk after injection, mice which had received TA3neo No. 1 and No. 8 displayed severe weight loss, and they were killed 1 wk later. However, animals which had received TA3sCD44v6-10 and v8-10 cells showed no signs of distress even several months after tumor cell injection. The mice injected with each TA3sCD44 transfectant were killed at the end of the fourth week and compared for metastatic tumor growth with that in TA3neo mice. All of the mice injected with TA3neo No. 1 and No. 8 displayed massive pulmonary metastases (Fig. 2 A, c and d). By contrast, mice injected with TA3sCD44s transfectants displayed either no tumor nodules (TA3sCD44v6-10 No. 17 and TA3sCD44v8-10 No. 13) or only a small number (<30) of tumor nodules per lung (TA3sCD33v6-10 No. 12 and TA3sCD44v8-10 No. 19, Fig. 2 A, a and b, and Table 2). No tumor nodules were found in other organs. RT-PCR on total RNA derived from several of the scarce tumor nodules from the lungs of mice injected with TA3sCD44v6-10 No. 12 revealed that these tumors had lost expression of transfected soluble CD44 while retaining endogenous cell surface CD44 expression (Fig. 2 B). This result provides an internal control that underscores the notion that local release of soluble, tumor-derived CD44 inhibits formation of lung metastasis by TA3 cells.
Table 2.
Effect of Soluble CD44 and ICAM-1 Expression on TA3/St Cell Metastasis
| Cell line | Massive metastasis | No visible metastasis | Few metastatic colonies (<30/lung) |
|---|---|---|---|
| TA3neo No. 1 | 10/10 | ||
| TA3neo No. 8 | 7/7 | ||
| TA3sCD44 v6-v10 No. 12 | 4/6 | 2/6 | |
| TA3sCD44 v6-v10 No. 17 | 7/7 | ||
| TA3sCD44 v8-v10 No. 13 | 6/6 | ||
| TA3sCD44 v8-v10 No. 19 | 3/6 | 3/6 | |
| TA3sCD44 v6-v10R43A No. 43 | 6/6 | ||
| TA3sCD44 v6-v10R43A No. 55 | 6/6 | ||
| TA3sICAM-1 No. 13 | 6/6 | ||
| TA3sICAM-1 No. 24 | 6/6 |
Figure 2.
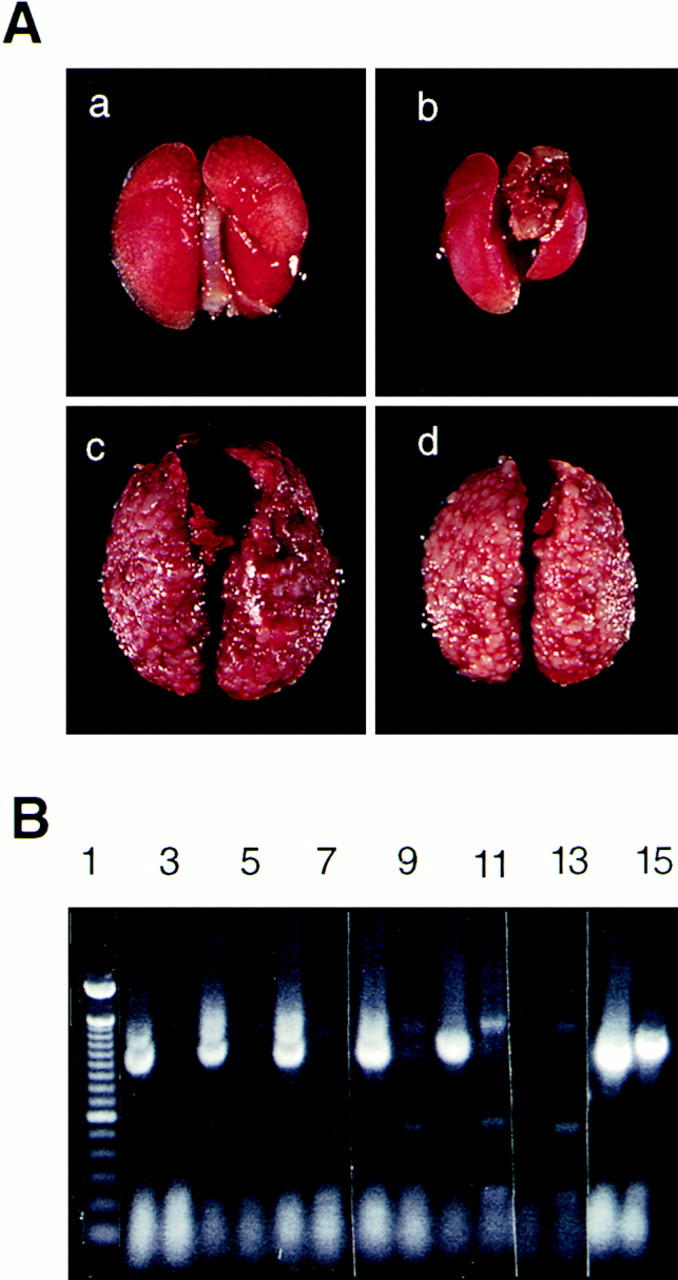
(A) Expression of soluble CD44 prevents lung metastasis of TA3 cells. Lungs from two representative mice injected with the TA3sCD44v6-10 No. 17 cells reveal no apparent tumor nodules (a and b); lungs from two representative mice injected with TA3neo No. 1 display innumerable tumor nodules throughout the lung parenchyma (c and d). (B) Tumors derived from lungs of mice injected with TA3sCD44v6-v10 No. 12 cells have lost soluble CD44 expression. Five independent tumor nodules were dissected out from the lung, and RT-PCR was performed using total RNA derived from each nodule (lanes 2–11). The primers used were: lanes 2, 4, 6, 8, 10, and 14, primer 1f and primer 20r, to detect expression of endogenous CD44; lanes 3, 5, 7, 9, 11, and 15, primer 1f and new v10r to detect expression of soluble CD44 (16); lanes 12–15: total RNA from cultured TA3sCD44v6-v10 No. 12 cells was used as template for lanes 12 and 13, negative controls, where PCR was performed with 1f primers only; lanes 14 and 15, positive controls where PCR was performed with primers 1f and 20r (lane 14), and 1f and new v10r (lane 15). Molecular weight markers (100-bp ladder) are shown in lane 1. RT-PCR analysis indicates that all five tumor nodules express endogenous but not soluble recombinant CD44.
To ensure that the observed inhibition of metastasis is not a non-specific effect of secreted protein, and to determine whether it is related to the ability of soluble CD44 to bind HA, we assessed the effect of expression of soluble ICAM-1 and a soluble CD44 mutant which has lost the ability to bind HA (Tables 1 and 2). The CD44 mutant contained a substitution of Arg43 to Ala which has been found to abrogate the ability of CD44 to bind HA (R41A in human CD44; reference 26). Two independent isolates of each transfectant were found to form lung metastasis after intravenous injection, similar to TA3neo cells (Table 2). Thus, secretion of an unrelated adhesion receptor did not interfere with the ability of TA3 cells to form tumors in vivo and the observed inhibitory effect of soluble CD44 was related to its ability to bind HA.
TA3 Cells Expressing Soluble CD44 Undergo Apoptosis after Penetration of Lung Tissue.
To address the fate of TA3sCD44 cells in vivo, and thereby gain insight into the stage of the metastatic process at which cell surface CD44 might be required, we labeled tumor cells with green CMFDA, which allows cell tracing for at least 48 h after injection. Animals injected with the labeled transfectants were killed at 1, 24, and 48 h after injection and lung tissue sections were examined by fluorescence microscopy. Both TA3neo and TA3sCD44 transfectants were found to arrest within lung blood vessels and extravasate into the interstitial stroma within 1 h after injection (Fig. 3 A). Between 1 and 24 h after injection, both TA3neo No. 1 and TA3sCD44v6-10 No. 17 cells were observed to invade lung stroma (Fig. 3 A, a–d, and B, a and b, and data not shown). However, at 48 h, when TA3neo transfectants were forming clusters, only isolated TA3sCD44v6-10 cells were visible (Fig. 3 A, e and f). To determine whether TA3sCD44v6-10 transfectants were undergoing apoptosis within the lung interstitium, TUNEL assays were performed on the tissue sections and TUNEL-positive cells were scored among 100 CMFDA-labeled cells. At 1 and 24 h after injection, minimal TUNEL staining was observed among TA3neo and TA3sCD44v6-10 cells alike (Fig. 3 B, a and b, and data not shown). However, at 48 h, 93 and 88% of the CMFDA-labeled TA3sCD44 cells tested positive for the TUNEL assay in contrast to 10 and 19% of the TA3neo transfectants in two independent experiments (Fig. 3 B, c and d, and data not shown).
Figure 3.
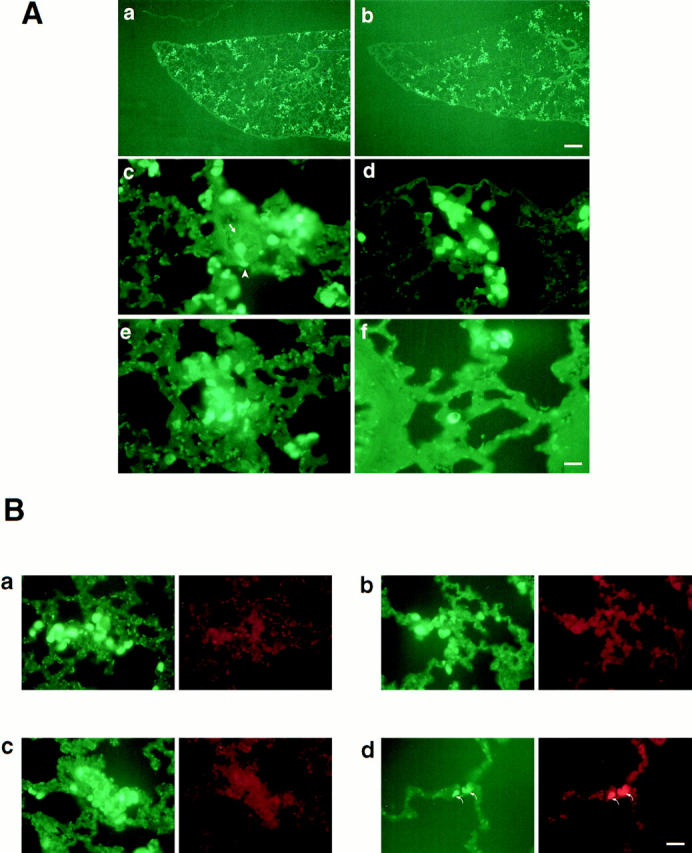
(A) Soluble CD44 inhibits events required for TA3/St cell growth in the lung tissue microenvironment. TA3neo No. 1 (a, c, and e) and TA3sCD44v6-10 No. 17 (b, d, and f) cells were labeled with green CMFDA and 5 × 106 cells in 0.2 ml HBSS buffer were injected into the tail vein of A/jax mice. The animals were killed at 1, 24, and 48 h after injection, the lungs were fixed and paraffin-embedded, and 5-μm-thick paraffin sections were mounted onto slides and examined by fluorescence microscopy. 1 h after injection, both TA3neo No. 1 and TA3sCD44 v6-v10 No. 17 cells were observed to be arrested in pulmonary blood vessels (a and b) and to penetrate the pulmonary interstitium (c and d). Occasional cells appeared to be in the process of extravasation (c, arrowhead and the arrow indicate an extravasating cell and the lumen of a blood vessel, respectively). 48 h after injection (e and f), TA3 neo cells display cluster formation (e), whereas only a few isolated TA3sCD44v6-10 No. 17 cells remain in the lung parenchyma (f). Bar in a and b = 304 μm; bar in c–f = 76 μm. (B) In situ detection of apoptosis by TUNEL assay. Lung sections showing TA3neo No. 1 cells (a and c) and TA3sCD44v6-10 No. 17 cells (b and d) labeled with CMFDA (green fluorescence) or detected with ApopTag (red fluorescence) 1 h (a and b) and 48 h (c and d) after intravenous injection. 1 h after injection, TA3neo No. 1 and TA3sCD44 v6-v10 No. 17 cells display similar distribution and no reactivity with ApopTAG (red fluorescence). 48 h after injection, the majority of TA3neo cells were ApopTAG-negative (c) whereas most (>80%) of the remaining TA3sCD44v6-v10 No. 17 cells tested positive for ApopTAG staining (d, arrows). Bar in a–d = 125 μm.
Expression of Soluble CD44 Reduces TA3 Cell Ability to Bind and Internalize HA and Abrogates their Ability to Invade HA-producing Monolayers.
To determine whether the reduction of TA3sCD44 cell ability to form metastases correlates with the suppression of known functions of cell surface CD44 by locally secreted CD44, TA3sCD44 transfectants were compared to parental cells for HA binding and internalization, invasion of cell monolayers, response to growth factors, and adhesion to a panel of ECM proteins. TA3neo and TA3sCD44 cells responded comparably to basic fibroblast growth factor (FGF) and heparin-binding epidermal growth factor (EGF), both of which bound v3-containing CD44 isoforms (15), and displayed similar adhesion to laminin, collagen type IV, and fibronectin in vitro (data not shown). By contrast, expression of soluble CD44 in TA3 cells, but not soluble ICAM-1 or CD44R43A, was found to reduce their ability to attach to HA substrata (Table 1). In addition, TA3sCD44 cells displayed a decreased ability to internalize radiolabeled HA (Fig. 4 A) and invade G8 myoblast monolayers, which produce abundant pericellular HA (Fig. 4 B and data not shown). The ability of TA3sCD44 transfectants to penetrate the G8 monolayers was restored after treatment of the monolayers with streptomyces HA (Fig. 4 B), indicating that pericellular HA may constitute a barrier to tissue invasion by cells whose CD44-dependent ability to bind and internalize HA is compromised. Treatment of G8 monolayers with heparinase, used as a control, could not restore TA3sCD44 cell invasiveness (data not shown).
Figure 4.
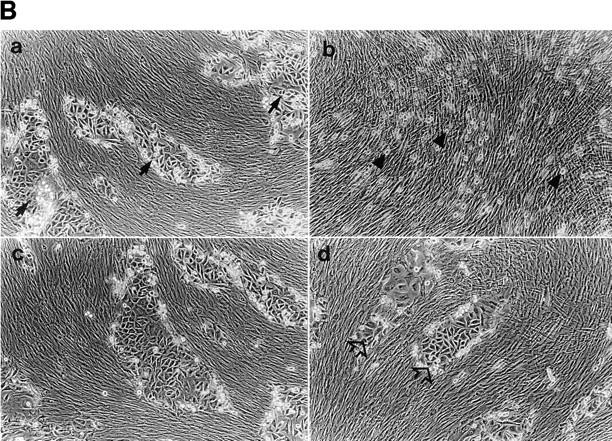

(A) [3H]-HA uptake and degradation. Soluble CD44 expression by the transfected TA3 cells results in a reduction of up to three-fold of 3H-HA uptake and degradation. (B) Invasion of G8 myoblast monolayers by TA3neo and TA3sCD44 cells. TA3neo (a and c) and TA3sCD44v6-10 (b and d) cells (5 × 103 cells/well) were seeded onto DMSO-fixed G8 monolayers which were untreated (a and b) or treated with streptomyces hyaluronidase (10 U/ml, ICN, Costa Mesa, CA; c and d) at 37°C for 3 h. 6 d after seeding of TA3 transfectants, TA3 neo cells are observed to penetrate and invade the monolayer, treated or not with hyaluronidase, and form tumor cell islets (arrows, TA3 neo cells). By contrast, TA3sCD44 cells were unable to penetrate the untreated G8 monolayer (b, arrowheads), whereas treatment of the fixed G8 cells with hyaluronidase restored the ability of TA3sCD44 cell to penetrate the monolayer (d, open arrows).
TA3 Cell Invasion of Lung Tissue Elicits HA Production by Stromal Cells.
The impaired ability of TA3sCD44 cells to attach to, internalize, and degrade HA and penetrate HA-coated cell monolayers in vitro may translate into their inability to penetrate ECM-associated HA in host tissue in vivo. Therefore, we addressed HA production in lung tissue after TA3neo and TA3sCD44 invasion. HA production has been shown to be increased at sites of tumor invasion in some tumor models (27), and is thought to be derived mainly from stromal cells. Stromal cell HA overproduction is proposed to result, at least in part, from physical interaction between tumor and stromal cells (28). To determine whether invasion of lung tissue by TA3 cells elicits local HA production, tissue sections of lung derived from mice injected with neo- or sCD44-transfected TA3 cells were probed with biotinylated HA-binding bPG at a series of time points after injection. Neither type of tumor cell induced detectable local HA production 1 h after injection (Fig. 5, a–d). However, at 48 h, marked HA production was observed at sites of both types of tumor invasion (Fig. 5, e and f). Although tumor cell clusters were clearly visible in lung tissue from animals injected with TA3neo cells, most of the foci of HA overproduction in lungs from sCD44-TA3 cell–injected animals were either devoid of tumor cells or contained tumor cells with small, condensed nuclei (Fig. 5, g and h), consistent with the observation that most of these cells were in the process of undergoing or had already undergone apoptosis.
Figure 5.
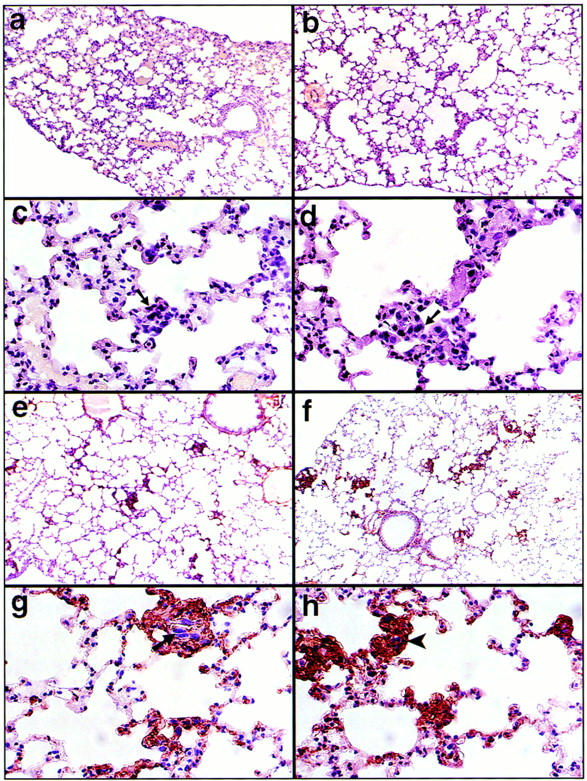
HA production at sites of tumor invasion. (a–d) Lung sections 1 h after tail vein injection of TA3neo No. 1 (a and c) and TA3sCD44v6-10 No. 17 (b and d) reveal no detectable HA production as assessed by staining with biotinylated HA-binding bPG. Arrows indicate invading tumor cells that display large hyperchromatic nuclei. 48 h after tail vein injection (e–h), both TA3 neo No. 1 (e and g) and TA3sCD44v6-10 No. 17 (f and h) induce stromal HA production. TA3neo No. 1 cells are visible at the sites of HA production (g, arrow) whereas TA3sCD44v6-10 No. 17 cells are no longer distinguishable (h, arrowhead).
Discussion
In this work we have shown that tumor-derived, truncated, soluble CD44 isoforms can block TA3/St tumor metastasis by impairing the ability of the tumor cells to survive and proliferate within the invaded tissue. The observed inhibition of metastatic growth was not soluble CD44 isoform–specific, as isoforms containing variant exons v6–v10 and v8–v10 had a comparable effect. Moreover, TA3 transfectants expressing standard soluble CD44, lacking variant exons, were also unable to form lung tumors after intravenous injection (Yu, Q., and I. Stamenkovic, manuscript in preparation). However, it appears clear that soluble CD44 is responsible for inhibition of TA3/St tumor formation in the lung, based on the observations that the few tumor nodules derived from some of the TA3sCD44 clones had lost soluble CD44 expression, and that neither soluble ICAM-1 nor the soluble CD44R43A mutant could block the metastatic growth. Therefore, functional cell surface CD44 expression provides a survival mechanism for TA3/St cells in the invaded tissue microenvironment.
Analysis of the precise mechanism by which tumor-derived soluble CD44 impairs endogenous cell surface CD44 function will require further work. An earlier study has suggested that CD44 released from the cell surface can partially inhibit CD44-mediated cell attachment to HA (29), while other studies have shown significantly increased soluble CD44 in the serum of tumor-bearing animals and patients (30, 31). However, CD44 released from the tumor cell surface, presumably by proteolytic shedding, appears to delay but not to abrogate local and metastatic tumor growth (29). Moreover, in naturally occurring tumors, it remains to be determined which tumor cell subsets display CD44 shedding, at what stage of tumor development the shedding occurs, and whether release of CD44 may be secondary to tumor cell death by necrosis, particularly when large tumor masses are present. In the model used here, tumor cells actively secrete soluble CD44. It has been shown that cell surface CD44 can form aggregates as a result of binding to HA, which may be important for subsequent internalization and processing (32). Therefore, it is possible that, using locally produced HA as a molecular bridge, soluble CD44 may aggregate with endogenous CD44 on the cell surface, disrupting normal receptor-mediated HA internalization. In addition, secreted CD44 may act as decoy receptors, reducing or blocking putative ligand-induced signal transduction by membrane-bound CD44.
Because CD44 isoforms have been proposed to recognize a variety of potential ligands, the inability of TA3sCD44 cells to form metastases could conceivably be attributed to the abrogation of binding to an as yet unidentified CD44 ligand, or to disruption of endogenous CD44 cooperation with another adhesion receptor expressed on the same cell surface. Nevertheless, the reduced ability to bind and internalize HA remains the most likely mechanism for the impaired metastatic proclivity of TA3sCD44 cells, and is supported by several observations. First, the observed inhibition of TA3 cell metastasis to lung was not soluble CD44 isoform–specific, but was similar whether standard or variant truncated CD44 was expressed and depended on the ability of the soluble CD44 to bind HA. Second, TA3sCD44 cells could not invade G8 myoblast monolayers in the presence of G8 cell–derived HA, but could do so when the HA was removed by hyaluronidase treatment. In contrast, wild-type TA3 cells could penetrate the G8 monolayer without requiring hyaluronidase treatment of the G8 cells. Third, the enhanced tumorigenesis of CD44-transfected lymphoma and melanoma cells has been shown to be dependent on the ability of CD44 to bind HA (23) and the aggressiveness of human mammary carcinoma cell lines in vivo has been found to correlate with their ability to internalize and degrade HA via CD44 (33). Finally, recent work has shown that a major function of CD44 in skin keratinocytes (which express multiple CD44 isoforms) is the regulation of local HA uptake and degradation (34), which in turn participates in the regulation of keratinocyte ability to proliferate in response to external stimuli (34).
Tumor cell interaction with host tissue stroma is known to stimulate stromal cell HA production (28). Consistent with these observations, staining of the lung sections with HA-binding bPG revealed a comparable increase in HA production at sites of infiltration of both TA3neo and TA3sCD44 cells. However, the role of the increased local HA production in tumor development remains uncertain. One widely held view is that stromal cell HA production facilitates tumor cell migration in tissues and possibly triggers HA receptor-mediated growth stimulatory signals (35, 36). However, excess HA can have an inhibitory effect on cell migration (37), raising the possibility that increased stromal cell HA production in response to infiltrating inflammatory or malignant cells may constitute a nonspecific tissue repair mechanism that serves to contain exogenous cell infiltration. Abundant local HA accumulation may reduce the diffusion of growth factors and cytokines to their cell surface receptors and prevent the infiltrating cells from adhering to ECM proteins. To survive in invaded tissues, tumor cells which elicit local HA production may therefore require CD44, other putative HA receptors, or extracellular hyaluronidase expression to penetrate the HA barrier and gain access to ECM proteins. In tumor cells whose survival depends on signals generated by interaction with ECM proteins and/or ECM-sequestered growth factors, an inability to efficiently remove locally produced HA may result in apoptosis, in much the same way that a disruption of adhesion to substrate leads to apoptosis in normal endothelial and epithelial cells (38, 39).
Taken all together, our results indicate that cell surface CD44 mediates events that are required for survival of at least some metastasizing tumor cell types within the host tissue microenvironment. These observations provide direct evidence that it may be possible to induce metastasizing tumor cell death in situ by disrupting specific receptor-dependent events required for interaction with host tissue.
Acknowledgments
We would like to thank Charles Underhill for gifts of biotinylated hyaluronan-binding proteoglycan and radiolabeled hyaluronan, Jing-wen Kuo of Anika Research Corp. for fluoresceinated hyaluronan, and Robert T. McCluskey for helpful discussions.
This study was supported by National Institutes of Health (NIH) grants CA-55735 and GM-48614 to I. Stamenkovic. I. Stamenkovic is a Scholar of the Leukemia Society of America. Q. Yu is supported by NIH training grant T32-CA-09216.
Footnotes
1
Abbreviations used in this paper: bPG, biotinylated proteoglycan; CMFDA, 5-chloromethyl-fluorescein diacetate; ECM, extracellular matrix; FBS, fetal bovine serum; FL-HA, fluorescein-labeled hyaluronan; HA, hyaluronan; ICAM, intracellular adhesion molecule; RT, reverse transcriptase; TUNEL, Tdt-mediated dUTP-biotin nick end labeling.
References
- 1.Ruoslahti E, Reed JC. Anchorage dependence, integrins and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 2.Hynes RO. Integrins: versatility, modulation and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 3.Juliano RL, Varner JA. Adhesion molecules in cancer: the role of integrins. Curr Opin Cell Biol. 1993;5:812–818. doi: 10.1016/0955-0674(93)90030-t. [DOI] [PubMed] [Google Scholar]
- 4.Gunthert U, Hofmann M, Rudy W, Reber S, Zoller M, Haussmann I, Matzku S, Wenzel A, Ponta H, Herrlich P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991;65:13–24. doi: 10.1016/0092-8674(91)90403-l. [DOI] [PubMed] [Google Scholar]
- 5.Sy M-S, Guo Y-J, Stamenkovic I. Distinct effects of two CD44 isoforms on tumor growth in vivo. J Exp Med. 1991;174:859–866. doi: 10.1084/jem.174.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sy M-S, Guo Y-J, Stamenkovic I. Inhibition of tumor growth in vivo with a soluble CD44–immunoglobulin fusion protein. J Exp Med. 1992;176:623–627. doi: 10.1084/jem.176.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lesley J, Hyman JR, Kincade P. CD44 and its interaction with extracellular matrix. Adv Immunol. 1993;54:271–335. doi: 10.1016/s0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]
- 8.Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI. Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc Natl Acad Sci USA. 1992;89:12160–12164. doi: 10.1073/pnas.89.24.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 10.Miyake K, Underhill CB, Lesley J, Kincade PW. Hyaluronate can function as a cell adhesion molecule and CD44 participates in hyaluronate recognition. J Exp Med. 1990;172:69–75. doi: 10.1084/jem.172.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stamenkovic I, Aruffo A, Amiot M, Seed B. The hematopoietic and epithelial forms of CD44 are distinct polypeptides with different adhesion potentials for hyaluronate bearing cells. EMBO (Eur Mol Biol Organ) J. 1991;10:343–348. doi: 10.1002/j.1460-2075.1991.tb07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas L, Byers HR, Vink J, Stamenkovic I. CD44H regulates tumor cell migration on hyaluronate-coated substrate. J Cell Biol. 1992;118:971–977. doi: 10.1083/jcb.118.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu Y, van Seventer GA, Siraganian R, Wahl L, Shaw S. Dual role of the CD44 molecule in T cell adhesion and activation. J Immunol. 1989;143:2457–2463. [PubMed] [Google Scholar]
- 14.Lesley J, Howes N, Perschl A, Hyman R. Hyaluronan binding function of CD44 is transiently activated on T cells during an in vivo immune response. J Exp Med. 1994;180:383–387. doi: 10.1084/jem.180.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett KL, Jackson DG, Simon JC, Tanczos E, Peach R, Modrell B, Stamenkovic I, Plowman G, Aruffo A. CD44 isoforms containing exon v3 are responsible for the presentation of heparin-binding growth factor. J Cell Biol. 1995;128:687–698. doi: 10.1083/jcb.128.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber GF, Ashkar S, Glimcher MJ, Cantor H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1) Science. 1996;271:509–512. doi: 10.1126/science.271.5248.509. [DOI] [PubMed] [Google Scholar]
- 17.Jalkanen S, Jalkanen M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J Cell Biol. 1992;116:817–825. doi: 10.1083/jcb.116.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Culty M, Nguyen HA, Underhill CB. The hyaluronan receptor (CD44) participates in the uptake and degradation of hyaluronan. J Cell Biol. 1992;116:1055–1062. doi: 10.1083/jcb.116.4.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hua Q, Knudson CB, Knudson W. Internalization of hyaluronan by chondrocytes occurs via receptor-mediated endocytosis. J Cell Sci. 1993;106:365–375. doi: 10.1242/jcs.106.1.365. [DOI] [PubMed] [Google Scholar]
- 20.Laurent TC, Fraser JR. Hyaluronan. FASEB (Fed Am Soc Exp Biol) J. 1992;6:2397–2404. [PubMed] [Google Scholar]
- 21.Toole BP. Hyaluronan and its binding proteins, the hyaladherins. Curr Opin Cell Biol. 1990;2:839–844. doi: 10.1016/0955-0674(90)90081-o. [DOI] [PubMed] [Google Scholar]
- 22.Toole, B.P. 1982. Glycosaminoglycans and morphogenesis. In Cell Biology of the Extracellular Matrix second edition. E. Hay, editor. Plenum Press, New York. pp. 259–294.
- 23.Bartolazzi A, Peach R, Aruffo A, Stamenkovic I. Interaction between CD44 and hyaluronate is directly implicated in the regulation of tumor development. J Exp Med. 1994;180:53–66. doi: 10.1084/jem.180.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Q, Toole BP. A new alternatively spliced exon between v9 and v10 provides a molecular basis for synthesis of soluble CD44. J Biol Chem. 1996;271:20603–20607. doi: 10.1074/jbc.271.34.20603. [DOI] [PubMed] [Google Scholar]
- 25.Green SJ, Tarone G, Underhill CB. Distribution of hyaluronate and hyaluronate receptors in the adult lung. J Cell Sci. 1988;90:145–156. doi: 10.1242/jcs.90.1.145. [DOI] [PubMed] [Google Scholar]
- 26.Peach RJ, Hollenbaugh D, Stamenkovic I, Aruffo A. Identification of hyaluronic acid binding sites in the extracellular domain of CD44. J Cell Biol. 1993;122:257–264. doi: 10.1083/jcb.122.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toole BP, Biswas C, Gross J. Hyaluronate and invasiveness of the rabbit V2 carcinoma. Proc Natl Acad Sci USA. 1979;76:6299–6303. doi: 10.1073/pnas.76.12.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knudson W, Toole BP. Membrane association of the hyaluronate stimulatory factor from LX-1 human lung carcinoma cells. J Cell Biochem. 1988;38:165–177. doi: 10.1002/jcb.240380304. [DOI] [PubMed] [Google Scholar]
- 29.Bartolazzi A, Jackson D, Bennett K, Aruffo A, Dickinson R, Shields J, Whittle N, Stamenkovic I. Regulation of growth and dissemination of a human lymphoma by CD44 splice variants. J Cell Sci. 1995;108:1723–1733. doi: 10.1242/jcs.108.4.1723. [DOI] [PubMed] [Google Scholar]
- 30.Katoh S, McCarthy JB, Kincaide PW. Characterization of soluble CD44 in the circulation of mice. J Immunol. 1994;153:3440–3449. [PubMed] [Google Scholar]
- 31.Guo Y-J, Liu G, Wang X, Jin D, Wu M, Ma J, Sy M-S. Potential use of soluble CD44 in serum as an indicator of tumor burden and metastasis in patients with gastric or colon cancer. Cancer Res. 1994;54:422–426. [PubMed] [Google Scholar]
- 32.Sleeman J, Rudy W, Hofmann M, Herrlich P, Ponta H. Regulated clustering of variant CD44 proteins increases their hyaluronate binding capacity. J Cell Biol. 1996;135:1139–1150. doi: 10.1083/jcb.135.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Culty M, Shizari M, Thompson EW, Underhill CB. Binding and degradation of hyaluronan by human breast cancer cell lines expressing different forms of CD44. J Cell Physiol. 1994;160:275–286. doi: 10.1002/jcp.1041600209. [DOI] [PubMed] [Google Scholar]
- 34.Kaya G, Rodriguez I, Jorcano JL, Vassalli P, Stamenkovic I. Selective suppression of CD44 in keratinocytes of mice bearing an antisense CD44 transgene driven by a tissue-specific promoter disrupts hyaluronate metabolism in the skin and impairs keratinocyte proliferation. Genes Dev. 1997;11:996–1007. doi: 10.1101/gad.11.8.996. [DOI] [PubMed] [Google Scholar]
- 35.Knudson W, Biswas C, Li X-Q, Nemec RE, Toole BP. The role and regulation of tumor associated hyaluronan. Ciba Found Symp. 1989;143:150–170. doi: 10.1002/9780470513774.ch10. [DOI] [PubMed] [Google Scholar]
- 36.McKee CM, Lowenstein CJ, Horton MR, Wu J, Bao C, Chin BY, Choi AM, Noble PW. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor kβ-dependent mechanism. J Biol Chem. 1997;272:8013–8018. doi: 10.1074/jbc.272.12.8013. [DOI] [PubMed] [Google Scholar]
- 37.Shannon BT, Love SH, Myrvik QN. Participation of hyaluronic acid in the macrophage disappearance reaction. Immunol Commun. 1980;9:357–370. doi: 10.3109/08820138009052982. [DOI] [PubMed] [Google Scholar]
- 38.Meredith JE, Fazeli B, Schwartz MA. The extracellular matrix as a survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frisch SM, Francis H. Disruption of epithelial cell–matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]