Recycling of E-Cadherin: A Potential Mechanism for Regulating Cadherin Dynamics (original) (raw)
. 1999 Jul 12;146(1):219–232.
Abstract
E-Cadherin plays critical roles in many aspects of cell adhesion, epithelial development, and the establishment and maintenance of epithelial polarity. The fate of E-cadherin once it is delivered to the basolateral cell surface, and the mechanisms which govern its participation in adherens junctions, are not well understood. Using surface biotinylation and recycling assays, we observed that some of the cell surface E-cadherin is actively internalized and is then recycled back to the plasma membrane. The pool of E-cadherin undergoing endocytosis and recycling was markedly increased in cells without stable cell-cell contacts, i.e., in preconfluent cells and after cell contacts were disrupted by depletion of extracellular Ca2+, suggesting that endocytic trafficking of E-cadherin is regulated by cell-cell contact. The reformation of cell junctions after replacement of Ca2+ was then found to be inhibited when recycling of endocytosed E-cadherin was disrupted by bafilomycin treatment. The endocytosis and recycling of E-cadherin and of the transferrin receptor were similarly inhibited by potassium depletion and by bafilomycin treatment, and both proteins were accumulated in intracellular compartments by an 18°C temperature block, suggesting that endocytosis may occur via a clathrin-mediated pathway. We conclude that a pool of surface E-cadherin is constantly trafficked through an endocytic, recycling pathway and that this may provide a mechanism for regulating the availability of E-cadherin for junction formation in development, tissue remodeling, and tumorigenesis.
Keywords: epithelial junctions, endocytosis, epithelial morphogenesis, clathrin-coated vesicles, biotinylation
The cadherins are a large family of cell surface glycoproteins which mediate cell-cell adhesion in most solid tissues of the body (Takeichi 1991, Takeichi 1995; Yap et al. 1997a). Cadherin-based adhesion is a central determinant of cell patterning during development and is necessary for the preservation of established tissue architecture in mature organisms. This is exemplified by E-cadherin, the prototypical epithelial cadherin. Genetic studies have shown that E-cadherin is essential for epithelial integrity during development in flies, Xenopus, and mice (Levine et al. 1994; Hermiston and Gordon 1995; Tepass 1996; Uemura et al. 1996). At the cellular level, E-cadherin facilitates assembly of other specialized intercellular junctions (desmosomes, gap, and tight junctions) necessary to link epithelial cells into functional monolayers (Gumbiner et al. 1988; Musil et al. 1990; Wheelock and Jensen 1992). Cadherin-based adhesive contacts also participate in establishing apical-basal polarity (Vega-Salas et al. 1987; Nelson et al. 1990; Yap et al. 1995), possibly through a role in the targeted delivery of membrane components to the basolateral cell surface (Mays et al. 1995; Grindstaff et al. 1998). Finally, cellular studies have suggested that E-cadherin may influence cell locomotion (Hermiston and Gordon 1995; Hermiston et al. 1996) and population dynamics (Watabe et al. 1994; Hermiston and Gordon 1995), properties which could contribute to its morphogenetic influence. Taken together, these findings indicate a central role for E-cadherin in many aspects of epithelial biogenesis.
It is becoming increasingly clear that cadherin function is a dynamic process. Morphogenetic events associated with cellular rearrangements, movements, and wound healing are reported to involve regulated changes in cadherin function. For example, compaction of blastomeres in the early mouse embryo is associated with the activation of E-cadherin by protein kinase C signaling pathways (Vestweber et al. 1987; Winkel et al. 1990), whereas physiological reduction in the adhesiveness of C-cadherin triggered by the mesoderm-inducing factor, activin, is necessary for morphogenetic movements to occur in early Xenopus development (Brieher and Gumbiner 1994; Zhong et al. 1999). In postnatal life as well, morphogenetic movements in tissues with rapid turnover rates, such as the gut epithelium, are likely to involve the continual breaking and reforming of cell-cell adhesive contacts (Gumbiner 1992).
In contrast to the signals which may regulate cadherins, much less is known about the effector mechanisms which cells utilize to modulate cadherin adhesive function (Yap et al. 1997a). One potential mechanism is dynamic turnover of cadherin molecules on the cell surface. The amount of cadherin protein expressed on the cell surface clearly influences cadherin adhesiveness (Angres et al. 1996; Yap et al. 1997b). Several observations suggest that cadherins may undergo regulated trafficking to and from the surface. Isolated epithelial cells commonly display a prominent intracellular pool of E-cadherin which appears to be recruited to the cell surface upon cell-cell contact (McNeill et al. 1993; Adams et al. 1996; Myat et al. 1998) implying the existence of a trafficking pathway for targeted delivery of E-cadherin. Conversely, chelation of extracellular calcium (Ca2+) induced the internalization of intact adherens plaques or large plaque fragments, including molecules such as E-cadherin (Kartenbeck et al. 1982; Duden and Franke 1988; Kartenbeck et al. 1991). The developmentally regulated uptake of cadherins from the surface, with accumulation within intracellular vesicles, has also been observed in some instances of epithelial-to-mesenchymal transformation (Miller and McClay 1997). These observations suggest that surface expression of E-cadherin may be influenced by the balance between transport processes that deliver proteins to the surface and endocytotic mechanisms that mediate uptake and recycling of membrane components.
In this study we have investigated the endocytosis and recycling of detergent-soluble surface E-cadherin in MDCK cells. Our studies indicate that E-cadherin at the cell surface is not automatically incorporated into stable junctional complexes. Instead, even at steady-state in confluent monolayers, at least one pool of surface E-cadherin remains subject to endocytosis and is recycled to the cell surface via a post-Golgi endosomal pathway. The proportion of E-cadherin in this recycling pool is increased in the absence of stable cell-cell contacts—in preconfluent cells and after cell-cell contacts are disrupted by chelation of extracellular Ca2+. We suggest that the regulated uptake and recycling of surface E-cadherin provides a mechanism for the dynamic modulation of cadherin expression and cell adhesion.
Materials and Methods
Cell Culture
MDCK cells, strain II, were grown and passaged as described previously (Narula et al. 1992) in DMEM with 10% FCS and 2 mM glutamine in 5% CO2 and 95% air. Cells used in experiments were plated on semipermeable polycarbonate filters (Transwell; Corning Costar) as confluent monolayers or plated on glass coverslips at different densities. Confluent monolayers were plated at confluent density and maintained for 1–3 d before being used for experiments. Preconfluent cells were seeded sparsely on coverslips and used at day 3 after plating, at which time the cultures contained discrete islands of cells which had not yet fused to form larger patches of polarized cells. For experiments requiring depletion of extracellular Ca2+, cell monolayers were washed twice with Ca2+-free PBS, and then incubated in serum-free DMEM supplemented with 2.5 mM EDTA. For some experiments, cells were incubated in 10 μM cycloheximide to block protein synthesis (Lever 1979). For some experiments on endocytosis and recycling, cells were incubated in normal DMEM containing 1 μM bafilomycin A1 from Streptomyces griseus (Sigma Chemical Co.) for 1 h at 37°C. Bafilomycin A1 is a specific inhibitor of vacuolar type H+-ATPase proton pumps and has been reported to cause a twofold retardation in the rate of recycling of human transferrin receptors (TfR)1 back to the cell surface in CHO cells (Presley et al. 1997).
Antibodies
A mouse E-cadherin antibody (3B8) raised against MDCK E-cadherin (a kind gift of Dr. Warren Gallin, University of Alberta, Edmonton, Alberta, Canada) was used for immunofluorescence experiments. For immunoblotting we used a mouse monoclonal antibody against human E-cadherin (Transduction Laboratories) or a rat monoclonal E-cadherin antibody (DECMA-1; Sigma Chemical Co.). Other primary antibodies used include mouse monoclonal against β-catenin (Transduction Laboratories), mouse monoclonal antibody 6H directed against the Na+K+ATPase α subunit (a generous gift of Dr. M. Caplan, Yale University, New Haven, CT), mouse anti-TfR (Zymed Laboratories), rabbit anti–rab 5, and rabbit anti–rab 7 (both provided by Dr. Chavrier, European Molecular Biology Laboratory, Heidelberg, Germany). Secondary antibody conjugates used were sheep anti–mouse IgG-Cy3 (Jackson ImmunoResearch Labs), goat anti–rabbit IgG-Cy3 (Sigma Chemical Co.), goat anti–rabbit IgG-ALEXA 488 (Molecular Probes), HRP-labeled goat anti–mouse IgG (Bio-Rad Laboratories), and HRP-labeled sheep anti–rat IgG (Amersham Life Science).
Immunofluorescence Staining
Cells were fixed in 4% paraformaldehyde in PBS for 90 min and then permeabilized for 5 min in PBS containing 0.1% Triton X-100. Cells were incubated with primary antibodies followed by incubation in secondary antibodies using PBS containing BSA as a blocking buffer. Cells were mounted in 50% glycerol/1% _n_-propyl-gallate in PBS and viewed by confocal microscopy on a Bio-Rad MRC-600 confocal laser scanning microscope mounted on a Zeiss Axioskop or by epifluorescence on an Olympus Provis X-70 microscope. Images were collected with an Olympus CCD300ET-RCX camera using NIH image software. Intensity measurement values were obtained from analysis of multiple fields in duplicate images on the confocal microscope. Image analysis was performed by measuring the total and relative fluorescence intensities using SOM software.
Cell Surface Biotinylation
MDCK cells grown on filters were incubated with 1.5 mg/ml sulfosuccinimidyl 2-(biotinamido) ethyl-dithioproprionate (sulfo-NHS-SS-biotin) (Pierce Chemical Co.) applied to the basal side of the filter, followed by washing with sulfo-NHS-SS-biotin blocking reagent (50 mM NH4Cl in PBS containing 1 mM MgCl2 and 0.1 mM CaCl2) to quench free sulfo-NHS-SS-biotin, followed by several further washes in PBS. Cells were then scraped off filters and lysed in 500 μl of RIPA buffer (20 mM Tris-HCl, pH 7.4, with 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 5 mM EDTA) with protease inhibitors. Cell extracts were centrifuged to obtain a detergent-insoluble pellet and a detergent-soluble supernatant which was incubated with streptavidin beads (Sigma Chemical Co.) to collect bound, biotinylated proteins. These samples were then analyzed by SDS-PAGE and immunoblotting to identify E-cadherin. In all cases, immunoblot membranes were stained with 0.1% Coomassie brilliant blue to ensure even protein transfer and protein loading. Different luminescence exposures were collected and exposures in the linear range were used.
Biotinylation Assay for Endocytosis and Recycling
Confluent MDCK cells grown on Transwell filters were biotinylated as above at 0°C followed by washing and quenching free biotin. Cells were then incubated in normal media at 18°C or 37°C. The 18°C temperature block has been used to accumulate internalized proteins in early or sorting endosomes by preventing them from progressing further into the endocytic or recycling pathways (Galloway et al. 1983; Dunn et al. 1989; Czekay et al. 1997). Monolayers were glutathione stripped essentially as described by Graeve et al. 1989. Cells were incubated in two 20-min washes of glutathione solution (60 mM glutathione, 0.83 M NaCl, with 0.83 M NaOH and 1% BSA added before use) at 0°C which removed all cell surface biotin groups. Remaining biotinylated proteins were sequestered inside cells by endocytosis and were therefore protected from glutathione stripping.
To measure recycling of endocytosed proteins accumulated at 18°C, cells were glutathione stripped at 0°C and then returned to 37°C for various times in normal medium. Cells were then washed quickly in PBS and incubated with 0.01% trypsin (type IV; Sigma Chemical Co.) in Ca2+-free PBS for 20 min followed by addition of 100-fold excess soybean trypsin inhibitor to inhibit further protease digestion. Trypsinized, biotinylated cell surface proteins were recovered from the supernatant by incubation with streptavidin beads, and the cells were then lysed in RIPA buffer and remaining cell-associated biotinylated proteins recovered on streptavidin beads. The extracellular fragment of E-cadherin released by trypsin and cell-associated E-cadherin were analyzed by SDS-PAGE and immunoblotting.
Potassium (K+) Depletion
Cells were depleted of K+ to selectively block clathrin-mediated endocytosis essentially as described by Larkin et al. 1983. Cultures were rinsed three times with K+-free buffer (140 mM NaCl, 20 mM Hepes, pH 7.4, 1 mM CaCl2, 1 mM MgCl2, and 1 mg/ml d-glucose), and hypotonically shocked by a brief rinse followed by incubation at 37°C for 5 min in K+-free buffer diluted 1:1 with distilled water. Next, cells were rinsed three times and incubated in K+-free buffer at 37°C for 15 min and finally incubated with 1.5 mg/ml biotin as described previously. Biotinylated E-cadherin was recovered with streptavidin beads and detected by immunoblotting.
Ricin Uptake
The uptake of FITC-ricin was used to measure non–clathrin-mediated endocytosis as described previously (Wilson and Colton 1997). Confluent MDCK cells grown on Transwell filters were incubated for 1 h at 4°C in 0.1 mg/ml ricin-FITC (Sigma Chemical Co.) in serum-free or under conditions of K+ depletion. Cells were warmed to 37°C for 30 min and rinsed 4 × 15 min in 0.2 M lactose to remove surface-bound ricin. Cells were then fixed at 4°C and examined by confocal microscopy to measure relative amounts of cell surface or intracellular labeling.
Results
Accumulation of an Intracellular Pool of E-Cadherin in Confluent MDCK Monolayers
E-Cadherin in confluent MDCK cells was localized mostly on the lateral plasma membrane. However, small amounts of staining were also seen intracellularly in a punctate, vesicular pattern close to the cell surface (Fig. 1 a). Nonpermeabilized cells showed no comparable punctate staining (data not shown), confirming that this staining pattern was intracellular. To see whether the internal pool of E-cadherin is possibly a result of endocytosis from the cell surface, we incubated cells at 18°C for 2 h before fixation and staining. An 18°C temperature block has been shown to cause the accumulation of endocytosed proteins in early or sorting endosomes (Czekay et al. 1997). The 18°C temperature block did not adversely affect cell morphology or cell-cell contacts as shown by the F-actin staining pattern (Fig. 1e and Fig. f), nor was there a detectable change in the cell surface staining of E-cadherin at 18°C. However, the vesicular staining of E-cadherin was more pronounced in cells incubated at 18°C (Fig. 1 b). Notably, at 18°C the location of the vesicular E-cadherin staining within the cells was now more prominent in the perinuclear region rather than being at the cell periphery. E-Cadherin staining was largely unaltered in cells pretreated with cycloheximide to stop protein synthesis (Fig. 1 d), indicating that both the cell surface staining and the intracellular staining represent stable pools of E-cadherin. Thus, while the majority of E-cadherin is present on the lateral plasma membrane, the presence of intracellular staining suggests that a pool of E-cadherin may be internalized from the cell surface, where it accumulates at 18°C.
Figure 1.
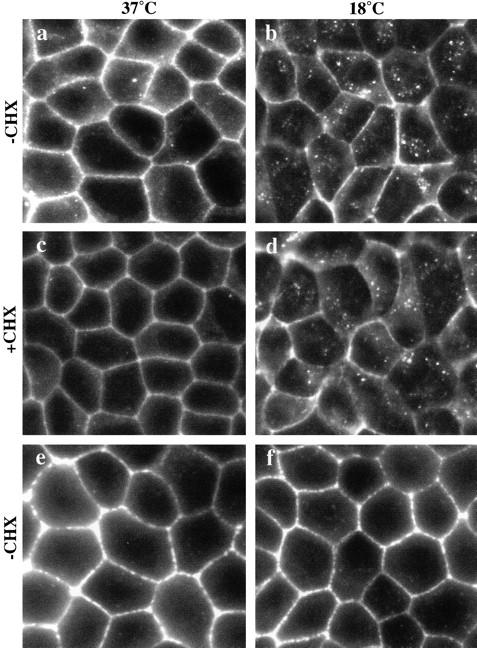
E-Cadherin distribution in confluent MDCK cells. (a) Immunofluorescence staining of E-cadherin is mostly localized to the lateral cell surfaces in confluent MDCK monolayers. Small amounts of intracellular, vesicular staining can also be seen scattered throughout the periphery of cells. (b) When cells were fixed and stained after incubation at 18°C, intracellular staining of E-cadherin was more intense. The vesicular staining relocated and was now clustered in a perinuclear position. (c and d) Preincubation of cells in cycloheximide (CHX) did not significantly alter the staining pattern; both cell surface and vesicular staining can still be seen. (e and f) Phalloidin staining of F-actin at the level of adhesion junctions shows that cell morphology was preserved in both control cultures and in monolayers incubated at 18°C. Representative fields of cells were photographed with similar exposures.
Endocytosis of Basolateral Surface E-Cadherin
To establish whether surface E-cadherin can be internalized we developed an assay to track the uptake of E-cadherin labeled by biotinylation on the basolateral surfaces of MDCK cells. As described in Materials and Methods, cells were surface-biotinylated at 0°C then returned to 37°C for 1 h to allow trafficking to resume. Cells were then incubated in several washes of glutathione solution at 0°C to remove covalently bound biotin groups from amines exposed on the cell surface. Biotinylated E-cadherin internalized at 37°C should be sequestered and, therefore, protected from glutathione stripping. Cycloheximide-treated cells were included in all experiments to eliminate the pool of newly synthesized E-cadherin from consideration. In control experiments, cells were surface-biotinylated for 1 h at 0°C, then immediately washed in glutathione at 0°C. No E-cadherin was recovered in the biotinylated fraction (Fig. 2 a, lane 3), confirming that under these conditions glutathione efficiently stripped all biotinyl groups from surface proteins. In contrast, after 1 h at 37°C a biotinylated pool of E-cadherin was detected in cells following glutathione stripping of surface proteins (Fig. 2 a, lanes 6 and 7). This pool represented ∼13% of the total E-cadherin biotinylated at the beginning of the experiment, indicating that upon return to physiological temperature, a small amount of surface E-cadherin was internalized and hence protected from glutathione stripping. In contrast, the basolateral membrane protein Na+K+ATPase did not undergo internalization under the same conditions, since a glutathione-resistant pool of Na+K+ATPase was not detected after incubating biotinylated cells at 37°C for 1 h (Fig. 2 a, lanes 6 and 7).
Figure 2.
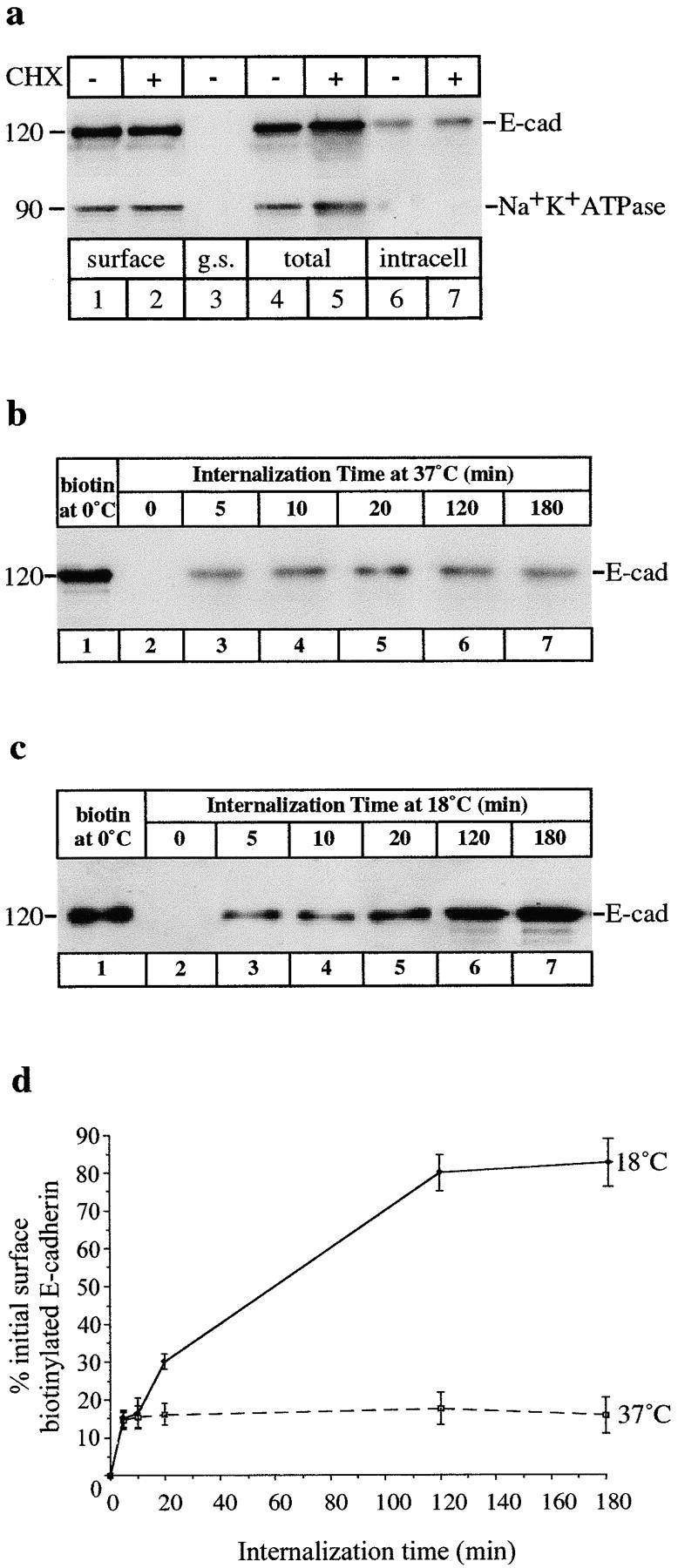
Internalization of surface-biotinylated E-cadherin. (a) Confluent MDCK cells treated with (+) or without (−) cycloheximide (CHX) were surface-biotinylated at 0°C. Detergent-soluble, surface-biotinylated proteins were recovered on streptavidin beads and analyzed by SDS-PAGE. E-Cadherin and Na+K+ATPase in these fractions were detected by immunoblotting as depicted. Biotinylated cell surface E-cadherin was recovered from the cell extracts (lanes 1 and 2). Glutathione stripping (g.s.) immediately after biotinylation at 0°C completely removed biotin from surface E-cadherin (lane 3). Surface-biotinylated cells were then incubated at 37°C and after 1 h, total biotinylated E-cadherin was recovered (lanes 4 and 5). E-Cadherin sequestered in an internal pool was recovered after glutathione stripping showing that some of the surface E-cadherin was endocytosed (lanes 6 and 7). Na+K+ATPase was biotinylated (lanes 1 and 2) but was not found in an internal pool under the same conditions (lanes 6 and 7). (b) A time course of E-cadherin uptake. CHX-treated cells were surface-biotinylated at 0°C (lane 1) and then incubated at 37°C for periods of 0–180 min (lanes 2–7). A constant amount of surface-biotinylated E-cadherin was sequestered from glutathione stripping at chase times from 5–180 min (lanes 3–7), consistent with a steady-state flux of E-cadherin into and out of this intracellular pool. (c) When the same experiments were carried out on MDCK cells incubated at 18°C, internalized E-cadherin now accumulated, resulting in increasing amounts of E-cadherin in the internal pool over 180 min (lanes 3–7). Fainter, lower molecular weight bands (lane 6 and 7) may be due to some degradation of internalized E-cadherin after prolonged incubations. (d) Relative amounts of internalized E-cadherin, quantitated by densitometry and expressed as a percentage of the total initial surface-biotinylated pool in cells incubated at 37°C or 18°C. Data are means ± SEM from three separate experiments.
To assess the kinetics of internalization, we allowed surface-biotinylated E-cadherin to internalize for various times (5–180 min) at 37°C in cells preincubated with cycloheximide (Fig. 2 b). An internalized pool of E-cadherin was detected after 5 min, but for the remainder of the 3-h chase period, the amount of intracellular biotinylated E-cadherin did not change, indicating that at physiological temperature, the relative size of the endocytosed pool of E-cadherin was kept constant in confluent monolayers (Fig. 2 b, lanes 3–7, and Fig. 2 d). The fact that this pool did not decrease in cycloheximide-blocked cells shows that the internalized E-cadherin is not generally fated for degradation after endocytosis, while the lack of accumulation suggests that there may be constant recycling of the internal pool of E-cadherin.
In light of the immunofluorescence observation that E-cadherin accumulates intracellularly at 18°C, surface biotinylation was also used to assay the effect of low temperature on the internalization and accumulation of E-cadherin. Whereas at 37°C there was a constant internalized pool of biotinylated E-cadherin (Fig. 2 b), at 18°C the internalized pool of E-cadherin showed progressive accumulation (Fig. 2c and Fig. d). After 20 min at 18°C, 35% of the surface-biotinylated E-cadherin was internalized (Fig. 2 c, lane 5) and by 2 h the majority (80%) of the surface-biotinylated E-cadherin had accumulated inside cells (Fig. 2 c, lane 6). After prolonged accumulation (3 h) some apparent degradation products of E-cadherin were noted on gels (Fig. 3, lane 7). The absence of such bands at 37°C (Fig. 2 b) further suggests that E-cadherin is normally recycled. Overall these results show that there is active internalization of E-cadherin from the cell surface and that its uptake is selective, since other basolateral cell surface proteins, such as Na+K+ATPase, are not undergoing the same process.
Figure 3.

Recycling of E-cadherin. Cells were surface-biotinylated on ice, then incubated at 18°C for 2 h to allow endocytosis and accumulation of E-cadherin. After glutathione stripping cells were then returned to 37°C. At each chase time (0–15 min) cells were trypsinized to release cell surface proteins. Glutathione stripping immediately after biotinylation effectively removed all surface-biotinylated proteins (g.s., lane 2). Biotinylated proteins from both the cell-associated (a, top) and trypsin-released fractions (a, bottom) were recovered on streptavidin beads and analyzed by SDS-PAGE. Intact E-cadherin (120 kD) or its trypsin-cleaved, 82-kD ecto-domain (a) and TfR (b) were detected by immunoblotting with specific antibodies. (a) The top shows that internalized E-cadherin accumulated at 18°C (lane 3) gradually disappeared from the internal pool over 15 min (lanes 4–7). The bottom shows that the 82-kD ectodomain of biotinylated E-cadherin was initially detected after 5 min at 37°C (lane 5) and maximal amounts were detected after 15 min at 37°C (lane 7) showing that the internalized E-cadherin was recycling and reappearing on the cell surface. (b) Surface-biotinylated (lane 1) TfR was also internalized (lane 4) but disappeared from the internal pool (lanes 5–7) more rapidly under the same conditions. Results are representative of four experiments.
Endocytosed E-Cadherin Is Actively Recycled to the Basolateral Membrane
To study possible recycling of E-cadherin, we then developed an assay to identify recycling of internalized biotinylated E-cadherin back to the cell surface. Surface proteins were biotinylated at 0°C, then cells were incubated at 18°C to allow the internalization and accumulation of E-cadherin. After treatment with glutathione to strip remaining biotinyl groups from cell surface proteins, cells were then released at 37°C to resume trafficking. Endocytosed E-cadherin that was returned to the cell surface was collected by surface trypsinization under conditions which cleave the ectodomain of E-cadherin, releasing a soluble fragment (Takeichi 1977). The supernatants of trypsin-treated cells were then incubated with streptavidin beads, separated by SDS-PAGE, and immunoblotted with an antibody to detect the cadherin ectodomain. No biotinylated E-cadherin fragments were detected in the medium of cells trypsinized immediately upon warming to 37°C (Fig. 3 a, lane 4). However, an 82-kD proteolytic fragment of biotinylated E-cadherin was detected in the medium 5 min after cells were rewarmed to 37°C (Fig. 3 a, lane 5). Maximal recovery of recycled E-cadherin was achieved after 15 min release (Fig. 3 a, lane 7), with a corresponding decrease in the internal pool of E-cadherin throughout the release period. It should be noted that cells were exposed to trypsin for identical durations and under identical conditions at all times after release of the 18°C block. The progressive accumulation of the biotinylated E-cadherin ectodomain in the medium is therefore unlikely to represent release of intracellular E-cadherin from cells damaged by prolonged exposure to trypsin. This indicates that internalized E-cadherin can be recycled back to the cell surface. As a control for endocytosis, we studied the kinetics of uptake and recycling of TfR by measuring depletion of the internalized biotinylated TfR (Fig. 3 b). Biotinylated TfR undergoes endocytosis, it is accumulated at 18°C (Fig. 3 b, lane 3), and then after returning cells to 37°C it is rapidly depleted from the internal pool at an apparently faster rate than E-cadherin (Fig. 3 b, lanes 4–7). Thus, biotinylated E-cadherin on the basolateral surface of MDCK cells can be followed through endocytic and recycling pathways.
To further test the influence of recycling on the internal pool of E-cadherin, we treated MDCK cells with bafilomycin A1. Bafilomycin A1 inhibits recycling by blocking transport of endocytosed material back to the cell surface at a late endosomal stage (Johnson et al. 1993; Presley et al. 1997). Cells were surface biotinylated at 0°C then incubated in the presence or absence of bafilomycin A1 (1 μM) at 37°C, followed by glutathione stripping. In the presence of bafilomycin A1 there was a threefold increase in the glutathione-resistant pool of E-cadherin (Fig. 4, lane 6) in comparison to the normal amount of internalized E-cadherin (Fig. 4, lane 5). Bafilomycin A1 was therefore effective in accumulating the endocytosed E-cadherin, consistent with a block in the recycling of E-cadherin.
Figure 4.

Endocytosis of E-cadherin in the presence of bafilomycin A1 (BAF). Cells were surface-biotinylated at 0°C and then incubated at 37°C in the presence (+) or absence (−) of 1 μM BAF. Surface-biotinylated E-cadherin (lane 1) was recovered from total cell extracts (lanes 3 and 4) and from the internal pool (lanes 5 and 6) sequestered from glutathione stripping. A greater amount of internalized biotinylated E-cadherin was recovered after BAF treatment (lane 6) in comparison to the normal amount of internal E-cadherin (lane 5), consistent with BAF-induced accumulation of E-cadherin. The result is representative of three independent experiments.
Preconfluent Cells Have a Large Pool of Internalized E-Cadherin
In light of reports that cell-cell contact may influence recruitment of E-cadherin to the cell surface, we compared the immunofluorescence localization of E-cadherin in MDCK cells grown and maintained at different densities. In confluent cell monolayers, E-cadherin staining was found predominantly at the cell surface, as shown in Fig. 1 a. In contrast, preconfluent cells which were not yet polarized and had not yet formed extensive adherens junctions showed relatively little E-cadherin staining at the cell surface but there was a concomitantly larger intracellular pool of labeled E-cadherin (Fig. 5 a). Some of the intracellular staining in the perinuclear Golgi region disappeared after cycloheximide treatment and is thus likely to represent newly synthesized E-cadherin in the biosynthetic pathway. There was also prominent vesicular staining of E-cadherin in the peripheries of preconfluent cells. As in confluent cells, staining of this vesicular pool was not altered by cycloheximide treatment suggesting that it represents E-cadherin in an endocytic pathway, a pool which is enhanced in preconfluent cells.
Figure 5.

Distribution of E-cadherin in preconfluent MDCK cells. (a) Immunofluorescence staining of E-cadherin in preconfluent cultures treated with or without cycloheximide (+/− CHX). At steady state (−CHX), there is a small amount of cell surface staining, prominent perinuclear staining over the Golgi complex, and bright vesicular staining throughout the cytoplasm. When protein synthesis is blocked (+CHX), the Golgi staining disappears but the vesicular staining remains. Representative fields of cells were photographed with similar exposures. (b) Surface biotinylation of E-cadherin. Preconfluent and confluent cells were surface-biotinylated; biotinylated E-cadherin in detergent-soluble cell extracts was recovered by streptavidin affinity, biotinylated and unbiotinylated (supernatant) fractions were analyzed by SDS-PAGE and immunoblotting. All of the biotinylated fraction and 20% of the total unbiotinylated fraction were loaded. Unbiotinylated E-cadherin from preconfluent cells (lane 1) and confluent cells (lane 3) was compared to the amounts of surface-biotinylated E-cadherin in preconfluent cells (lane 2) and confluent cells (lane 4). There is an increased amount of biotinylated cell surface E-cadherin in confluent cells with a concomitant decrease in the unbiotinylated fraction, which includes the intracellular pool. (c) The relative amounts of E-cadherin in detergent-soluble (biotinylated and unbiotinylated) and detergent-insoluble (TX-insoluble) fractions were compared by immunoblotting and densitometry in preconfluent and confluent cells. Detergent-insoluble E-cadherin, which is likely to represent protein incorporated into cytoskeleton-stabilized junctional complexes, increases in confluent cells. Within the detergent-soluble pool, biotinylated cell surface E-cadherin increases (from ∼10% to ∼50%) in confluent monolayers. Data are means ± SEM from three separate experiments.
Surface biotinylation experiments confirmed the relative difference in E-cadherin distribution between confluent and preconfluent cells. In preconfluent cells, ∼10% of the detergent-soluble E-cadherin was biotinylated on the basolateral surface, whereas in confluent cells, ∼47% of the E-cadherin was biotinylated on the cell surface (Fig. 5b and Fig. c). Taken together, these results suggest that E-cadherin redistributes from a predominantly intracellular pool to sites of cell-cell contact as MDCK cells grow to confluence. Insofar as a significant proportion of the intracellular E-cadherin in preconfluent cells represents a stable cycloheximide-resistant pool that is capable of undergoing recycling, this suggested that cell-cell contact may influence the recycling of E-cadherin.
E-Cadherin Recycling Mediates Restoration of Cell-Cell Contact after Chelation of Extracellular Ca2+
To further investigate the influence of cell-cell contact on E-cadherin recycling, we examined the effect of EDTA on epithelial morphology and E-cadherin localization in confluent MDCK monolayers. Chelation of extracellular Ca2+ disrupts epithelial cohesion, at least partly through inhibition of the adhesive binding activity of the E-cadherin ectodomain (Takeichi et al. 1981; Kemler et al. 1989; Kartenbeck et al. 1991). As shown in Fig. 6, exposure of MDCK cells to EDTA (2.5 mM) induces a rapid and reversible change in monolayer organization. Within 10–15 min cells began to retract from one another, and by 45 min there were mostly isolated cells lacking cell-cell contacts (Fig. 6 b). Restoration of extracellular Ca2+ rapidly restored cell-cell contacts, and by 1 h most cells had spread to reform extensive regions of confluence (Fig. 6 d). Staining for E-cadherin showed that EDTA induced the rapid internalization of surface E-cadherins with intense intracellular punctate labeling apparent after Ca2+ chelation (Fig. 7 a and Fig. 8 b) compared to the predominant cell surface staining of E-cadherin in contact zones in untreated cells (Fig. 7 a and Fig. 8 a). Quantitative immunofluorescence analysis indicated that whereas in control cells the majority of E-cadherin staining was at the cell surface, after treatment with EDTA E-cadherin was now predominantly intracellular (Fig. 7 b). Upon restoration of extracellular Ca2+, E-cadherin staining reappeared at sites of cell-cell contact, with a concomitant reduction in intracellular staining (Fig. 8 d).
Figure 6.
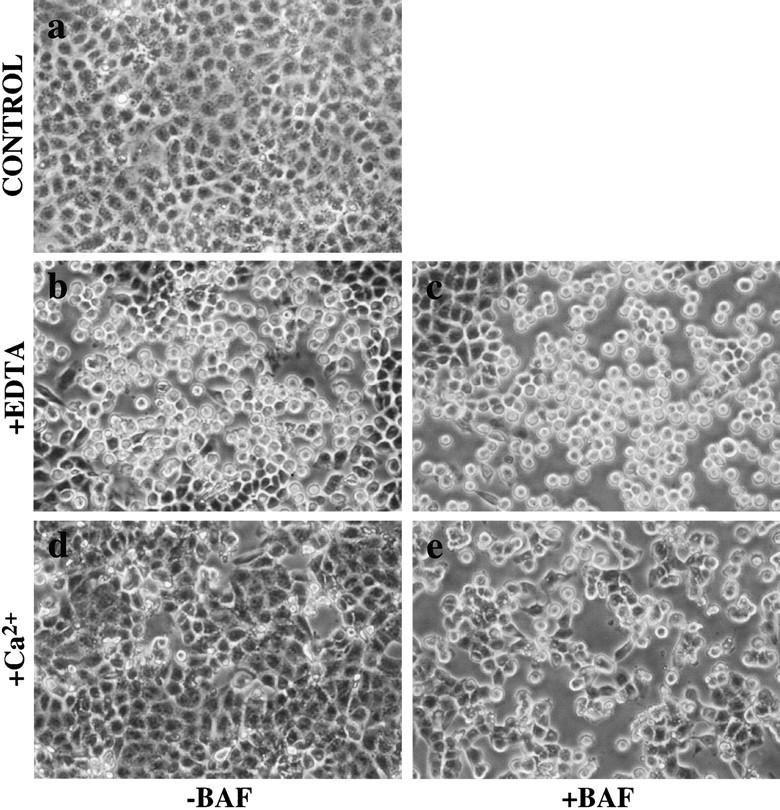
Restoration of cell-cell contact after Ca2+ depletion: bafilomycin A1 (BAF) inhibits restoration of monolayer integrity following Ca2+ depletion. Phase-contrast images of confluent MDCK monolayers exposed to 2.5 mM EDTA for 45 min to chelate extracellular Ca2+ and then following restoration of Ca2+ in the presence or absence of bafilomycin A1 (BAF). Cells in untreated monolayers (a) have intact cell contacts. In the presence (c) or absence (b) of BAF (1 μM) cells are rounded and retracted from each other upon chelation of Ca2+ with EDTA. Within 1 h of restoring extracellular Ca2+ control cells had spread, covering most of the substrate (d); in contrast, cells treated with BAF (e) spread to a much lesser degree, leaving extensive regions of bare substrate exposed. Representative fields of cells show the results of three separate experiments.
Figure 7.
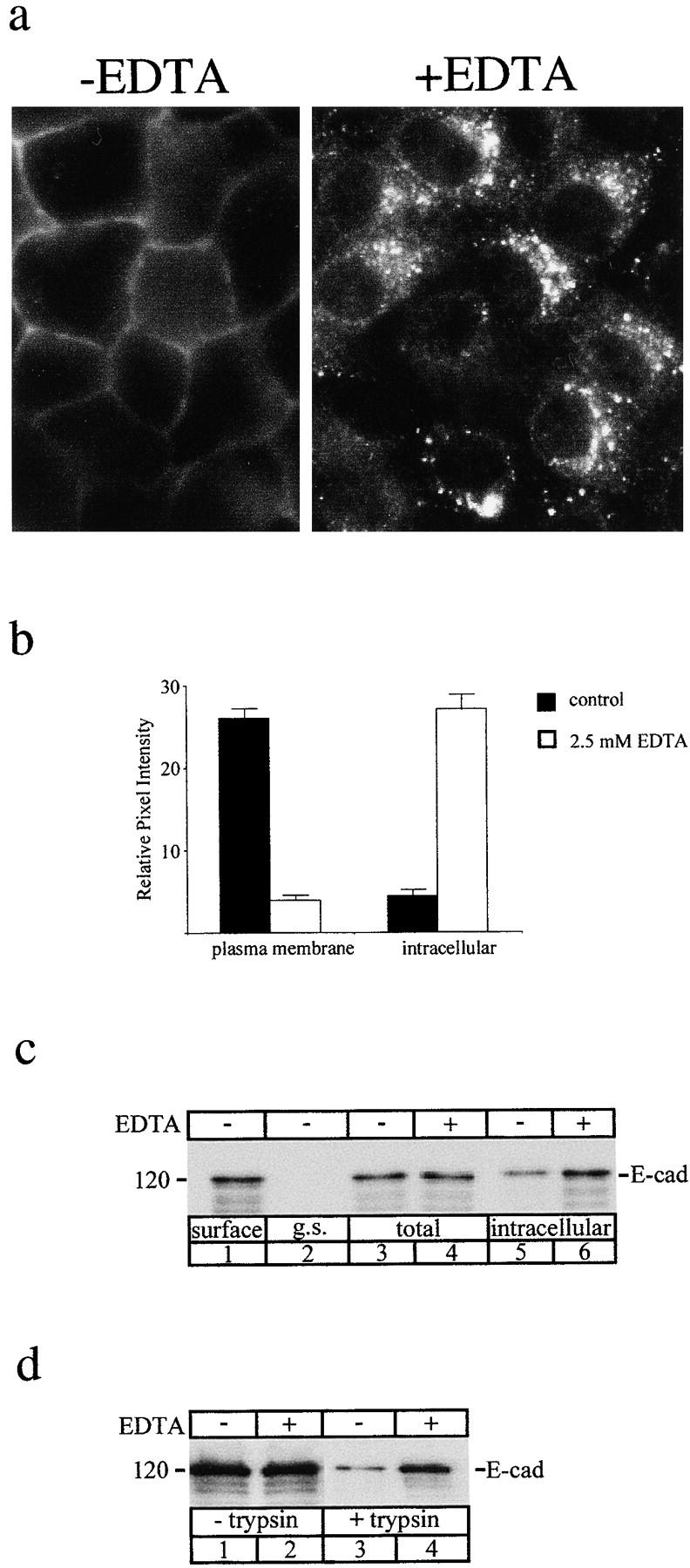
Endocytosis of E-cadherin after depletion of extracellular Ca2+. Internalization of E-cadherin was compared in untreated confluent MDCK cells and in cells incubated in medium containing EDTA to chelate extracellular Ca2+. (a) Immunofluorescence localization of E-cadherin in control MDCK cells (−EDTA) and MDCK cells incubated in DMEM containing 2.5 mM EDTA (+EDTA) and cycloheximide (10 μM) for 30 min. In the presence of EDTA cells show prominent intracellular vesicular staining of E-cadherin. (b) Quantification of relative fluorescence intensities at the cell surface and inside cells measured by SOM software showed that chelation of Ca2+ resulted in a dramatic increase in intracellular staining accompanied by a concomitant decrease in plasma membrane staining, indicative of E-cadherin endocytosis stimulated by EDTA. Data are means ± SEM  . (c) Surface biotinylation. Cells were surface-biotinylated at 0°C (lane 1) and then incubated at 37°C in normal medium (lanes 3 and 5) or in medium containing EDTA (lanes 4 and 6) for 30 min to allow for internalization. Total biotinylated E-cadherin was unchanged in the total cell extracts under both these conditions (lanes 3 and 4). After glutathione stripping there was a significantly increased pool of internalized biotinylated E-cadherin after Ca2+ depletion (lane 6) compared to control cells (lane 5). Thus, EDTA treatment increased internalization of surface-biotinylated E-cadherin. (d) Surface trypsinization. Cells were incubated in normal media or in medium containing 2.5 mM EDTA for 30 min. Cell surface proteins were removed by trypsinization and the remaining E-cadherin in cell extracts was analyzed by SDS-PAGE and immunoblotting with a NH2 terminus antibody (3B8). Total cellular E-cadherin remained unchanged in the absence (lane 1) or presence (lane 2) of Ca2+ chelation. A small pool of internalized E-cadherin was detected after trypsin treatment in cells incubated in normal media (lane 3), but this pool was dramatically increased in the presence of EDTA (lane 4), showing increased internalization of surface E-cadherin. Results shown are representative of three independent experiments.
. (c) Surface biotinylation. Cells were surface-biotinylated at 0°C (lane 1) and then incubated at 37°C in normal medium (lanes 3 and 5) or in medium containing EDTA (lanes 4 and 6) for 30 min to allow for internalization. Total biotinylated E-cadherin was unchanged in the total cell extracts under both these conditions (lanes 3 and 4). After glutathione stripping there was a significantly increased pool of internalized biotinylated E-cadherin after Ca2+ depletion (lane 6) compared to control cells (lane 5). Thus, EDTA treatment increased internalization of surface-biotinylated E-cadherin. (d) Surface trypsinization. Cells were incubated in normal media or in medium containing 2.5 mM EDTA for 30 min. Cell surface proteins were removed by trypsinization and the remaining E-cadherin in cell extracts was analyzed by SDS-PAGE and immunoblotting with a NH2 terminus antibody (3B8). Total cellular E-cadherin remained unchanged in the absence (lane 1) or presence (lane 2) of Ca2+ chelation. A small pool of internalized E-cadherin was detected after trypsin treatment in cells incubated in normal media (lane 3), but this pool was dramatically increased in the presence of EDTA (lane 4), showing increased internalization of surface E-cadherin. Results shown are representative of three independent experiments.
Figure 8.
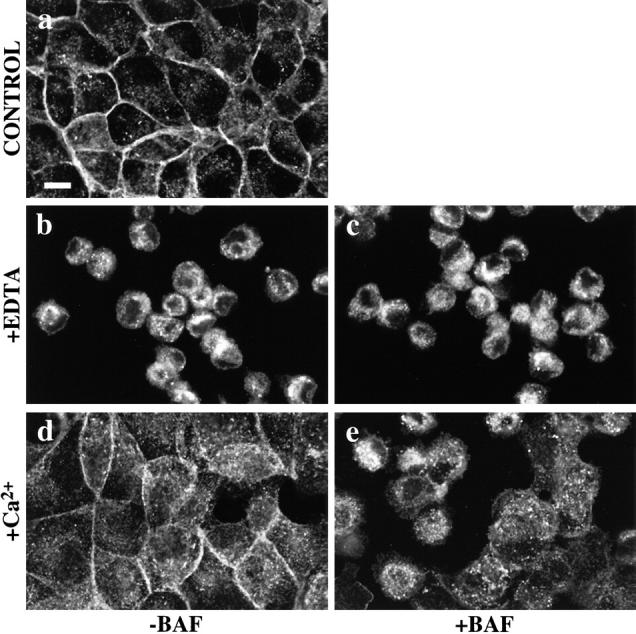
Bafilomycin A1 (BAF) inhibits the reaccumulation of E-cadherin in cell-cell contacts following Ca2+ depletion. Confluent MDCK monolayers were exposed to DMEM containing 2.5 mM EDTA for 45 min before restoration of normal extracellular Ca2+ concentrations before fixation and staining for E-cadherin. Before Ca2+ chelation, monolayers showed E-cadherin staining in a typical circumferential pattern of cell-cell contacts, in addition to scattered intracellular punctate deposits (a). Upon chelation of Ca2+, E-cadherin staining was concentrated in a prominent perinuclear vesicular pattern both in the presence (c) or absence (b) of BAF (1 μM). Upon restoration of extracellular Ca2+, control cells showed redistribution of E-cadherin to reforming cell-cell junctions (d). In contrast, upon restoration of extracellular Ca2+, BAF-treated cells retained significant amounts of intracellular E-cadherin in peripheral cytoplasmic vesicles as well as in the perinuclear area (e).
Exposure to EDTA also significantly increased the pool of biotinylated E-cadherin resistant to surface stripping with glutathione, consistent with increased internalization of surface cadherins (Fig. 7 c). Because biotinylation may not label all surface proteins (Gottardi et al. 1995), we also assessed the effect of EDTA on the pool of E-cadherin accessible to cleavage by extracellular trypsin (Takeichi 1977; Yap et al. 1997b). Whereas in control cells the majority of E-cadherin was sensitive to digestion by extracellular trypsin (Fig. 7 d), after treatment with EDTA, E-cadherin was predominantly resistant to trypsin, consistent with internalization to a protected site. Taken together, these biochemical and immunofluorescence findings confirm earlier reports (Kartenbeck et al. 1982; Duden and Franke 1988; Kartenbeck et al. 1991) that chelation of extracellular Ca2+ destabilizes cell junctions and induces endocytosis of E-cadherin. Furthermore, since these results were performed in cycloheximide-treated cells, they further suggest that regulated recycling of the endocytosed pool may be responsible for reestablishing stable cadherin-based cell-cell contacts upon restoration of extracellular Ca2+.
To test the potential role of E-cadherin recycling in restoration of epithelial integrity we used bafilomycin A1 to block recycling in EDTA-treated cells. Bafilomycin A1 did not affect the disruption of epithelial integrity (Fig. 6 c) nor the endocytosis of E-cadherin induced by Ca2+ chelation (Fig. 8 c). However, bafilomycin-treated cells failed to restore epithelial contacts upon replacement of extracellular Ca2+. Cells tended to spread upon replacement of Ca2+, but showed only limited cell-cell contacts that were markedly less extensive than those seen in control cultures (Fig. 6 e). Immunofluorescence staining showed that, in bafilomycin-treated cells, E-cadherin remained concentrated in cytoplasmic vesicles despite replacement of extracellular Ca2+ (Fig. 8 e). Therefore, inhibition of E-cadherin recycling to the cell surface by bafilomycin A1 has associated with failure of cells to restore epithelial integrity, suggesting that the recycling of endocytosed cadherins back to the cell surface was necessary to restore cell-cell contacts following correction of extracellular Ca2+.
E-Cadherin May Be Endocytosed via a Clathrin-dependent Pathway
Endocytosis of E-cadherin could occur through either clathrin-mediated or clathrin-independent pathways. To study whether E-cadherin is internalized via clathrin-dependent endocytosis, we used K+ depletion combined with hypotonic shock, a maneuver which has been shown to specifically inhibit clathrin-coated pit uptake of the low density lipoprotein receptor (Larkin et al. 1983) and other receptors. MDCK cells were preincubated with K+-free media, then gently rinsed and exposed to hypotonic K+-free media followed by incubation in K+-free media. Cells were then surface-biotinylated, incubated at 37°C for internalization, and then the surface was glutathione stripped. In control cells some of the surface-biotinylated E-cadherin was recovered in an internal pool (Fig. 9 a, lane 5; see also Fig. 2 a). A similar proportion of E-cadherin was internalized after hypotonic shock alone (Fig. 9 a, lane 6) which did not arrest E-cadherin internalization. However in cells treated with hypotonic shock followed by incubation in K+-free media there was no biotinylated E-cadherin internalized (Fig. 9 a, lane 7). Under the same conditions, the internalization of biotinylated TfR, which is known to be taken up by a clathrin-dependent pathway, was also blocked (Fig. 9 a, lane 7), whereas the surface residence of Na+K+ATPase, which in our hands is not internalized, was unchanged. In contrast, the uptake of FITC-labeled ricin, which occurs via a clathrin-independent mechanism (for review see Sandvig and van Deurs 1996), was unaffected by hypotonic shock and K+ depletion (Fig. 9 b). The same levels of FITC-ricin staining were measured in treated and untreated monolayers. Thus, K+ depletion effectively and specifically blocked clathrin-dependent uptake, implicating such a pathway in the internalization of E-cadherin.
Figure 9.

Endocytosis of E-cadherin is inhibited by K+ depletion. (a) Control cells (lane 1), cells pretreated with hypotonic shock alone (lane 2), or cells which were pretreated with hypotonic shock for 5 min then also incubated in K+-free medium for 15 min (lane 3) were surface-biotinylated. Biotinylated proteins were collected and analyzed by SDS-PAGE and immunoblotted to detect E-cadherin, transferrin receptor, and Na+K+ATPase. A set of duplicate cells was then treated and surface-biotinylated and then incubated at 37°C in either K+-free medium (lanes 6 and 7) or normal medium (lane 5) to allow for internalization of surface proteins. Cells were glutathione-stripped and the internal pool recovered with streptavidin beads. Biotinylated E-cadherin and TfR were both internalized in control cells (lane 5) and in cells treated with hypotonic shock alone (lane 6), but internalization of both proteins was effectively blocked in cells depleted of K+ using hypotonic shock and K+-free medium (lane 7). Na+K+ATPase did not internalize under either control or K+ depletion conditions (lanes 5–7). The results are representative of four separate experiments. (b) Clathrin-independent endocytosis of FITC-ricin. Cells were incubated in either normal media or were hypotonically shocked and incubated in K+-free media as outlined above. Cells were then surface-labeled with FITC-ricin at 4°C for 1 h and then incubated in normal or K+-free media for 30 min at 37°C to allow for internalization. Nonendocytosed FITC-ricin was removed by several washes with 0.2 M lactose in PBS. Cells were then fixed and the amount of internalized FITC-ricin was viewed and analyzed by confocal microscopy using SOM software. Similar amounts of intracellular FITC-ricin staining were obtained in both sets of cells. Inhibition of clathrin-mediated endocytosis by K+ depletion under the shown conditions in a and b, did not affect FITC-ricin uptake. Data are means ± SEM  .
.
Double labeling was also carried out by immunofluorescence staining to colocalize internalized E-cadherin with known markers of compartments in clathrin-mediated pathways. Confluent cell monolayers were temperature-blocked at 18°C in order to accumulate intracellular E-cadherin. In cells at 18°C, internalized E-cadherin was colocalized in some, but not all, vesicles stained for the early endosomal marker, rab 5 (Fig. 10, a and b). In contrast, internalized E-cadherin did not colocalize with the late endosomal protein, rab 7 (Fig. 10c and Fig. d). The vesicular staining pattern of E-cadherin and its partial overlap with rab 5 shows that at 18°C endocytosed E-cadherin accumulates in early endosomal or recycling compartments. The lack of colocalization with rab 7 in late endosomes is further evidence that this pool of endocytosed E-cadherin is not destined for lysosomal degradation.
Figure 10.
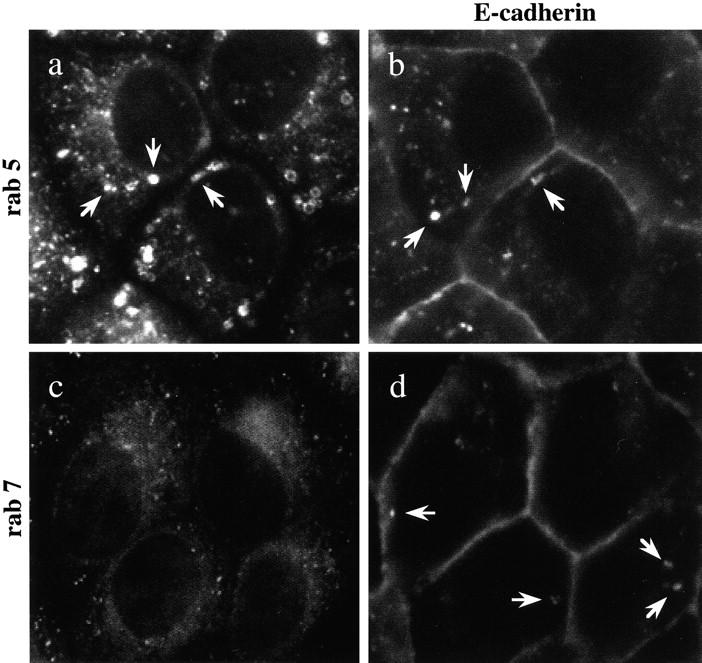
Double labeling of E-cadherin and rab proteins. MDCK cells were incubated at 18°C, fixed, and double labeled by immunofluorescence with antibodies to E-cadherin (b and d) and rab 5 (a) or E-cadherin and rab 7 (c). Vesicular staining of rab 5 (a) is consistent with its presence on early endosomes. E-Cadherin is colocalized with rab 5 in some endosomes (a and b). Punctate staining of rab 7 on late endosomes in the perinuclear area (c) did not coincide with any of the vesicles labeled for E-cadherin (d). Representative fields of cells were photographed.
Internalization of E-Cadherin in Complexes with β-Catenin
At the cell surface classical cadherins exist in macromolecular complexes with cytoplasmic catenins (Takeichi 1991, Takeichi 1995; Yap et al. 1997a). As a preliminary investigation into whether catenins are internalized we probed for β-catenin in surface-biotinylated fractions containing E-cadherin. β-Catenin was coisolated with surface-biotinylated E-cadherin (Fig. 11). Then we allowed surface-biotinylated E-cadherin to be internalized at 18°C, collected the internalized pool on streptavidin beads after surface stripping with glutathione, and probed for β-catenin (Fig. 11). As shown previously (Fig. 2 c), the majority of surface-biotinylated E-cadherin is internalized under these conditions (Fig. 11). Immunoblotting showed that β-catenin is also present in the internalized, biotinylated fraction under these conditions. Insofar as E-cadherin is the major surface protein known to associate with β-catenin in MDCK cells, we assume that β-catenin and E-cadherin are being internalized as a complex. Interestingly, only a relatively small amount of β-catenin is internalized compared to the initial surface-associated pool. Thus, internalization may result in altered stoichiometry of cadherin-catenin complexes. Further studies are now required to extend these observations.
Figure 11.

Endocytosis of β-catenin. Confluent MDCK cells treated with (+) or without (−) CHX were surface-biotinylated at 0°C, biotinylated proteins were collected with streptavidin beads, and the bound fraction was analyzed by SDS-PAGE and immunoblotting. Typically, E-cadherin was detected in this fraction, with β-catenin also present (lane 1). Glutathione stripping (g.s.) immediately after biotinylation completely removed biotin from surface E-cadherin; neither E-cadherin nor β-catenin was then collected on streptavidin beads (lane 2). Surface-biotinylated cells were then incubated at 18°C, and after 3 h E-cadherin sequestered in an internal pool after glutathione stripping (lanes 3 and 4) was recovered on streptavidin beads. A small amount of β-catenin was also recovered in the biotinylated internalized fraction suggesting that it too was endocytosed (lanes 3 and 4). The results are representative of three separate experiments.
Discussion
In this paper we have studied the trafficking movements of cell surface E-cadherin. Our findings indicate that, rather than being a uniformly stable resident on the cell surface, some pools of surface E-cadherin are endocytosed and recycled back to the surface. Experiments designed to follow the fate of surface-biotinylated proteins revealed that a portion of surface-labeled E-cadherin is endocytosed into an intracellular compartment. This corresponds to a vesicular pool of intracellular E-cadherin that was detected by immunofluorescence staining, a significant proportion of which persisted after inhibition of protein synthesis by cycloheximide and therefore represents a stable pool that is not in the biosynthetic pathway. The internalization process was somewhat selective for E-cadherin rather than reflecting bulk clearance of membrane proteins, since under conditions where E-cadherin showed clear uptake, no similar internalization of another basolateral membrane protein, Na+K+ATPase, could be detected. Moreover, at steady-state the biotinylated E-cadherin was not degraded after endocytosis, but was instead recycled back to the cell surface where it could be detected by surface trypsinization. Thus, there appears to be a rapid recycling pathway responsible for constantly internalizing and recycling a portion of E-cadherin on the cell surface.
What proportion of cell surface cadherin is endocytosed and recycled? Our data indicate that this is influenced by cell-cell contact. In confluent monolayers with stable cellular junctions, only a small pool of E-cadherin appears to recycle. At physiological temperature, the internalized pool of surface-labeled E-cadherin was consistently ∼13% of the total biotinylated pool. When recycling was inhibited by bafilomycin A1 or by an 18°C temperature block, the internalized pool accumulated, finally representing up to 80% of the total biotinylated pool after 2 h at 18°C. However, under the same conditions, immunofluorescence showed that the majority of cellular E-cadherin remained at the cell surface in contact zones. Therefore, it seems clear that biotinylation detects a relatively small subset of the total surface E-cadherin in stable monolayers, albeit one that includes the pool capable of undergoing selective recycling. In stable epithelial monolayers, E-cadherin exists in multiple pools, including classical adherens junctions as well as in extrajunctional regions of the lateral cell surface (Wang et al. 1990; Nathke et al. 1994; Yap et al. 1995) that may be differentially extractable in nonionic detergents (McCrea and Gumbiner 1991; Shore and Nelson 1991). Therefore, while previous studies have demonstrated uptake of junctional plaques or large fragments (Kartenbeck et al. 1991), it is tempting to speculate that in mature monolayers the extrajunctional cadherins are more likely to be free to migrate within the plane of the membrane and hence be endocytosed.
In contrast, recycling is significantly increased when cells are unable to make stable cell-cell contacts; i.e., in preconfluent cultures and when productive cadherin-based contacts are disrupted by depletion of extracellular Ca2+. The proportion of stable intracellular E-cadherin was found to be considerably greater in preconfluent cells than in confluent monolayers, and as reported by earlier studies (Kartenbeck et al. 1991; Alexander et al. 1998), surface E-cadherin was rapidly internalized when cells were subjected to chelation of extracellular Ca2+. Indeed, assessment by quantitative immunofluorescence and surface trypsin susceptibility suggested that the majority (∼80%) of total cellular E-cadherin is now internalized upon depletion of extracellular Ca2+.
The simplest explanation of these findings is that cell-cell contact regulates E-cadherin trafficking by downregulating the endocytosis of surface E-cadherin. We envisage that in the absence of stable contacts, a large proportion of E-cadherin is constantly recycled to and from the cell surface. Upon formation of productive contacts endocytosis would be downregulated and participating E-cadherin molecules withdrawn from the recycling pathway. Other examples of regulated endocytosis and recycling pathways exist for cellular control of cell surface events; the insulin-responsive glucose transporter, GLUT-4 (Rea and James 1997), the CFTR chloride channel (Bradbury et al. 1992), and aquaporin water channels (Brown et al. 1998) are all sequestered inside cells under baseline conditions, being released to the cell surface via regulated recycling pathways in response to cytoplasmic signaling events. E-Cadherin may thus be another example of how a repertoire of recycling pathways are used to regulate the function of cell surface proteins.
Our experiments do not yet allow us to identify the molecular mechanism by which cell contact regulates cadherin recycling. It is attractive to speculate that cadherin ligation itself might influence the cytoplasmic machinery responsible for endocytosis. This would be consistent with the central role of E-cadherin in establishing cell-cell contacts (Gumbiner et al. 1988; Musil et al. 1990; Wheelock and Jensen 1992; McNeill et al. 1993; Yap et al. 1995) and with evidence that ligand binding can influence the clustering activity of the cadherin cytoplasmic tail (Yap et al. 1998). The observation that β-catenin association may be decreased in the internalized pool of E-cadherin further suggests that cytoplasmic interactions mediated by the E-cadherin tail may differ depending on whether E-cadherin is undergoing recycling or is stabilized at the cell surface. However, changes in extracellular Ca2+ can also perturb cell junctions via activation of protein kinases (Citi 1992; Alexander et al. 1998). Various signaling molecules, including heterotrimeric G proteins (de Almeida et al. 1994), are also recruited to sites of contact as cells grow to confluence (Ostman et al. 1994; Gebbink et al. 1995). Contact-dependent regulation of E-cadherin recycling may therefore reflect not only changes in the state of E-cadherin binding but also cytoplasmic signaling events associated with, and/or independent of, the E-cadherin complex itself.
Irrespective of the precise molecular mechanism involved, contact-dependent regulation of recycling has potential implications for understanding the dynamics of E-cadherin expression at the cell surface. For example, contact-dependent inhibition of endocytosis may contribute to the stabilization of E-cadherin expression at the cell surface that has been commonly documented to occur as cells grow to confluence (Shore and Nelson 1991). This model may also account for the observation that when migrating MDCK cells make nascent productive contacts, cytoplasmic E-cadherin appears to be recruited specifically to the regions of contact (McNeill et al. 1993; Adams et al. 1996; Adams et al. 1998). In light of our findings, it is plausible that this site-specific accumulation of cadherins is due to inhibition of endocytosis and consequent stabilization of cadherins at the cell surface in regions of contact, rather than solely through directed transport to the site of contact.
Regulated cadherin recycling may also act to remodel adhesive contacts in dynamic situations where contacts must be rapidly broken and remade, such as during gastrulation movements or wound healing (Gumbiner 1992; Miller and McClay 1997). Thus, we found that when cellular contacts were experimentally disrupted by removal of extracellular Ca2+, recycling of preexisting surface E-cadherin was sufficient and necessary to restore epithelial monolayers. Even when protein synthesis was blocked by cycloheximide, cells were able to reform stable contacts and monolayers within 1–2 h after restoration of extracellular Ca2+. Importantly, however, restitution of epithelial integrity was severely compromised when recycling of E-cadherin was blocked with bafilomycin. Therefore, despite the replacement of extracellular Ca2+, the E-cadherins remaining on the surface of the isolated cells did not suffice to restore stable contacts; instead recycling of endocytosed E-cadherin to the surface was necessary for stable contacts to be reestablished. By extension, in tissues undergoing dynamic remodeling, endocytosis could act to rapidly clear cadherin molecules from regions where adhesion must be reduced or broken, with these endocytosed cadherins then redeployed by the recycling pathway to sites where new contacts are being formed. Indeed, endocytosis of cadherin was observed to accompany some instances of epithelial-to-mesenchymal transformation during sea urchin gastrulation (Miller and McClay 1997), a context where cell-cell contacts are expected to be undergoing rapid remodeling. Furthermore, integrin recycling has been documented in locomoting neutrophils (Lawson and Maxfield 1995), suggesting that cadherins may exemplify more general mechanisms for modulating surface adhesive activity.
Cellular proteins may be endocytosed via clathrin-mediated or non–clathrin-mediated pathways. Several lines of evidence in the present study point to a clathrin-dependent pathway for selective E-cadherin recycling: (a) E-cadherin uptake was disrupted by hypotonic shock and K+ depletion, a maneuver which has been previously used to specifically inhibit clathrin-coated pit uptake of the low density lipoprotein receptor (Larkin et al. 1983) and other receptors; (b) some of the intracellular pool of stable E-cadherin colocalizes with rab 5, a marker which is typically located in early or recycling endosomes of clathrin-mediated trafficking pathways (Chavrier et al. 1990); (c) recycling of surface E-cadherin was significantly reduced by bafilomycin A1, which is characteristic of a pH-dependent, clathrin-mediated recycling pathway (Johnson et al. 1993; Presley et al. 1997); and (d) recycling was also inhibited by an 18°C temperature block, which has been observed to inhibit trafficking beyond early or sorting endosomes (Galloway et al. 1983; Dunn et al. 1989; Czekay et al. 1997). In addition, it is interesting to note that the published sequence of mouse E-cadherin contains several signal sequences in its cytoplasmic domain which are known to specify clathrin-coated endocytosis. A di-leucine motif is found in the juxtamembrane region of the mouse E-cadherin cytoplasmic tail at amino acid position 743–744 (SWISS-PROT. accession number PO9803) (Ringwald et al. 1987), 10 amino acids from the predicted border with the transmembrane region. Also a motif (YDSLL) is found at amino acids 829–833 of the cytoplasmic tail. This corresponds to a classical tyrosine-based targeting signal (YXXØ) overlapping with a di-leucine motif. Identical sequences are found in the cytoplasmic tails of human and Xenopus E-cadherin. Tyrosine-based signals are required for clathrin-coated pit recruitment (Trowbridge et al. 1993) and di-leucine motifs act as potential endocytic signals (Johnson and Kornfeld 1992; Letourneur and Klausner 1992). Although mutation of these sites did not affect exocytic trafficking of E-cadherin (Chen et al. 1999), their roles in endocytosis have not yet been assessed. Taken together these findings suggest that E-cadherin is endocytosed via a clathrin-coated pathway and subsequently recycled to the surface principally via an early endosomal compartment. Immunoelectron microscopy studies are currently underway in our laboratories to definitively confirm this possibility.
In conclusion, our findings identify a post-Golgi recycling pathway for E-cadherin that is regulated by cell-cell contact. Recycling appears to be capable of influencing cadherin expression and, by implication, adhesive function, at the cell surface in both stable cell monolayers and during dynamic remodeling. Such a pathway has broad functional implications. Recycling could be utilized, or corrupted, to alter adhesion and tissue patterning during development or in tumorigenesis. Internalization of E-cadherin may also be relevant to its pathogenetic role as a cell surface receptor for cellular invasion by Listeria monocytogenes (Mengaud et al. 1996), a pathway which has yet to be fully elucidated. Understanding the cellular pathway for cadherin recycling and its regulation is therefore likely to provide important insights into the morphogenetic mechanisms of cadherins in development and disease.
Acknowledgments
We thank Mr. Darren Brown and Mr. Colin Macqueen for their assistance in microscopy, Dr. Rob Parton for helpful discussions, Mr. Danny Thomas and Ms. Rowan Allison for their technical support, and Drs. Warren Gallin and Mike Caplan for kind gifts of antibodies.
This work was supported by grants from the National Health and Medical Research Council (J.L. Stow and A.S. Yap) and by the Royal Australasian College of Physicians (A.S. Yap). J.L. Stow is a Wellcome Trust Senior Medical Research Fellow.
Footnotes
1.used in this paper: sulfo-NHS-SS-biotin, sulfosuccinimidyl 2-(biotinamido) ethyl-dithioproprionate; TfR, transferrin receptor
References
- Adams C.L., Nelson W.J., Smith S.J. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J. Cell Biol. 1996;135:1899–1911. doi: 10.1083/jcb.135.6.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams C.L., Chen Y.T., Smith S.J., Nelson W.J. Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin–green fluorescent protein. J. Cell Biol. 1998;142:1105–1119. doi: 10.1083/jcb.142.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J.S., Jackson S.A., Chaney E., Kevil C.G., Haselton F.R. The role of cadherin endocytosis in endothelial barrier regulationinvolvement of protein kinase C and actin-cadherin interactions. Inflammation. 1998;22:419–433. doi: 10.1023/a:1022325017013. [DOI] [PubMed] [Google Scholar]
- Angres B., Barth A., Nelson W.J. Mechanism for transition from initial to stable cell-cell adhesionkinetic analysis of E-cadherin-mediated adhesion using a quantitative adhesion assay. J. Cell Biol. 1996;134:549–557. doi: 10.1083/jcb.134.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury N.A., Jilling T., Berta G., Sorscher E.J., Bridges R.J., Kirk K.L. Regulation of plasma membrane recycling by CFTR. Science. 1992;256:530–532. doi: 10.1126/science.1373908. [DOI] [PubMed] [Google Scholar]
- Brieher W.M., Gumbiner B.M. Regulation of C-cadherin function during activin induced morphogenesis of Xenopus animal caps. J. Cell Biol. 1994;126:519–527. doi: 10.1083/jcb.126.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D., Katsura T., Gustafson C.E. Cellular mechanisms of aquaporin trafficking. Am. J. Physiol. 1998;275:F328–F331. doi: 10.1152/ajprenal.1998.275.3.F328. [DOI] [PubMed] [Google Scholar]
- Chavrier P., Parton R.G., Hauri H.P., Simons K., Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- Chen Y.T., Stewart D.B., Nelson W.J. Coupling assembly of the E-cadherin/beta-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J. Cell Biol. 1999;144:687–699. doi: 10.1083/jcb.144.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citi S. Protein kinase inhibitors prevent junction dissociation induced by low extracellular calcium in MDCK epithelial cells. J. Cell Biol. 1992;117:169–178. doi: 10.1083/jcb.117.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czekay R.P., Orlando R.A., Woodward L., Lundstrom M., Farquhar M.G. Endocytic trafficking of megalin/RAP complexesdissociation of the complexes in late endosomes. Mol. Biol. Cell. 1997;8:517–532. doi: 10.1091/mbc.8.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida J.B., Holtzman E.J., Peters P., Ercolani L., Ausiello D.A., Stow J.L. Targeting of chimeric Gαi proteins to specific membrane domains. J. Cell Sci. 1994;107:507–515. doi: 10.1242/jcs.107.3.507. [DOI] [PubMed] [Google Scholar]
- Duden R., Franke W.W. Organization of desmosomal plaque proteins in cells growing at low calcium concentrations. J. Cell Biol. 1988;107:1049–1063. doi: 10.1083/jcb.107.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K.W., McGraw T.E., Maxfield F.R. Iterative fractionation of recycling receptors from lysosomally destined ligands in an early sorting endosome. J. Cell Biol. 1989;109:3303–3314. doi: 10.1083/jcb.109.6.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway C.J., Dean G.E., Marsh M., Rudnick G., Mellman I. Acidification of macrophage and fibroblast endocytic vesicles in vitro. Proc. Natl. Acad. Sci. USA. 1983;80:3334–3338. doi: 10.1073/pnas.80.11.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebbink M.F., Zondag G.C., Koningstein G.M., Feiken E., Wubbolts R.W., Moolenaar W.H. Cell surface expression of receptor protein tyrosine phosphatase RPTP mu is regulated by cell-cell contact. J. Cell Biol. 1995;131:251–260. doi: 10.1083/jcb.131.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottardi C.J., Dunbar L.A., Caplan M.J. Biotinylation and assessment of membrane polaritycaveats and methodological concerns. Am. J. Physiol. 1995;268:F285–F295. doi: 10.1152/ajprenal.1995.268.2.F285. [DOI] [PubMed] [Google Scholar]
- Graeve L., Drickamer K., Rodriguez-Boulan E. Functional expression of the chicken liver asialoglycoprotein receptor in the basolateral surface of MDCK cells. J. Cell Biol. 1989;109:2809–2816. doi: 10.1083/jcb.109.6.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindstaff K.K., Bacallao R.L., Nelson W.J. Apiconuclear organization of microtubules does not specify protein delivery from the trans-Golgi network to different membrane domains in polarized epithelial cells. Mol. Biol. Cell. 1998;9:685–699. doi: 10.1091/mbc.9.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B., Stevenson B., Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintainance of the epithelial junctional complex. J. Cell Biol. 1988;107:1575–1587. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B.M. Epithelial morphogenesis. Cell. 1992;69:385–387. doi: 10.1016/0092-8674(92)90440-n. [DOI] [PubMed] [Google Scholar]
- Hermiston M.L., Gordon J.I. In vivo analysis of cadherin function in the mouse intestinal epitheliumessential roles in adhesion, maintainance of differentiation, and regulation of programmed cell death. J. Cell Biol. 1995;129:489–506. doi: 10.1083/jcb.129.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermiston M.L., Wong M.H., Gordon J.I. Forced expression of E-cadherin in the mouse intestinal epithelium slows cell migration and provides evidence for nonautonomous regulation of cell fate in a self-renewing system. Genes Dev. 1996;10:985–996. doi: 10.1101/gad.10.8.985. [DOI] [PubMed] [Google Scholar]
- Johnson K.F., Kornfeld S. A His-Leu-Leu sequence near the carboxyl terminus of the cytoplasmic domain of the cation-dependent mannose 6-phosphate receptor is necessary for the lysosomal enzyme sorting function. J. Biol. Chem. 1992;267:17110–17115. [PubMed] [Google Scholar]
- Johnson L.S., Dunn K.W., Pytowski B., McGraw T.E. Endosome acidification and receptor traffickingbafilomycin A1 slows receptor externalization by a mechanism involving the receptor's internalization motif. Mol. Biol. Cell. 1993;4:1251–1266. doi: 10.1091/mbc.4.12.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartenbeck J., Schmid E., Franke W.W., Geiger B. Different modes of internalization of proteins associated with adherens junctions and desmosomesexperimental separation of lateral contacts induces endocytosis of desmosomal plaque material. EMBO (Eur. Mol. Biol. Organ.) J. 1982;1:725–732. doi: 10.1002/j.1460-2075.1982.tb01237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartenbeck J., Schmelz M., Franke W.W., Geiger B. Endocytosis of junctional acadherins in bovine kidney epithelial (MBCK) cells cultured in low Ca2+ ion medium. J. Cell Biol. 1991;113:881–892. doi: 10.1083/jcb.113.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemler R., Ozawa M., Ringwald M. Calcium-dependent cell adhesion molecules. Curr. Opin. Cell. Biol. 1989;1:892–897. doi: 10.1016/0955-0674(89)90055-0. [DOI] [PubMed] [Google Scholar]
- Larkin J.M., Brown M.S., Goldstein J.L., Anderson R.G.W. Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell. 1983;33:273–285. doi: 10.1016/0092-8674(83)90356-2. [DOI] [PubMed] [Google Scholar]
- Lawson M.A., Maxfield F.R. Ca(2+)- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- Letourneur F., Klausner R.D. A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell. 1992;69:1143–1157. doi: 10.1016/0092-8674(92)90636-q. [DOI] [PubMed] [Google Scholar]
- Lever J.E. Inducers of mammalian cell differentiation stimulate dome formation in a differentiated kidney epithelial cell line (MDCK) Proc. Natl. Acad. Sci. USA. 1979;76:1323–1327. doi: 10.1073/pnas.76.3.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine E., Lee C.H., Kintner C., Gumbiner B.M. Selective disruption of E-cadherin function in early Xenopus embryos by a dominant negative mutant. Development. 1994;120:901–909. doi: 10.1242/dev.120.4.901. [DOI] [PubMed] [Google Scholar]
- Mays R.W., Siemers K.A., Fritz B.A., Lowe A.W., van Meer A.W., Nelson W.J. Hierarchy of mechanisms involved in generating Na/K-ATPase polarity in MDCK epithelial cells. J. Cell Biol. 1995;130:1105–1115. doi: 10.1083/jcb.130.5.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea P.D., Gumbiner B.M. Purification of a 92-kDa cytoplasmic protein tightly associated with the cell-cell adhesion molecule E-cadherin (uvomorulin). Characterization and extractability of the protein complex from the cell cytostructure. J. Biol. Chem. 1991;266:4514–4520. [PubMed] [Google Scholar]
- McNeill H., Ryan T.A., Smith S.J., Nelson W.J. Spatial and temporal dissection of immediate and early events following cadherin-mediated epithelial cell adhesion. J. Cell Biol. 1993;120:1217–1226. doi: 10.1083/jcb.120.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J., Ohayon H., Gounon P., Mege R.-M., Cossart P. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell. 1996;84:923–932. doi: 10.1016/s0092-8674(00)81070-3. [DOI] [PubMed] [Google Scholar]
- Miller J.R., McClay D.R. Characterization of the role of cadherin in regulating cell adhesion during sea urchin development. Dev. Biol. 1997;192:323–339. doi: 10.1006/dbio.1997.8740. [DOI] [PubMed] [Google Scholar]
- Musil L.S., Cunningham B.A., Edelman G.M., Goodenough D.A. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J. Cell Biol. 1990;111:2077–2088. doi: 10.1083/jcb.111.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myat M.M., Chang S., Rodriguez-Boulan E., Aderem A. Identification of the basolateral targeting determinant of a peripheral membrane protein, MacMARCKS, in polarized cells. Curr. Biol. 1998;8:677–683. doi: 10.1016/s0960-9822(98)70273-8. [DOI] [PubMed] [Google Scholar]
- Narula N., McMorrow I., Plopper G., Doherty J., Matlin K.S., Burke B., Stow J.L. Identification of a 200-kD, brefeldin-sensititve protein on Golgi membranes. J. Cell Biol. 1992;117:27–38. doi: 10.1083/jcb.117.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathke I.S., Hinck L., Swedlow J.R., Papkoff J., Nelson W.J. Defining interactions and distributions of cadherin and catenin complexes in polarized epithelial cells. J. Cell Biol. 1994;125:1341–1352. doi: 10.1083/jcb.125.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson W.J., Shore E.M., Wang A.Z., Hammerton R.W. Identification of a membrane-cytoskeletal complex containing the cell adhesion molecule uvomorulin (E-cadherin), ankyrin, and fodrin in Madin-Darby canine kidney epithelial cells. J. Cell Biol. 1990;110:349–357. doi: 10.1083/jcb.110.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostman A., Yang Q., Tonks N.K. Expression of DEP-1, a receptor-like protein-tyrosine-phosphatase, is enhanced with increasing cell density. Proc. Natl. Acad. Sci. USA. 1994;91:9680–9684. doi: 10.1073/pnas.91.21.9680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley J.F., Mayor S., McGraw T.E., Dunn K.W., Maxfield F.R. Bafilomycin A1 treatment retards transferrin receptor recycling more than bulk membrane recycling. J. Biol. Chem. 1997;272:13929–13936. doi: 10.1074/jbc.272.21.13929. [DOI] [PubMed] [Google Scholar]
- Rea S., James D.E. The biogenesis and trafficking of GLUT4 storage vesicles. Diabetes. 1997;46:1667–1677. doi: 10.2337/diab.46.11.1667. [DOI] [PubMed] [Google Scholar]
- Ringwald M., Schuh R., Vestweber D., Eistetter H., Lottspeich F., Engel J., Dolz R., Jahnig F., Epplen J., Mayer S. The structure of cell adhesion molecule uvomorulin. Insights into the molecular mechanism of Ca2+-dependent cell adhesion. EMBO (Eur. Mol. Biol. Organ.) J. 1987;6:3647–3653. doi: 10.1002/j.1460-2075.1987.tb02697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K., van Deurs B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996;76:949–966. doi: 10.1152/physrev.1996.76.4.949. [DOI] [PubMed] [Google Scholar]
- Shore E.M., Nelson W.J. Biosynthesis of the cell adhesion molecule uvomorulin (E-cadherin) in Madin-Darby canine kidney epithelial cells. J. Cell Biol. 1991;266:19672–19680. [PubMed] [Google Scholar]
- Takeichi M. Functional correlation between cell adhesive properties and some cell surface proteins. J. Cell Biol. 1977;75:464–474. doi: 10.1083/jcb.75.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 1995;7:619–627. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- Takeichi M., Atmusi T., Yoshida C., Uno K., Okada T.S. Selective adhesion of embryonal carcinoma cells and differentiated cells by Ca2+-dependent sites. Dev. Biol. 1981;87:340–350. doi: 10.1016/0012-1606(81)90157-3. [DOI] [PubMed] [Google Scholar]
- Tepass U. Crumbs, a component of the apical membrane, is required for zonula adherens formation in primary epithelia of Drosophila . Dev. Biol. 1996;177:217–225. doi: 10.1006/dbio.1996.0157. [DOI] [PubMed] [Google Scholar]
- Trowbridge I.S., Collawn J.F., Hopkins C.R. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu. Rev. Cell Biol. 1993;9:129–161. doi: 10.1146/annurev.cb.09.110193.001021. [DOI] [PubMed] [Google Scholar]
- Uemura T., Oda H., Kraut R., Hayashi S., Kotaoka Y., Takeichi M. Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the Drosophila embryo. Genes Dev. 1996;10:659–671. doi: 10.1101/gad.10.6.659. [DOI] [PubMed] [Google Scholar]
- Vega-Salas D.E., Salas P.J.I., Gundersen D., Rodriguez-Boulan E. Formation of the apical pole of epithelial (Madin-Darby canine kidney) cells. Polarity of an apical protein is independent of tight junctions while segregation of a basolateral marker requires cell-cell interactions. J. Cell Biol. 1987;104:905–1007. doi: 10.1083/jcb.104.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D., Gossler A., Boller K., Kemler R. Expression and distribution of cell adhesion molecule uvomorulin in mouse preimplantation embryos. Dev. Biol. 1987;124:451–456. doi: 10.1016/0012-1606(87)90498-2. [DOI] [PubMed] [Google Scholar]
- Wang A.Z., Ojakian G.K., Nelson W.J. Steps in the morphogenesis of a polarized epithelium. I. Uncoupling the roles of cell-cell and cell-substratum contact in establishing plasma membrane polarity in multicellular epithelial (MDCK) cysts. J. Cell Sci. 1990;95:137–151. doi: 10.1242/jcs.95.1.137. [DOI] [PubMed] [Google Scholar]
- Watabe M., Nagafuchi A., Tsukita S., Takeichi M. Induction of polarized cell-cell association and retardation of growth by activation of the E-cadherin–catenin adhesion system in a dispersed carcinoma line. J. Cell Biol. 1994;127:247–256. doi: 10.1083/jcb.127.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock M.J., Jensen P.J. Regulation of keratinocyte intercellular junction organization and epidermal morphogenesis by E-cadherin. J. Cell Biol. 1992;117:415–425. doi: 10.1083/jcb.117.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J.M., Colton T.L. Targeting of an intestinal apical endosomal protein to endosomes in nonpolarized cells. J. Cell Biol. 1997;136:319–330. doi: 10.1083/jcb.136.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkel G.K., Ferguson J.E., Takeichi M., Nuccitelli R. Activation of protein kinase C triggers premature compaction in the four-cell stage mouse embryo. Dev. Biol. 1990;138:1–15. doi: 10.1016/0012-1606(90)90171-e. [DOI] [PubMed] [Google Scholar]
- Yap A.S., Stevenson B.R., Keast J.R., Manley S.W. Cadherin-mediated adhesion and apical membrane assembly define distinct steps during thyroid epithelial polarization and lumen formation. Endocrinology. 1995;136:4672–4680. doi: 10.1210/endo.136.10.7664688. [DOI] [PubMed] [Google Scholar]
- Yap A.S., Brieher W.M., Gumbiner B.M. Molecular and functional analysis of cadherin-based adherens junctions Annu. Rev. Cell. Dev. Biol. 13 1997. 119 146a [DOI] [PubMed] [Google Scholar]
- Yap A.S., Brieher W.M., Pruschy M., Gumbiner B.M. Lateral clustering of the adhesive ectodomaina fundamental determinant of cadherin function Curr. Biol. 7 1997. 308 315b [DOI] [PubMed] [Google Scholar]
- Yap A.S., Niessen C.M., Gumbiner B.M. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y., Brieher W.M., Gumbiner B.M. Analysis of C-cadherin regulation during tissue morphogenesis with an activating antibody. J. Cell Biol. 1999;144:351–359. doi: 10.1083/jcb.144.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]