Structure of M11L: A myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2 (original) (raw)
Abstract
Apoptosis of virally infected cells is an innate host mechanism used to prevent viral spread. However, viruses have evolved a number of proteins that function to modulate the apoptotic cascades and thereby favor productive viral replication. One such antiapoptotic protein, myxoma virus M11L, has been shown to inhibit mitochondrial-dependent apoptosis by binding to and blocking the two executioner proteins Bak and Bax. Since M11L has no obvious sequence homology with Bcl-2 or Bcl-xL, the normal cellular inhibitors for Bak and Bax, and the structure of M11L has not been solved, the mode of binding to Bak and Bax is not known. In order to understand how M11L functions, the crystal structure of M11L was solved to 2.91 Å. Despite the lack of sequence similarity, M11L is a structural homolog of Bcl-2. Studies using a peptide derived from Bak indicate that M11L binds to Bak with a similar affinity (4.9 ± 0.3 μM) to the published binding affinities of Bcl-2 and Bcl-xL to the same peptide (12.7 μM and 0.5 μM, respectively), indicating that M11L inhibits apoptosis by mimicking and competing with host proteins for the binding of Bak and Bax. The structure provides important insight into how myxoma virus and other poxviruses facilitate viral dissemination by inhibiting mitochondrial dependent apoptosis.
Keywords: M11L, poxvirus, apoptosis inhibitor, X-ray crystallography, Bcl-2 homology, fluorescence polarization, immunomodulation
Apoptosis is thought to be an ancient innate cellular response to infection by intracellular pathogens. In order to circumvent this antiviral process, the majority of successful viruses have evolved to express a wide variety of immunomodulatory proteins (Benedict et al. 2002). Many viruses modulate the death receptor pathway by secreting soluble viroreceptors, as well as by modulating the mitochondrial pathway through a number of strategies, including the expression of Bcl-2 mimics (vBcl-2s). In addition, the executioner caspases can be downregulated by viruses through the expression of caspase inhibitor molecules such as the IAP family of proteins. It is interesting that in the case of poxviruses, whose large genomic size permits the expression of immunomodulatory proteins, only fowlpox virus and canarypox virus encode a clear homolog of Bcl-2 (Cuconati and White 2002; Taylor and Barry 2006). However, a number of poxviruses encode proteins that modulate the mitochondrial checkpoint of apoptotic signaling (Everett and McFadden 2001; Barry et al. 2004; Boya et al. 2004). Two such proteins are the M11L protein of myxoma virus and the F1L protein of vaccinia virus. Both M11L and F1L localize to the mitochondria and block pro-apoptotic signals that are propagated via this organelle (Everett et al. 2000; Wasilenko et al. 2003; Stewart et al. 2005), yet these proteins show no sequence homology with each other or to any member of the Bcl-2 family of antiapoptotic proteins.
M11L is critical for the virulence of myxoma virus, a member of the leporipoxvirus genus and the causative agent of a lethal rabbit-specific disease known as myxomatosis (Kerr and McFadden 2002). Rabbits infected with an M11L knockout virus develop an attenuated form of disease and ultimately make a full recovery, whereas infection with wild-type virus leads to complete mortality (Opgenorth et al. 1992). This led to the discovery that M11L is also capable of blocking apoptosis in a broad spectrum of mammalian cells, including human cells (Everett et al. 2000, 2002). The M11L protein is a small (166 residue) protein with a C-terminal transmembrane domain that localizes it to the outer mitochondrial membrane, where it exhibits antiapoptotic activity (Everett et al. 2000, 2002). M11L forms a complex with the peripheral benzodiazepine receptor (PBR), and this interaction protects against the loss of mitochondrial membrane potential, a key point in the apoptotic pathway. M11L and F1L also interact with members of the Bak/Bax family of pro-apoptotic regulators to block upstream signals that commit the cell to the death program (Wang et al. 2004; Wasilenko et al. 2005; Fischer et al. 2006; Postigo et al. 2006; Su et al. 2006; Taylor et al. 2006). Although it seems that M11L and F1L are mimicking the function of the Bcl-2 family of antiapoptotic proteins, how they manage to do so without any obvious homology with Bcl-2, has remained a mystery.
The Bcl-2 family of proteins act as gatekeepers for mitochondria-mediated apoptosis (Reed et al. 1998). Members are defined by the presence of at least one of four Bcl-2 homology (BH) motifs (Petros et al. 2004). The family is divided into two separate classes based on their functions: pro-apoptotic and antiapoptotic. The antiapoptotic proteins Bcl-2 and Bcl-xL contain all four BH motifs and are responsible for protecting the integrity of the mitochondrial outer membrane. The pro-apoptotic proteins and their functions can be divided into two classes depending on the BH motifs present. Bax-like proteins (e.g., Bax, Bak), directly responsible for disrupting the membrane, contain the BH1, BH2, and BH3 motifs, while BH3-only proteins, as their name implies, contain only a BH3 motif and promote apoptosis by inhibiting Bcl-2 and Bcl-xL as well as activating Bax. Bcl-2 and Bcl-xL inhibit apoptosis by dimerizing with Bak and Bax (Borner 2003; Sharpe et al. 2004). During apoptosis, BH3-only proteins interfere with the binding of these complexes releasing Bak and Bax, which are then able to form oligomers. The oligomers form a pore in the mitochondrial outer membrane leading to the loss of membrane potential and the release of apoptotic agents from the mitochondria (e.g., cytochrome c and Smac/DIABLO) (Marsden and Strasser 2003). Viral Bcl-2 homologs can act either by binding and sequestering the BH3-only proteins or by directly binding to Bak and Bax, forming complexes that are resistant to BH3-only protein interference (Cuconati and White 2002; Polster et al. 2004). The vBcl-2s are able to accomplish this because of their sequence and structural similarity to Bcl-2/Bcl-xL (Huang et al. 2002), but how viral inhibitors like M11L and F1L accomplish the same feat is unknown.
Sequence alignments show that there is no significant amino acid sequence similarity between M11L and Bcl-2/Bcl-xL. Although secondary structure predictions indicate that, like Bcl-2, M11L is entirely helical, threading analysis (3DPSSM) produced no significant hits. In order to determine exactly how M11L is able to bind Bak and Bax, the structure of M11L was solved. This communication reports the structural features of M11L that facilitate its interaction with Bak and Bax as well as the significance of its overall structure.
Results and Discussion
Expression and initial characterization of M11L
Initial expression tests of full-length M11L resulted in low expression levels of insoluble protein. Therefore, a truncated version of M11L (M11LΔ143) was produced to remove a predicted C-terminal transmembrane region. M11LΔ143 also expressed in an insoluble form, but when fused to the C terminus of His-tagged maltose binding protein (His6–MBP), it was soluble. Expression levels of the soluble His6–MBP–M11LΔ143 fusion remained low even when expanded codon usage cells (BL21[DE3] Rosetta, Stratagene) were used. In order to increase the yield, an Escherichia coli codon optimized M11L gene was constructed. The codon optimized His6–MBP–M11LΔ143 construct produced significantly higher levels of soluble protein, typically ∼28 mg/L of fusion protein which corresponds to ∼7.5 mg/L of cleaved M11LΔ143.
Consistent with secondary structure predictions, a circular dichroism (CD) spectrum of M11L showed that the protein is folded and helical (Fig. 1). Initial mass spectrometry results indicated that M11LΔ143 contains one intramolecular disulfide (data not shown), which was not entirely unexpected as the protein contains six cysteine residues. To explore whether the disulfide was important for the structure, a CD spectrum was taken in the presence of 1 mM Tris(2-carboxyethyl phosphine) (TCEP), a potent reducing agent. The addition of TCEP had no effect on the overall spectrum, indicating that either the disulfide is not important for the structure/stability of M11L, or that the disulfide is resistant to TCEP, and therefore likely important to the structure (data not shown).
Figure 1.

Far-UV circular dichroism spectrum of M11L▵143.
Structure determination of M11L
The structure was solved using selenomethionine derivatized protein and single wavelength anomalous diffraction (SAD) to locate the heavy atoms. Multiple wavelength anomalous diffraction (MAD) was used to refine the heavy atom sites after which the phases were extended using high-resolution native data. The structure was manually built and refined to 2.91 Å resolution with final R work and R free values of 24.9% and 29.0%, respectively. The crystal was found to have two M11L monomers in the asymmetric unit (chain A and chain B), resulting in a 71.96% solvent content (Matthews 1968; Kantardjieff and Rupp 2003). Crystallographic data is summarized in Table 1. Apart from a short loop from Asp15 to Met24, which lacks density in chain B, the two monomers adopt an identical confirmation (Cα-RMSD = 0.436 Å). An important crystal contact is made by the interaction of the C-terminal helix of chain B with a surface region on chain A. Likewise, the C-terminal helix of chain A binds the same surface on chain B in an adjacent unit cell. This interaction is critical to the crystal packing, and is discussed in more depth below. An intramolecular disulfide was found between Cys33 and Cys127, despite the crystals being grown in the presence of 1 mM TCEP, suggesting that it is structurally relevant; this is interesting, since most intracellular proteins do not contain disulfides because of the reducing environment in the cytoplasm, although some other examples have been reported (Prinz et al. 1997).
Table 1.
Data collection, refinement, and stereochemistry

The structure of M11L is made up of six amphipathic helices packed around a single core helix (Fig. 2A). The overall topology of the structure is similar to that of Bcl-2 (Petros et al. 2001) and Bcl-xL (Muchmore et al. 1996), which is surprising given the negligible sequence homology, but not completely unexpected since M11L binds to both Bak and Bax (Wang et al. 2004; Su et al. 2006). The surface of M11L contains a few charged patches, but whether they are functionally important is unknown at this time (Fig. 2B). There is a patch of positive charge found above the hydrophobic groove (Fig. 2B, left), as well as a patch of negative charge on the opposite side of the protein from the groove (Fig. 2B, right). There is also a wide, shallow hydrophobic groove found on the surface (Fig. 3).
Figure 2.
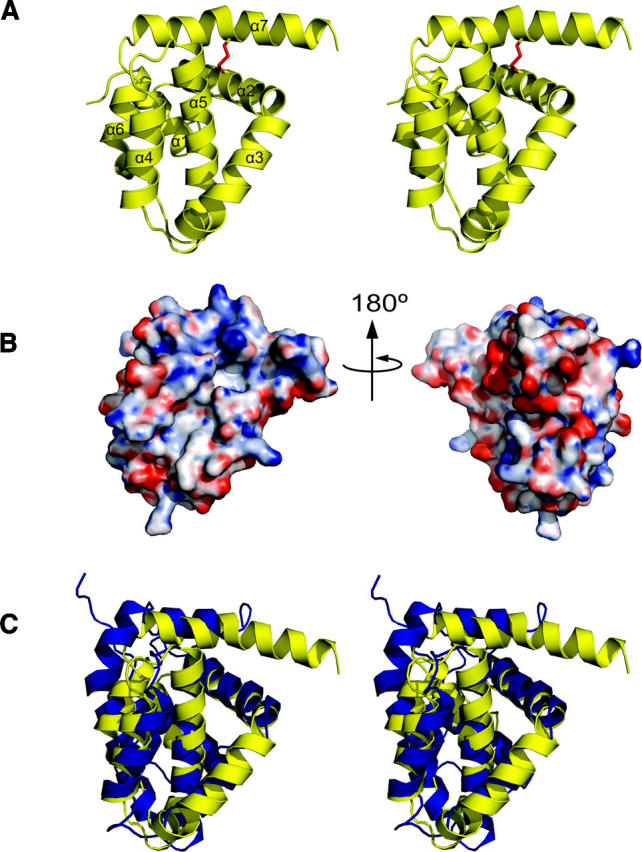
Structural representations of M11L. (A) Stereo cartoon depiction of the structure of M11L. Helices are numbered sequentially from the N terminus. The disulfide between Cys33 and Cys127 is shown in red. (B) Electrostatic surface of M11L. Red and blue indicate electronegative and electropositive surface, respectively. The surface was calculated using APBS (Baker et al. 2001) for PyMOL and is contoured from −20 to 20. (C) Stereo superposition of M11L (yellow) onto Bcl-xL (blue). Structures were aligned using LSQMAN. Figures were generated with PyMol (De Lano 2002).
Figure 3.

Solvent-accessible surface comparisons of M11L with Bcl-2 and Bcl-xL. (A) M11L (PDB ID: 2O42), (B) Bcl-xL (1R2D), and (C) Bcl-2 (1G5M). Views of the hydrophobic groove are from the same perspective from LSQMAN aligned structures. Leucine, isoleucine, valine, methionine, tyrosine, phenylalanine, and tryptophan are colored yellow. Aspartate and glutamate are colored red. Lysine, arginine, and histidine are colored blue. Figures were generated with PyMol (De Lano 2002).
Comparison of M11L to Bcl-2 family structures
The overall structure of M11L is highly similar to that of the Bcl-2 family proteins Bcl-2 and Bcl-xL. They share the same number of α-helices, the same fold, and they all contain a hydrophobic groove on their surface. Structural alignments of the 138 Cα atoms of M11L to those of Bcl-2 and Bcl-xL using LSQMAN (Kleywegt 1996) give an RMSD of 2.044 Å and 1.984 Å, respectively. Figure 2C shows an overlay of M11L (shown in yellow) onto Bcl-xL (blue). An alignment based on the overlay of M11L with Bcl-2 and Bcl-xL can be seen in Figure 4.
Figure 4.

Structural alignment of M11L, Bcl-xL, and Bcl-2. Sequence alignment based on the alignment of the helices from M11L, Bcl-xL, and Bcl-2. Alignment after helix-7 is based on sequence and functional alignments, due to a lack of structural data. BH domains from Bcl-2 and Bcl-xL are highlighted. Secondary structure elements are shown as cylinders.
Helix 1 and the loop between helix 1 and helix 2 are both shorter in M11L than those found in either Bcl-2 or Bcl-xL. However, the lengths of both helix 1 and the first loop are similar in length to those found in the Kaposi Sarcoma-associated herpes virus (KSHV) Bcl-2 homolog (Huang et al. 2002). There is some evidence that this loop region is important in regulating the pro-survival character of Bcl-2 and Bcl-xL (Clem et al. 1998; Ojala et al. 2000). With a shortened α-helix and loop region, M11L is still able to make structural interactions with helix 6 and 7, but may not be subject to the same regulation as Bcl-2/Bcl-xL. The NWGR sequence motif found at the beginning of helix 5 in the BH1 domain is highly conserved across all Bcl-2 family members, but it is noticeably absent from M11L (instead of the conserved sequence, M11L contains SPSV in the same position; Fig. 4). In Bcl-2 and Bcl-xL the tryptophan makes extensive contacts with residues in helix 7 and 8, whereas in M11L, the corresponding proline interacts much less with helix 7. Instead, it seems that M11L employs an intramolecular disulfide bond between Cys33 and Cys127 to help hold helix 7 in place. In addition there is a hydrogen bond between Lys80 and a backbone amide found in the loop between helices 6 and 7. These two interactions likely take the place of the interactions lost by the mutation of the conserved tryptophan.
Structural studies of Bcl-2 family proteins have led to a better understanding of how Bax and Bak may function and how various members of the family may interact. All of the members of the Bcl-2 family have the same fold, characterized by an α-helical bundle arranged around a single central α-helix (Muchmore et al. 1996; Sattler et al. 1997; Suzuki et al. 2000; Petros et al. 2001) . The structure of Bcl-xL also showed that the conserved BH1, BH2, and BH3 domains are all in close spatial proximity and form a shallow hydrophobic groove on the surface (Muchmore et al. 1996). It was proposed, and later confirmed via crystal structures, that this was the site for binding of Bak. When the crystal structure of Bcl-xL and a peptide fragment containing the Bak–BH3 domain was solved, the peptide was found bound in this elongated hydrophobic groove (Sattler et al. 1997). It is thought then that the hydrophobic grooves present on Bcl-2 and Bcl-xL mediate their binding to both Bak and Bax.
Based on similarities between the folds of M11L and Bcl-2 proteins, the hydrophobic groove present on the surface of M11L may be analogous to the groove found in both Bcl-2 and Bcl-xL. This idea is strengthened by a fortuitous crystal contact, in which the C-terminal helix of one molecule is found binding directly into this region of another molecule (Fig. 5). When the M11L structure is aligned with the Bcl-xL/Bak-peptide structure (PDB ID: 1BXL), the helix bound in the M11L structure overlays with the BH3-domain found bound in the Bcl-xL/Bak-peptide structure. However, the groove found on the surface of M11L is wider and slightly more shallow than the groove found in either Bcl-2 or Bcl-xL. This can be explained by the helix that is found bound in the pocket. The groove on M11L is similar in shape to the groove in the Bcl-xL/Bak-peptide structure (Fig. 6). In M11L, Tyr41 protrudes into the groove, creating a bump (Fig. 6A). An analogous large protrusion is not present in Bcl-2 or Bcl-xL, and its effect on binding is unknown at this time. As was found in the Bcl-xL/Bak-peptide structure, there is a hydrophobic pocket that accommodates the highly conserved leucine found in the Bak and Bax BH3 domains (Fig. 6B). In M11L, this pocket is formed by Tyr41, Tyr45, Leu48, Ala82, and Leu86, whereas in Bcl-xL it is formed by Phe97, Tyr101, Leu130, Ala142, and Phe146. The electrostatic distribution around the binding groove in M11L is also very different from that of Bcl-2 and Bcl-xL. As shown in Figure 5, the binding groove of Bcl-xL is immediately lined with Arg100 and Arg103 on one side and Glu129, Arg132, and Arg139 on the other. In contrast, M11L has Lys43 and Asp50 on one side and Arg73 and Asp74 on the other, but these charged residues are further removed from the binding pocket and are unlikely to interact with a bound peptide. The majority of the charge differences are unlikely to affect binding since analysis of Bcl-xL binding to the Bak BH3 domain showed that the only charged residue making an important stabilizing interaction with the peptide was Arg139 (Sattler et al. 1997). There is no homologous residue to Arg139 found in the hydrophobic groove of M11L, so it is unclear how M11L makes up for this loss of binding energy. It was previously reported that M11L contained a putative BH3 domain (Wang et al. 2004). Analysis of this region indicates that the conserved residues are all buried in M11L as they are in Bak and Bcl-xL. It is unclear at this time whether this domain is functional in binding other Bcl-2 proteins or simply important for the overall structure of M11L.
Figure 5.

C-terminal helix of M11L bound in the hydrophobic groove. The C-terminal helix of one M11L monomer (chain B) (purple) is bound into the hydrophobic groove of another M11L monomer (chain A). The Bak-BH3 peptide (green) is shown based on structural alignment of Bcl-xL/Bak-peptide structure (PDB ID: 1BXL) using LSQMAN (Kleywegt 1996). M11L is shown as a solvent-accessable surface area. Leucine, isoleucine, valine, methionine, tyrosine, phenylalanine, and tryptophan are colored yellow. Aspartate and glutamate are colored red. Lysine, arginine, and histidine are colored blue.
Figure 6.

Comparison of hydrophobic grooves from M11L and Bcl-xL. (A) M11L (PDB ID: 2O42) and (B) Bcl-xL (1BXL). Leucine, isoleucine, valine, methionine, tyrosine, phenylalanine, and tryptophan are colored yellow. Aspartate and glutamate are colored red. Lysine, arginine, and histidine are colored blue. Specific amino acid residues are indicated in blue or black.
Binding of Bak to M11L
Binding of M11L to Bak and Bax, its intracellular targets, was previously demonstrated (Wang et al. 2004; Su et al. 2006), but the affinity was not known (Table 2). The use of full length Bak or Bax in binding studies has proven difficult as these proteins seem to be highly toxic to cells, even when expressed as fusions. Therefore, short peptides containing the BH3 regions of Bcl-2 family members have been used instead to approximate binding constants between Bcl-2 family members using fluorescence polarization (Kelekar et al. 1997; Ottilie et al. 1997; Huang et al. 2002; Zhang et al. 2002) (Table 2). We used a 16-residue peptide containing the BH3 domain of Bak (GQVGRQLAIIGDCINR) to approximate the affinity of M11L for Bak by fluorescence polarization. The peptide was synthesized and subsequently derivatized with fluorescein using cysteine chemistry. Figure 7 shows the binding curve of M11LΔ143 for the peptide. The Kd was determined to be 4.86 ± 0.334 μM. This affinity is 10-fold lower than the affinity of Bak for Bcl-xL (Petros et al. 2000), but it is similar to the affinity of Bak for Bcl-2 (Petros et al. 2001), suggesting that binding to M11L is physiologically relevant. The ability of M11L to bind the peptide confirms that the purified protein is natively folded, and supports the model that M11L acts directly as a structural mimic of the Bcl-2 pro-survival proteins.
Table 2.
Affinities of apoptosis inhibitors for Bak/Bax peptides

Figure 7.

Binding curve of M11L for the Bak peptide. Fluorescence polarization binding curve for the binding of M11L to the Bak(BH3) peptide. The peptide concentration was 10 nM.
Summary and Conclusions
In this study, the crystal structure of M11L was solved to 2.91 Å. The structure shows that, despite a lack of sequence homology, M11L is a structural homolog of the Bcl-2 family of apoptotic inhibitors. M11L lacks the large unstructured loop that is present in both Bcl-2 and Bcl-xL (Fig. 4); this loop is thought to contain sites involved in regulating the antiapoptotic function of these proteins, so removal of this loop might allow M11L to function under conditions where Bcl-2 and Bcl-xL would be inactivated. The presence of a hydrophobic groove on the surface of M11L provides an explanation of how binding to pro-apoptotic Bak and Bax may occur. It is interesting to note that the hydrophobic groove found on M11L is quite different from the grooves found on either Bcl-2 or Bcl-xL. The differences are likely due to M11L's necessity to bind to the BH3 domains of both Bak and Bax. Previous studies have shown that Bcl-2 and Bcl-xL have different binding affinities to peptides derived from the BH3 domains of Bak and Bax (Table 2). In the case of M11L, both of these BH3 domains have to be bound with high affinity, even in the presence of BH3-only proteins. This requirement may have shaped the character of the groove to maximize binding to both substrates, and not to other BH3 domains. Future structural studies of M11L with BH3 domains from other Bcl-2 family members are needed to determine which residues in the binding pocket are important for the binding to only Bak and Bax.
The discovery that M11L is a Bcl-2 homolog sheds some light on the possible structures of a number of other poxvirus proteins (Table 3). Although it has no sequence homology with M11L, it is likely that F1L and similar proteins are also Bcl-2 homologs. This is further strengthened by the recent structure of the vaccinia virus N1L protein (Aoyagi et al. 2007). N1L has no sequence homology with Bcl-2, but adopts a similar fold and has been shown to bind the BH3 domain from Bak. According to phylogenetic analysis, pox viruses can be broken into four distinct groups (Gubser et al. 2004). Avipoxvirus genus (FPV), Molluscipoxvirus genus (MCV), and the Orthopox genus (OPV) each form separate groups while the Yatapoxvirus, Capripoxvirus, Suipoxvirus, and Leporipoxvirus genera all cluster together (Fig. 8). Interestingly, each of the groups seems to contain its own Bcl-2 homolog. FPVs are the only poxviruses that possess “classic” Bcl-2 sequence homologs, but many of the OPVs, including camelpox, cowpox, variola, and monkeypox, contain a protein that is similar to F1L from the vaccinia virus. In the last group, rabbit fibroma virus has a very close homolog to M11L, while sheeppox, goatpox, lumpy skin disease virus, swinepox, and deerpox all express a more distant relative. It is quite interesting that the poxvirus family, unlike Herpes viruses that express clear Bcl-2 homologs, expresses multiple different proteins with very little homology with Bcl-2, yet they all inhibit apoptosis by binding to Bak, and in some cases to Bax. How many other virus families possess cryptic Bcl-2 homologs is currently unknown. Further structural analysis of both M11L and F1L may lead to the identification of new sequence motifs that can be used to search out these unknown Bcl-2 homologs.
Table 3.
Poxviral mitochondrial inhibitors of apoptosiss

Figure 8.

Poxvirus phylogeny. Poxvirus family phylogeny (data adapted from Gubser et al. 2004).
Materials and Methods
Cloning
The sequence of the codon-optimized M11L gene was generated using an in-house computer program which replaces the natural codons of a given protein with codons observed at high frequency in bacterial cells. Repetitive sequences, high GC content, and mRNA structure are then eliminated with cycles of Monte Carlo optimization (N. Pokala, unpubl.). The optimized gene was then synthesized using seven sets of overlapping primers (MWG Biotech). A fragment containing resides 1–143 of M11L, lacking the C-terminal transmembrane anchor (M11LΔ143), was cloned into the pSV272 expression vector (N. Pokala and T.M. Handel, unpubl.) to produce a fusion of M11L to the C terminus of His6–MBP (His6–MBP–M11LΔ143). A tobacco etch virus (TEV) protease site was placed between the MBP and M11LΔ143 to allow for cleavage of the fusion protein, leaving a Gly–Ala on the N terminus of M11LΔ143 (Kapust and Waugh 1999).
Protein expression and purification
The His6–MBP–M11LΔ143 fusion protein was expressed in E. coli BL21(DE3)pLysS cells (Stratagene) by inducing with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at OD600 = 0.6 for 4 h. Cells were harvested and spun down at 6000_g_ at 4°C and resuspended in buffer (50 mM potassium phosphate [pH 8.0], 300 mM NaCl, 20 mM imidazole). Protease inhibitors (Complete: EDTA-free, Roche Diagnostics) were added and the cells were then frozen in liquid nitrogen.
For purification, cells were ruptured using an Emulsiflex C5 (Avestin Inc.) at 12,000 PSI and centrifuged at 25,000_g_ for 20 min at 4°C. The supernatant was run over Ni-NTA resin (Qiagen) and the peak fractions pooled. Pooled protein was then concentrated via ultrafiltration (Centriprep YM-10, Millipore) and cleaved for 1 h at room temperature using a 1:100 molar ratio of TEV protease:M11L-fusion protein. The cleavage reaction was dialyzed against 50 mM potassium phosphate pH 8.0, 300 mM NaCl at 4°C overnight and run over a Ni-Sepharose column (GE Healthcare). The flow-through was concentrated and passed over an S75 gel filtration column (GE Healthcare). The peak fractions were then pooled and concentrated via ultrafiltration (Centriprep YM-3, Millipore). The protein sequence was verified via electrospray mass spectrometry (courtesy of Dr. David King, UC: Berkeley and HHMI).
Selenomethionine-labeled protein was expressed via the method of Van Duyne et al. (1993) and purified as described for native His6–MBP–M11LΔ143 with the addition of 1 mM β-mercaptoethanol at all steps up to gel filtration when 1 mM Tris(2-carboxyethyl phosphine) (TCEP) was used instead.
Circular dichroism
M11LΔ143 was dialyzed into 20 mM potassium phosphate (pH 7.5), 100 mM NaCl. Circular dichroism (CD) data was collected on an Aviv 62DS spectropolarimeter (25°C, 1-cm path length).
Crystallization and structure determination
Purified selenomethionine-derivatized M11LΔ143 was dialyzed against 20 mM potassium phosphate (pH 7.4), 100 mM NaCl, and 1 mM TCEP overnight. The protein was then concentrated to 13.5 mg/mL. Crystals were grown via hanging drop vapor diffusion by mixing 1 μL M11LΔ143 with 1 μL of well solution containing 100 mM sodium citrate (pH 5.5), 700 mM ammonium sulfate, and 200 mM KCl. For harvesting, crystals were transferred to a cryoprotectant solution containing well solution plus 30% xylitol before flash freezing in liquid nitrogen.
All data sets were collected on Beamline 8.3.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory (MacDowell et al. 2004). Data were indexed using HKL2000 (Otwinowski et al. 1997), and heavy atom sites were determined from SAD data using Solve (Terwilliger 2004). Heavy atom sites were refined and phases solved using MLPHARE (Collaborative Computational Project 1994) implemented by ELVES (Holton and Alber 2004). Initial density modification was undertaken by RESOLVE (Terwilliger 2004), and manual model building was performed with O (Jones et al. 1991). The model was refined using Refmac5 (Collaborative Computational Project 1994), followed by TLS refinement in Refmac5 (Winn et al. 2001). The model was refined, along with several rounds of manual rebuilding, to a resolution of 2.91 Å, and a final working _R_-factor of 24.9% and R free of 29.0%. The final model consisted of residues 1–138 of M11L. A total of 90.1% of nonglycine residues are in the most favored regions of Ramachandran space, with 8.3% in additional allowed areas and 1.6% in generously allowed areas (Table 1).
Fluorescence polarization
Unmodified Bak(BH3) peptide (GQVGRQLAIIGDCINR) was obtained from Elim Biopharmaceuticals, Inc. The peptide was derivatized using fluorescein-5-maleimide from Molecular Probes (Invitrogen), purified via reverse-phase HPLC on a C-18 column (Vydac), and verified by electrospray mass spectrometry. Binding experiments were carried out in 120 mM sodium phosphate (pH 7.4), 0.1 mg/mL bovine serum albumin, using a peptide concentration of 10 nM and M11LΔ143 concentrations ranging from 1 nM to 325 mM. Data was collected on a Molecular Devices M5 fluorescence plate reader, and fit nonlinearly to an independent binding site model (Clarke 1996) using KaleidaGraph (Synergy Software).
Data deposition
Atomic coordinates and diffraction data have been deposited in the Protein Data Bank. (Accession code 2O42).
Acknowledgments
This work was supported by the National Institutes of Health Grant NIH RO1-AI37113 to T.M.H. We also acknowledge David King for providing the mass spectrometry data, Susan Marqusee for generously allowing the use of her CD, and James Holton and his staff for the excellent conditions at beamline 8.3.1. G.M. holds a Howard Hughes Medical Institute International Scholarship. A.E.D. thanks Emmanuel Skordalakes for all his sage advice and council on protein crystallization.
Footnotes
Reprint requests to: Tracy M. Handel, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California–San Diego, 9500 Gilman Drive MC 0684, La Jolla, CA 92093, USA; e-mail: thandel@ucsd.edu; fax: (858) 822-6655.
References
- Aoyagi M., Zhai, D., Jin, C., Aleshin, A.E., Stec, B., Reed, J.C., and Liddington, R.C. 2007. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 16**:** 118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N.A., Sept, D., Joseph, S., Holst, M.J., and McCammon, J.A. 2001. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 98**:** 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry M., Wasilenko, S.T., Stewart, T.L., and Taylor, J.M. 2004. Apoptosis regulator genes encoded by poxviruses. Prog. Mol. Subcell. Biol. 36**:** 19–37. [DOI] [PubMed] [Google Scholar]
- Benedict C.A., Norris, P.S., and Ware, C.F. 2002. To kill or be killed: Viral evasion of apoptosis. Nat. Immunol. 3**:** 1013–1018. [DOI] [PubMed] [Google Scholar]
- Borner C. 2003. The Bcl-2 protein family: Sensors and checkpoints for life-or-death decisions. Mol. Immunol. 39**:** 615–647. [DOI] [PubMed] [Google Scholar]
- Boya P., Pauleau, A.L., Poncet, D., Gonzalez-Polo, R.A., Zamzami, N., and Kroemer, G. 2004. Viral proteins targeting mitochondria: Controlling cell death. Biochim. Biophys. Acta 1659**:** 178–189. [DOI] [PubMed] [Google Scholar]
- Clarke A. 1996. Analysis of ligand binding by enzymes. In Enzymology labfax (ed. P.C. Engel), pp. 199–221. BIOS Scientific, Oxford UK.
- Clem R.J., Cheng, E.H., Karp, C.L., Kirsch, D.G., Ueno, K., Takahashi, A., Kastan, M.B., Griffin, D.E., Earnshaw, W.C., Veliuona, M.A., et al. 1998. Modulation of cell death by Bcl-XL through caspase interaction. Proc. Natl. Acad. Sci. 95**:** 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, N 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50**:** 760–763. [DOI] [PubMed] [Google Scholar]
- Cuconati A. and White, E. 2002. Viral homologs of BCL-2: Role of apoptosis in the regulation of virus infection. Genes & Dev. 16**:** 2465–2478. [DOI] [PubMed] [Google Scholar]
- De Lano W. 2002. The PyMOL molecular graphics system. Delando Scientific, San Carlos, CA.
- Everett H. and McFadden, G. 2001. Viruses and apoptosis: Meddling with mitochondria. Virology 288**:** 1–7. [DOI] [PubMed] [Google Scholar]
- Everett H., Barry, M., Lee, S.F., Sun, X., Graham, K., Stone, J., Bleackley, R.C., and McFadden, G. 2000. M11L: A novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med. 191**:** 1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett H., Barry, M., Sun, X., Lee, S.F., Frantz, C., Berthiaume, L.G., McFadden, G., and Bleackley, R.C. 2002. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J. Exp. Med. 196**:** 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S.F., Ludwig, H., Holzapfel, J., Kvansakul, M., Chen, L., Huang, D.C., Sutter, G., Knese, M., and Hacker, G. 2006. Modified vaccinia virus Ankara protein F1L is a novel BH3-domain-binding protein and acts together with the early viral protein E3L to block virus-associated apoptosis. Cell Death Differ. 13**:** 109–118. [DOI] [PubMed] [Google Scholar]
- Gubser C., Hue, S., Kellam, P., and Smith, G.L. 2004. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 85**:** 105–117. [DOI] [PubMed] [Google Scholar]
- Holton J. and Alber, T. 2004. Automated protein crystal structure determination using ELVES. Proc. Natl. Acad. Sci. 101**:** 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Petros, A.M., Virgin, H.W., Fesik, S.W., and Olejniczak, E.T. 2002. Solution structure of a Bcl-2 homolog from Kaposi sarcoma virus. Proc. Natl. Acad. Sci. 99**:** 3428–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47**:** 110–119. [DOI] [PubMed] [Google Scholar]
- Kantardjieff K.A. and Rupp, B. 2003. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein–nucleic acid complex crystals. Protein Sci. 12**:** 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapust R.B. and Waugh, D.S. 1999. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8**:** 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelekar A., Chang, B.S., Harlan, J.E., Fesik, S.W., and Thompson, C.B. 1997. Bad is a BH3 domain-containing protein that forms an inactivating dimer with Bcl-XL. Mol. Cell. Biol. 17**:** 7040–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr P. and McFadden, G. 2002. Immune responses to myxoma virus. Viral Immunol. 15**:** 229–246. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J. 1996. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. D Biol. Crystallogr. 52**:** 842–857. [DOI] [PubMed] [Google Scholar]
- MacDowell A.A., Celestre, R.S., Howells, M., McKinney, W., Krupnick, J., Cambie, D., Domning, E.E., Duarte, R.M., Kelez, N., Plate, D.W., et al. 2004. Suite of three protein crystallography beamlines with single superconducting bend magnet as the source. J. Synchrotron Radiat. 11**:** 447–455. [DOI] [PubMed] [Google Scholar]
- Marsden V.S. and Strasser, A. 2003. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 21**:** 71–105. [DOI] [PubMed] [Google Scholar]
- Matthews B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33**:** 491–497. [DOI] [PubMed] [Google Scholar]
- Muchmore S.W., Sattler, M., Liang, H., Meadows, R.P., Harlan, J.E., Yoon, H.S., Nettesheim, D., Chang, B.S., Thompson, C.B., Wong, S.L., et al. 1996. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381**:** 335–341. [DOI] [PubMed] [Google Scholar]
- Ojala P.M., Yamamoto, K., Castanos-Velez, E., Biberfeld, P., Korsmeyer, S.J., and Makela, T.P. 2000. The apoptotic v–cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat. Cell Biol. 2**:** 819–825. [DOI] [PubMed] [Google Scholar]
- Opgenorth A., Graham, K., Nation, N., Strayer, D., and McFadden, G. 1992. Deletion analysis of two tandemly arranged virulence genes in myxoma virus, M11L and myxoma growth factor. J. Virol. 66**:** 4720–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottilie S., Diaz, J.-L., Horne, W., Chang, J., Wang, Y., Wilson, G., Chang, S., Weeks, S., Fritz, L.C., and Oltersdorf, T. 1997. Dimerization properties of human BAD. Identification of a BH-3 domain and analysis of its binding to mutant BCL-2 and BCL-XL proteins. J. Biol. Chem. 272**:** 30866–30872. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor, W., and Carter Jr, C.W. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Methods in enzymology, pp. 307–326. Academic Press, San Diego, CA. [DOI] [PubMed]
- Petros A.M., Nettesheim, D.G., Wang, Y., Olejniczak, E.T., Meadows, R.P., Mack, J., Swift, K., Matayoshi, E.D., Zhang, H., Thompson, C.B., et al. 2000. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 9**:** 2528–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros A.M., Medek, A., Nettesheim, D.G., Kim, D.H., Yoon, H.S., Swift, K., Matayoshi, E.D., Oltersdorf, T., and Fesik, S.W. 2001. Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. 98**:** 3012–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros A.M., Olejniczak, E.T., and Fesik, S.W. 2004. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 1644**:** 83–94. [DOI] [PubMed] [Google Scholar]
- Polster B.M., Pevsner, J., and Hardwick, J.M. 2004. Viral Bcl-2 homologs and their role in virus replication and associated diseases. Biochim. Biophys. Acta 1644**:** 211–227. [DOI] [PubMed] [Google Scholar]
- Postigo A., Cross, J.R., Downward, J., and Way, M. 2006. Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell Death Differ. 13**:** 1651–1662. [DOI] [PubMed] [Google Scholar]
- Prinz W.A., Aslund, F., Holmgren, A., and Beckwith, J. 1997. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272**:** 15661–15667. [DOI] [PubMed] [Google Scholar]
- Reed J.C., Jurgensmeier, J.M., and Matsuyama, S. 1998. Bcl-2 family proteins and mitochondria. Biochim. Biophys. Acta 1366**:** 127–137. [DOI] [PubMed] [Google Scholar]
- Sattler M., Liang, H., Nettesheim, D., Meadows, R.P., Harlan, J.E., Eberstadt, M., Yoon, H.S., Shuker, S.B., Chang, B.S., Minn, A.J., et al. 1997. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 275**:** 983–986. [DOI] [PubMed] [Google Scholar]
- Sharpe J.C., Arnoult, D., and Youle, R.J. 2004. Control of mitochondrial permeability by Bcl-2 family members. Biochim. Biophys. Acta 1644**:** 107–113. [DOI] [PubMed] [Google Scholar]
- Stewart T.L., Wasilenko, S.T., and Barry, M. 2005. Vaccinia virus F1L protein is a tail-anchored protein that functions at the mitochondria to inhibit apoptosis. J. Virol. 79**:** 1084–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J., Wang, G., Barrett, J.W., Irvine, T.S., Gao, X., and McFadden, G. 2006. Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of Bax at the mitochondria. J. Virol. 80**:** 1140–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M., Youle, R.J., and Tjandra, N. 2000. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell 103**:** 645–654. [DOI] [PubMed] [Google Scholar]
- Taylor J.M. and Barry, M. 2006. Near death experiences: Poxvirus regulation of apoptotic death. Virology 344**:** 139–150. [DOI] [PubMed] [Google Scholar]
- Taylor J.M., Quilty, D., Banadyga, L., and Barry, M. 2006. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J. Biol. Chem. 281**:** 39728–39739. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. 2004. SOLVE and RESOLVE: Automated structure solution, density modification and model building. J. Synchrotron Radiat. 11**:** 49–52. [DOI] [PubMed] [Google Scholar]
- Van Duyne G.D., Standaert, R.F., Karplus, P.A., Schreiber, S.L., and Clardy, J. 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229**:** 105–124. [DOI] [PubMed] [Google Scholar]
- Wang G., Barrett, J.W., Nazarian, S.H., Everett, H., Gao, X., Bleackley, C., Colwill, K., Moran, M.F., and McFadden, G. 2004. Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J. Virol. 78**:** 7097–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilenko S.T., Stewart, T.L., Meyers, A.F., and Barry, M. 2003. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. 100**:** 14345–14350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilenko S.T., Banadyga, L., Bond, D., and Barry, M. 2005. The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. J. Virol. 79**:** 14031–14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.D., Isupov, M.N., and Murshudov, G.N. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57**:** 122–133. [DOI] [PubMed] [Google Scholar]
- Zhang H., Nimmer, P., Rosenberg, S.H., Ng, S.C., and Joseph, M. 2002. Development of a high-throughput fluorescence polarization assay for Bcl-x(L). Anal. Biochem. 307**:** 70–75. [DOI] [PubMed] [Google Scholar]