β-Catenin Is Dispensable for Hematopoiesis and Lymphopoiesis (original) (raw)
Abstract
β-catenin–mediated Wnt signaling has been suggested to be critically involved in hematopoietic stem cell maintenance and development of T and B cells in the immune system. Unexpectedly, here we report that inducible Cre-loxP–mediated inactivation of the β-catenin gene in bone marrow progenitors does not impair their ability to self-renew and reconstitute all hematopoietic lineages (myeloid, erythroid, and lymphoid), even in competitive mixed chimeras. In addition, both thymocyte survival and antigen-induced proliferation of peripheral T cells is β-catenin independent. In contrast to earlier reports, these data exclude an essential role for β-catenin during hematopoiesis and lymphopoiesis.
Keywords: Wnt signaling, T cells, B cells, development, gene targeting
Introduction
All mature blood cells are derived from multipotent hematopoietic stem cells (HSCs) present in the BM (1). HSCs have the capacity to self-renew to maintain hematopoiesis throughout life. Both self-renewal capacity and multipotency are lost upon differentiation to committed progenitors of lymphoid, erythroid, and myeloid lineages. Stem cell maintenance and cell fate decisions of the hematopoietic system are controlled by a handful of highly conserved developmentally regulated signal transduction pathways. One of these is the Wnt cascade, which has been shown to control early lymphopoiesis (2) and more recently was suggested to play a crucial role in self-renewal of HSCs (3, 4). Wnts are secreted glycoproteins that mediate cell to cell communication during development. They bind to the family of Frizzled receptors (5) in a complex with the low density lipoprotein receptor–related protein 5 and 6 (6–8). Activation of these receptor complexes results in stabilization and accumulation of a protein called β-catenin (9). In the absence of Wnt signaling, β-catenin is retained in the cytoplasm in a multiprotein complex comprising the tumor suppressor adenomatous polyposis coli (APC), the scaffold protein Axin (10, 11), and GSK-3β. In this complex β-catenin is phosphorylated by GSK-3β at its NH2 terminus (12) and thereby tagged for ubiquitination and proteosomal degradation (13). Upon activation of the Wnt pathway GSK3-β is inhibited by Dishevelled (14–16) and β-catenin can no longer be phosphorylated at its NH2 terminus. Unphosphorylated β-catenin is more stable and accumulates in the cytoplasm and translocates to the nucleus where it binds transcription factors of the TCF/LEF family thereby activating transcription of downstream target genes.
Evidence that Wnt proteins influence hematopoiesis and lymphopoiesis comes from both gain and loss of function approaches. Feeder or stromal cells overexpressing Wnt proteins induce proliferation of cocultured human CD34+ HSCs (17) and mouse pro–B cells (18), whereas purified Wnt3A promotes the self-renewal of HSCs (4). Moreover retroviral expression of Wnt1 and Wnt4 in fetal thymocytes results in increased cell numbers in suspension culture (19). In reciprocal experiments, Wnt1 and Wnt4 gene-targeted mice show decreased thymus cellularity due to reduced proliferation of immature thymocytes (20). In addition, retroviral expression of the extracellular Wnt-binding domain of Frizzled receptors acts in a dominant-negative fashion to inhibit fetal liver stem cells from developing into T cells (19). Taken together, these data show that Wnt proteins can modulate proliferation of hematopoietic cells and suggest that they may also influence T cell differentiation.
Recent retroviral overexpression studies suggest that β-catenin plays a key role in HSC homeostasis (3). Thus, overexpression of a dominant active form of β-catenin (lacking the NH2-terminal phosphorylation domain) in HSCs led to enhanced self-renewal capacity in vitro with preservation of stem cell markers. HSCs expressing this construct also had increased reconstitution potential in vivo compared with control HSCs. In contrast, HSCs transduced with Axin, which negatively regulates β-catenin by enhancing its degradation, had reduced growth potential in vitro as well as a drastic reduction in the ability to reconstitute the hematopoietic compartment of irradiated mice. The reciprocal outcome of these experiments led the authors to suggest that β-catenin–mediated Wnt signaling is critical for normal HSC homeostasis.
Additional data supporting a role for Wnt signaling in early lymphopoiesis comes from TCF1 and LEF1 gene-targeted mice. T lineage cells express both TCF1 and LEF1, whereas developing B cells express only LEF1. TCF1 gene-targeted mice have a progressive impairment in thymocyte proliferation and differentiation with increasing age, whereas fetal thymocyte development appears normal (21–23). LEF1-deficient mice show multiple nonhematopoietic defects during organogenesis and die shortly after birth (24). The lymphoid compartment of these mice exhibits normal T cell development but reduced proliferation and increased apoptosis of pro–B cells (18). Interestingly TCF1/LEF1 double knockout mice have a much more severe defect in T cell development compared with TCF1−/− mice, suggesting that TCF1 and LEF1 have partially redundant functions in the thymus (23).
Although these data clearly show that components of the Wnt signaling pathway can influence hematopoiesis and lymphopoiesis, the presumed critical role of β-catenin in this process has not been tested directly under physiological conditions. Studying the role of β-catenin in hematopoiesis by a conventional loss of function approach is hampered by the fact that β-catenin gene-targeted mice are early embryonic lethal (25, 26). To circumvent this problem an inducible Cre-_loxP_–mediated gene targeting approach was used. Surprisingly, inactivation of β-catenin in BM progenitors does not lead to any detectable perturbation in hematopoiesis or lymphopoiesis.
Materials and Methods
Activation of the Cre Recombinase.
Adult mice received five i.p. injections of 250 μg polyI-polyC (pI-pC; Sigma-Aldrich) at 2-d intervals. 2 d after the last injection, mice were killed and genomic DNA was prepared from BM cells using standard protocols. The deletion efficiency was assessed by Southern blot analysis of EcoRI-digested genomic DNA hybridized with a 7,000-bp EcoRI probe (see Fig. 1; reference 27) and quantified using a PhosphorImager (BAS-1000; Fuji).
Figure 1.

Inducible targeting of the β-catenin gene in the BM by the Mx-Cre recombinase. (a) Schematic representation of the β-catenin protein containing an NH2-terminal region (N-term), 12 Armadillo repeats, and a carboxy terminus harboring the transactivation domain (C-term). The genomic organization of the β-cateninlox/lox locus is shown with cylinders indicating coding exons and small black rectangles indicating noncoding exons. Exons 2–6 are flanked by two loxP sequences (triangles). β-cateninlox/lox mice were crossed to mice carrying the IFN-α–inducible Mx-Cre transgene to inducibly inactivate β-catenin in the BM after pI-pC treatment. Arrows indicate approximate positions of the PCR primers used to verify the deletion of the floxed β-catenin locus. EcoRI restriction sites and the probe for Southern blot analysis are indicated. (b) Southern blot analysis of Eco-RI–digested genomic DNA from BM of WT, β-cateninlox/lox, or β-catenin−/− mice. The probe indicated in (a) reveals a 7-kb fragment for the WT allele, two fragments of 3.8 and 3.2 kb for the homozygously floxed β-catenin locus, and one 5.2-kb fragment for the β-catenin−/− allele after successful deletion of the loxP flanked gene segment. (c) Survival curve of β-cateninlox/lox and β-catenin−/− mice after injection of pI-pC as indicated by arrows. Death of β-catenin−/− mice starts 2 wk after the last injection. n = 8 for both β-cateninlox/lox (controls) and β-catenin−/−.
BM Chimeras.
CD45.1+ C57BL/6 female mice were purchased from The Jackson Laboratory. BM chimeras were prepared as previously described (29). In brief, lethally irradiated mice (1,000 rads 24 h before transfer) that had been treated i.p. 48 h previously with 100 μg anti-NK1.1 Monoclonal antibodies were reconstituted with 107 CD45.2+ β-cateninlox/lox or β-catenin−/− BM for straight chimeras, or with a 1:2 mixture (5 × 106:10 × 106) of CD45.1+ WT and either CD45.2+ β-cateninlox/lox or β-catenin−/− BM for mixed chimeras. Mice were maintained on antibiotic (Bactrim) containing water and long-term reconstitution of BM and lymphoid organs by donor-derived cells was analyzed 3–6 mo later.
5- and 6-Carboxyfluorescein Diacetate Succinimidyl Ester (CFSE) Staining.
Single cell suspensions were made from the spleens of mixed BM chimeras. Cells were filtered, centrifuged, and resuspended at 107/ml in PBS/0.1% BSA at 37°C. A total of 10 × 107 splenocytes were labeled with CFSE (Molecular Probes) at a final concentration of 5 μM and then incubated at 37°C for 10 min. At the end of the incubation period, the cells were immediately washed three times in cold PBS/0.1% BSA. A total of 10 × 107 cells were transferred i.v. into the tail vein. 20 μg staphylococcal enterotoxin B (SEB; Toxin Technology) was injected i.p. 1 d after cell transfer. 2 d after SEB injection the mice were killed and splenocytes were analyzed.
Immunoblot Analysis.
Total thymocytes derived from nonmixed BM chimeras of either control or β-catenin−/− mice were lysed in 50 μl lysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 1% Triton X-100, and 1 mM DTT containing a mixture of protease inhibitors) for 30 min on ice and debris was removed by centrifugation. 100 μg protein extracts were separated on polyacrylamide gels, transferred to nitrocellulose, and probed with a monoclonal antibody specific for the COOH terminus of the mouse β-catenin protein (BD Transduction Laboratories). Bound antibodies were detected with horseradish peroxidase–conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). To ensure that equal amounts of protein were loaded, the membrane was reprobed with a monoclonal antibody to α-tubulin (clone no. B-5-1-2; Sigma-Aldrich).
Monoclonal Antibodies and Flow Cytometry.
Single cell suspensions of lymphocytes from BM, thymus, and spleen were prepared and stained using standard protocols for FACS® analysis as previously described (29). Dead cells and debris were eliminated by gating on forward scatter (FSC) and side scatter (SSC). The following monoclonal antibody conjugates were purchased from eBioscience: CD117 (c-kit R, ACK2)-PE; CD127 (IL-7Rα chain, A7R34)-PE-Cy5; CD11b (M1/70)-PE-Cy5; Sca-1 (Ly-6A/E, D7)-PE and -PE-Cy5; Ter 119-PE and PE-Cy5; B220 (RA3-6B2)-PE-Cy5; and anti–TCRβ-PE and anti–IgM-PE. Anti-CD21 (7G6)-FITC, CD43 (S7)-FITC, CD41 (MWReg30)-FITC, and CD23 (B3B4)-PE were purchased from BD Biosciences. Gr-1 (Ly-6G, RB6-8C5)-FITC and -Alexa 647, Ter 119-FITC, B220 (RA3-6B2)-FITC, CD11b-FITC, CD4 (GK1.5)-FITC, PE and APC, CD8α (53.6.7)-FITC and Alexa 647, CD45.2 (ALI-4A2)-FITC, -PE, and -Alexa 647, CD161 (NK1.1, PK136)-FITC and PE, CD3ɛ (145-2C11)-FITC, CD45.1 (A20.1)-FITC, -PE, and -Alexa 647, and TCRVβ 8.1,2,3 (F23.1)-PE were purified from hybridoma supernatants and conjugated in this laboratory according to standard protocols. Alexa 647 conjugates were prepared using the appropriate Alexa protein labeling kits (Molecular Probes). APC and PE conjugates were prepared using kits purchased from Prozyme. Streptavidin-APC (Molecular Probes), streptavidin PE-Cy5 (eBioscience), and streptavidin-PE (Caltag) were used to reveal biotin conjugates. Four-color FACS® analysis (FITC, PE, PE-Cy5, and APC or Alexa 647) was performed using a FACSCalibur™ Flow Cytometer (Becton Dickinson) and data was analyzed using CELLQuest™ software (Becton Dickinson). FACS® sorting was performed using a FACStar™ flow cytometer (Becton Dickinson).
Tissue Culture and Analysis of Thymocyte Sensitivity to Glucocorticoids.
Cells were cultured in DMEM containing 10% FCS, 2 mM glutamine, 25 mM Hepes, 100 U/ml penicillin, and 100 μg/ml streptomycin. Thymocytes from mixed BM chimeras containing both β-catenin−/− (CD45.2+) and WT (CD45.1+) cells were incubated at 3 × 106 cells/ml in 24-well plates in medium alone or in medium supplemented with various concentrations (10−10–10−6 M) of dexamethasone (Sigma-Aldrich). Cells were collected 12 h after the addition of dexamethasone and stained with antibodies against CD45.2, CD4, and CD8. Dead cells were identified and gated out by 7AAD (BD Biosciences) staining. A fixed number of beads of 6 μm in diameter (microsphere standard from Bacteria Counting Kit for flow cytometry; Molecular Probes) was added to the samples before analysis by FACS®. A viable cell gate was established based on FSC and SSC and another gate was set on the beads that could be distinguished from cells by their different FSC and SSC. The number of viable cells was calculated by comparing the ratio of beads to viable cells in a given file of 2 × 105 events to a standard ratio of a sample of known cell and bead numbers.
Results
Experimental Strategy.
To directly address the role of β-catenin–mediated signaling during lymphocyte development and for hematopoiesis in general we inducibly inactivated the β-catenin gene in BM cells. Transgenic mice expressing the Cre recombinase under the control of the IFN-inducible Mx promoter (28) were crossed to mice in which essential portions of the β-catenin gene are flanked by loxP sequences (β-catenin lox/lox; Fig. 1 a; reference 27). β-catenin lox/lox (control) and β-catenin lox/lox and Mx-Cre mice were treated with the IFN-α inducer pI-pC five times at 2-d intervals. To assess the β-catenin deletion efficiency, genomic DNA of induced β-catenin−/− (hereafter β-catenin−/−) and control BM was isolated 2 d after the last injection and analyzed by Southern blot. The deletion efficiency in β-catenin−/− BM was close to 100% (Fig. 1 b), as expected from previous studies(29).
Mx-Cre–induced β-catenin Inactivation Results in Mortality.
Interestingly, during the time of pI-pC treatment we noted that the β-catenin−/− mice had already started to lose weight after the first injection, whereas the weight of the injected littermate control mice remained constant (unpublished data). All β-catenin−/− but none of the control mice died within 23 d after the first pI-pC injection (Fig. 1 c). Analysis of the hematopoietic compartment of a group of β-catenin−/− mice just before death did not reveal any severe anomalies (unpublished data), making the consequences of inactivation of β-catenin in BM cells an unlikely cause of death. Although induction of the Mx-Cre transgene is very efficient in the BM, its expression is not restricted to the BM compartment as efficient inactivation of floxed target genes is also observed in other tissues such as the liver and the gut, which may account for the mortality of the β-catenin−/− mice. However, because no obvious abnormalities were observed in these tissues the cause of death of β-catenin−/− mice is currently unknown.
β-catenin–deficient BM Precursors Can Reconstitute All Major Blood Lineages in BM Chimeras.
The early mortality of the β-catenin−/− mice precluded longitudinal analysis of the hematopoietic compartment. However, they survived long enough to obtain close to 100% deletion efficiency in BM precursor cells. Therefore, CD45.1+ WT lethally irradiated mice were injected with CD45.2+ control or β-catenin−/− BM to determine whether β-catenin–deficient BM precursors can reconstitute all major blood lineages. Interestingly, BM chimeras reconstituted with β-catenin–deficient BM survived without showing any signs of hematopoietic failure. Control or β-catenin–deficient BM chimeras were analyzed 4 and 6 mo after transplantation. Surprisingly, all major blood lineages in the BM such as granulocytes, macrophages, megakaryocytes, early erythroblasts, or B cells were generated from β-catenin–deficient BM at comparable levels to control BM (Fig. 2 a). Similarly all major thymus subsets (Fig. 2 b) as well as mature T and B cells in the spleen (Fig. 2 c) were generated normally from β-catenin−/− BM progenitors.
Figure 2.

β-catenin–deficient BM reconstitutes all major hematopoietic lineages. BM chimeras were analyzed 4–7 mo after reconstitution. (a) Absolute donor cell numbers for total BM and different subsets: early erythroblasts (Early Eryth., Ter119+), granulocytes (Gran., Gr1+), B cells (B220+), megakaryocytes (Mega., CD41+), and macrophages (Mac, Gr1− CD11b+) of either β-cateninlox/lox (open bars) or β-catenin−/− (shaded bars) mice. The bars represent mean ± SD values (n = 5). (b) Absolute donor cell numbers for total thymocytes and thymocyte subsets: CD4− CD8− (DN), CD4+ CD8+ (DP), CD4+ CD8− TCRβ+ (CD4), and CD8+ CD4− TCRβ+ (CD8). The bars represent mean ± SD values for β-cateninlox/lox (open bars) or β-catenin−/− (filled bars) mice, where n = 5 for both β-cateninlox/lox and β-catenin−/− mice. (c) Absolute donor cell numbers for total splenocytes, B cells (B220+), and T cells (CD3+). n = 5 for control and β-catenin−/− mice. (d) Southern blot analysis of Eco-RI–digested genomic DNA from donor thymocytes of control (β-cateninlox/lox) or β-catenin−/− BM chimeric mice (refer to Fig.1 a for details). (e) Immunoblot analysis of β-catenin protein expression in total donor thymocytes from control or β-catenin−/− BM chimeras. The blots were probed with an antiserum specific to the COOH terminus of the β-catenin protein. To verify that equal amounts of protein were loaded, the membrane was reprobed with a monoclonal antibody against α-tubulin (bottom).
To exclude the possibility that reconstitution of the BM chimeras is due to a few BM cells that have escaped deletion, Southern blot analysis and immunoblotting using an antibody against the carboxy-terminal portion of β-catenin was performed on total thymocytes from control and β-catenin−/− chimeras. The Southern blot shows the expected pattern for the inactivated β-catenin allele (Fig. 2 d), and β-catenin protein is only detected in thymocytes derived from control BM cells (Fig. 2 e). No full-length nor any truncated form of β-catenin is detected in thymocytes derived from β-catenin−/− BM cells (Fig. 2 e), confirming that the loss of function approach for β-catenin was successful and that hematopoiesis can occur in the absence of any detectable β-catenin protein.
Self-Renewal and Differentiation of β-catenin–deficient BM Precursors in a Competitive Situation.
Although the BM reconstitution suggests that all major blood lineages can in principle be generated from β-catenin–deficient progenitor cells, it does not address the question of the reconstitution efficiency. Thus, competitive mixed bone chimeras were set up in which lethally irradiated WT hosts (CD45.1+) were reconstituted with either CD45.2+ β-catenin−/− BM or control BM, each mixed at a 2:1 ratio with CD45.1+ WT BM. Chimeras were analyzed 3–5 mo later for hematopoietic progenitor cells, myeloid lineages, and lymphoid lineages derived from each donor population.
Surprisingly, the relative number of cells in phenotypically defined progenitor subsets including HSC (lin− CD117+ Sca1+), common myeloid progenitor (CMP; lin− CD117+ Sca1−), and common lymphoid progenitor (CLP; lin− CD117lo Sca1lo Il7Rα+) within donor-derived BM was similar in both control and β-catenin–deficient chimeras (Fig. 3 , a and b). Furthermore, no differences were observed in the percentages of more mature BM cell types (including early erythroblasts, granulocytes, and macrophages) in β-catenin−/− chimeras versus control (Fig. 3 c). The results indicate that β-catenin–deficient BM progenitors can self-renew and differentiate normally in the BM, even in a competitive situation.
Figure 3.

Normal development of β-catenin−/− hematopoietic lineages in a competitive situation. Mixed BM chimeric mice were analyzed 4–6 mo after reconstitution with a 1:2 mixture of WT (CD45.1+) and either β-cateninlox/lox or β-catenin−/− (CD45.2+) BM-derived populations. (a) A representative FACS® analysis of BM stained with anti-CD117 and anti-Sca1 gated on lin− CD45.2+ donor cells of either β-cateninlox/lox or β-catenin−/− mixed chimeras. Numbers indicate relative percentage of HSCs (CD117+ Sca1+) CLPs (CD117low Sca1low), and CMPs (CD117+ Sca1−) in each indicated region. (b) Percentage HSCs, CMPs, and CLPs gated on lin− CD45.2+ cells from either β-cateninlox/lox (open bars) or β-catenin−/− mixed chimeras (shaded bars). The bars represent mean ± SD (n = 8). (c) Percentage early erythroblasts (Ter119+), granulocytes (Gr1+), and macrophages (Gr1− CD11b+) after gating on CD45.2+ cells from either β-cateninlox/lox or β-catenin−/− mixed chimeras. The bars represent mean ± SD (n = 8).
Competitive Lymphocyte Development from β-catenin–deficient BM Precursors.
As defects in both B and (more severely) T cell development have been reported in LEF1 and TCF1 gene-targeted mice, respectively, these two lymphoid lineages were analyzed in more detail in mixed BM chimeras. Flow cytometric analysis of B cell subsets in the BM shows that the relative numbers of B220+ IgM+ and B220+ IgM− cells within the CD45.2+ donor population is similar between control and β-catenin–deficient chimeras (Fig. 4 a). Furthermore, the generation of the more immature B220+ CD43+ cells defined as the pro–B cell compartment (Hardy fraction A–C′) in mixed BM chimeras seems not to be perturbed by the absence of β-catenin (Fig. 4 b).
Figure 4.
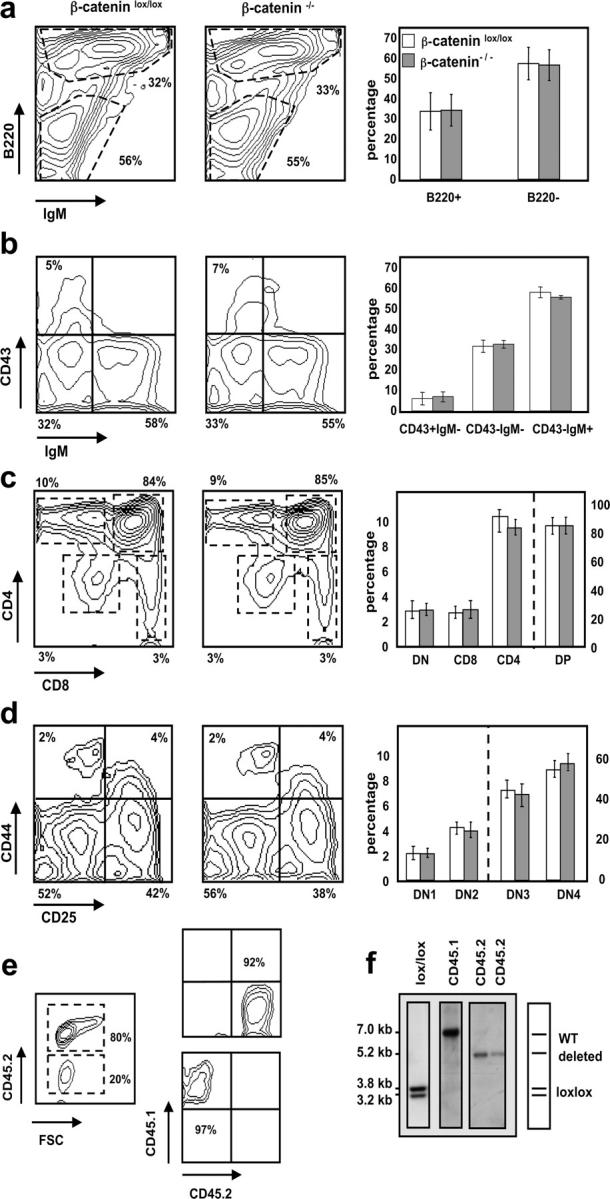
Normal development of β-catenin–deficient B and T cells in mixed BM chimeras. Representative FACS® analyses (left and middle panels) gated on CD45.2+ donor cells. Bar diagrams (right panels) indicate relative number ± SD of the different subsets where n = 12 for both β-cateninlox/lox (open bars) and β-catenin−/− (shaded bars) mice. (a) BM stained with anti-B220 and anti-IgM antibodies. (b) BM cells stained with anti-CD43 and anti-IgM antibodies after gating on B220+ cells. (c) Total thymocytes stained with anti-CD4 and anti-CD8 antibodies. DN (CD4− CD8−), DP (CD4+ CD8+), CD4 (CD4+ CD8− TCRβ+), and CD8 (CD4− CD8+ TCRβ+). (d) Thymocytes stained with anti-CD44 and anti-CD25 antibodies after gating on donor (CD45.2+)-derived lineage-negative cells. DN1 (CD44+ CD25−), DN2 (CD44+ CD25+), DN3 (CD44− CD25+), and DN4 (CD44− CD25−). (e) CD45.2+ (donor) and CD45.1+ (WT) cells derived from chimeric spleens were purified by FACS® sorting to verify deletion of the floxed β-catenin alleles after reconstitution. FACS® analysis before (left) and after the sort (right). Relative purity of the sorted populations are indicated. (f) Southern blot analysis of Eco-RI–digested genomic DNA from sorted CD45.1+ and CD45.2+ donor splenocytes. CD45.1+ cells were pooled from three different mice and CD45.2+ cells are derived from two individual mice.
T cell differentiation is progressively inhibited with increasing age in the absence of TCF1. TCF1−/− mice show multiple developmental blocks (21) at the double negative (DN)2 (CD44+ CD25+), DN4 (CD44− CD25−), and immature single positive (ISP; CD8+ TCR−) stages during thymocyte development. Interestingly, control and β-catenin−/− BM were able to reconstitute the thymus of mixed BM chimeras to a similar extent (the chimerism for control and β-catenin−/− CD45.2+ thymocytes was 62 ± 14% and 69 ± 19%, respectively). After gating on CD45.2+ cells, all major thymic subpopulations (DN, double positive [DP], CD4 single positive, and CD8 single positive) were present in similar percentages in both control and β-catenin−/− chimeras (Fig. 4 c). Similarly, the more immature thymocyte populations DN1–DN4 (defined by CD44 and CD25 expression) were generated in similar numbers in β-catenin−/− compared with control mixed BM chimeras (Fig. 4 d). None of the expected developmental blocks at DN2, DN4, or ISP stages were observed. To again rule out the possibility that reconstitution in the β-catenin−/− mixed BM chimeras is mediated by cells that have escaped deletion, CD45.2+ and CD45.1+ splenocytes of these mice were sorted and analyzed for the deletion efficiency of the β-catenin alleles. The purity of the sorted CD45.1+ and CD45.2+ cells was 97 and 92%, respectively (Fig. 4 e). Southern blot analysis shows the expected bands characterizing the WT and inactivated alleles for the sorted CD45.1+ and CD45.2+ cells, respectively (Fig. 4 f), confirming that T and B cells were indeed generated from β-catenin−/− BM progenitors in mixed BM chimeras. These results indicate that β-catenin–deficient B cells and thymocytes do not have a developmental disadvantage in a competitive situation in the BM or the thymus.
Because TCF1−/− thymocytes exhibit increased apoptosis probably due to reduced levels of Bcl-xL proteins (30), spontaneous and glucocorticoid-induced cell death of β-catenin–deficient and WT thymocytes were analyzed. Total thymocytes from β-catenin−/− and control BM chimeras were cultured at 37°C in the presence or absence of different concentrations of dexamethasone. The rate of spontaneous apoptosis as well as glucocorticoid-mediated death of DP thymocytes was similar between β-catenin−/− and WT chimeras (Fig. 5) .
Figure 5.

Absence of β-catenin does not affect spontaneous or glucocorticoid-induced death of thymocytes. (a) Percentage of surviving thymocytes gated on CD45.1+ (WT donor) or CD45.2+ (β-catenin−/− donor) derived from mixed BM chimeras after 12 h of culture at 37°C. The percentage of surviving cells is normalized to control cells maintained at 4°C. Data represent mean ± SD of five experiments. (b) Glucocorticoid-induced cell death in WT (CD45.1+) and β-catenin−/− (CD45.2+) CD4+ CD8+ DP thymocytes after 12 h of culture at 37°C. Data are normalized to untreated controls and represent mean ± SD from five individual experiments.
Antigen-induced Proliferation of β-catenin−/− Peripheral T Cells.
Because β-catenin–mediated Wnt signaling influences proliferation of many different cell types, we wanted to test whether the absence of β-catenin in peripheral T cells influences their ability to proliferate in response to antigen. For this purpose peripheral T cells were isolated from either control or β-catenin−/− mixed BM chimeras, labeled with the fluorescent dye CSFE, and then transferred into recipient mice. 24 h after transfer of the labeled cells the mice were injected with SEB and proliferation of the antigen-specific T cells was monitored 2 d after antigen challenge. Transferring a mixed population of CD45.1+ WT and CD45.2+ β-catenin−/− T cells derived from mixed BM chimeras permits a direct comparison of antigen-induced proliferation of CD8+ and CD4+ T cells within the same mouse. As shown in Fig. 6 , antigen-specific proliferation of CD8+ Vβ8+ and CD4+ Vβ8+ cells was indistinguishable between CD45.1+ WT and CD45.2+ β-catenin–deficient populations. The results indicate that β-catenin is also dispensable for antigen-induced T cell proliferation.
Figure 6.

Loss of β-catenin does not perturb antigen-induced T cell proliferation. CFSE-labeled splenocytes isolated from mixed BM chimeras were transferred i.v. into WT mice. The next day mice were injected i.p. with 20 μg SEB. 2 d later the draining popliteal LNs were removed and analyzed by FACS®. After gating on CD45.1+ (WT donor) or CD45.2+ (β-catenin−/− donor), the number of cell divisions in Vβ8− (negative control), CD8+ Vβ8+, and CD4+ Vβ8+ populations are shown. One representative experiment out of four is shown.
Discussion
To investigate the potential role of β-catenin in hematopoiesis we made use of floxed β-catenin mice (27) that express the Cre recombinase under the IFN-inducible Mx promoter (28). Unexpectedly, β-catenin–deficient BM progenitors were able to generate all myeloid and erythroid lineages in BM chimeras. Most surprisingly, T and B cell development appeared normal in the absence of β-catenin, suggesting that the protein is dispensable for lymphopoiesis as well as for hematopoiesis. Loss of function of the β-catenin gene was directly confirmed by the absence of β-catenin protein in donor thymocytes of β-catenin–deficient BM chimeras. Even when β-catenin−/− BM progenitor cells were placed in a competitive situation by generating mixed BM chimeras, they did not exhibit any detectable developmental disadvantage.
Our data are in apparent contrast to previous studies implicating β-catenin as a critical protein in HSC homeostasis (3) and in T and B cell development (for review see reference 2). The lack of any detectable phenotype in the hematopoietic compartment of β-catenin−/− mice is not due to rare cells that may have escaped deletion of the floxed β-catenin gene because donor-reconstituted hematopoietic cells in BM chimeras showed the expected pattern of deleted alleles. Although our floxed gene targeting strategy could in theory allow the production of a NH2-terminal truncated β-catenin protein, we believe this to be highly unlikely for several reasons. First, no truncated form of β-catenin was detected by Western blot in β-catenin−/− hematopoietic cells. Furthermore, inactivation of the same floxed β-catenin allele by other tissue-specific Cre recombinases (such as Wnt1-Cre, K19-Cre, or Msx2-Cre) results not only in the lack of any detectable β-catenin protein, but also in severe developmental phenotypes. Thus, inactivation of β-catenin by the Wnt1-Cre transgene results in the absence of part of the midbrain and all of the cerebellum, in the lack of generation of melanoblasts and neurogenin 2–dependent sensory neurons, as well as craniofacial defects (27, 31). K19-Cre–mediated inactivation of β-catenin in the definitive endoderm results in the formation of multiple hearts during embryogenesis (32) and conditional removal of β-catenin by Msx-2-Cre in ventral ectodermal cells as well as in the apical ectodermal ridge of the developing limb leads to severe limb defects (33). These earlier reports together with the rapid mortality reported here for mice in which β-catenin is inducibly inactivated by the Mx-Cre transgene provide compelling evidence that successful deletion of this floxed β-catenin gene segment results in a true loss of function allele.
Why then does inactivation of β-catenin in hematopoietic progenitors not lead to the expected phenotypes? The recently proposed role for β-catenin in HSC homeostasis (3) is based on studies using enriched Bcl-2 transgenic HSCs transduced with retroviruses overexpressing either a dominant active form of β-catenin or axin, a known inhibitor of the Wnt signaling pathway. Enforced expression of β-catenin results in an increased expansion of the HSC pool in vitro as well as in a better reconstitution efficiency in vivo. Overexpression of axin in HSCs leads to a reduction in both HSC growth and reconstitution efficiency in vivo (3). This is in contrast to our results with β-catenin–deficient BM progenitors that are able to self-renew and reconstitute the hematopoietic compartment of lethally irradiated mice as efficiently as WT cells in mixed BM chimeras. In attempting to reconcile these apparent discrepancies it should be noted that overexpression of β-catenin or axin in HSCs may activate (or repress) signaling pathways and target genes that are normally not controlled by β-catenin–mediated Wnt signaling under physiological conditions. Similar arguments apply to the recently described effects of constitutive β-catenin expression on early thymocyte differentiation (34).
The best documented role of β-catenin–mediated Wnt signaling in the hematopoietic system is deduced from gene targeting experiments of the two most downstream components of the Wnt pathway, TCF1 and LEF1. These proteins are HMG-box transcription factors that function as repressors via their association with transcriptional corepressors of the Groucho family (35) in the absence of Wnt signaling. TCF1-deficient mice show a progressive age-dependent impairment of thymocyte differentiation at multiple developmental stages. In particular, the highly proliferating immature thymocyte subsets such as DN2 (CD44+ CD25+), DN4 (CD44− CD25−), and ISP (CD8+ TCRβ−) are affected by the loss of TCF1 (21), and DP (CD4+ CD8+) thymocytes undergo increased apoptosis (30). In addition, adult TCF1−/− BM cells fail to reconstitute the T cell compartment of lethally irradiated WT hosts, whereas the development of all other blood lineages appears to be normal (21). LEF1−/− mice exhibit multiple organogenic defects during development including lack of pelage hair, whiskers, teeth, and functional mammary glands (24). Proliferation of LEF1−/− pro–B cells is reduced and an increased proportion of these cells undergo apoptosis (18). TCF1/LEF1 double knockout mice display a more severe defect in T cell development that is characterized by a complete block at the ISP stage and additional defects in the DN thymocyte subsets. Transfer of TCF1−/−/LEF1−/− fetal liver cells into lethally irradiated hosts fails to reconstitute the thymus (23, 36). From these studies one would expect that β-catenin–deficient BM progenitor cells should not be able to reconstitute the T cell compartment of BM chimeras, or at least should display severe defects during lymphocyte development. However, β-catenin−/− BM progenitors reconstitute the T and B cell compartments of mixed and nonmixed BM chimeras without obvious developmental defects, indicating that β-catenin–mediated Wnt signaling is dispensable for lymphopoiesis.
Another protein that can interact with transcription factors of the TCF/LEF1 family is plakoglobin, a close relative of β-catenin. Plakoglobin shares multiple structural features with β-catenin such as the ability to bind to E-cadherin, APC, and Axin, as well as possessing putative NH2-terminal GSK3β phosphorylation sites and a strong COOH-terminal transactivation domain (37). Despite these similarities the question of whether plakoglobin is involved in Wnt signaling in vivo is very controversial. Under certain experimental conditions plakoglobin has been shown to have similar functions to β-catenin. For example, plakoglobin can activate reporter constructs driven by promoters containing TCF/LEF binding sites when overexpressed in cell culture (for review see reference 37). Injection of Xenopus embryos with either β-catenin or plakoglobin mRNA mimics the phenotype obtained by injection of Wnt1 mRNA (38), suggesting that plakoglobin can transduce Wnt signals. However, later studies have shown that cells expressing high levels of exogenous plakoglobin have increased levels of endogenous β-catenin, suggesting that the effects mediated by plakoglobin overexpression are a consequence of up-regulating the pool of signaling competent β-catenin (39). Importantly, plakoglobin cannot compensate for β-catenin function during embryogenesis as conventional and tissue-specific inactivation of β-catenin results in early embryonic death or severe abnormalities in the targeted tissues (25, 27, 31–33, 40). Furthermore, expression of β-catenin and plakoglobin under the same skin-specific promoter (keratin 14 promoter) results in completely different phenotypes (41, 42). In light of our current knowledge we cannot formally exclude the possibility that plakoglobin, which is present at normal levels in β-catenin−/− thymocytes (unpublished data), might be able to compensate for β-catenin in hematopoietic cells and thus restore Wnt signaling. Nevertheless, such a scenario would most likely represent a particularity of the hematopoietic system because β-catenin deficiency results in severe phenotypes in many other tissues despite the fact that plakoglobin is ubiquitously expressed.
Even if plakoglobin cannot substitute for β-catenin, it remains possible that Wnt signaling in the hematopoietic compartment might be mediated by the so-called noncanonical (β-catenin–independent) pathway (for review see reference 43). The mechanisms by which Wnts transduce signals in a β-catenin–independent manner are not fully understood. However, Ca2+ signaling, activation of heterotrimeric G proteins, or the JNK pathway have been shown to be involved. Whether such noncanonical Wnt pathways are implicated in regulating hematopoiesis and/or lymphopoiesis in β-catenin–deficient mice remains to be investigated.
If, on the other hand, Wnt signaling is truly abolished in the absence of β-catenin, other Wnt-independent functions of TCF1 and LEF1 during lymphopoiesis must be considered. In this scenario the functions of TCF and LEF1 would differ between hematopoietic cells and epithelial cells. In epithelial cells they would mediate Wnt-induced signaling via the canonical β-catenin–dependent pathway, whereas in hematopoietic cells TCF1 and LEF1 would rather exert β-catenin–independent functions such as (but not restricted to) spatial organization of T cell–specific enhancers (44) or transcriptional repression through their interaction with Groucho proteins (35). Based on the phenotypes of TCF1- and LEF1-deficient mice, these putative novel functions of TCF1 and LEF1 would presumably intersect preferentially with pathways that are important for lymphocyte proliferation and/or survival. The fact that transgenic expression of Bcl-2 in thymocytes partially rescues TCF1 deficiency (30) is consistent with such a model.
In conclusion, and in contrast to prevailing views, our data indicate that β-catenin is dispensable for hematopoiesis and lymphopoiesis under physiological conditions. Nevertheless, they in no way preclude the possibility that the Wnt signaling pathway might be manipulated to achieve therapeutic goals in the hematopoietic system, as suggested by others (3, 4, 45).
Acknowledgments
We thank Pierre Zaech for cell sorting, Rosemary Lees for providing the SEB superantigen, and Alain Hirschy for providing the anti–β-catenin antibody.
This work was supported in part by the Swiss National Science Foundation (Foerderungsprofessur to F. Radtke), the Swiss Cancer League, the Leenaards Foundation, and the EMBO Young Investigator Program.
Abbreviations used in this paper: APC, adenomatous polyposis coli; CFSE, 5- and 6-carboxyfluorescein diacetate succinimidyl ester; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; DN, double negative; DP, double positive; FSC, forward scatter; HSC, hematopoietic stem cell; ISP, immature single positive; pI-pC, polyI-polyC; SEB, staphylococcal enterotoxin B; SSC, side scatter.
References
- 1.Morrison, S.J., N. Uchida, and I.L. Weissman. 1995. The biology of hematopoietic stem cells. Annu. Rev. Cell Dev. Biol. 11:35–71. [DOI] [PubMed] [Google Scholar]
- 2.van de Wetering, M., W. de Lau, and H. Clevers. 2002. WNT signaling and lymphocyte development. Cell. 109:S13–S19. [DOI] [PubMed] [Google Scholar]
- 3.Reya, T., A.W. Duncan, L. Ailles, J. Domen, D.C. Scherer, K. Willert, L. Hintz, R. Nusse, and I.L. Weissman. 2003. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 423:409–414. [DOI] [PubMed] [Google Scholar]
- 4.Willert, K., J.D. Brown, E. Danenberg, A.W. Duncan, I.L. Weissman, T. Reya, J.R. Yates III, and R. Nusse. 2003. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 423:448–452. [DOI] [PubMed] [Google Scholar]
- 5.Bhanot, P., M. Brink, C.H. Samos, J.C. Hsieh, Y. Wang, J.P. Macke, D. Andrew, J. Nathans, and R. Nusse. 1996. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 382:225–230. [DOI] [PubMed] [Google Scholar]
- 6.Tamai, K., M. Semenov, Y. Kato, R. Spokony, C. Liu, Y. Katsuyama, F. Hess, J.P. Saint-Jeannet, and X. He. 2000. LDL-receptor-related proteins in Wnt signal transduction. Nature. 407:530–535. [DOI] [PubMed] [Google Scholar]
- 7.Wehrli, M., S.T. Dougan, K. Caldwell, L. O'Keefe, S. Schwartz, D. Vaizel-Ohayon, E. Schejter, A. Tomlinson, and S. DiNardo. 2000. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 407:527–530. [DOI] [PubMed] [Google Scholar]
- 8.Pinson, K.I., J. Brennan, S. Monkley, B.J. Avery, and W.C. Skarnes. 2000. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 407:535–538. [DOI] [PubMed] [Google Scholar]
- 9.Behrens, J., J.P. von Kries, M. Kuhl, L. Bruhn, D. Wedlich, R. Grosschedl, and W. Birchmeier. 1996. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 382:638–642. [DOI] [PubMed] [Google Scholar]
- 10.Ikeda, S., S. Kishida, H. Yamamoto, H. Murai, S. Koyama, and A. Kikuchi. 1998. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17:1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kishida, S., H. Yamamoto, S. Ikeda, M. Kishida, I. Sakamoto, S. Koyama, and A. Kikuchi. 1998. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta-catenin. J. Biol. Chem. 273:10823–10826. [DOI] [PubMed] [Google Scholar]
- 12.Behrens, J., B.A. Jerchow, M. Wurtele, J. Grimm, C. Asbrand, R. Wirtz, M. Kuhl, D. Wedlich, and W. Birchmeier. 1998. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 280:596–599. [DOI] [PubMed] [Google Scholar]
- 13.Aberle, H., A. Bauer, J. Stappert, A. Kispert, and R. Kemler. 1997. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16:3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noordermeer, J., J. Klingensmith, N. Perrimon, and R. Nusse. 1994. Dishevelled and armadillo act in the wingless signalling pathway in Drosophila. Nature. 367:80–83. [DOI] [PubMed] [Google Scholar]
- 15.Kishida, S., H. Yamamoto, S. Hino, S. Ikeda, M. Kishida, and A. Kikuchi. 1999. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol. Cell. Biol. 19:4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smalley, M.J., E. Sara, H. Paterson, S. Naylor, D. Cook, H. Jayatilake, L.G. Fryer, L. Hutchinson, M.J. Fry, and T.C. Dale. 1999. Interaction of axin and Dvl-2 proteins regulates Dvl-2-stimulated TCF-dependent transcription. EMBO J. 18:2823–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Den Berg, D.J., A.K. Sharma, E. Bruno, and R. Hoffman. 1998. Role of members of the Wnt gene family in human hematopoiesis. Blood. 92:3189–3202. [PubMed] [Google Scholar]
- 18.Reya, T., M. O'Riordan, R. Okamura, E. Devaney, K. Willert, R. Nusse, and R. Grosschedl. 2000. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity. 13:15–24. [DOI] [PubMed] [Google Scholar]
- 19.Staal, F.J., J. Meeldijk, P. Moerer, P. Jay, B.C. van de Weerdt, S. Vainio, G.P. Nolan, and H. Clevers. 2001. Wnt signaling is required for thymocyte development and activates Tcf-1 mediated transcription. Eur. J. Immunol. 31:285–293. [DOI] [PubMed] [Google Scholar]
- 20.Mulroy, T., J.A. McMahon, S.J. Burakoff, A.P. McMahon, and J. Sen. 2002. Wnt-1 and Wnt-4 regulate thymic cellularity. Eur. J. Immunol. 32:967–971. [DOI] [PubMed] [Google Scholar]
- 21.Schilham, M.W., A. Wilson, P. Moerer, B.J. Benaissa-Trouw, A. Cumano, and H.C. Clevers. 1998. Critical involvement of Tcf-1 in expansion of thymocytes. J. Immunol. 161:3984–3991. [PubMed] [Google Scholar]
- 22.Verbeek, S., D. Izon, F. Hofhuis, E. Robanus-Maandag, H. te Riele, M. van de Wetering, M. Oosterwegel, A. Wilson, H.R. MacDonald, and H. Clevers. 1995. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature. 374:70–74. [DOI] [PubMed] [Google Scholar]
- 23.Okamura, R.M., M. Sigvardsson, J. Galceran, S. Verbeek, H. Clevers, and R. Grosschedl. 1998. Redundant regulation of T cell differentiation and TCRalpha gene expression by the transcription factors LEF-1 and TCF-1. Immunity. 8:11–20. [DOI] [PubMed] [Google Scholar]
- 24.van Genderen, C., R.M. Okamura, I. Farinas, R.G. Quo, T.G. Parslow, L. Bruhn, and R. Grosschedl. 1994. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 8:2691–2703. [DOI] [PubMed] [Google Scholar]
- 25.Haegel, H., L. Larue, M. Ohsugi, L. Fedorov, K. Herrenknecht, and R. Kemler. 1995. Lack of beta-catenin affects mouse development at gastrulation. Development. 121:3529–3537. [DOI] [PubMed] [Google Scholar]
- 26.Huelsken, J., R. Vogel, V. Brinkmann, B. Erdmann, C. Birchmeier, and W. Birchmeier. 2000. Requirement for beta-catenin in anterior-posterior axis formation in mice. J. Cell Biol. 148:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brault, V., R. Moore, S. Kutsch, M. Ishibashi, D.H. Rowitch, A.P. McMahon, L. Sommer, O. Boussadia, and R. Kemler. 2001. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 128:1253–1264. [DOI] [PubMed] [Google Scholar]
- 28.Kuhn, R., F. Schwenk, M. Aguet, and K. Rajewsky. 1995. Inducible gene targeting in mice. Science. 269:1427–1429. [DOI] [PubMed] [Google Scholar]
- 29.Radtke, F., A. Wilson, G. Stark, M. Bauer, J. van Meerwijk, H.R. MacDonald, and M. Aguet. 1999. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 10:547–558. [DOI] [PubMed] [Google Scholar]
- 30.Ioannidis, V., F. Beermann, H. Clevers, and W. Held. 2001. The beta-catenin–TCF-1 pathway ensures CD4(+)CD8(+) thymocyte survival. Nat. Immunol. 2:691–697. [DOI] [PubMed] [Google Scholar]
- 31.Hari, L., V. Brault, M. Kleber, H.Y. Lee, F. Ille, R. Leimeroth, C. Paratore, U. Suter, R. Kemler, and L. Sommer. 2002. Lineage-specific requirements of beta-catenin in neural crest development. J. Cell Biol. 159:867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lickert, H., S. Kutsch, B. Kanzler, Y. Tamai, M.M. Taketo, and R. Kemler. 2002. Formation of multiple hearts in mice following deletion of beta-catenin in the embryonic endoderm. Dev. Cell. 3:171–181. [DOI] [PubMed] [Google Scholar]
- 33.Barrow, J.R., K.R. Thomas, O. Boussadia-Zahui, R. Moore, R. Kemler, M.R. Capecchi, and A.P. McMahon. 2003. Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 17:394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gounari, F., I. Aifantis, K. Khazaie, S. Hoeflinger, N. Harada, M.M. Taketo, and H. von Boehmer. 2001. Somatic activation of beta-catenin bypasses pre-TCR signaling and TCR selection in thymocyte development. Nat. Immunol. 2:863–869. [DOI] [PubMed] [Google Scholar]
- 35.Roose, J., M. Molenaar, J. Peterson, J. Hurenkamp, H. Brantjes, P. Moerer, M. van de Wetering, O. Destree, and H. Clevers. 1998. The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature. 395:608–612. [DOI] [PubMed] [Google Scholar]
- 36.Held, W., H. Clevers, and R. Grosschedl. 2003. Redundant functions of TCF-1 and LEF-1 during T and NK cell development, but unique role of TCF-1 for Ly49 NK cell receptor acquisition. Eur. J. Immunol. 33:1393–1398. [DOI] [PubMed] [Google Scholar]
- 37.Barker, N., and H. Clevers. 2000. Catenins, Wnt signaling and cancer. Bioessays. 22:961–965. [DOI] [PubMed] [Google Scholar]
- 38.Karnovsky, A., and M.W. Klymkowsky. 1995. Anterior axis duplication in Xenopus induced by the over-expression of the cadherin-binding protein plakoglobin. Proc. Natl. Acad. Sci. USA. 92:4522–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller, J.R., and R.T. Moon. 1997. Analysis of the signaling activities of localization mutants of beta-catenin during axis specification in Xenopus. J. Cell Biol. 139:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huelsken, J., R. Vogel, B. Erdmann, G. Cotsarelis, and W. Birchmeier. 2001. Beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 105:533–545. [DOI] [PubMed] [Google Scholar]
- 41.Gat, U., R. DasGupta, L. Degenstein, and E. Fuchs. 1998. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell. 95:605–614. [DOI] [PubMed] [Google Scholar]
- 42.Charpentier, E., R.M. Lavker, E. Acquista, and P. Cowin. 2000. Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J. Cell Biol. 149:503–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veeman, M., J. Axelrod, and R. Moon. 2003. A second canon. Functions and mechanisms of beta-catenin-independent Wnt. Dev. Cell. 5:367–377. [DOI] [PubMed] [Google Scholar]
- 44.Giese, K., J. Cox, and R. Grosschedl. 1992. The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell. 69:185–195. [DOI] [PubMed] [Google Scholar]
- 45.Eaves, C.J. 2003. Manipulating hematopoietic stem cell amplification with Wnt. Nat. Immunol. 4:511–512. [DOI] [PubMed] [Google Scholar]