CD8 T Cell Recognition of Endogenously Expressed Epstein-Barr Virus Nuclear Antigen 1 (original) (raw)
Abstract
The Epstein-Barr virus (EBV) nuclear antigen (EBNA)1 contains a glycine-alanine repeat (GAr) domain that appears to protect the antigen from proteasomal breakdown and, as measured in cytotoxicity assays, from major histocompatibility complex (MHC) class I–restricted presentation to CD8+ T cells. This led to the concept of EBNA1 as an immunologically silent protein that although unique in being expressed in all EBV malignancies, could not be exploited as a CD8 target. Here, using CD8+ T cell clones to native EBNA1 epitopes upstream and downstream of the GAr domain and assaying recognition by interferon γ release, we show that the EBNA1 naturally expressed in EBV-transformed lymphoblastoid cell lines (LCLs) is in fact presented to CD8+ T cells via a proteasome/peptide transporter–dependent pathway. Furthermore, LCL recognition by such CD8+ T cells, although slightly lower than seen with paired lines expressing a GAr-deleted EBNA1 protein, leads to strong and specific inhibition of LCL outgrowth in vitro. Endogenously expressed EBNA1 is therefore accessible to the MHC class I pathway despite GAr-mediated stabilization of the mature protein. We infer that EBNA1-specific CD8+ T cells do play a role in control of EBV infection in vivo and might be exploitable in the control of EBV+ malignancies.
Keywords: Epstein-Barr virus, cytotoxic T lymphocytes, antigen presentation, EBNA1
Introduction
CD8 T cells combat viral infection through recognition of peptides produced by proteasomal cleavage of endogenously expressed viral proteins and presented at the infected cell surface as a complex with MHC class I molecules (1). Although several viruses have evolved strategies that interfere with this presentation pathway at a postproteasomal stage (2), the best example of proteasomal targeting involves the EBV, a γ herpesvirus associated with several human tumors. This virus persists in vivo through establishing latent infection in B lymphocytes and in vitro can transform such cells into permanent lymphoblastoid cell lines (LCLs; 3). EBV encodes a nuclear antigen, EBV nuclear antigen (EBNA)1, which is essential for virus genome maintenance in latently infected cells and is a potentially important immunological target in being the only viral protein present in all EBV-associated malignancies. Interestingly, EBNA1 contains an internal glycine-alanine repeat (GAr) domain that is not required for genome maintenance or growth transformation (4), but is thought to have an immune evasion function. Thus, when the GAr domain was transferred into a known target antigen for CD8 T cells (in this case another EBV-latent cycle protein, EBNA3B), cells expressing the chimaeric antigen were not killed by EBNA3B-specific effectors in short-term chromium release assays (5). Subsequent work inserting GAr sequences into other indicator proteins and measuring protein stability by biochemical and immunofluorescence assays showed that GAr conferred resistance in cis to proteasomal digestion, albeit to degrees that depended upon the precise chimaeric construct (6–9). Although this effect appeared to provide an explanation for the earlier immunological findings, the relationship between GAr-mediated protection from proteasomal breakdown and the originally observed protection from CD8 T cell recognition has remained largely unexplored.
The first hint that EBNA1 might be an immunologically silent protein came from experiments in a mouse tumor rejection model where, unlike certain other EBV-latent proteins, EBNA1 failed to confer immunogenicity to a nonimmunogenic tumor line (10). Stronger support for this hypothesis came from work with human CD8+ T cell preparations reactivated in vitro from EBV-immune donors. Such cells never showed specific killing of targets expressing the native EBNA1 protein from a vaccinia vector, in contrast to the frequent detection of responses that mapped to other latent cycle antigens, in particular EBNAs 3A, 3B, and 3C (11, 12). However, once the effect of the GAr domain was known, screening in vitro–reactivated CD8+ T cell clones for lysis of vaccinia-infected targets expressing a GAr-deleted form of EBNA1 clearly showed that EBNA1-specific reactivities were present in some individuals and could be mapped to defined EBNA1 peptide epitopes presented in the context of particular HLA alleles. Nevertheless, such epitope-specific clones still did not kill targets expressing the full-length EBNA1 protein (13, 14). This implied that such responses to an antigen that in its endogenously expressed form appeared to be protected from T cell recognition, must arise through cross-priming by antigen-loaded dendritic cells in vivo. Furthermore, such responses were unlikely to be biologically effective in vivo because they would be incapable of recognizing naturally infected cells or indeed EBV+ tumors.
Here we cast doubt on these inferences by showing that when assayed by IFN-γ release, EBNA1 epitope–specific CD8+ T cells can indeed recognize LCLs endogenously expressing the native EBNA1 protein and, in coculture assays, such effectors specifically inhibit LCL outgrowth. Thus, GAr+ EBNA1 is not fully protected from the MHC class I presentation pathway and may yet prove to be an effective target for T cell–based tumor therapy.
Materials and Methods
Target Cell Lines.
LCLs were established using the reference EBV strains B95.8, Akata, or Sal (15), or the B95.8-based recombinant EBV strains d17 (GAr-deleted; 4), 2828 (EBNA1-deleted), or 2089 (control; 16). The transporter associated with antigen processing (TAP)-deficient T2:B35-LCL is as previously described (17). The EBV+ Burkitt lymphoma (BL)-derived Sal-BL, Chep-BL, and Akata-BL lines used in CD40 ligand induction experiments are as previously described (18). Although these lines show differential responses to antibody-mediated CD40 ligation, they are all strongly responsive to the more potent stimulus provided by CD40 ligand–expressing mouse L cells (19). All cell lines were maintained in RPMI 1640 medium supplemented with 2 mM glutamine and 10% fetal calf serum (standard medium). In LCL mixing experiments, the “donor” and “acceptor” LCLs were seeded together in a 1:1 ratio and the coculture was left undisturbed for 7 d.
CD8+ T Cell Clones.
The CD8+ T cell clones were generated from selected EBV-immune donors in equal number by in vitro reactivation of peripheral blood mononuclear cells with the autologous B95.8-transformed LCL or with the epitope peptide as previously described (14), followed by limiting dilution cloning of the polyclonal population in 30% MLA 144 supernatant and 100 IU/ml recombinant IL-2. Epitope-specific clones were identified by screening for IFN-γ and cytotoxic responses to peptide-loaded target cells, and their antigen specificity was confirmed in cytotoxicity assays on target cells expressing the relevant cognate antigen (GAr-deleted EBNA1 or EBNA3A) from vaccinia vectors, as previously described (14). Similar reactivities were generated by LCL or by peptide stimulation.
Cytotoxicity and IFN-γ Release Assays.
Cytotoxicity was assessed in standard 5-h chromium release assays at known effector/target ratios. IFN-γ release was measured by ELISA. In this assay, T cells were tested in triplicate microwell cultures against target cells cultured in Iscove's modified Dulbecco's medium (Life Technologies) containing 10% fetal calf serum and 25 IU/ml recombinant IL-2. Numbers of T cells and target cells used per well are indicated in the figure legends. In some experiments, target cells were prepulsed with 10−7 M epitope peptide followed by washing three times with RPMI. After overnight incubation, culture supernatants were harvested and IFN-γ was measured by ELISA using antibody reagents from Endogen.
Inhibition Experiments.
Target cells were washed with PBS and pellets were gently resuspended in citrate buffer phosphate (0.131 M citric acid, 0.066 M Na2HPO4), pH3, for 3 min on ice. After neutralization by washing with excess standard medium, target cells were incubated in standard medium for 2 h at 37°C with or without proteasome inhibitors at the indicated concentration. The cells were then washed, resuspended in standard medium, and then added as targets to appropriate CD8+ T cell effectors. To check the efficiency with which acid stripping had removed cell surface peptide, some cells were fixed immediately after acid stripping by incubating for 1 min with 0.05% glutaraldehyde followed by quenching with 0.2 M glycine and washing with PBS, and then resuspended in standard medium and used as targets as described above. To check the viability of cells exposed to proteasome inhibitors, some cells were removed immediately after the 2-h drug treatment and, along with untreated cells from the same acid-stripped culture, were washed and then loaded with exogenous epitope peptide (at a range of 100-fold dilutions from 10−6 to 10−14 M) before use as targets in the assay.
Outgrowth Assays.
Target cell lines were seeded into round-bottom microtest plate wells at one of a range of doubling dilutions from 2 × 104–313 cells/well, with duplicate wells at each seeding density. Before seeding, some target cells were prepulsed with 10−7 M epitope peptide followed by washing three times with RPMI. T cells were added at 104 cells/well to target cell cultures and as a control to wells without targets. All cultures were set up in standard medium without cytokine supplements and were refed weekly by a half change of medium. Outgrowth was scored visually at 4 wk and the B cell identity of the growing cultures was confirmed by monoclonal antibody staining for CD19.
Results
Effect of the GAr Domain on EBNA1 Processing: Cytotoxicity Assays.
This work used CD8+ effector T cell clones against the four EBNA1 epitopes shown in Fig. 1 A and referred to as HPV (B*3501-restricted), RPQ and IPQ (both B*0701-restricted), and VLK (A*0203-restricted). The EBNA1 specificity of these CD8+ clones was in each case confirmed in cytotoxicity assays against targets exogenously loaded with epitope peptide and targets infected with a vaccinia expressing GAr-deleted EBNA1 (14; not depicted). Target LCLs were generated from selected donors by transformation with the reference B95.8 EBV strain and with a recombinant B95.8 virus, dl7, carrying a GAr-deleted EBNA1 gene (4). Fig. 1 B shows an immunoblot of SDS-PAGE–separated proteins from two such LCL pairs, probed with the IH4 monoclonal antibody against the COOH-terminal region of EBNA1 downstream of the GAr domain. Both wild-type transformants express the standard 80-kD EBNA1 protein as does the reference B95.8 cell line itself, whereas the dl7 transformants express the 52-kD GAr-deleted protein. These LCLs are identical in their expression of the other EBV-latent cycle proteins (unpublished data).
Figure 1.
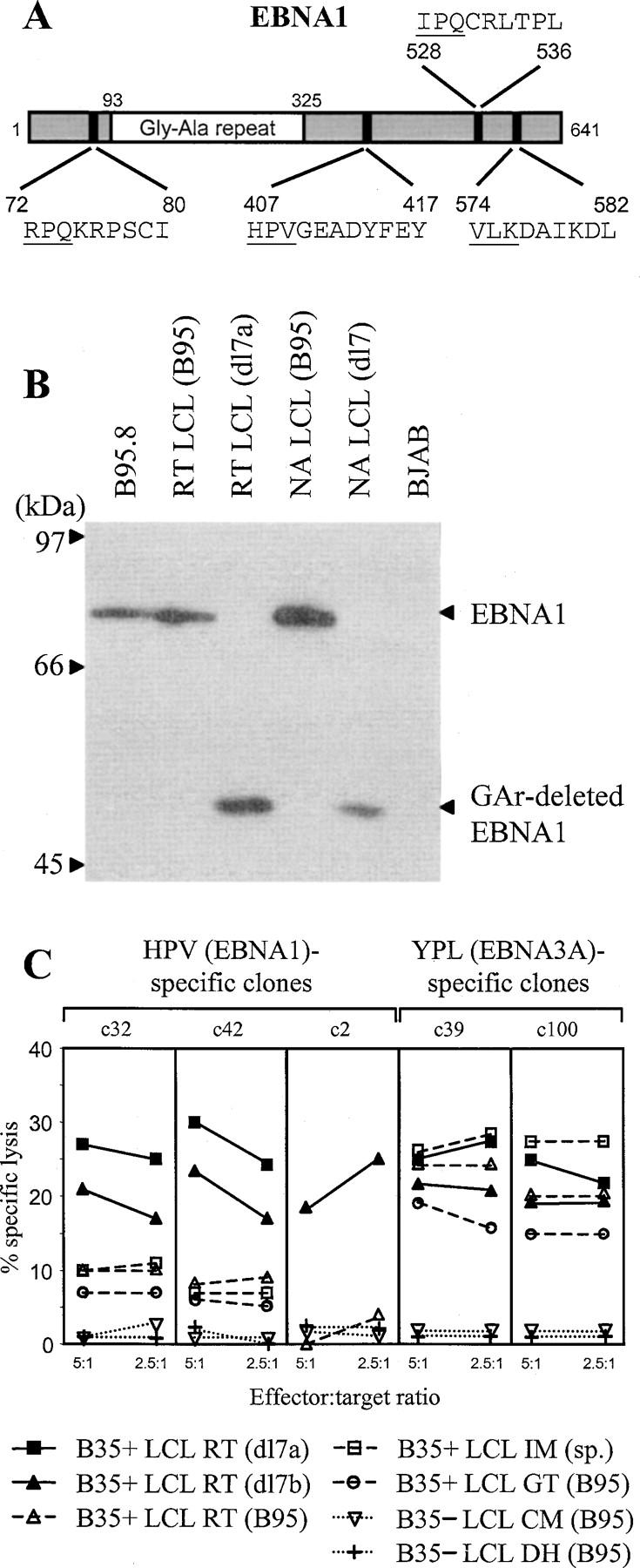
EBNA1-specific CD8+ T cell recognition of LCLs measured by cytotoxicity. (A) Schematic diagram of wild-type EBNA1 protein showing the sequence of CD8+ epitopes. Numbers refer to coordinates in the 641–amino acid sequence of the B95.8 strain EBNA1 protein. (B) Immunoblot of SDS-PAGE–separated protein extracts from the B95.8 cell line, B95.8 virus-transformed LCLs, RT (B95), and NA (B95), expressing wild-type EBNA1 and from the corresponding dl7 virus transformants, RT (dl7a) and NA (dl7), expressing GAr-deleted EBNA1. BJAB is an EBV− B lymphoma cell line. The blot is probed with the EBNA1-specific monoclonal antibody IH4. (C) Results of 5-h chromium release assays in which B*3501-restricted CD8+ T cell clones specific for the HPV (EBNA1) epitope or for the YPL (EBNA3A) epitope were used as effectors on a common panel of LCL targets. Targets included two dl7 virus transformants from B*3501+ donor RT, RT-LCL (dl7a), and RT-LCL (dl7b) expressing GAr-deleted EBNA1, three B*3501+ LCLs from donors RT, GT, and IM transformed either with B95.8 virus (for RT and GT) or by spontaneous outgrowth with the donor's own EBV strain (for IM), and two B95.8 LCLs from B*3501− donors CM and DH. Results are shown as percent-specific lysis at effector/target ratios of 5:1 and 2.5:1.
Recognition of such LCL target pairs by EBNA1-specific effectors was then tested in conventional 5-h chromium release assays. Fig. 1 C shows data from three CD8+ T cell clones, c32, c42, and c2, illustrating the overall range of results seen with effectors specific for the B*3501-restricted HPV epitope. These clones mediated 20–30% specific lysis of two LCLs, RT-dl7a and RT-dl7b, independently established from a B*3501+ donor and expressing the GAr-deleted form of EBNA1. By contrast, lysis of the paired RT-B95 line and other B*3501+ LCLs expressing wild-type EBNA1 was either indistinguishable from the very low backgrounds seen with HLA-mismatched LCLs or just above that background but in a range (5–10%) that is not usually considered significant in chromium release assays. Fig. 1 C also shows data from two representative CD8+ T cell clones, YPL c39 and c100, specific for a different B*3501-restricted epitope, YPLHEQHGM, derived from the EBNA3A protein. These effectors killed all B*3501+ LCLs at similar levels whether they carried wild-type virus or the dl7 recombinant, showing that the two types of LCLs were not inherently different in their susceptibility to T cell–mediated lysis.
Similar assays were conducted with CD8+ T cell clones to the two HLA-B*0701–restricted EBNA1 epitopes, RPQ and IPQ, for which HLA-matched dl7 and wild-type target lines were again available. Such clones showed only low level killing of the dl7 transformant and no specific lysis of a number of wild-type B*07.01+ LCLs. Likewise, CD8+ clones specific for the HLA-A*0203–restricted epitope VLK showed no detectable killing of A*0203+ wild-type LCLs (unpublished data).
Effect of the GAr Domain on EBNA1 Processing: IFN-γ Release Assays.
In earlier work, we described CD8+ T cell clones reactivated in vitro against EBV-latent cycle antigens other than EBNA1, which were clearly EBV epitope and antigen specific in their ability to kill target cells exogenously loaded with synthetic epitope peptide or overexpressing the relevant target antigen from a vaccinia vector, yet which failed to kill naturally infected LCL targets in conventional cytotoxicity assays (20). While studying the basis of this phenomenon, we found that such clones do show specific, HLA class I–restricted recognition of unmanipulated LCL targets when measured by ELISA for IFN-γ release (unpublished data). Fig. 2 A shows data obtained when three HPV-specific CD8+ clones, again representative of the complete range, were retested on similar target cell panels using this more sensitive assay. Now all clones showed significant recognition of three different wild-type B*3501+ LCLs at levels that though below those seen for the RT-LCL (dl7a) transformant, were clearly well above the background shown by HLA-mismatched LCL controls. We went on to compare the efficiency of dl7 and wild-type LCL recognition by representative EBNA1-specific clones in titration assays, varying the numbers of either effector or target cells added. Fig. 2 B shows data from such an experiment assaying two HPV-specific clones, c32 and c33, against the RT (dl7a) and RT (B95) LCL pair. Assaying different numbers of effectors against a standard number of targets (Fig. 2 B, left), levels of wild-type LCL recognition by the HPV-specific clones were consistently 40–70% of those seen against the dl7 transformant. A similar pattern was observed when assaying a standard number of HPV-specific effectors against different numbers of targets (Fig. 2 B, right). Interestingly, when we repeated these experiments using ELISPOT assays (21) as a read out, the different levels of response were apparent not in the numbers of T cells that responded but in the intensity of the IFN-γ produced per responding cell (not depicted). Fig. 2 B also shows the results obtained in the same titration assays with a CD8+ T cell clone (c98) specific for the YPL epitope. Like other YPL-specific effectors tested, this clone did not discriminate between the two types of target LCLs.
Figure 2.

EBNA1-specific CD8+ T cell recognition of LCLs measured by IFN-γ release. (A) Three CD8+ T cell clones specific for the HPV/B*3501 epitope were tested against a panel of B*3501+ and B*3501− LCL targets transformed either with wild-type B95.8 virus or with the dl7 strain expressing GAr-deleted EBNA1. T cells and LCL targets were both seeded at 104 cells/well. As controls, T cells alone were tested in the presence of 10−7 M HPV epitope peptide or with no peptide. (B) The efficiency of RT-LCL (B95) and RT-LCL (dl7a) target recognition was assessed in titration assays using two HPV (EBNA1)–specific CD8+ T cell clones and one YPL (EBNA3A)–specific clone. The effect of titrating numbers of effector and target cells are shown on the left and right, respectively. When titrating numbers of effectors, LCL targets were added at 104 cells/well, and when titrating numbers of targets, effectors were added at 5 × 103 cells/well. Results represent the mean values of IFN-γ release measured from triplicate cultures (+SD).
The greater sensitivity of IFN-γ release assays now allowed us to examine target cell recognition by CD8+ T cell clones to the other EBNA1 epitopes. As shown in Fig. 3 A, three clones specific for the B*0701-restricted RPQ epitope, though giving different absolute levels of response, all showed reproducible recognition of the wild-type LCL from a B*0701+ donor NA, again at levels around 50–70% of that seen against the paired dl7 transformant and clearly well above the background shown by HLA-mismatched LCLs. This assay also includes another pair of target LCLs established from a B*0701+ donor MD, one (MD-B95) transformed with recombinant virus containing the complete B95.8 genomic sequence, the other (MD-dE1) transformed with a recombinant from which the entire EBNA1 gene has been deleted, the genome maintenance function of EBNA1 having been rendered redundant by integration of the virus genome into host cell DNA (16). There was clear recognition of the wild-type but not the EBNA1-deleted LCL, further confirming that recognition is absolutely dependent upon EBNA1 being expressed in the target cells. Similar results to the above were observed with the one available CD8+ T cell clone specific for a second B*0701-restricted EBNA1 epitope, IPQ (unpublished data). Fig. 3 B shows data from three representative clones specific for the A*0203-restricted EBNA1 epitope, VLK. Though there were no dl7 transformants available as internal standards in this case, all three VLK-specific clones clearly recognized wild-type EBNA1-expressing LCLs from A*0203+ donors but not from HLA-mismatched donors, even from donors with other A*02 subtype alleles.
Figure 3.
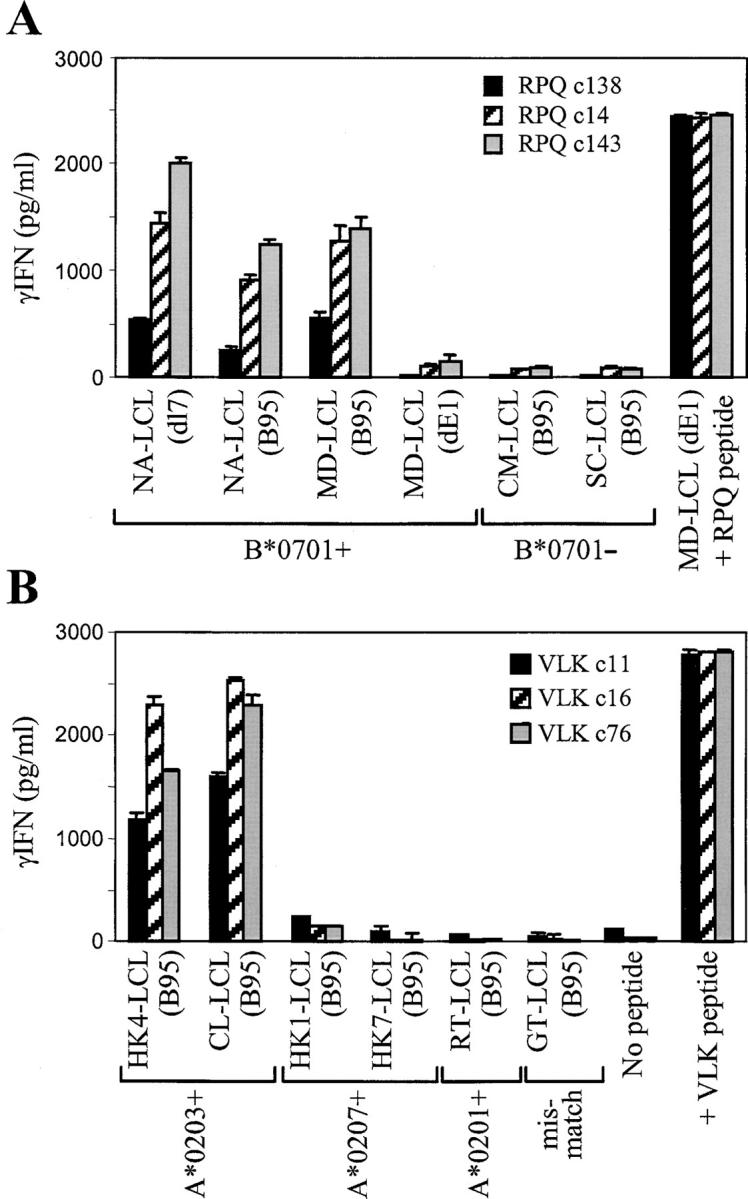
B*0701- and A*0203-restricted CD8+ T cell recognition of endogenously expressed EBNA1. (A) Three CD8+ T cell clones specific for the RPQ/B*0701 epitope were tested against a panel of B*0701+ and B*0701− LCL targets transformed with the wild-type EBV strain B95.8, against the B*0701+ NA-LCL (dl7) carrying an EBNA1 gene deleted for the GAr-coding sequence, or against the B*0701+ MD-LCL (dE1) carrying an EBV genome from which the EBNA1 gene has been completely deleted. As a positive control, T cells were tested against MD-LCL (dE1) cells that had been precoated with the target epitope peptide RPQ. (B) Three CD8+ T cell clones specific for the VLK/A*0203 epitope were tested against a panel of HLA-matched and -mismatched LCL targets transformed with the wild-type EBV strain B95.8. As a positive control, T cells were tested with 10−7 M VLK epitope peptide alone. T cells were tested at 104 cells/well and LCL targets were tested at 2 × 104 cells/well. Results expressed as in Fig. 2.
Requirements for Processing of Endogenously Expressed GAr+ EBNA1.
Subsequent experiments aimed to identify the pathway whereby endogenously expressed wild-type EBNA1 was presented to CD8 T cells. We first examined the situation in EBV+ BL cell lines that have retained the original tumor-like “group I” phenotype in vitro. Compared with LCLs, such lines show more limited patterns of EBV-latent antigen expression usually restricted to EBNA1 only and, most importantly, are deficient in their capacity to process endogenously expressed antigen via the conventional proteasome/TAP-dependent pathway (22). Here we used two EBV+ BL cell lines that are known to have this processing deficiency, the B*3501+ Sal-BL line and the B*0701+ Chep-BL line. As controls, we used LCLs generated from the normal B cells of these same patients by in vitro transformation either with their own resident EBV strain (in the case of Sal-LCL) or with the B95.8 strain (in the case of Chep-LCL). The EBNA1 immunoblot of protein extracts from these BL/LCL pairs is shown in Fig. 4 A, confirming that the BL lines express the native EBNA1 protein at levels at least equal to those seen in the matching LCLs. Furthermore, sequencing the EBNA1 gene in the Sal-BL and Chep-BL virus strains confirmed that the relevant epitope sequences, HPV and RPQ, respectively, were conserved (not depicted). As shown in Fig. 4 A, three CD8+ T cell clones specific for the HPV/B*3501 epitope recognized the Sal-LCL as expected, but did not recognize the EBNA1-expressing Sal-BL target unless this was exogenously coated with synthetic epitope peptide. Likewise, three clones specific for the RPQ/B*0701 epitope recognized the Chep-LCL but not Chep-BL cells unless the latter were peptide coated. As a specificity control, in each case there was no recognition of a peptide-coated HLA-mismatched BL or LCL target.
Figure 4.
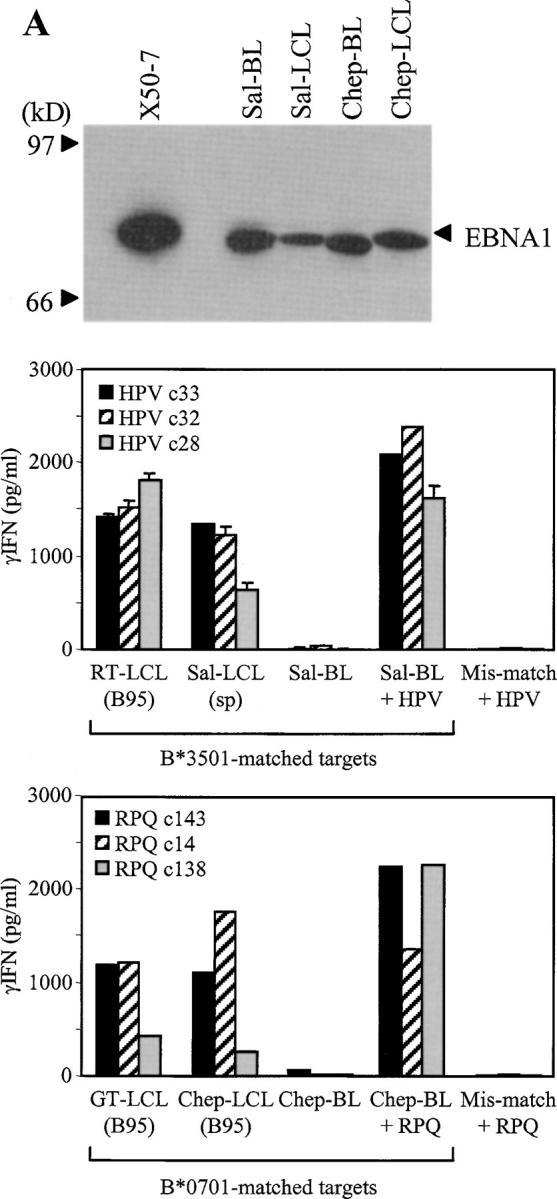
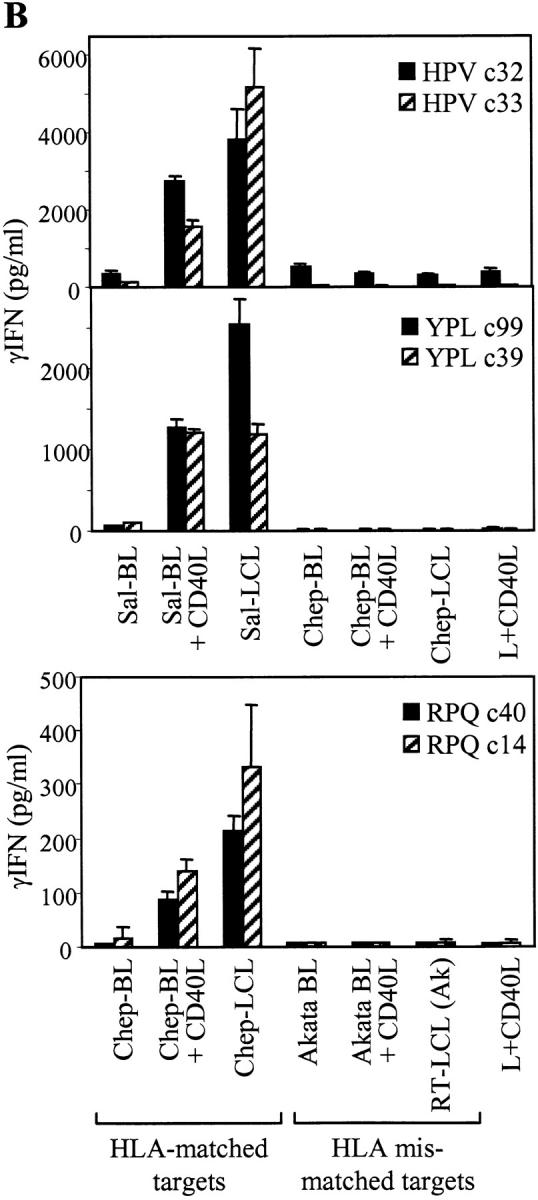

Presentation of EBNA1 to CD8+ T cells does not occur in cells with defects in the proteasome/TAP-dependent processing pathway. (A) Top: EBNA1 immunoblot (as in Fig. 1 B) of protein extracts from the Sal-BL/Sal-LCL and Chep-BL/Chep-LCL pairings. X50-7 is a standard B95.8 virus-transformed LCL. Middle: Three CD8+ T cell clones specific for the HPV/B*3501 epitope were tested against the B*3501+ Sal-BL/Sal-LCL pair of targets and against Sal-BL cells precoated with HPV peptide. Control targets were the B*3501+ RT-LCL (B95) and an HLA-mismatched LCL precoated with HPV peptide. Bottom: Three T cell clones specific for the RPQ/B*0701 epitope were tested against the B*0701+ Chep-BL/Chep-LCL pair of targets and against Chep-BL cells precoated with RPQ peptide. Control targets were the B*0701+ GT-LCL (B95) and an HLA-mismatched LCL precoated with RPQ peptide. (B) Top panels: CD8+ T cell clones specific for the B*3501-restricted epitopes HPV (EBNA1) or YPL (EBNA3A) were tested against the Sal-BL/Sal-LCL pair of targets and against Sal-BL cells preexposed for 2 d to CD40 ligand–expressing mouse L cells (L+CD40L). Control targets included the Chep-BL/Chep-LCL pair, Chep-BL cells similarly exposed to L+CD40L cells, and L+CD40L cells themselves. Bottom: CD8+ T cell clones specific for the RPQ/B*0701 epitope were tested against the Chep-BL/Chep-LCL pair and against Chep-BL cells preexposed as described above to L+CD40L cells. Control targets included the HLA-mismatched Akata BL line, with and without preexposure to L+CD40L cells, the HLA-mismatched RT-LCL (Ak) transformed with the Akata virus strain, and L+CD40L cells themselves. (C) CD8+ T cell clones against the HPV and YPL epitopes were tested against the TAP− T2:B35 LCL target line precoated or not with the relevant epitope peptide. Control targets included the B*3501+ RT-LCL (B95) and an HLA-mismatched LCL precoated with the relevant peptide. In all the above assays, T cells were tested at 1,000–10,000 cells per well and targets at 25,000 cells per well. Results expressed as in Fig. 2.
The deficiency in proteasome/TAP-dependent antigen processing can be transiently reversed in BL cell lines by CD40 ligation, restoring processing function toward LCL-like levels (23). Accordingly, Fig. 4 B shows data obtained on target lines used with or without previous exposure for 2 d to CD40 ligand–transfected mouse L cells. CD40 ligation clearly rendered the Sal-BL recognizable by HPV-specific T cell clones and the Chep-BL line recognizable by RPQ-specific clones, in each case to levels approaching that seen for the paired LCL. Such recognition was specific because responses to CD40-ligated but HLA-mismatched BL cells remained completely negative. Normally in such assays involving BL cells, there is no other viral protein available as a source of reference epitopes because most group I BL lines limit their EBV-latent protein expression to EBNA1. However, recent work identified Sal-BL as an unusual line in which viral antigen expression extends to include the immunodominant EBNA3A, 3B, and 3C proteins (15). Therefore, this allowed us to use the B*3501-restricted YPL as a reference epitope whose processing and presentation from endogenously expressed EBNA3A is known to involve the proteasome/TAP pathway (17). As expected, unmanipulated Sal-BL cells were unable to present endogenously expressed EBNA3A to YPL-specific effectors but acquired the capacity to do so after CD40 ligation (Fig. 4 B), confirming that the conventional HLA class I processing pathway was indeed inoperative in Sal-BL cells but could be activated by CD40 ligation.
To further examine the TAP dependence of EBNA1 processing, we tested the capacity of EBNA1-specific effectors to recognize endogenously expressed antigen in a B*3501 transfectant of the TAP1/TAP2− T2-LCL. As shown in Fig. 4 C, HPV-specific clones recognized a wild-type EBNA1-expressing LCL but not the T2:B35 line. As controls, T2:B35 cells were recognized if loaded with epitope peptide, whereas a peptide-coated HLA-mismatched LCL was not. This is the result expected if EBNA1 is being processed by the conventional HLA class I pathway because we already know that the HPV epitope, when processed and presented from GAr-deleted EBNA1 in vaccinia assays, is TAP dependent (13). Results obtained in parallel with two YPL-specific clones followed a similar pattern in that unmanipulated T2:B35 cells were again not recognized, confirming the known TAP dependence of this epitope's presentation from EBNA3A (17).
To further examine the proteasome dependence of EBNA1 processing, we performed a series of assays in which, according to a previously described protocol (24), LCL cells were first stripped of surface HLA class I–bound peptides by brief acid treatment at pH3, washed, and then cultured for 2 h in the presence or absence of proteasome inhibitors before being washed and added as targets in overnight assays with EBNA1-specific CD8+ T cells. Therefore, the levels of IFN-γ release observed reflect the reappearance of specific HLA–peptide complexes at the LCL surface within the 16-h assay period, and can be compared with those induced by untreated (nonstripped) cells included in these assays as positive control targets. As a further control, we checked how well acid stripping had removed cognate HLA–peptide complexes by glutaraldehyde fixing some cells immediately after stripping and including these cells as targets. Fig. 5 A, top, shows typical results from one such experiment using the wild-type RT-LCL as a target and two HPV-specific CD8+ T cell clones as effectors. The acid-stripped target cells that had not been exposed to inhibitors showed a complete recovery of epitope peptide display to positive control levels. However, acid-stripped targets then exposed to either of two irreversible proteasome inhibitors, lactacystin (25) and epoxomicin (26), showed very poor recovery, stimulating levels of IFN-γ release only just above the low baseline induced by target cells fixed immediately after stripping. A third proteasome inhibitor, MG132 (27), also impaired recovery but to a lesser extent (not depicted), reflecting the fact that its action is reversible. The effects of lactacystin and epoxomicin were not due to some nonspecific drug toxicity because acid-stripped cells reloaded with exogenous epitope peptide after drug treatment remained efficient stimulators of an IFN-γ response (Fig. 5, legend). The processing of wild-type EBNA1 to CD8+ T cells appeared to be just as sensitive to proteasome inhibition as that of GAr-deleted EBNA1 because similar results were observed in a parallel acid stripping assay using the RT-dl7a LCL as the target cell line (Fig. 5 A, bottom).
Figure 5.
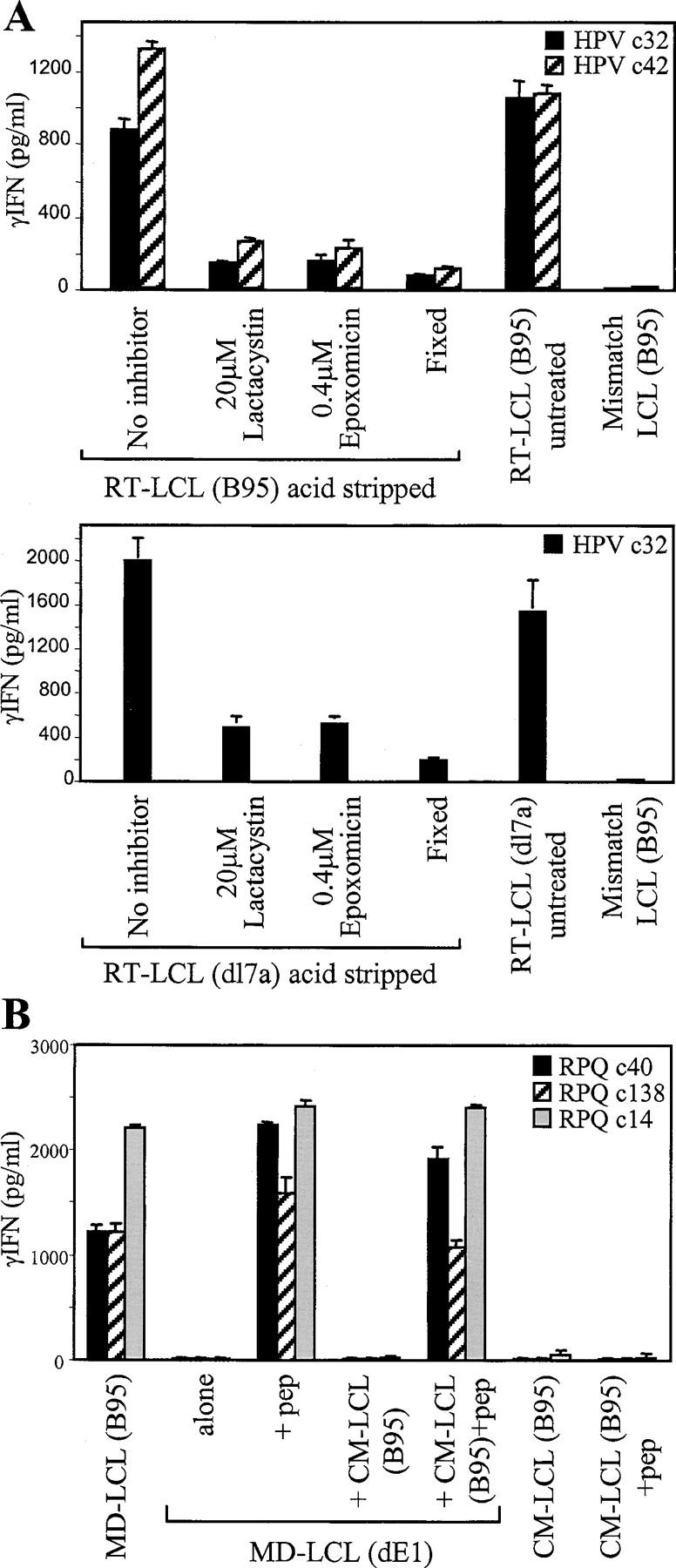
Presentation of EBNA1 to CD8+ T cells is blocked by inhibitors of proteasome function and requires endogenously expressed antigen. (A) The B*3501+ RT-LCL (B95) and RT-LCL (dl7a) target cells were acid stripped to remove surface HLA class I–bound peptides, washed, and incubated either in standard medium or in the presence of 20 μM lactacystin or 0.4 μM epoxomicin. The target cells were then washed extensively and mixed with CD8+ T cell clones specific for the HPV/B*3501 epitopes. Control targets included cells of the same line that had been acid stripped and then immediately fixed with glutaraldehyde (Fixed), cells of the same line that had not been acid stripped or exposed to inhibitors (untreated), and cells of an HLA-mismatched B95.8 virus–transformed LCL. In a parallel experiment to check for nonspecific toxicity of the inhibitors, the above acid-stripped LCL cells were exposed to epitope peptide dilutions after their 2-h treatment with proteasome inhibitors, and then washed and tested as targets. These cells stimulated IFN-γ release after exposure to peptide concentrations as low as 10−10 M just as efficiently as did acid-stripped cells that had not been inhibitor treated. (B) The acceptor B*0701+/EBNA1− MD-LCL (dE1) was mixed with an equal number of cells from the donor B*0701−/EBNA1+ CM-LCL (B95), and the mixture was cocultivated for 7 d before being used as targets for CD8+ T cell clones specific for the RPQ/B*0701 epitope. Control targets included the donor and acceptor LCLs alone, assayed pulsed or not pulsed with the RPQ peptide, the donor + acceptor cocultures pulsed with RPQ peptide, and the B*0701+ EBNA1+ cell line MD-LCL (B95). Results expressed as in Fig. 2.
An involvement of the proteasome in EBNA1 processing can still be reconciled with GAr-mediated protection if the protein is being handled by a cross-priming route, i.e., if EBNA1 were released from dying cells, phagocytosed by neighboring cells, and partially broken down by phagosomal proteases to GAr-free fragments that are then retro-transported to the proteasome (28, 29). Therefore, we set up experiments to look for evidence of such cross-priming by mixing a B*0701− donor LCL-expressing wild-type EBNA1 with the B*0701+ but EBNA1− acceptor LCL, MD (dE1), and looking for cross-presentation of EBNA1 using CD8+ T cells specific for the B*0701-restricted RPQ epitope. Results from one representative experiment (Fig. 5 B) show that even leaving donor and acceptor LCLs undisturbed in coculture for 7 d did not lead to detectable recognition by any of three RPQ-specific CD8+ clones. Yet clearly MD (dE1) acceptor cells were still present in the cocultures because the mixed population could present exogenously loaded RPQ peptide almost as efficiently as the MD (dE1) LCL itself. As controls in the same assays, these clones did not recognize the B*0701− donor line, CM-LCL (B95), with or without peptide, but did recognize the wild-type MD-LCL (B95) included as an EBNA1+ B*0701+ target.
Effect of EBNA1-specific T Cell Recognition on EBV-infected B Cell Growth.
A final set of experiments asked whether these EBNA1-specific CD8+ T cells could affect the long-term growth and survival of EBNA1-expressing B cells. Various EBV+ B cell lines were seeded in microtest plate wells at a range of input cell numbers (20,000–313 per well), either alone or in the presence of added CD8+ T cells (10,000 per well) from clones of known specificity. Cultures were maintained by weekly refeeding and were assayed for B cell line outgrowth after 4 wk. By this time, outgrowth was immediately apparent from microscopic inspection of the wells, and we confirmed by CD19 staining that this reflected growth of the target B cell line and not surviving T cells. Control cultures set up with T cells alone contained no viable cells at 4 wk.
Fig. 6 presents the results of one such experiment involving a variety of target B cell lines each seeded alone and in cocultures with three CD8+ T cell clones specific for the B*3501-restricted EBNA1 epitope HPV and with two clones specific for the B*3501-restricted EBNA3A epitope YPL. Results are expressed as the minimum target cell seeding giving successful outgrowth in each case compared with the growth of target cells alone (dotted line) showing the effects of T cell addition. The key findings are shown in Fig. 6 A, where the three HPV-specific clones all significantly inhibited outgrowth not just of the B*3501+ LCL, RT (dl7a), expressing a GAr-deleted EBNA1 protein, but also of two B*3501+ LCLs, RT (B95) and PB (B95), expressing wild-type EBNA1. Indeed, the degree of inhibition seen in these cocultures was similar to that mediated in the same assay by the two YPL-specific clones. Note that in all cocultures where the addition of T cells had prevented B cell outgrowth, there were no viable cells of either type detectable at 4 wk. These effects were not nonspecific consequences of coculture because three B*3501− LCLs, SC (B95), YC (B95), and CM (B95), showed no significant inhibition of outgrowth from either HPV- or YPL-specific clones. Indeed, with some of these HLA-mismatched combinations we even observed a slight enhancement of LCL outgrowth from low target cell seedings. Fig. 6 B shows outgrowth data from processing-deficient target lines that again parallel the IFN-γ results seen earlier with these same lines (Fig. 4). Thus, the T2:B35-LCL was not growth inhibited by HPV- or YPL-specific effectors, whereas the same targets were strongly inhibited if preexposed to the relevant epitope peptide. Likewise, both HPV- and YPL-specific effectors did not affect outgrowth of the B*3501+ Sal-BL line, unless these cells were first epitope loaded.
Figure 6.
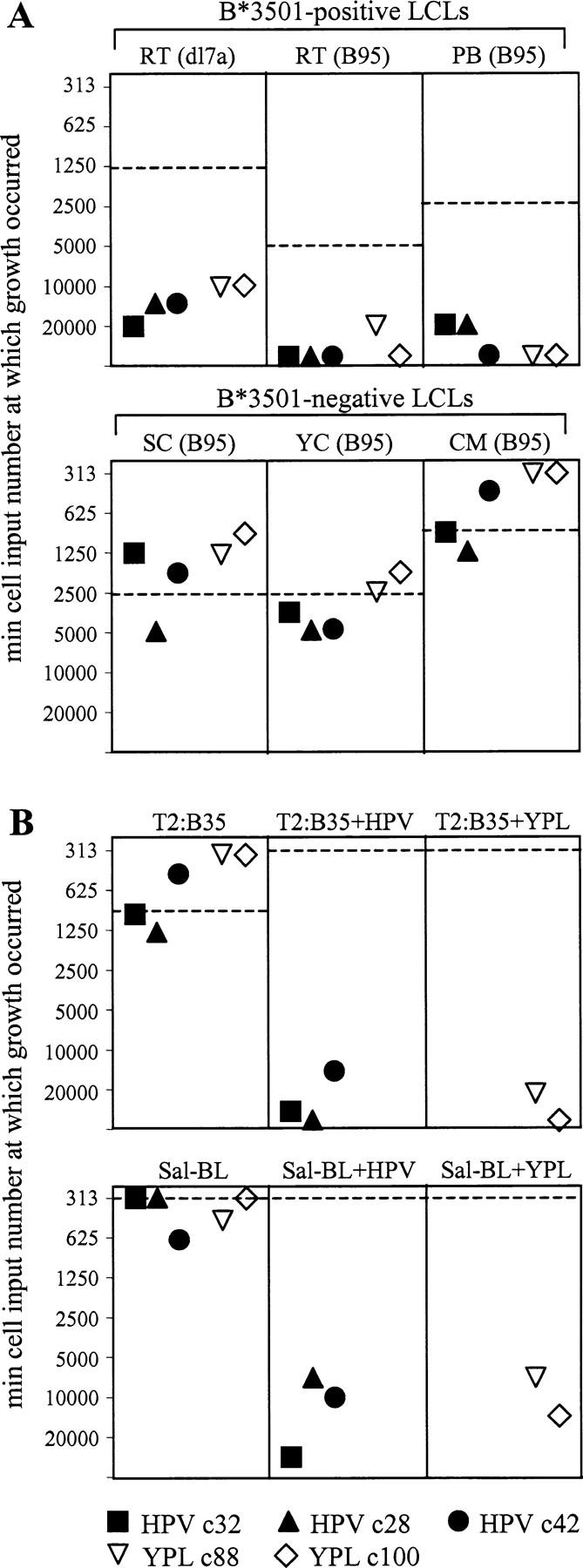
EBNA1-specific CD8+ T cells inhibit the in vitro outgrowth of EBV-transformed LCLs of the appropriate HLA type. EBV+ target B cell lines were seeded in microtest plate wells at a range of input cell numbers (20,000–313 per well), either alone or in the presence of added CD8+ T cell clones (10,000 per well) specific for the HPV/B*3501 or YPL/B*3501 epitopes. For each target, results are expressed as the minimum target cell seeding required for successful outgrowth from the different types of coculture. The corresponding value for target cells in the absence of T cells is shown as a dotted horizontal line. Targets included B*3501+ and B*3501− LCLs transformed either with B95.8 virus or with the dl7 strain as well as the TAP− LCL T2:B35 and the processing-deficient B*3501+ Sal-BL line, both either pulsed or not pulsed with the appropriate epitope peptide HPV or YPL. Cultures were assessed for cell growth after 4 wk. Control cultures set up with T cells alone contained no viable cells by that stage.
Importantly, the ability to inhibit target cell outgrowth through recognition of endogenously expressed EBNA1 was not restricted to HPV-specific T cells. Thus, we repeated the coculture experiments using two VLK-specific clones, VLK c16 and c76, which had clearly shown specific LCL recognition by IFN-γ release (Fig. 3) but, unlike the HPV effectors, had never given any hint of target cell killing in chromium release assays. When tested on a range of wild-type (B95.8) LCLs, these clones strongly inhibited outgrowth of A*0203+ lines without affecting that of HLA-mismatched controls (not depicted).
Discussion
All previous studies of GAr-mediated protection from cytotoxic T cell recognition have either looked at the processing of indicator epitopes from GAr- or epitope-inserted chimaeric proteins (5, 30), or have examined native EBNA1 epitope processing using nonphysiologic systems for EBNA1 expression (13, 14, 31). Here, to assay the effect of GAr in its natural context, we used a range of CD8+ T cell clones specific for native EBNA1 epitopes and pairs of EBV-transformed target LCLs expressing either full-length EBNA1 or an EBNA1 from which all but 13 of the original 239 GAr residues had been deleted (4). We consider it very unlikely that this residual 13–amino acid sequence has any immunologic effect because GAr functions are known to be length dependent (32) and short GAr inserts are only active in particular circumstances (7). As a read out, we switched from cytotoxicity to cytokine release because, unlike the experience of others with influenza-specific human T cells (33), we found that IFN-γ production was a more sensitive indicator of target cell recognition by CD8+ T cell clones reactive to EBV-latent cycle antigens. The IFN-γ release assays clearly show that LCL cells expressing a wild-type GAr+ EBNA1 protein are recognized by CD8+ T cell clones against four different EBNA1 epitopes situated both upstream and downstream of the GAr domain. Comparing clones to the upstream RPQ and downstream HPV epitopes, recognition of the wild-type EBNA1 protein was in both cases within 40–80% of that seen for the GAr-deleted protein. We conclude that for EBNA1 expressed in its natural setting in LCL cells, the GAr domain affords the protein only partial protection from CD8+ T cell recognition. In light of this, we must qualify our original proposition that the EBNA1-specific CD8+ responses found in infected individuals must necessarily have been induced by cross-priming in vivo (13, 14). Because EBNA1 epitopes are presented on the infected cell surface, it is conceivable that the response to EBNA1 (and to other EBV-latent antigens) is primed via a direct interaction between the CD8+ T cell repertoire and naturally infected B cells.
The inability of EBNA1-specific CD8+ T cell clones to recognize either EBV+ BL cells or the TAP-deficient T2:B35 LCL was consistent with the view that in standard LCLs, wild-type EBNA1 is being processed via the conventional proteasome/TAP-dependent pathway. This was further supported by experiments in which three known inhibitors of proteasomal function, lactacystin (25, 34), epoxomicin (26), and MG132 (27), all blocked the reappearance of EBNA1 peptide–HLA class I complexes on the surface of wild-type LCLs after the removal of existing complexes by acid stripping. Similar results were obtained whether the wild-type GAr+ or the dl7 GAr-deleted EBNA1 was the epitope source, so the processing of both antigens appears equally proteasome dependent. We asked whether the target for this processing might actually be antigen acquired by intercellular transfer within LCL cultures, a route which for wild-type EBNA1 could involve an initial breakdown by phagosomal proteases into GAr-free fragments that after retrotransport to the cytosol would be susceptible to proteasomal digestion (35, 36). However, under standard conditions of LCL culture, mixtures of appropriate donor and acceptor LCLs gave no evidence of such antigen transfer and subsequent processing. We believe that this is a significant negative result because the same LCL mixing protocol has successfully demonstrated the intercellular transfer of EBNAs into the HLA class II pathway in such cells (unpublished data). Although we do not rule out that intercellular transfer of antigen into the class I pathway may occur at some level in LCLs, our results clearly show that this is not the main source of EBNA1 epitope display. We infer that endogenously expressed EBNA1 is indeed targeted intracellularly by the proteasome despite the presence of the GAr domain. Two studies contemporaneous with this work have come to similar conclusions, although in both cases the evidence comes from experiments where EBNA1 fusion proteins have been expressed by transfection in epithelial cell lines (38, 39).
In the same context, another recent study has identified a second function of the GAr domain, whether in native EBNA1 or fused at the NH2 or C terminus of an indicator protein (OVA), which is to specifically inhibit translation of its own mRNA (37). Although the mechanism of this effect remains unknown, GAr was a more effective inhibitor of translation when fused to OVA at the NH2 than at the C terminus, whereas the two fusion proteins, once synthesized, appeared to be equally well protected from proteasomal digestion. When these fusion proteins were expressed by transient transfection in mouse cells, the N terminally tagged fusion was completely protected from recognition by CD8+ T cells to an H2-Kb–restricted OVA epitope, whereas the C terminally tagged fusion was only partially protected. The authors argued that the effect of GAr on antigen presentation reflected more its capacity to inhibit translation than to stabilize the protein once it was synthesized. This is consistent with the view that defective ribosomal products (DriPs), recently synthesized and immediately marked for proteasomal digestion, are a more important source of peptides entering the MHC class I pathway than is the steady state turnover of mature proteins (28, 29).
Our findings on the processing of native EBNA1 can also be accommodated within this view. Thus, in EBV-transformed LCLs, although the GAr domain may regulate EBNA1 mRNA translation to some extent, full-length EBNA1 has to be synthesized de novo with each cell cycle to maintain the levels of this important protein partitioned to daughter cells. Defective products from such synthesis could therefore provide new substrates for the proteasome. If this is indeed the case, the fact that EBNA1 epitopes located upstream and downstream of GAr are equally well presented implies that the DriPs in question are misfolded full-length products and not N-terminal fragments whose extension has been stalled by the GAr domain. It remains to be seen whether GAr+ DriPs are directly accessible to the proteasome or whether they require initial cleavage by another cytosolic protease. Whatever the detail, the available evidence now strongly suggests that the GAr domain is more effective as a stabilizing influence on the mature EBNA1 protein than as a protective influence against CD8+ T cell recognition. This would support our earlier contention, made from the study of EBNA1 homologues in simian γ herpeviruses (40), that the principal evolutionary force driving acquisition of the GAr domain has not been immune pressure per se but the requirement that EBNA1, the virus genome maintenance protein that is centrally important for virus persistence in cells, be protected from proteolysis.
Finally, although EBNA1 epitope-specific CD8 clones did not kill wild-type LCL targets in short-term cytotoxicity assays, we found clear evidence that they could prevent the longer term outgrowth of these cells in vitro_._ These inhibitory effects, culminating in death of the LCL, were specific and correlated precisely with the results of IFN-γ release assays in that recognition was only observed if target cells expressed the relevant HLA-restricting allele and could process antigens via the proteasome/TAP pathway. The mechanism whereby these T cells inhibit LCL outgrowth is clearly of interest and is now the subject of further investigations. However, given these results, we infer that EBNA1-induced CD8 responses will be biologically effective against natural EBV infection in vivo in the same way as CD8+ T cells against other latent cycle proteins, namely in controlling the virus-driven expansions of latently infected B cells that are a major feature of primary infection and may also recur at any time during virus persistence (3). Furthermore, our results imply that EBNA1-specific CD8+ T cells might be exploited therapeutically, possibly along with EBNA1-specific CD4+ T cells (41), to target EBV+ malignancies. This is particularly relevant in the context of EBV+ Hodgkin's disease and nasopharyngeal carcinoma where, in the absence of the immunodominant EBNA3 antigens, EBNA1 is one of the few virus proteins expressed and where evidence to date suggests that the proteasome/TAP-dependent pathway of antigen presentation remains intact in tumor cells (42–44). This contrasts with the situation in BL where EBNA1 is usually the only available antigen but where EBNA1-specific CD8+ T cells could only be effective if combined with protocols designed to restore antigen-presenting capacity in the tumor. These findings also have important implications for strategies that seek to silence potentially immunogenic therapeutic proteins by GAr domain insertion (45). Such fusion proteins will not only be capable of inducing CD8+ T cell responses in vivo, but cells expressing those proteins are likely to be recognized by the effectors thus induced.
Acknowledgments
This work is supported by a Medical Research Council Programme Grant to A.B. Rickinson and by Cancer Research UK Project Grants to A.B. Rickinson and N.W. Blake. S.P. Lee is supported by a Senior Cancer Research Fellowship from Cancer Research UK.
S.P. Lee and J.M. Brooks contributed equally to this work.
Abbreviations used in this paper: BL, Burkitt lymphoma; DriPs, defective ribosomal products; EBNA, EBV nuclear antigen; GAr, glycine-alanine repeat; LCL, lymphoblastoid cell line; TAP, transporter associated with antigen processing.
References
- 1.Pamer, E., and P. Cresswell. 1998. Mechanisms of MHC class I–restricted antigen processing. Annu. Rev. Immunol. 16:323–358. [DOI] [PubMed] [Google Scholar]
- 2.Koszinowski, U.H., and H. Hengel. 2002. Viral Proteins Counteracting Host Defences. Springer-Verlag, Berlin. 325 pp.
- 3.Rickinson, A.B., and E. Kieff. 2001. Epstein-Barr virus. Fields Virology. D.M. Knipe, and P.M. Howley, editors. Lippincott Williams and Wilkins, Philadelphia. 2575–2627.
- 4.Lee, M.A., M.E. Diamond, and J.L. Yates. 1999. Genetic evidence that EBNA-1 is needed for efficient, stable latent infection by Epstein-Barr virus. J. Virol. 73:2974–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levitskaya, J., M. Coram, V. Levitsky, S. Imreh, P.M. Steigerwald-Mullen, G. Klein, M.G. Kurilla, and M.G. Masucci. 1995. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen 1. Nature. 375:685–688. [DOI] [PubMed] [Google Scholar]
- 6.Levitskaya, J., A. Shapiro, A. Leonchiks, A. Ciechanover, and M.G. Masucci. 1997. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA. 94:12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharipo, A., M. Imreh, A. Leonchiks, S. Imreh, and M.G. Masucci. 1998. A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat. Med. 4:939–944. [DOI] [PubMed] [Google Scholar]
- 8.Heessen, S., A. Leonchiks, N. Issaeva, A. Sharipo, G. Selivanova, M.G. Masucci, and N.P. Dantuma. 2002. Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc. Natl. Acad. Sci. USA. 99:1532–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dantuma, N.P., A. Sharipo, and M.G. Masucci. 2002. Avoiding proteasomal processing: the case of EBNA1. Curr. Top. Microbiol. Immunol. 269:23–36. [DOI] [PubMed] [Google Scholar]
- 10.Trivedi, P., M.G. Masucci, G. Winberg, and G. Klein. 1991. The Epstein-Barr-virus-encoded membrane protein LMP but not the nuclear antigen EBNA-1 induces rejection of transfected murine mammary carcinoma cells. Int. J. Cancer. 48:794–800. [DOI] [PubMed] [Google Scholar]
- 11.Murray, R.J., M.G. Kurilla, J.M. Brooks, W.A. Thomas, M. Rowe, E. Kieff, and A.B. Rickinson. 1992. Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV-positive malignancies. J. Exp. Med. 176:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khanna, R., S.R. Burrows, M.G. Kurilla, C.A. Jacob, I.S. Misko, T.B. Sculley, E. Kieff, and D.J. Moss. 1992. Localization of Epstein-Barr virus cytotoxic T cell epitopes using recombinant vaccinia - implications for vaccine development. J. Exp. Med. 176:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blake, N., S. Lee, I. Redchenko, W. Thomas, N. Steven, A. Leese, P. Steigerwald-Mullen, M.G. Kurilla, L. Frappier, and A. Rickinson. 1997. Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity. 7:791–802. [DOI] [PubMed] [Google Scholar]
- 14.Blake, N., T. Haigh, G. Shaka'a, D. Croom-Carter, and A. Rickinson. 2000. The importance of exogenous antigen in priming the human CD8+ T cell response: lessons from the EBV nuclear antigen EBNA1. J. Immunol. 165:7078–7087. [DOI] [PubMed] [Google Scholar]
- 15.Kelly, G., A. Bell, and A. Rickinson. 2002. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat. Med. 8:1098–1104. [DOI] [PubMed] [Google Scholar]
- 16.Humme, S., G. Reisbach, R. Feederle, H.J. Delecluse, K. Bousset, W. Hammerschmidt, and A. Schepers. 2003. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc. Natl. Acad. Sci. USA. 100:10989–10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lautscham, G., S. Mayrhofer, G. Taylor, T. Haigh, A. Leese, A. Rickinson, and N. Blake. 2001. Processing of a multiple membrane spanning Epstein-Barr virus protein for CD8+ T cell recognition reveals a proteasome-dependent, transporter associated with antigen processing–independent pathway. J. Exp. Med. 194:1053–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henriquez, N.V., E. Floettmann, M. Salmon, M. Rowe, and A.B. Rickinson. 1999. Differential responses to CD40 ligation among Burkitt lymphoma lines that are uniformly responsive to Epstein-Barr virus latent membrane protein 1. J. Immunol. 162:3298–3307. [PubMed] [Google Scholar]
- 19.Schultze, J.L., S. Michalak, M.J. Seamon, G. Dranoff, K. Jung, J. Daley, J.C. Delgado, J.G. Gribben, and L.M. Nadler. 1997. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J. Clin. Invest. 100:2757–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill, A.B., S.P. Lee, J.S. Haurum, N. Murray, Q.Y. Yao, M. Rowe, N. Signoret, A.B. Rickinson, and A.J. McMichael. 1995. Class I major histocompatibility complex–restricted cytotoxic T lymphocytes specific for Epstein-Barr virus (EBV) nuclear antigens fail to lyse the EBV-transformed B lymphoblastoid lines against which they were raised. J. Exp. Med. 181:2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan, L.C., N. Gudgeon, N.E. Annels, P. Hansasuta, C.A. O'Callaghan, S. Rowland-Jones, A.J. McMichael, A.B. Rickinson, and M. Callan. 1999. A re-evaluation of the frequency of CD8(+) T cells specific for EBV in healthy virus carriers. J. Immunol. 162:1827–1835. [PubMed] [Google Scholar]
- 22.Khanna, R., S.R. Burrows, V. Argaet, and D.J. Moss. 1994. Endoplasmic reticulum signal sequence facilitated transport of peptide epitopes restores immunogenicity of an antigen processing defective tumor cell line. Int. Immunol. 6:639–645. [DOI] [PubMed] [Google Scholar]
- 23.Khanna, R., L. Cooper, N. Kienzle, D.J. Moss, S.R. Burrows, and K.K. Khanna. 1997. Engagement of CD40 antigen with soluble CD40 ligand up-regulates peptide transporter expression and restores endogenous processing function in Burkitt's lymphoma cells. J. Immunol. 159:5782–5785. [PubMed] [Google Scholar]
- 24.Gavioli, R., S. Vertuani, and M.G. Masucci. 2002. Proteasome inhibitors reconstitute the presentation of cytotoxic T-cell epitopes in Epstein-Barr virus-associated tumors. Int. J. Cancer. 101:532–538. [DOI] [PubMed] [Google Scholar]
- 25.Fenteany, G., R.F. Standaert, W.S. Lane, S. Choi, E.J. Corey, and S.L. Schreiber. 1995. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 268:726–731. [DOI] [PubMed] [Google Scholar]
- 26.Meng, L., R. Mohan, B.H. Kwok, M. Elofsson, N. Sin, and C.M. Crews. 1999. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA. 96:10403–10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee, D.H., and A.L. Goldberg. 1996. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 271:27280–27284. [DOI] [PubMed] [Google Scholar]
- 28.Schubert, U., L.C. Anton, J. Gibbs, C.C. Norbury, J.W. Yewdell, and J.R. Bennink. 2000. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 404:770–774. [DOI] [PubMed] [Google Scholar]
- 29.Reits, E.A., J.C. Vos, M. Gromme, and J. Neefjes. 2000. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature. 404:774–778. [DOI] [PubMed] [Google Scholar]
- 30.Tellam, J., M. Sherritt, S. Thomson, R. Tellam, D.J. Moss, S.R. Burrows, E. Wiertz, and R. Khanna. 2001. Targeting of EBNA1 for rapid intracellular degradation overrides the inhibitory effects of the Gly-Ala repeat domain and restores CD8+ T cell recognition. J. Biol. Chem. 276:33353–33360. [DOI] [PubMed] [Google Scholar]
- 31.Mukherjee, S., P. Trivedi, D.M. Dorfman, G. Klein, and A. Townsend. 1998. Murine cytotoxic T lymphocytes recognize an epitope in an EBNA-1 fragment, but fail to lyse EBNA-1–expressing mouse cells. J. Exp. Med. 187:445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dantuma, N.P., S. Heessen, K. Lindsten, M. Jellne, and M.G. Masucci. 2000. Inhibition of proteasomal degradation by the gly-Ala repeat of Epstein-Barr virus is influenced by the length of the repeat and the strength of the degradation signal. Proc. Natl. Acad. Sci. USA. 97:8381–8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valitutti, S., S. Muller, M. Dessing, and A. Lanzavecchia. 1996. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J. Exp. Med. 183:1917–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Craiu, A., M. Gaczynska, T. Akopian, C.F. Gramm, G. Fenteany, A.L. Goldberg, and K.L. Rock. 1997. Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J. Biol. Chem. 272:13437–13445. [DOI] [PubMed] [Google Scholar]
- 35.Guermonprez, P., L. Saveanu, M. Kleijmeer, J. Davoust, P. Van Endert, and S. Amigorena. 2003. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 425:397–402. [DOI] [PubMed] [Google Scholar]
- 36.Houde, M., S. Bertholet, E. Gagnon, S. Brunet, G. Goyette, A. Laplante, M.F. Princiotta, P. Thibault, D. Sacks, and M. Desjardins. 2003. Phagosomes are competent organelles for antigen cross-presentation. Nature. 425:402–406. [DOI] [PubMed] [Google Scholar]
- 37.Yin, Y., B. Manoury, and R. Fahraeus. 2003. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science. 301:1371–1374. [DOI] [PubMed] [Google Scholar]
- 38.Voo, K.S., T. Fu, H.Y. Wang, J. Tellam, H.E. Heslop, M.K. Brenner, C.M. Rooney, and R.F. Wang. 2004. Evidence for the presentation of major histocompatibility complex class I–restricted Epstein-Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J. Exp. Med. 199:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tellam, J., G. Connolly, K.J. Green, J.J. Miles, D.J. Moss, S.R. Burrows, and R. Khanna. 2004. Endogenous presentation of CD8+ T cell epitopes from Epstein-Barr virus–encoded nuclear antigen 1. J. Exp. Med. 199:1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blake, N.W., A. Moghaddam, P. Rao, A. Kaur, R. Glickman, Y.G. Cho, A. Marchini, T. Haigh, R.P. Johnson, A.B. Rickinson, et al. 1999. Inhibition of antigen presentation by the glycine/alanine repeat domain is not conserved in simian homologues of Epstein-Barr virus nuclear antigen 1. J. Virol. 73:7381–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikiforow, S., K. Bottomly, G. Miller, and C. Munz. 2003. Cytolytic CD4(+)-T-cell clones reactive to EBNA1 inhibit Epstein-Barr virus-induced B-cell proliferation. J. Virol. 77:12088–12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee, S.P., C.M. Constandinou, W.A. Thomas, D. Croom-Carter, N.W. Blake, P.G. Murray, J. Crocker, and A.B. Rickinson. 1998. Antigen presenting phenotype of Hodgkin Reed-Sternberg cells: analysis of the HLA class I processing pathway and the effects of interleukin 10 on Epstein-Barr virus-specific cytotoxic T cell recognition. Blood. 92:1020–1030. [PubMed] [Google Scholar]
- 43.Lee, S.P., A.T. Chan, S.T. Cheung, W.A. Thomas, D. Croomcarter, C.W. Dawson, C.H. Tsai, S.F. Leung, P.J. Johnson, and D.P. Huang. 2000. CTL control of EBV in nasopharyngeal carcinoma (NPC): EBV-specific CTL responses in the blood and tumors of NPC patients and the antigen-processing function of the tumor cells. J. Immunol. 165:573–582. [DOI] [PubMed] [Google Scholar]
- 44.Khanna, R., P. Busson, S.R. Burrows, C. Raffoux, D.J. Moss, J.M. Nicholls, and L. Cooper. 1998. Molecular characterization of antigen-processing function in Nasopharyngeal Carcinoma (NPC): evidence for efficient presentation of Epstein-Barr virus cytotoxic T-cell epitopes by NPC cells. Cancer Res. 58:310–314. [PubMed] [Google Scholar]
- 45.Ossevoort, M., B.M. Visser, D.J. van den Wollenberg, E.I. van der Voort, R. Offringa, C.J. Melief, R.E. Toes, and R.C. Hoeben. 2003. Creation of immune “stealth” genes for gene therapy through fusion with the Gly-Ala repeat of EBNA-1. Gene Ther. 10:2020–2028. [DOI] [PubMed] [Google Scholar]