Interleukin (IL)-1 Receptor–associated Kinase (IRAK) Requirement for Optimal Induction of Multiple IL-1 Signaling Pathways and IL-6 Production (original) (raw)
Abstract
Interleukin (IL)-1 is a proinflammatory cytokine with pleiotropic effects in inflammation. IL-1 binding to its receptor triggers a cascade of signaling events, including activation of the stress-activated mitogen-activated protein (MAP) kinases, c-Jun NH2-terminal kinase (JNK) and p38 MAP kinase, as well as transcription factor nuclear factor κB (NF-κB). IL-1 signaling results in cellular responses through induction of inflammatory gene products such as IL-6. One of the earliest events in IL-1 signaling is the rapid interaction of IL-1 receptor–associated kinases, IRAK and IRAK-2, with the receptor complex. The relative roles of IRAK and IRAK-2 in IL-1 signaling pathways and subsequent cellular responses have not been previously determined. To evaluate the importance of IRAK in IL-1 signaling, IRAK-deficient mouse fibroblast cells were prepared and studied. Here we report that IL-1–mediated activation of JNK, p38, and NF-κB were all reduced in embryonic fibroblasts deficient in IRAK expression. In addition, IL-6 production in response to IL-1 was also dramatically reduced in IRAK-deficient embryonic fibroblasts and in skin fibroblasts prepared from IRAK-deficient mice. Our results demonstrate that IRAK plays an essential proximal role in coordinating multiple IL-1 signaling pathways for optimal induction of cellular responses.
Keywords: IRAK, IL-1, JNK, p38, NF-κB
Interleukin 1 (IL-1α and IL-1β) plays an important role in inflammation, acting locally and systemically to induce other proinflammatory cytokines, chemotactic factors, adhesion molecules, acute phase proteins, and fever (1). Animals that lack expression of IL-1 or IL-1 receptor have reduced inflammatory responses (2–5). Cellular responses to IL-1 are mediated by a cascade of intracellular signaling events including activation of the stress-activated mitogen-activated protein (MAP)1 kinases, c-Jun NH2-terminal kinase (JNK) and p38, as well as transcription factor nuclear factor κB (NF-κB) (6). Upon IL-1 binding, IL-1 receptor type I forms a complex with IL-1 receptor accessory protein (7–9). IL-1 receptor–associated serine/threonine kinase (IRAK), and a recently identified homologue, IRAK-2, are rapidly recruited to this receptor complex via the adaptor protein MyD88 (10–13). IRAK becomes phosphorylated and subsequently interacts with TRAF6, a member of the TNF receptor–associated factor (TRAF) family (13, 14). TRAF6 has been implicated in activation of both JNK and NF-κB (14, 15). TRAF6 associates with NF-κB–inducing kinase (NIK), a MAP 3 kinase–related protein that is essential for TNF-α– and IL-1–mediated NF-κB activation but has no effect on the activation of JNK or p38 (15–17). NIK interacts with and may directly activate the recently identified kinases of NF-κB inhibitor (IκB; 18–20). IκB kinases are responsible for activation of NF-κB via phosphorylation of its inhibitory partners, the IκB proteins, leading to their degradation by proteasomes (21).
IRAK and IRAK-2 are homologous to Pelle, a Drosophila protein kinase identified genetically to be important in dorsal–ventral pattern formation and in pathogen resistance (22, 23). Pelle is essential for the activation of Dorsal, an NF-κB–like protein which is mediated by Toll, an IL-1 receptor homologue in Drosophila (24). The rapid IL-1–dependent association of IRAK and IRAK-2 with the IL-1 receptor complex and their homology to Pelle suggest that IRAK and/or IRAK-2 may serve important functions in initiating IL-1 signaling. However, the roles of IRAK and IRAK-2 in activation of the multiple downstream IL-1 signaling pathways have not previously been determined. To dissect the role of IRAK in IL-1 signaling pathways, we disrupted the IRAK gene by homologous recombination and prepared IRAK-deficient fibroblasts. IL-1-induced activation of JNK, p38, and transcription factor NF-κB, and subsequent induction of IL-6 was analyzed in IRAK-deficient cells.
Materials and Methods
Preparation of IRAK Antibody.
Polyclonal rabbit antiserum to IRAK was raised against a peptide (Bio-Synthesis, Inc., Lewisville, TX) corresponding to the COOH-terminal amino acids (657–677) of mouse IRAK protein (25).
Disruption of the IRAK Gene and Generation of IRAK-deficient Mice.
Mouse IRAK clones were isolated from a 129/Ola mouse genomic library. A 7-kb BamHI–EcoRI DNA fragment covering the first 9 exons of the mouse IRAK gene was used to prepare the knockout construct. A cassette containing a neomycin resistance gene was used to replace a 940-bp region covering exon 5 to exon 7 of the gene. A herpes simplex–thymidine kinase cassette was placed at the 3′ end of the construct. The DNA construct was introduced into E14 embryonic stem cells by electroporation. Cells were cultured in the presence of 400 μg/ml G418 and 0.2 μM gancyclovir. Embryonic stem cells with the disrupted gene were detected by PCR and then confirmed by Southern hybridization using a DNA probe flanking the 3′ end of the construct. Chimeric mice were generated from embryos injected with embryonic stem cells. Germline mice were obtained from breeding of chimeric male mice with C57BL/6J females. Germline female mice heterozygous for the disrupted IRAK gene were identified by PCR. IRAK-deficient male mice carrying only the disrupted IRAK gene were obtained from cross-breeding of heterozygous female mice with wild-type littermates.
Preparation of Embryonic Fibroblasts and Skin Fibroblasts.
To prepare embryonic fibroblasts (EFs), embryonic stem cells with the disrupted IRAK gene were injected into C57BL/6J blastocysts and transferred to pseudopregnant mice. Embryos at day 15 of gestation were harvested. Fibroblast cell suspensions were prepared by trypsin treatment of the minced embryonic tissues. Fibroblasts derived from embryonic stem cells were enriched by culturing in DMEM with 10% FCS and 1 mg/ml G418. Fibroblasts after 3–4 wk of culture with G418 were used in these studies. Control EF cells with the wild-type IRAK gene were prepared from embryos of CD8-deficient mice (26). To prepare skin fibroblasts (SFs), mouse body skin was shaved, cut into small pieces, and then subjected to trypsin treatment. SFs in cell suspensions were cultured in DMEM with 10% FCS.
Cell Stimulation.
Control and IRAK-deficient cells (9 × 105/ plate) were plated overnight in 100-mm cell culture plates with DMEM containing 5% FCS. EF cells were kept in the presence of 200 μg/ml G418. Before each experiment, cells were starved in serum-free DMEM for 4 h. The cells were then stimulated with IL-1β (R&D Systems, Minneapolis, MN) or TNF-α (Genzyme Corp., Cambridge, MA) at 37°C. After stimulation, cells were scraped from the plates in ice-cold PBS and used immediately.
In Vitro Kinase Assay.
After stimulation, cells were lysed in NP-40 lysis buffer (20 mM Hepes, pH 7.5, 150 mM NaCl, 1% NP-40, and 1 mM Na3VO4) containing EDTA-free complete protease inhibitor cocktail (Boehringer Mannheim Corp., Indianapolis, IN), centrifuged at 16,000 g for 10 min, and precleared twice with 50 μl of GammaBind G–Sepharose slurry (Pharmacia Biotech, Piscataway, NJ). MAP kinases were immunoprecipitated with 50 μl GammaBind G–Sepharose slurry and 2 μg polyclonal rabbit antibody, specific for the 20 COOH-terminal residues of p38α or the 17 COOH-terminal residues of JNK1 (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoprecipitates were washed twice with NP-40 lysis buffer and twice with kinase reaction buffer (25 mM Hepes, pH 7.5, 10 mM MgCl2, 10 mM MnCl2, 20 mM β-glycerophosphate, and 1× EDTA-free complete protease inhibitor cocktail). Kinase reactions were performed for 20 min at 30°C in kinase reaction buffer containing 50 μM ATP and 5 μCi γ-[32P]ATP (3,000 Ci/mmol; Amersham Corp., Arlington Heights, IL) with 0.5 μg GST–MAP kinase– activated protein kinase 2 (MAPKAPK2; Upstate Biotechnology, Lake Placid, NY) as p38 substrate or 2 μg GST–c-JUN (BioMol, King of Prussia, PA) as JNK1 substrate. Samples were boiled in SDS sample buffer, electrophoresed in 10% Tris-Glycine polyacrylamide gels, and transferred to PVDF membranes (Novex, San Diego, CA). Bands were quantified on a PhosphorImager System (Storm 840; Molecular Dynamics Inc., Sunnyvale, CA). Membranes were stained for p38 and JNK1 protein using the p38 and JNK1 antibodies described above and the Vistra ECF Western blotting system (Amersham Corp.).
Western Blotting.
Western blot analyses were carried out as previously described (27). For detection of IRAK and IκB-α protein levels, NP-40 cell lysates (105 cells /sample) were separated on SDS-PAGE and transferred to nitrocellulose (MSI, Westboro, MA). The filters were immunoblotted with IRAK antiserum (1: 1,000 dilution) or rabbit antibody to IκB-α (Santa Cruz Biotechnology). The bands corresponding to specific proteins were detected by horseradish-peroxidase–conjugated rabbit IgG and enhanced chemiluminescence (Amersham Corp.).
NF-κB Mobility Shift Assay.
After stimulation, the cells from 100-mm plates were suspended in 500 μl of Buffer A (10 mM Hepes, pH 7.9, 10 mM KCl, 100 μM EDTA, 100 μM EGTA, 1 mM dithiothreitol (DTT), and 500 μM PMSF) and incubated on ice for 15 min. After adding 30 μl of 10% NP-40, the samples were centrifuged at 14,000 rpm at 4°C for 30 s. The pellet was washed once with buffer A and resuspended in 50 μl of buffer B (20 mM Hepes, pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1 mM PMSF) for 30 min at 4°C. Nuclear extracts were obtained by centrifugation of the samples at 14,000 rpm for 10 min. Protein concentrations were measured by the BCA method (Pierce, Rockford, IL). The consensus double-stranded oligonucleotide for NF-κB binding (Santa Cruz Biotech) was labeled by T4 polynucleotide kinase. The nuclear binding reaction was carried out with 5 μl of nuclear extract (10 μg), 1 μl of Poly(dI-dC), 1 μl of labeled probe (100,000 cpm), 5 μl 4× buffer (20 mM Hepes, pH 7.9, 20 mm MgCl2, and 1 mM EDTA) and 8 μl of 25% glycerol for 30 min at room temperature. The binding complexes were separated on 6% polyacrylamide gel in 0.5× Tris-buffered EDTA (Novex) and detected by autoradiography.
Northern Blot Analysis and IL-6 ELISA.
For IL-6 mRNA detection, EF cells cultured in 100-mm plates were treated with IL-1β or TNF-α for 3 h at 37°C. Total RNA was extracted from EF cells with RNAzol (Tel-Test, Inc., Friendswood, TX). Northern blot analysis was performed as previously described (28) using a mouse IL-6 cDNA probe prepared by PCR from commercial mouse IL-6 cDNA template and oligonucleotides (Clontech, Palo Alto, CA). For detection of secreted IL-6 protein, EF (5 × 104 /well) and SF (104/well) cells were incubated overnight in flat-bottomed microtiter plates in 10% FCS-DMEM. After a 2–4-h incubation with low endotoxin 1% FCS-RPMI-1640 medium (Sigma Chemical Co., St. Louis, MO), cells were treated for 8 h with mouse IL-1β in fresh medium. Supernatants were then collected and analyzed for IL-6 using commercial ELISA kits (from Endogen, Inc., Woburn, MA, or Genzyme Corp.), with recombinant mouse IL-6 as a standard.
Results and Discussion
IRAK-deficient Cells.
To investigate whether IRAK is indispensable for IL-1 signaling pathway, IRAK-deficient mouse primary EF cells were prepared from mouse embryos injected with embryonic stem cells carrying a disrupted IRAK gene as described in Materials and Methods. The IRAK gene was disrupted by deletion of exons 5–7, which encode the NH2-terminal portion of the kinase domain, subdomains I–V, including conserved amino acids involved in ATP binding (Fig. 1 A). The deletion was confirmed by Southern hybridization (Fig. 1 B). Detection of only the disrupted IRAK allele in male embryonic stem cells suggests that the mouse IRAK gene is located on the X chromosome. Consistent with our finding, the human IRAK gene was also reported to be on the X chromosome (EMBL/GenBank'DDBJ accession number U52112). The absence of IRAK protein in EF cells containing the disrupted IRAK allele was demonstrated by immunoblotting with an anti-IRAK antiserum (Fig. 1 C). The lack of IRAK expression in IRAK-deficient EF cells enabled us to evaluate the importance of IRAK in IL-1 signaling.
Figure 1.



Lack of IRAK expression in IRAK-deficient cells. (A) Map of the mouse IRAK gene and construct. A genomic DNA fragment containing the 5′ portion of the mouse IRAK gene was used as the homologous region for recombination. Exon 5 to exon 7 of the gene was replaced by a neomycin resistant gene cassette (NEO). A herpes simplex–thymidine kinase gene (HSV-TK) was placed 3′ of the construct. Restriction sites are BamHI (B), EcoRI (E) and SpeI (S). (B) Disruption of the single IRAK allele in male embryonic stem cells. Genomic DNAs from embryonic stem cells with the wild-type (+) or disrupted (−) IRAK gene were digested with EcoRI and hybridized to the DNA probe shown in A. The 12-kb DNA band of the wild-type allele and the 2.6-kb band of the disrupted allele detected in hybridization are also shown in A. (C) Absence of IRAK protein in IRAK-deficient cells. Primary embryonic EF cells were prepared as described in the Materials and Methods. Proteins from control (+) and IRAK-deficient (−) EF cells were separated by 10% SDS-PAGE and IRAK was detected by immunoblotting using an IRAK-specific antibody. Equal loading of proteins was demonstrated by subsequent blotting with an antibody to extracellular signal–regulated kinase 2 (ERK-2).
Defective Activation of JNK and p38 in IRAK-deficient Cells.
JNK and p38 are strongly activated by IL-1, TNF-α, cell stress, and LPS (29–33). JNK phosphorylates the transactivation domain of c-Jun, which is involved in the activation of activator protein 1–dependent genes, including genes involved in inflammatory responses (29–31). p38 is also involved in activation of gene expression and protein synthesis during inflammation, including IL-1–induced IL-6 and prostaglandin synthesis (34). The proximal events responsible for IL-1–induced JNK and p38 activation are only partially understood. We therefore compared IL-1–induced activation of JNK and p38 in IRAK-deficient and control EF cells. JNK activity was measured by in vitro kinase assays of JNK immunoprecipitates, using c-Jun as substrate. Activation of JNK was reduced in IRAK-deficient cells by two- to threefold at all concentrations of IL-1 tested, although not completely eliminated (Fig. 2). This defect in JNK activation was IL-1 specific, since TNF-α–induced JNK activity was comparable in control and IRAK-deficient cells. Activation of p38 was measured in p38 immunoprecipitates using MAPKAPK-2 as a substrate. IL-1–induced activation of p38 was also reduced in IRAK-deficient cells compared with control cells, by three- to fivefold at all concentrations of IL-1 tested. In contrast, TNF-α–induced p38 activity was similar in control and IRAK-deficient cells (Fig. 2). These results demonstrate that IL-1–induced JNK and p38 activities are both specifically reduced in IRAK-deficient cells, and implicate IRAK as the most proximal signaling component in the activation of the JNK and p38 pathways.
Figure 2.

Defective IL-1–induced p38 and JNK activation in IRAK-deficient EF cells. IRAK-deficient (−) or control (+) EF cells were stimulated with IL-1β (0.1–10 ng/ml) or TNF-α (100 ng/ml) for 10 min at 37°C, and immune complex kinase assays for p38 and JNK1 activity were performed. (A) Phosphorylation of MAPKAPK2 by immunoprecipitated p38 and of c-Jun by immunoprecipitated JNK1. Western blotting for p38 and JNK1 was performed as a loading control. (B) Quantitation of p38 and JNK1 activation. p38 and JNK1 kinase assays were quantitated by PhosphorImager (Molecular Dynamics), and normalized to the amount of p38 or JNK1 detected by Western blotting for each sample and to the activity in unstimulated IRAK-deficient EF cells for each assay. The p38 data are representative of three experiments, and the JNK1 data are expressed as an average of two experiments.
Defective Activation of NF-κB.
IL-1 and TNF-α are the two most efficient activators of the NF-κB family of transcription factors (21, 35). NF-κB remains in an inactive state when sequestered by IκB in the cytoplasm. Phosphorylation of IκB by IκB kinases leads to ubiquitination and subsequent degradation of IκB by proteasomes (18–21). Released NF-κB is then translocated to the nucleus, where it binds to regulatory sites in NF-κB–induced genes (21). IκB levels in IRAK-deficient cells and control cells treated with IL-1 were determined by immunoblotting with an IκB-α–specific antibody. At concentrations of IL-1 ranging from 10 pg/ml to 1 ng/ml, IκB degradation was significantly less in IRAK-deficient cells as compared with control cells (Fig. 3 A). However, at 10 ng/ml IL-1, IκB-α was degraded almost completely in both IRAK-deficient and control cells. IL-1 induction of the NF-κB pathway was further investigated by examining NF-κB DNA-binding activity in nuclear extracts (Fig. 3 B). Significant reduction in NF-κB activation was observed in IRAK-deficient cells at low IL-1 concentrations. Consistent with the results in IκB degradation, NF-κB activation was comparable in control and IRAK-deficient cells at high IL-1 concentrations.
Figure 3.


Reduced activation of the NF-κB pathway by IL-1 in IRAK-deficient cells. (A) Inefficient IκB degradation in IRAK-deficient EF cells. Control (+) and IRAK-deficient (−) cells were stimulated with IL-1 for 15 min at 37°C. Proteins in cell lysates were separated on SDS-PAGE and IκB was detected with an IκB-α–specific antibody. Equal amounts of sample loading were demonstrated by subsequent blotting of the same filter with an antibody specific to extracellular signal–regulated kinase 2 (ERK-2). The intensity of the protein bands was quantitated by densitometry. The amount of IκB in the IL-1–stimulated cells is expressed as the percentage of that in unstimulated cells. (B) Decreased activation of NF-κB by IL-1 in IRAK-deficient cells. EF cells were stimulated with IL-1 or TNF-α for 30 min at 37°C. Nuclear extracts were prepared and NF-κB DNA-binding activity was determined with the electrophoretic mobility shift assay. The data are representative of two independent experiments with similar results.
Defective Induction of IL-6.
IL-1 signaling triggers cellular responses through the induction of various inflammatory gene products (1). IL-6 is strongly induced by IL-1 via activation of transcription factors including NF-κB and activator protein 1, which bind to the enhancer elements of the IL-6 gene promoter (6). IL-6 in turn plays a crucial role in inflammation by mediating acute phase reactions (36). To determine whether the observed defects in IL-1–activated signaling result in impaired cellular responses, IL-6 induction in response to IL-1 was measured. Levels of IL-6 mRNA induced by various concentrations of IL-1 were significantly lower in IRAK-deficient EF cells compared with control cells (Fig. 4 A). Similar decreases were also found in IL-6 secreted from IRAK-deficient cells compared with control cells (Fig. 4 B, top), and at various times after IL-1 treatment (data not shown). In three independent experiments, IL-6 induction by IL-1 was reduced by three- to sixfold. Thus, the reduced IL-1 signaling in IRAK-deficient cells results in decreased IL-6 production. To confirm the defects in IL-1 signaling and cellular response observed in IRAK-deficient EF cells, SF cells were obtained from mice deficient in IRAK expression, as described in Materials and Methods. Consistent with the observation in EF cells, IL-1–induced IL-6 production was significantly decreased in IRAK-deficient SF cells compared with SF cells prepared from wild-type mice (Fig. 4 B, bottom). IL-1–induced JNK activation and IκB degradation were also found to be defective in IRAK-deficient SF cells (data not shown).
Figure 4.

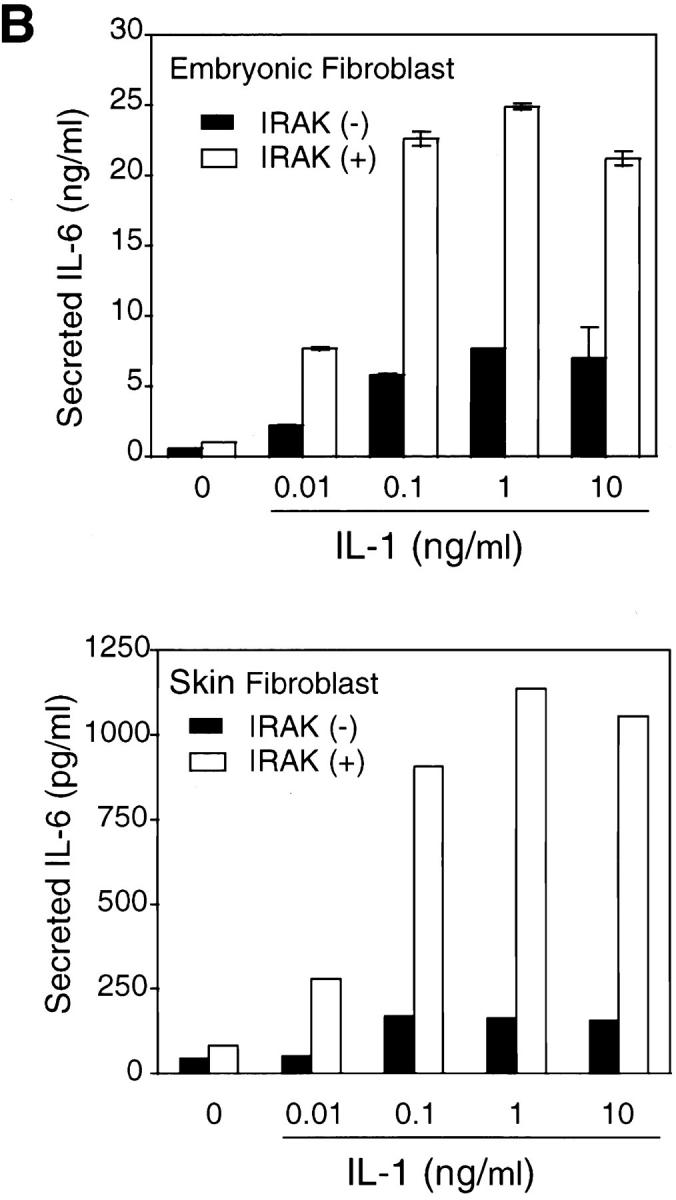
Decreased induction of IL-6 by IL-1 in IRAK-deficient cells. (A) Decreased induction of IL-6 mRNA in IRAK-deficient cells. Control (+) and IRAK-deficient (−) EF cells were treated with IL-1 or TNF-α for 3 h at 37°C. IL-6 mRNA expression in different samples was detected by Northern hybridization and quantitated on a PhosphorImager. (B) Decreased induction of secreted IL-6 in IRAK-deficient cells. Control (+) and IRAK-deficient (−) EF (top) and SF (bottom) cells were stimulated with IL-1 for 8 h at 37°C. The amount of secreted IL-6 in culture supernatants was determined by ELISA.
Role of IRAK in Regulating Multiple IL-1 Signaling Pathways.
Taken together, our data demonstrate that IRAK plays an important role in activating multiple IL-1 signaling pathways that lead to the induction of IL-1–responsive gene products such as IL-6. In the absence of IRAK, induction of JNK and p38 activities was significantly reduced at all concentrations of IL-1 tested, although not completely eliminated. Our results suggest that IRAK is required for optimal induction of JNK and p38 kinases and that loss of its function cannot be completely compensated for by IRAK-2 or other related kinases. Activation of JNK and p38 have been reported to be mediated by cascades of upstream kinases including MAP-2, MAP-3, and MAP-4 kinases (29). It remains to be determined at which levels within these cascades IRAK may act to induce JNK and p38 activity.
In contrast to the effects on JNK and p38, defects in NF-κB activation in IRAK-deficient cells could be overcome by high concentrations of IL-1. Our interpretation is that related kinases such as IRAK-2 may be able to fully activate the NF-κB pathway under these conditions. However, induction of cellular response to IL-1, such as IL-6 production, is dramatically reduced in IRAK-deficient cells even at high IL-1 concentrations. This suggests that activation of NF-κB alone is not sufficient for optimal induction of IL-1–mediated cellular responses. Induction of IL-6 and other IL-1–responsive gene products may be mediated synergistically by multiple pathways, including activation of NF-κB, and JNK/p38-mediated pathways. In the case of IL-6 secretion, the dramatic decrease in its induction by IL-1 in IRAK-deficient cells, even in the presence of full activation of NF-κB DNA-binding activity, may result from decreased activation of JNK/p38-dependent signaling pathways. This possibility is supported by a recent report that p38 and other MAP kinases are involved in TNF-induced IL-6 gene expression via modulation of transactivation potential of NF-kB without affecting its DNA binding activity (37).
Our observation of significant reduction in multiple signaling pathways and in IL-6 production induced by IL-1 in IRAK-deficient cells suggests that inhibitors of IRAK or IRAK-related kinases may be therapeutically useful for the treatment of IL-1–mediated inflammatory diseases.
Acknowledgments
We thank G. Olini, Julie Culver, and Michelle Courtney for their excellent technical assistance. We also thank Jonathan Sprent, Avery August, Linda Joliffe, Lars Karlsson, and Michael Jackson for helpful discussions.
Abbreviations used in this paper
EF
embryonic fibroblast
IκB
inhibitor of NF-κB
IRAK
interleukin 1 receptor–associated kinase
JNK
c-Jun NH2-terminal kinase
MAP
mitogen-activated protein kinase
MAPKAPK2
MAP kinase–activated protein kinase 2
NF-κB
nuclear factor κB
SF
skin fibroblast
TRAF
TNF receptor–associated factor
References
- 1.Dinarello CA. Biologic basis for interleukin–1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 2.Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol. 1997;159:3364–3371. [PubMed] [Google Scholar]
- 3.Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB, Bartfai T, Solorzano C, Moldawer LL, Chizzonite R, Mclntyre KW. Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor–deficient mice. J Immunol. 1997;159:2452–2461. [PubMed] [Google Scholar]
- 4.Leon LR, Conn CA, Glaccum M, Kluger MJ. IL-1 type I receptor mediates acute phase response to turpentine, but not lipopolysaccharide, in mice. Am J Physiol. 1996;271:R1668–R1675. doi: 10.1152/ajpregu.1996.271.6.R1668. [DOI] [PubMed] [Google Scholar]
- 5.Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann KJ, Conn CA, Soszynski D, Grabiec C, Trumbauer ME, Shaw A, et al. Resistance to fever induction and impaired acute phase response in interleukin-1β–deficient mice. Immunity. 1995;3:9–19. doi: 10.1016/1074-7613(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 6.Bankers-Fulbright JL, Kalli KR, McKean DJ. Interleukin-1 signal transduction. Life Sci. 1996;59:61–83. doi: 10.1016/0024-3205(96)00135-x. [DOI] [PubMed] [Google Scholar]
- 7.Wesche H, Korherr C, Kracht M, Falk W, Resch K, Martin MU. The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1–induced activation of interleukin-1 receptor–associated kinase (IRAK) and stress-activated protein kinases (SAP kinases) J Biol Chem. 1997;272:7727–7731. doi: 10.1074/jbc.272.12.7727. [DOI] [PubMed] [Google Scholar]
- 8.Korher C, Hofmeister R, Wesche H, Falk W. A critical role for interleukin-1 receptor accessory protein in interleukin-1 signaling. Eur J Immunol. 1997;27:262–267. doi: 10.1002/eji.1830270139. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Gao X, Li S, Cao Z. Recruitment of IRAK to the interleukin 1 receptor complex requires interleukin 1 receptor accessory protein. Proc Natl Acad Sci USA. 1997;94:12829–12832. doi: 10.1073/pnas.94.24.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 11.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 12.Croston E, Cao Z, Goeddel DV. NF-κB activation by interleukin-1 (IL-1) requires an IL-1 receptor–associated protein kinase activity. J Biol Chem. 1995;270:16514–16517. doi: 10.1074/jbc.270.28.16514. [DOI] [PubMed] [Google Scholar]
- 13.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 14.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 15.Song HY, Regnier CH, Kirschning CJ, Goeddel DV, Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor–κB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor– associated factor 2. Proc Natl Acad Sci USA. 1997;94:9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 17.Natoli G, Costanzo A, Moretti F, Fulco M, Balsano C, Levrero M. Tumor necrosis factor (TNF) receptor 1 signaling downstream of TNF receptor–associated factor 2. Nuclear factor κB (NFκB)-inducing kinase requirement for activation of activating protein 1 and NFκB but not of c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 1997;272:26079–26082. doi: 10.1074/jbc.272.42.26079. [DOI] [PubMed] [Google Scholar]
- 18.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 19.Woronicz D, Gao X, Cao Z, Rothe M, Goeddel DV. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 20.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li JW, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 21.Baldwin AS., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 22.Shelton CA, Wasserman SA. Pelle encodes a protein kinase required to establish dorsoventral polarity in the drosophila embryo. Cell. 1993;72:515–525. doi: 10.1016/0092-8674(93)90071-w. [DOI] [PubMed] [Google Scholar]
- 23.Lemaitre B, Nicolas E, Michaut L, Reichhart J-M, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/toll/cactus control the potent antifungal response in drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 24.Belvin MP, Anderson KV. A conserved signaling pathway: the Drosophila toll-dorsal pathway. Ann Rev Cell Dev Biol. 1996;12:393–416. doi: 10.1146/annurev.cellbio.12.1.393. [DOI] [PubMed] [Google Scholar]
- 25.Trofimova M, Sprenkle AB, Green M, Sturgill TW, Goebl MG, Harrington MA. Developmental and tissue-specific expression of mouse Pelle-like protein kinase. J Biol Chem. 1996;271:17609–17612. doi: 10.1074/jbc.271.30.17609. [DOI] [PubMed] [Google Scholar]
- 26.Fung-Leung W-P, Schilham MW, Rahemtulla A, Kudig TM, Vollenweider M, Potter J, van Ewijk W, Mak TW. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 1991;65:443–449. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- 27.Kanakaraj P, Duckworth B, Azzoni L, Kamoun M, Cantley LC, Perussia B. Phosphatidylinositol-3 kinase activation induced upon FcγRIIIA-ligand interaction. J Exp Med. 1994;179:551–558. doi: 10.1084/jem.179.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trotta R, Kanakaraj P, Perussia B. FcγR-dependent mitogen-activated protein kinase activation in leukocytes: a common signal transduction event necessary for expression of TNF-α and early activation genes. J Exp Med. 1996;184:1027–1035. doi: 10.1084/jem.184.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su B, Karin M. Mitogen activated protein kinase cascade and regulation of gene expression. Curr Opin Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- 30.Pulverer, B.J., J.M. Kyriakis, J. Avruch, E. Nikolakaki, and J. Woodgett. 1991. Phosphorylation of c-jun mediated by MAP kinases. _Natur_e. 353:670–674. [DOI] [PubMed]
- 31.Westwick JK, Weitzel C, Minden A, Karin M, Brenner DA. Tumor necrosis factor-α stimulates AP-1 activity through prolonged activation of the c-Jun kinase. J Biol Chem. 1994;269:26396–26401. [PubMed] [Google Scholar]
- 32.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 33.Bird TA, Kyriakis JM, Tyshler L, Gayle M, Milne A, Virca GD. Interleukin-1 activates p54 mitogen-activated protein (MAP) kinase/stress-activated protein kinase by a pathway that is independent of p21ras, Raf-1, and MAP kinase kinase. J Biol Chem. 1994;269:31836–31844. [PubMed] [Google Scholar]
- 34.Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, DeWitt DL, Saklatvala J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase. Regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]
- 35.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor α and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor κB. Proc Natl Acad Sci USA. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akira S, Kishimoto T. IL-6 and NF–IL-6 in acute-phase response and viral infection. Immunol Rev. 1992;127:25–50. doi: 10.1111/j.1600-065x.1992.tb01407.x. [DOI] [PubMed] [Google Scholar]
- 37.Berghe WV, Plaisance S, Boone E, De Bosscher K, Scmitz ML, Fiers W, Haegeman G. p38 and extracellular signal regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-κB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]