Urokinase Receptor (CD87) Regulates Leukocyte Recruitment via β2 Integrins In Vivo (original) (raw)
Abstract
The urokinase receptor (CD87; uPAR) is found in close association with β2 integrins on leukocytes. We studied the functional consequence of this association for leukocyte adhesion and migration. In vivo, the β2 integrin–dependent recruitment of leukocytes to the inflamed peritoneum of uPAR-deficient mice was significantly reduced as compared with wild-type animals. In vitro, β2 integrin–mediated adhesion of leukocytes to endothelium was lost upon removal of uPAR from the leukocyte surface by phosphatidyl-inositol–specific phospholipase C. Leukocyte adhesion was reconstituted when soluble intact uPAR, but not a truncated form lacking the uPA-binding domain, was allowed to reassociate with the cell surface. uPAR ligation with a monoclonal antibody induced adhesion of monocytic cells and neutrophils to vascular endothelium by six- to eightfold, whereas ligation with inactivated uPA significantly reduced cell-to-cell adhesion irrespective of the β2 integrin–stimulating pathway. These data indicate that β2 integrin–mediated leukocyte–endothelial cell interactions and recruitment to inflamed areas require the presence of uPAR and define a new phenotype for uPAR-deficient mice. Moreover, uPAR ligation differentially modulates leukocyte adhesion to endothelium and provides novel targets for therapeutic strategies in inflammation-related vascular pathologies.
Keywords: leukocyte, endothelial cells, urokinase receptor, β2 integrin, inflammation
Leukocyte activation and adhesion to the endothelium and the subsequent transendothelial migration are pivotal steps in the recruitment of cells to inflamed tissue. This highly coordinated multistep process requires tight regulation (1, 2). This includes the induction of genes coding for adhesion molecules and the modification of ligand-binding affinities of adhesion receptors on leukocytes as well as their change in avidity due to adhesion receptor clustering of leukocytes. Uncontrolled activation of leukocytes or endothelial cells leads to pathological chronic inflammation causing atherosclerosis, rheumatoid arthritis, and other disease states.
The β2 integrin family of adhesion receptors consists of the four members, LFA-1 (αLβ2, CD11a/CD18),1 Mac-1 (αMβ2, CD11b/CD18, CR3), p150,95 (αXβ2, CD11c/ CD18), and αDβ2 (CD11d/CD18) (3, 4). In acute inflammation, LFA-1 and Mac-1 are the predominant β2 integrins mediating leukocyte adhesion to vascular endothelium. Mac-1 is constitutively expressed on neutrophils and monocytes, whereas LFA-1 is predominantly expressed on lymphocytes, but recent data underline its important contribution in neutrophil recruitment (5, 6). Leukocyte activation via cytokines, chemoattractants, or PMA induces both conformational changes in β2 integrins necessary for enhanced ligand recognition and translocation of Mac-1 to the cell surface (2, 7). Likewise, integrin activation is achieved extracellularly in vitro by the divalent cation Mn2+ (8). After activation, Mac-1 and LFA-1 firmly bind to intercellular adhesion molecule (ICAM)-1 (CD54) expressed on vascular endothelial (1) and smooth muscle cells (9). β2 integrins have been reported to form complexes with other plasma membrane proteins such as CD63 (10), the immunoglobulin receptor FcγRIIIB (CD16b) (11) and the urokinase receptor (uPAR, CD87) (12, 13) suggesting possible functional interaction.
uPAR consists of three homologous domains and is anchored to the plasma membrane by a glycolipid moiety that is susceptible to dissociation by phosphatidyl-inositol– specific phospholipase C (piPLC) (14). Intact uPAR binds the protease uPA (urinary-type plasminogen activator) as well as the adhesive protein vitronectin with high affinity (15), and thereby plays a critical role in pericellular proteolysis and modulation of cellular contacts in adhesion and migration (16, 17). Although uPAR lacks its own transmembrane and cytoplasmic domain, uPA binding has been reported to transduce signals to the cell interior in leukocytes resulting in calcium mobilization (18), protein kinase phosphorylation (12, 19–21), and other cellular effects (18, 21, 22). For some of these functions, it has been suggested that uPAR uses related transmembrane integrins as signal transduction devices (23). In fact, uPAR has been localized together with different integrins in focal adhesion areas (24), and increased uPAR expression and localization of the receptor to the leading edge of migrating monocytes appears to be essential for locomotion or invasiveness of cells independent of uPA activity (20, 25). It has been shown in vitro that uPAR crucially influences integrin function (26): the presence of uPAR inhibited β1 integrin– mediated cell binding to fibronectin, whereas uPAR favored β2 integrin–dependent monocyte adhesion to fibrinogen, one of the adhesive ligands of Mac-1. Moreover, using uPA antisense or ligation with uPA inhibited Mac-1 binding to and degradation of fibrinogen (27, 28). Based on these studies, uPAR has been proposed to form a functional unit with integrins on the cell surface.
These relationships prompted us to investigate the contribution of uPAR in β2 integrin–mediated cell-to-cell interactions in vivo using uPAR knockout mice, as well as in vitro, to elute the mechanism of receptor cross-talk. Evidence is provided that uPAR is required for β2 integrin– dependent leukocyte adhesion and recruitment.
Materials and Methods
Materials
Manganese chloride was obtained from Sigma Chemical Co. (Munich, Germany) and PMA from GIBCO BRL (Paisley, Scotland). piPLC was from Oxford Glyco-Systems (Abingdon, UK). Intact recombinant soluble uPAR as well as the chymotrypsin-cleaved truncated form lacking domain 1 were produced as previously described (29, 30) and were provided by Dr. Niels Behrendt (Finsen Laboratory, Copenhagen, Denmark). uPA (Medac, Hamburg, Germany) was inactivated by diisopropyl-fluorophosphate (Serva, Heidelberg, Germany) as previously described (31).
Antibodies
The following mouse anti–human uPAR mAbs were used in vitro. mAb no. 3936 (IgG2a-type), provided by Dr. Richard Hart (American Diagnostica, Greenwich, CT), is known to block uPA binding by recognizing an epitope of uPAR that has not been clearly identified yet (32). (Fab′)2 fragments were generated using digestion by immobilized pepsin followed by protein A–Sepharose affinity chromatography (Pierce Chemical Co., Rockford, IL). Purity was controlled by polyacrylamide gel-electrophoretic analysis. mAbs R3 and R9, which recognize domain 1 and interfere with uPA binding, and mAbs R2, R4 and R8, which recognize domain 2 and 3 without influencing uPA binding, were provided by Dr. Gunilla Hoyer-Hansen (Finsen Laboratory, Copenhagen, Denmark). The characteristics of mAbs R2, R3, R4, R8, and R9 have been described previously (33). Mouse anti–human β2 chain (CD18) mAb 60.3 (IgG2a-type) blocks β2 integrin–mediated leukocyte adhesion to endothelium (34) and was provided by Dr. John Harlan (Harborview Medical Center, Seattle, WA). Mouse anti–human IgG2a (Sigma Chemical Co.) was used as isotype-matched control antibody. When necessary, fluorescein-conjugated mouse mAbs anti-CD11a (no. 25.3), anti-CD11b (Bear 1), and anti-CD18 (no. 7E4) (Immunotech, Hamburg, Germany) were used for flow cytometry.
The following reagents were used in animal experiments. R-PE-labeled streptavidin (Southern Biotechnology Associates, Inc., Birmingham, AL) and biotin-conjugated rat anti–mouse CD11b (M1/70), Ly-6G (Gr-1) (RB6-8C5), and CD3e (145-2C11) (all from PharMingen, San Diego, CA). Anti-αLβ2 integrin (CD11a) (FD 441.8) (35) and YN1.1 (36) (American Type Culture Collection, Rockville, MD) were affinity purified and dialyzed under endotoxin-free conditions.
Cells
Human myelo-monocytic HL60 and U937 cell lines (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) were cultured in RPMI 1640 supplemented with 10% (vol/vol) fetal calf serum, 1% sodium pyruvate, 1 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from GIBCO BRL). 24 h before experiments, monocytic differentiation was induced by addition of 50 ng/ml 1α,25-dihydroxyvitamin D3 and 1 ng/ml transforming growth factor β1 (Biomol, Hamburg, Germany). Peripheral blood PMNs were isolated by discontinuous density gradient centrifugation using Histopaque-1119 and -1077 (Sigma Chemical Co.) as described by the manufacturer. An enrichment of at least 95% neutrophils was obtained as controlled by flow cytometry using forward and side scatter analysis and staining for CD15. Human umbilical vein endothelial cells (HUVECs), provided by Dr. Bernd Pötzsch (Kerckhoff-Klinik), were isolated as previously described (37) and cultured (for 2–4 passages) in low serum endothelial cell growth medium (PromoCell, Heidelberg, Germany) on gelatin-coated tissue-culture plastic. Human vascular smooth muscle cells (HVSMCs) were isolated from aorta or saphenous vein by the explant method and characterized as previously described (38). Early passage cells were cultured in smooth muscle cell medium (PromoCell).
Peritonitis Assay
Peritonitis was induced in female uPAR−/− (39) or wild-type mice by intraperitoneal injection of a solution of 4% (wt/vol) in Bacto Fluid Thioglycollate Medium (Difco Labs., Detroit, MI). Endotoxin-free stock solutions were prepared at 100°C and subsequently heat-sterilized.
Mice obtained by shipping were generally kept (at the Basel Institute for Immunology) for at least 2 wk before the start of experiments to relieve stress. PBS alone or antibodies against ICAM-1, LFA-1 (200 μl of a 1 mg/ml solution each) were injected intravenously into the tail vein 30 min before induction of peritonitis. All reagents were endotoxin-free. After 4 or 24 h, respectively, mice were killed and peritoneal cells were suspended by injection of 5 ml PBS containing 2 mM EDTA and 50 μg/ml heparin into the peritoneum. The loaded mouse was shortly massaged and 4 ml of this lavage were collected and leukocytes were counted on a Coulter counter ZM equipped with a channelizer 256 (Coulter Corp., Miami, FL). Animal studies were approved by the Institutional Review Board.
Adhesion Assays
HUVECs were seeded in gelatin-coated 48-well plates (Costar, Badhoevedorp, The Netherlands) 48 h before the experiment. Confluency was confirmed by microscopic inspection before each experiment. HL60 or U937 cells were radiolabeled with 1 μCi/ml methyl-[3H]thymidine (Nycomed Amersham, Little Chalfont, Buckinghamshire, UK) for 24 h and differentiated to monocytic cells (see above) for another 24 h. These monocytic cells or freshly isolated neutrophils were washed twice in adhesion medium (serum-free RPMI 1640/Hepes 25 mM), followed by different pretreatments (see figure legends for details), and were added (7 × 105/ml adhesion medium) to the prewashed HUVEC monolayers in the presence or absence of the blocking mAb anti–β2 integrin or isotype IgG (final concentration 10 μg/ ml). After 30 min of coincubation (37°C, 5% CO2, 90% humidity), the plates were gently washed twice with adhesion buffer to remove nonadherent cells. Remaining adherent cells were lysed with 1 M NaOH and quantitated in a beta counter. When flow cytometry was performed in parallel to neutrophil adhesion, the number of adherent cells in 10 high power fields was counted using light microscopy. At least triplicate wells were run per test substance, and results are expressed as mean values ± SEM. The experimental protocol for cell adhesion to HVSMCs, which were seeded 7–9 d before the assay, was identical.
Flow Cytometry
Animal Model.
The mouse leukocyte subpopulations were further analyzed by flow cytometry (Becton Dickinson, Heidelberg, Germany) using the biotin-coupled anti–mouse CD11b (integrin αM chain, Mac-1), anti–Gr-1 (anti-myeloid differentiation antigen Gr-1), or anti-CD3 (anti-CD3 TCR-associated complex). Fc receptors of leukocytes were blocked by preincubation of the cells for 30 min with 5% (vol/vol) rat and 5% (vol/ vol) mouse serum in PBS. The secondary reagent PE-coupled streptavidin (dilution of 1:2,500) was incubated with the cells for 30 min.
In Vitro System.
Cells (2.5 × 105) were washed twice with Hepes-buffered saline and incubated with primary mouse anti– human antibodies for 30 min on ice (for CD11b detection at 21°C). Cells were washed again and resuspended in Hepes buffer containing fluorescein-conjugated (Fab′)2 fragment of goat anti– mouse IgG (Dianova, Hamburg, Germany). To test the effect of mouse antibodies on β2 integrin expression, fluorescein-conjugated mAbs at a dilution of 1:50 were used. Mean fluorescence of 5,000 cells was measured in a flow cytometer (Becton Dickinson). Nonspecific fluorescence was determined using an isotype-matched mouse IgG as the primary antibody.
Statistical Analysis
Comparisons between group means were performed using multivariant analysis (ANOVA). Data represent mean ± SEM; P < 0.05 was regarded as significant.
Results
Leukocyte Emigration in uPAR-deficient Mice.
Transendothelial migration of leukocytes to inflamed tissue depends on the interaction of the leukocyte with the vascular endothelium by β2 integrins and ICAM-1. Thioglycollate- induced peritonitis is a reliable model to test leukocyte emigration into sites of acute inflammation. Disruption of the mouse ICAM-1–β2 integrin interactions resulted in reduced leukocyte emigration in this model when compared with wild-type animals (40). Both uPAR-deficient and wild-type animals of the identical genotype (129 × C57/ BL6 F1) were compared for leukocyte emigration in the peritonitis model. The number and types of leukocytes in the peripheral blood were identical in both sets of mice (data not shown). Lavages performed 4 (Fig. 1) and 24 h (data not shown) after induction of peritonitis showed ∼50% reduction in counts of the total leukocyte population in uPAR-deficient mice when compared with wild-type animals (Fig. 1). When animals were treated with anti–ICAM-1 or anti–LFA-1 antibodies at the time of induction of peritonitis, the number of emigrating leukocytes was further reduced by 50% in wild-type mice, but by only 30% in uPAR-deficient animals, suggesting that a major part of the initial lack of emigration was due to a perturbed β2 integrin/ICAM-1 function. Analysis of the leukocyte subpopulations by flow cytometry using specific markers as indicated in Materials and Methods revealed that in uPAR-deficient mice granulocytes almost totally lost their ability to migrate into the peritoneum after 4 and 24 h of inflammation (Fig. 2). Myeloid lineage cells showed significant reduction in recruitment after 4 h (∼55%) and 24 h (∼70%), whereas T lineage cells were hardly affected by the absence of uPAR after 4 h, but showed significant inhibition in emigration (∼60%) after 24 h (Fig. 2). Consistently, administration of mAbs demonstrated that lymphocyte recruitment after 4 h was largely independent of LFA-1–ICAM-1 interactions in contrast to recruitment after 24 h of inflammation.
Figure 1.

Leukocyte emigration in thioglycollate-induced peritonitis. Wild-type mice (white bars) and uPAR-deficient mice (black bars) were injected intraperitoneally with buffer alone (Control) or with thioglycollate solution in the absence or presence of the indicated antibodies. After leukocyte emigration for 4 h, mice were killed and the leukocytes were counted in lavages from the peritoneum. SE was calculated from four animals for each condition and the experiment was repeated twice. # P < 0.02; *P < 0.01.
Figure 2.
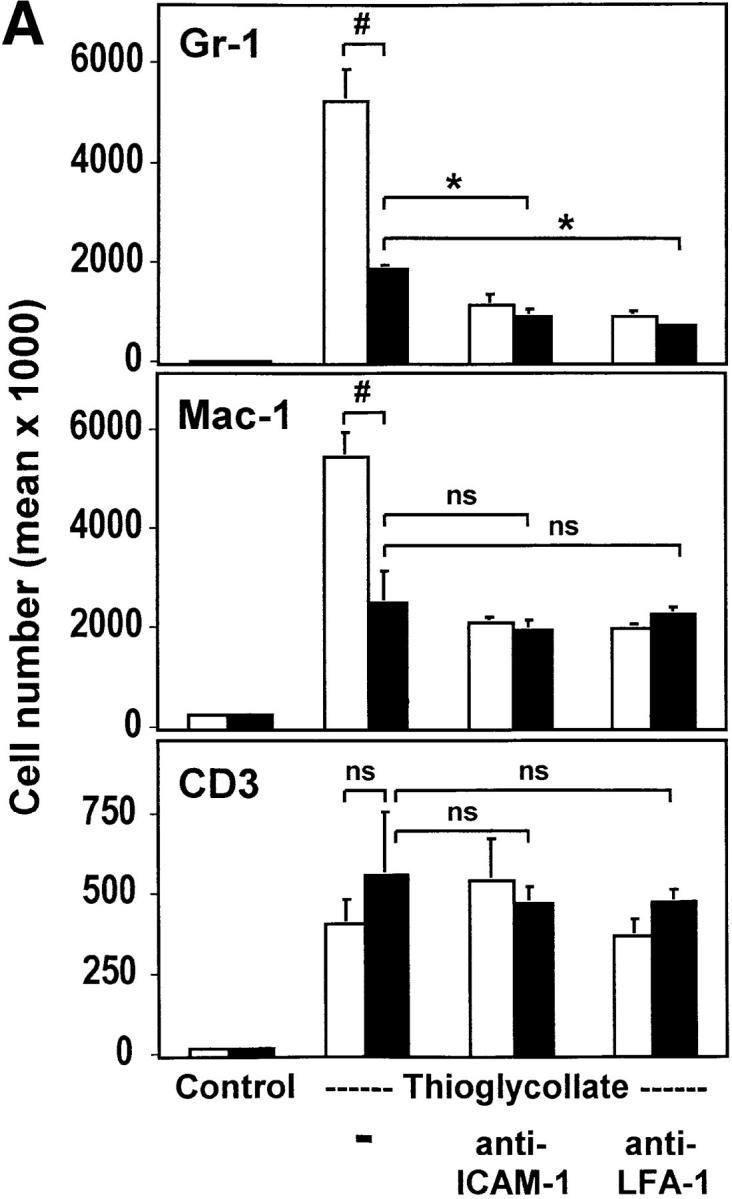

Analysis of subpopulations of emigrated leukocytes in the peritoneal lavage. Leukocytes obtained in peritoneal lavages after induction of peritonitis for 4 (A) or 24 h (B) from wild-type mice (white bars) and uPAR-deficient mice (black bars) were analyzed by flow cytometry for the expression of Gr-1 (anti-granulocytes), Mac-1 (anti-myeloid cells), or CD3 (anti-CD3 TCR complex). Absolute numbers of cells were calculated from the percentage of stained cells and the number of total emigrated cells shown in Fig. 1. # P < 0.01; *P < 0.002; § P < 0.005.
To further specify those granulocytic subpopulations that were mostly affected, a differential cell staining (May-Grünwald-Giemsa) was performed (Fig. 3). In uPAR-deficient mice, after 4 h, neutrophil and eosinophil recruitment was inhibited by >70% or 90%, respectively, and residual emigration was marginally affected by the administration of mAbs against ICAM-1 or LFA-1, respectively. In contrast, in wild-type mice the recruitment of these two cell types was effectively blocked by these antibodies down to the level of emigrated cells in uPAR−/− mice, suggesting that leukocyte recruitment in uPAR-deficient mice is diminished through impaired function of the β2 integrin/ICAM-1 system. Basophil emigration into the inflamed peritoneum was not significantly affected in uPAR-deficient mice but was comparable to that in wild-type mice receiving anti– ICAM-1 or anti–LFA-1 mAb, respectively. Thus, the definitive role of uPAR for basophil recruitment is not yet clear and requires further investigation. Comparable findings for granulocyte subpopulations were noted after 24 h of inflammation (data not shown). These data provide in vivo evidence for a functional consequence of the uPAR/β2 integrin system in leukocyte adhesion/migration and present a new phenotype for uPAR-deficient mice.
Figure 3.
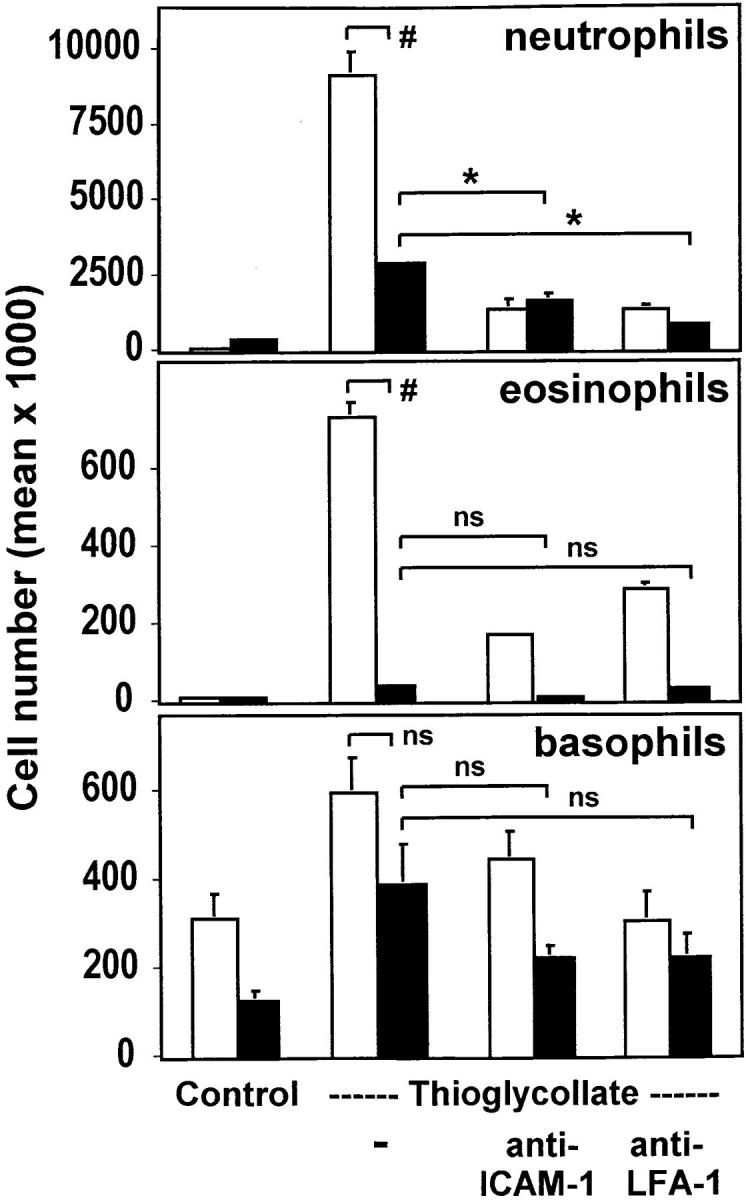
Quantitation of granulocyte subpopulations in the peritoneal lavage. The migrated leukocytes from wild-type mice (white bars) and uPAR-deficient mice (black bars) were cytocentrifuged and stained with May-Grünwald-Giemsa solution. The quantitation of cell numbers of leukocyte subpopulations was performed by light microscopy. SE was calculated from four animals for each condition and the experiment was repeated twice. # P < 0.02; *P < 0.005.
Requirement of uPAR for Leukocyte Adhesion to Endothelium In Vitro.
uPAR is expressed on circulating human blood cells, such as granulocytes, monocytes, and activated T cells. The human myelo-monocytic cell lines HL60 and U937 differentiate into mononuclear phagocyte–like cells after treatment with vitamin D3 and transforming growth factor β1 (31). This in vitro differentiation induced the expression of a monocyte-specific antigen pattern including the molecules CD14, CD11b, CD18, and CD87 (uPAR) as measured by flow cytometry (data not shown). Adhesion of these differentiated monocytic cells to endothelial cell monolayers was induced six- to eightfold by PMA or Mn2+, both reagents known for their activation of β2 integrins. The mAb 60.3 directed against the β2 integrin chain blocked adhesion by 75–85% in both cases, indicating that leukocyte adhesion to endothelium was β2 integrin dependent (Fig. 4).
Figure 4.

Requirement of uPAR for leukocyte adhesion to endothelial cells. Myelo-monocytic HL60 cells were either not treated (hatched bars) or pretreated with piPLC (0.5 U/ml, 90 min, 37°C; black bars), washed, and incubated for 10 min without or with soluble intact uPAR (0.8 μg/ml), followed by stimulation for 20 min with (A) PMA (10 ng/ml) or (B) Mn2+ (1 mM). After another washing step, the adhesion assay was performed in the absence or presence of anti–β2 integrin mAb 60.3 (10 μg/ml). Values are displayed as percent of PMA- or Mn2+-inducible adhesion and represent the mean ± SEM of three independent experiments. *P < 0.001 as compared with control.
Pretreatment of leukocytes with piPLC removed ∼80% of uPAR from the cell surface as assessed by flow cytometry (data not shown). This resulted in a 70–80% reduction of PMA-induced or a 65–70% reduction of Mn2+-induced leukocyte adhesion (Fig. 4). Reconstitution of the uPAR-depleted cells with intact soluble uPAR for 10 min restored adhesion to the level as observed with untreated cells. This restoration of uPAR-induced adhesion was abrogated by anti-CD18 antibody, indicating that the β2 integrin–dependent adhesion was inducible by uPAR.
In a dose-dependent manner, intact soluble uPAR increased adhesion of piPLC-treated leukocytes to endothelium to maximal levels, whereas addition of the truncated form of uPAR lacking the uPA binding domain D1 did not, indicating a specific structural requirement for β2 integrin activation (Fig. 5). In contrast to PMA, which induces both increased surface expression and activation of β2 integrins, flow cytometric analysis revealed no change in integrin expression after Mn2+ treatment (data not shown), as previously described (8). Likewise, removal of uPAR did not affect surface expression of the αL, αM, or β2 chain of the integrins in resting or stimulated cells as analyzed by flow cytometry (data not shown). Thus, the presence of uPAR appears to support adhesion by regulating integrin function rather than by quantitatively changing β2 integrin levels on the cell surface.
Figure 5.

Reconstitution of leukocyte adhesion to endothelium by soluble uPAR. Adhesion of monocytic cells to HUVECs was performed as described in the legend to Fig. 4. piPLC-pretreated myelo-monocytic HL60 cells were incubated with increasing concentrations of soluble intact (D1/D2/D3) uPAR or with its truncated form (D2/D3) lacking domain 1, respectively, and were subsequently stimulated by PMA. Values are displayed as percentage of PMA-inducible adhesion in non–piPLC-pretreated cells and one representative experiment out of three is shown. # P < 0.01 and *P < 0.001 as compared with piPLC-pretreated cells without soluble uPAR.
Anti-uPAR mAb No. 3936 Induces Leukocyte Adhesion to Endothelial and Smooth Muscle Cells via β2 Integrins.
Since uPAR is directly involved in β2 integrin–mediated leukocyte adhesion to the endothelium, we examined the consequences of uPAR occupancy on leukocyte adhesion to endothelial cells using different mAbs against uPAR. Preincubation with the anti-uPAR mAb no. 3936 resulted in the six- to eightfold increase of cell adhesion reaching the same maximal level as achieved with PMA or Mn2+ (Fig. 6). For undifferentiated HL60 or U937 cells, which show low surface expression of β2 integrins and uPAR, respectively, the activating anti-uPAR mAb increased adhesion only 1.5–2-fold (data not shown). Boiling of the antibody totally abrogated its adhesion-stimulating effect. In addition, (Fab′)2 fragments of mAb no. 3936 had a very similar proadhesive effect as compared with the intact mAb. Control isotype– matched mAb IgG2a as well as other anti-uPAR mAbs directed against different epitopes of uPAR did not induce leukocyte adherence. The dose- and time-dependent kinetics of antibody ligation suggested that the uPAR–mAb complex serves as a fast and effective trigger of β2 integrin activation (Fig. 7). In contrast, the anti-uPAR mAb R3 that has the same inhibitory effect on uPA binding to the receptor as mAb no. 3936 was not able to promote leukocyte adhesion. The anti-uPAR mAbs did not significantly change β2 integrin expression (data not shown), indicating that a functional conformational change of the adhesion receptor is responsible for the proadhesive effect.
Figure 6.

Effect of different anti-uPAR mAbs on leukocytic cell adhesion to endothelium. Myelo-monocytic HL60 cells were incubated for 30 min with different anti-uPAR mAbs (20 μg/ml each) representing uPA-blocking (no. 3936, R3, R9) or -nonblocking (R2, R4, R8) activity, with (Fab′)2 of mAb no. 3936 (12 μg/ml), boiled (5 min) mAb no. 3936, or control IgG2a as indicated. After washing, the adhesion assay was performed. Values are displayed as percentage of control (no antibody added) and represent the mean (± SEM) of at least three independent experiments. *P < 0.001 as compared with control.
Figure 7.

Induction of leukocyte adhesion to endothelium by anti-uPAR mAb no. 3936. Anti-uPAR mAb no. 3936 (▪) or R3 (□) were added to myelo-monocytic HL60 cells for 1 h at different concentrations (A), or at a concentration of 10 μg/ml for different time intervals (B). After washing, the adhesion assay was performed as described. Values (mean ± SEM, n = 3) are displayed as percentage of control (no antibody added). One representative experiment out of three is shown. *P < 0.001 as compared with control (no antibody added or time 0).
Analogous to PMA- or Mn2+-induced β2 integrin–dependent cell adhesion, mAb 60.3 could abrogate mAb no. 3936–induced leukocyte adhesion to cultured endothelial and smooth muscle cells, indicating β2 integrin dependency (Fig. 8). In addition, vitamin D3/transforming growth factor β1 differentiated U937 cells and freshly isolated peripheral blood neutrophils responded to mAb no. 3936 in an identical manner, emphasizing that this functional interaction of uPAR with β2 integrins occurs in different cell populations such as monocytes and neutrophils.
Figure 8.

Induction of leukocyte adhesion to endothelial and smooth muscle cells by anti-uPAR mAb no. 3936. Myelo-monocytic HL60 cells (black bars), monocytic U937 cells (hatched bars) and freshly isolated neutrophils (white bars) were pretreated with anti-uPAR mAb no. 3936 (20 μg/ml) for 30 min. After washing, the adhesion assay was performed in the absence or presence of anti–β_2_ integrin mAb 60.3 or isotype control mAb (10 μg/ml) as indicated. Values (mean ± SEM, n = 3) are displayed as percentage of control (no antibody added), and one representative experiment out of three is shown. *P < 0.001 as compared with anti-uPAR-induced adhesion.
Occupancy of uPAR by uPA Reduces β2 Integrin Activation– dependent Leukocyte Adhesion to Endothelium.
Although the addition of exogenous uPA, the natural ligand of uPAR, had no effect on background adhesion, uPA significantly inhibited β2 integrin–mediated adhesion in response to any of the three described stimulating pathways (Fig. 9); adhesion of leukocytes to endothelium achieved by cell activation via PMA, direct extracellular integrin activation via Mn2+, or uPAR ligation by the activating mAb no. 3936 was inhibited by active (data not shown) as well as by enzymatically inactive uPA. Thus, uPA binding to uPAR independent of its catalytic activity appears to control β2 integrin activation irrespective of the integrin-activating stimulus. Since uPA or its inactivated isoform did not alter the surface expression of the integrin αL, αM, or β2 chain in either resting or stimulated cells (data not shown), we propose that the binding of uPA to uPAR prevents the induction of conformational change(s) leading to β2 integrin activation.
Figure 9.

Interference of uPA with leukocyte adhesion to endothelium. Myelo-monocytic HL60 cells were incubated in the absence (hatched bars) or the presence (black bars) of inactivated uPA (50 nM) for 30 min (37°C, 5% CO2) before addition of medium, PMA (10 ng/ml), Mn2+ (1.0 mM), or anti-uPAR mAb no. 3936 (10 μg/ml) for another 30 min, as indicated. After washing, the adhesion assay was performed. Values are displayed as percent of untreated control (medium) and represent the mean ± SEM of three independent experiments. *P < 0.01.
Discussion
The major contribution of β2 integrins in immune defense and inflammatory processes relates to their pivotal role in mediating cellular contacts between leukocytes and endothelium as a prerequisite for subsequent transmigration towards a chemotactic stimulus. Previous in vitro studies have demonstrated that uPAR forms complexes with integrins (12, 13) and thereby modulates integrin-mediated binding to extracellular matrix proteins (26–28).
This study demonstrates that uPAR is needed for β2 integrin–dependent leukocyte recruitment into sites of acute inflammation. Migration of neutrophils and monocytes into the inflamed peritoneum was drastically reduced after 4 h in uPAR-deficient mice. Consistently, β2 integrin– dependent cell adhesion of leukocytes to endothelial cells was abrogated after depletion of uPAR from the cell surface, whereas reconstitution with soluble intact, but not truncated, uPAR could totally rescue β2 integrin–mediated adhesion. Regulation of β2 integrin activity probably involves uPAR domain 1, since occupancy by a mAb that blocks uPA-binding to this domain strongly induced leukocyte adhesion to vascular endothelial cells, whereas uPA itself inhibited β2 integrin–dependent adhesion.
The adhesion of leukocytes to inflamed vascular endothelium and the transendothelial migration largely depend on the activation of β2 integrins and binding to its counter-receptor ICAM-1 (41, 42), as evidenced by inhibition and gene-targeting studies (40, 43, 44). Although β2 integrin complexes are essential to neutrophil emigration, recent results from β2-deficient mice demonstrated that Mac-1/ LFA-1–independent pathways for cell recruitment can be used during acute inflammation in the peritoneum and the lung (45).
In this study, a new phenotype for uPAR-deficient mice is described that is similar to that of wild-type animals after treatment with inhibiting antibodies against the vascular β2 integrin LFA-1 and its ligand ICAM-1. Further blockade with mAbs of leukocyte migration in uPAR−/− mice was marginal, suggesting that the function of the β2 integrin/ ICAM-1 adhesion system was blocked by the absence of uPAR. This strongly suggests that leukocyte recruitment to the acutely inflamed peritoneum requires the uPAR. In fact, under conditions of acute inflammation the phenotype of uPAR mice resembles that of ICAM-1– or αL-deficient mice (40, 46).
In accordance with the known role of β2 integrins for the acute and early inflammatory responses, reduced leukocyte infiltration in uPAR−/− mice was observed after short-term inflammation for 4 or 24 h. It was shown previously that the total number of migrated leukocytes in uPAR−/− mice was not affected after long-term inflammation for 3 d (47). This may be due to the fact that prolonged inflammation upregulates and activates several other adhesion receptor systems on leukocytes and vascular cells apart from β2 integrins, such as β1 and β7 integrins, vascular cell adhesion molecule 1, or addressins, all of which contribute to leukocyte recruitment (1). Especially after long-term inflammation, the α4β1 integrin may substitute for β2 integrins. The finding that migration of granulocytes that do not express α4β1 integrin was mainly affected by the absence of the uPAR further supports this concept.
Recent in vitro studies have shown that the presence of uPAR is needed for Mac-1 binding to fibrinogen (27), and it also regulates fibrinogen degradation by forming a functional unit with the β2 integrin (28). Consistently, β2 integrin–dependent cell-to-cell adhesion required the presence of endogenous or exogenously added intact (soluble) uPAR in vitro. In the absence of uPAR, neither PMA nor Mn2+ induction of piPLC-treated leukocytes was sufficient to allow adhesion, indicating a superior regulatory function for uPAR regardless of the stimulatory pathway. Moreover, the intact (soluble) uPAR appears to be crucial in the cross-talk with β2 integrins (Fig. 10), implying direct binding interactions between these two receptors as was demonstrated in an isolated system for Mac-1 (26). In contrast, the truncated form of uPAR was ineffective, and uPA itself significantly inhibited cell-to-cell adhesion, suggesting that domain 1 of uPAR is predominantly involved in β2 integrin activation. A similar inhibitory effect of uPA on β2 integrin function has previously been proposed with regard to Mac-1–mediated fibrinogen binding and degradation (27, 28).
Figure 10.

Hypothetical model for the uPAR–β2 integrin cross-talk. The presence of uPAR on leukocyte subpopulations is required for their adhesion to endothelium and their subsequent recruitment into the inflamed tissue as documented here for a peritonitis model. In vitro, ligation of uPAR by a specific mAb no. 3936 induces leukocyte adhesion to endothelial cells, whereas uPA greatly inhibits cell-to-cell interactions independent of the stimulatory pathway.
Although uPAR-dependent proteolytic activity did not seem to be critical for leukocyte recruitment (47), the focalized proteolytic activity of the uPAR–uPA complex is tightly correlated with the migratory or invasive potential of cells in a variety of biological systems (48). On the other hand, active uPA has been found to support cell migration in vitro via proteolysis or cell-activating processes and was required for adequate leukocyte recruitment into the lungs and a subsequent inflammatory response after 3 wk of fungal infection (49). Collectively, our combined in vivo and in vitro studies together with previous reports strongly indicate that proteolysis-independent cross-talk of uPAR with β2 integrins occurs in the initial phase of leukocyte interaction with the vessel wall, whereas subsequent recruitment into inflamed tissue may require proteolysis-dependent mechanisms including plasmin action.
uPAR appeared to induce and facilitate β2 integrin activation on its own when stimulated by the specific anti-uPAR mAb no. 3936 or its (Fab′)2 fragment. Although cross-linking of uPAR cannot be ruled out, this antibody provides an additional stimulatory pathway for β2 integrin engagement. In accordance with this concept, uPA was found to interfere by either competing for mAb no. 3936 binding or by not allowing integrin activation to occur as pointed out before. Since removal of cell surface–associated uPA by acidic wash (data not shown) and the use of other antibodies that also block uPA binding to uPAR (such as R3) did not affect cell adhesion, we attribute the proadhesive capability of mAb no. 3936 to its specific ligation of uPAR rather than to competition with endogenous uPA. In contrast to our findings that the mAb no. 3936 induces β2 integrin–dependent cell-cell adhesion, Mac-1–mediated leukocyte adhesion to fibrinogen has been shown to be inhibited by the anti-uPAR mAb 3B10 (27). These diverse findings may be due to the experimental systems used and/or to the different integrins or ligands involved. Although vitronectin and PAI-1 are known to regulate uPAR as well as αv integrin–dependent cell-substrate adhesion (50–52), both factors did not interfere with β2 integrin–dependent cell–cell adhesion (data not shown), underlining the specificity of the uPAR system in this regard.
Our findings indicate that uPAR plays a major role for leukocyte adhesion. Nevertheless, patients with paroxysmal nocturnal hemoglobinuria (PNH) lacking cell-bound uPAR and other cell surface proteins due to a defective production of glycolipid anchors do not necessarily show an enhanced susceptibility for infections, whereas isolated neutrophils from these patients are impaired in their transendothelial migration in vitro (53). Possible explanations might be that (a) in PNH, which is an acquired clonal disease, a portion of the cells remains unaffected and may therefore be sufficient for host defense as demonstrated in the experiments with uPAR-deficient mice; or (b) circulating soluble uPAR is enhanced three- to fourfold in PNH patients (54) and might be sufficient for β2 integrin function in vivo in these patients, as documented here in vitro. The present observations indicate that uPAR controls integrin-mediated interactions in vitro as well as in vivo, and these findings might have therapeutic consequences for the treatment of hyperinflammatory or invasive processes related to vascular or immune diseases.
Acknowledgments
We are grateful to Barbara Eccabert (Basel Institute for Immunology) for her skilful expert assistance in the animal experiments and Drs. Niels Behrendt and Gunilla Hoyer-Hansen for their generous supply of soluble intact and truncated uPAR as well as anti-uPAR antibodies. We appreciate the kind gift of anti-β2 integrin antibody 60.3 from Dr. John Harlan. We also thank our colleagues Dr. Bernd Pötzsch and Bettina Kropp for providing HUVEC and Drs. Matthias Germer and Karl-Dieter Wohn for very helpful discussions.
Abbreviations used in this paper
HUVEC
human umbilical vein endothelial cells
HVSMC
human vascular smooth muscle cells
ICAM-1
intercellular adhesion molecule 1
LFA-1
αLβ2 integrin (CD11a/CD18)
Mac-1
αMβ2 integrin (CD11b/CD18)
ns
not significant
piPLC
phosphatidyl-inositol–specific phospholipase C
uPA
urokinase (urinary-type plasminogen activator)
uPAR
urokinase receptor
Footnotes
A.E. May was supported by a postdoctoral fellowship grant from the Deutsche Gesellschaft für Kardiologie, Herz- und Kreislaufforschung. Part of this work was supported by a grant (Pr 327/1-3) from the Deutsche Forschungsgemeinschaft and by a grant (no. 31-49241.96) from the Swiss Science Foundation.
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 3.Larson RS, Springer TA. Structure and function of leukocyte integrins. Immunol Rev. 1990;114:181–217. doi: 10.1111/j.1600-065x.1990.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 4.Van der Vieren M, Le Trong H, Wood CL, Moore PF, St. John T, Staunton DE, Gallatin WM. A novel leukointegrin, αdβ2, binds preferentially to ICAM-3. Immunity. 1995;3:683–690. doi: 10.1016/1074-7613(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 5.Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989;83:2008–2017. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu H, Smith CW, Perrard J, Bullard D, Tang LP, Shappell SB, Entman ML, Beaudet AL, Ballantyne CM. LFA-1 is sufficient in mediating neutrophil emigration in MAC-1–deficient mice. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller LJ, Bainton DF, Borregaard N, Springer TA. Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. J Clin Invest. 1987;80:535–544. doi: 10.1172/JCI113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altieri DC. Occupancy of CD11b/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J Immunol. 1991;147:1891–1898. [PubMed] [Google Scholar]
- 9.Couffinhal T, Duplaa C, Moreau C, Lamaziere J-MD, Bonnet J. Regulation of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 in human vascular smooth muscle cells. Circ Res. 1994;74:225–234. doi: 10.1161/01.res.74.2.225. [DOI] [PubMed] [Google Scholar]
- 10.Skubitz KM, Campbell KD, Iida J, Skubitz APN. CD63 associates with tyrosine kinase activity and CD11/CD18, and transmits an activation signal in neutrophils. J Immunol. 1996;157:3617–3626. [PubMed] [Google Scholar]
- 11.Zhou M, Todd RF, III, van de Winkel JG, Petty HR. Cocapping of the leukoadhesin molecules complement receptor type 3 and lymphocyte function–associated antigen-1 with Fcγ receptor III on human neutrophils. Possible role of lectin-like interactions. J Immunol. 1993;150:3030–3038. [PubMed] [Google Scholar]
- 12.Bohuslav J, Horejsí V, Hansmann C, Stöckl J, Weidle UH, Majdic O, Bartke I, Knapp W, Stockinger H. Urokinase plasminogen activator receptor, β2-integrins, and Src-kinases within a single receptor complex of human monocytes. J Exp Med. 1995;181:1381–1390. doi: 10.1084/jem.181.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue W, Kindzelskii AL, Todd RF, III, Petty HR. Physical association of complement receptor type 3 and urokinase-type plasminogen activator receptor in neutrophil membranes. J Immunol. 1994;152:4630–4640. [PubMed] [Google Scholar]
- 14.Ploug M, Ronne E, Behrendt N, Jensen AL, Blasi F, Dano K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J Biol Chem. 1991;266:1926–1933. [PubMed] [Google Scholar]
- 15.Hoyer-Hansen G, Ronne E, Solberg H, Behrendt N, Ploug M, Lund LR, Ellis V, Dano K. Urokinase plasminogen activator cleaves its cell surface receptor releasing the ligand-binding domain. J Biol Chem. 1992;267:18224–18229. [PubMed] [Google Scholar]
- 16.Kanse SM, Kost C, Wilhelm OG, Andreasen PA, Preissner KT. The urokinase receptor is a major vitronectin binding protein on endothelial cells. Exp Cell Res. 1996;224:344–353. doi: 10.1006/excr.1996.0144. [DOI] [PubMed] [Google Scholar]
- 17.Chapman HA. Plasminogen activators, integrins, and the coordinated regulation of cell adhesion and migration. Curr Opin Cell Biol. 1997;9:714–724. doi: 10.1016/s0955-0674(97)80126-3. [DOI] [PubMed] [Google Scholar]
- 18.Cao D, Mizukami IF, Garni-Wagner BA, Kindzelskii AL, Todd RF, III, Boxer LA, Petty HR. Human urokinase-type plasminogen activator primes neutrophils for superoxide anion release. Possible roles of complement receptor type 3 and calcium. J Immunol. 1995;154:1817–1829. [PubMed] [Google Scholar]
- 19.Dumler I, Petri T, Schleuning W-D. Interaction of urokinase-type plasminogenactivator (u-PA) with its cellular receptor (u-PAR) induces phosphorylation on tyrosine of a 38 kDA protein. FEBS Lett. 1993;322:37–40. doi: 10.1016/0014-5793(93)81106-a. [DOI] [PubMed] [Google Scholar]
- 20.Busso N, Masur SK, Lazega D, Waxman S, Ossowski L. Induction of cell migration by pro-urokinase binding to its receptor: possible mechanism for signal transduction in human epithelial cells. J Cell Biol. 1994;126:259–270. doi: 10.1083/jcb.126.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Resnati M, Guttinger M, Valcamonica S, Sidenius N, Blasi F, Fazioli F. Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. EMBO (Eur Mol Biol Organ) J. 1996;15:1572–1582. [PMC free article] [PubMed] [Google Scholar]
- 22.Rao NK, Shi G-P, Chapman HA. Urokinase receptor is a multifunctional protein: influence of receptor occupancy on macrophage gene expression. J Clin Invest. 1995;96:465–474. doi: 10.1172/JCI118057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petty HR, Todd RF., III Integrins as promiscuous signal transduction devices. Immunol Today. 1996;17:209–212. doi: 10.1016/0167-5699(96)30013-3. [DOI] [PubMed] [Google Scholar]
- 24.Pöllänen J, Stephens RW, Vaheri A. Directed plasminogen activation at the surface of normal and malignant cells. Adv Cancer Res. 1991;57:273–328. doi: 10.1016/s0065-230x(08)61002-7. [DOI] [PubMed] [Google Scholar]
- 25.Gyetko MR, Todd RF, III, Wilkinson CC, Sitrin RG. The urokinase receptor is required for human monocyte chemotaxis in vitro. J Clin Invest. 1994;93:1380–1387. doi: 10.1172/JCI117114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- 27.Sitrin RG, Todd RF, III, Petty HR, Brock TG, Shollenberger SB, Albrecht E, Gyetko MR. The urokinase receptor (CD87) facilitates CD11B/CD18-mediated adhesion of human monocytes. J Clin Invest. 1996;97:1942–1951. doi: 10.1172/JCI118626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon DI, Rao NK, Xu H, Wei Y, Majdic O, Ronne O, Kobzik L, Chapman HA. Mac-1 (CD11b/CD18) and the urokinase receptor (CD87) form a functional unit on monocytic cells. Blood. 1996;88:3185–3194. [PubMed] [Google Scholar]
- 29.Behrendt N, Ronne E, Ploug M, Petri T, Lober D, Nielsen LS, Schleuning W-D, Blasi F, Appella E, Dano K. The human receptor for urokinase plasminogen activator. NH2-terminal amino acid sequence and glycosylation variants. J Biol Chem. 1990;265:6453–6460. [PubMed] [Google Scholar]
- 30.Behrendt N, Ploug M, Patthy L, Houen G, Blasi F, Dano K. The ligand-binding domain of the cell surface receptor for urokinase-type plasminogen activator. J Biol Chem. 1991;266:7842–7847. [PubMed] [Google Scholar]
- 31.Waltz DA, Sailor LZ, Chapman HA. Cytokines induce urokinase-dependent adhesion of human myeloid cells. A regulatory role for plasminogen activator inhibitors. J Clin Invest. 1993;91:1541–1552. doi: 10.1172/JCI116360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizukami IF, Vinjamuri SD, Trochelman RD, Todd RF., III A structural characterization of the uPA-R activation antigen expressed on the plasma membrane of human mononuclear phagocytes. J Immunol. 1990;146:1841–1848. [PubMed] [Google Scholar]
- 33.Ronne E, Behrendt N, Ellis V, Ploug M, Dano K, Hoyer-Hansen G. Cell-induced potentiation of the plasminogen activation system is abolished by a monoclonal antibody that recognizes the NH2-terminal domain of the urokinase receptor. FEBS Lett. 1991;288:1–2. doi: 10.1016/0014-5793(91)81042-7. [DOI] [PubMed] [Google Scholar]
- 34.Wallis WJ, Hickstein DD, Schwartz BR, June CH, Ochs HD, Beatty PG, Klebanoff SJ, Harlan JM. Monoclonal antibody–defined functional epitopes on the adhesion-promoting glycoprotein complex (CDw18) of human neutrophils. Blood. 1986;67:1007–1013. [PubMed] [Google Scholar]
- 35.Sanchez-Madrid F, Simon P, Thompson S, Springer TA. Mapping of antigenic and functional epitopes on the α and β subunits of two related mouse glycoproteins involved in cell interactions, LFA-1 and Mac-1. J Exp Med. 1983;158:586–602. doi: 10.1084/jem.158.2.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prieto J, Takei F, Gendelman R, Christenson B, Biberfeld P, Patarroyo M. MALA-2, mouse homologue of human adhesion molecule ICAM-1 (CD54) Eur J Immunol. 1989;19:1551–1557. doi: 10.1002/eji.1830190906. [DOI] [PubMed] [Google Scholar]
- 37.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanse SM, Wijelath E, Kanthou C, Newman P, Kakkar VV. The proliferative responsiveness of human vascular smooth muscle cells to endothelin correlates with endothelin receptor density. Lab Invest. 1995;72:376–382. [PubMed] [Google Scholar]
- 39.Bugge TH, Suh TT, Flick MJ, Daugherty CC, Romer J, Solberg H, Ellis V, Dano K, Degen JL. The receptor for urokinase-type plasminogen activator is not essential for mouse development or fertility. J Biol Chem. 1995;270:16886–16894. doi: 10.1074/jbc.270.28.16886. [DOI] [PubMed] [Google Scholar]
- 40.Sligh JE, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oppenheimer-Marks N, Lipsky PE. The adhesion and trans-endothelial migration of human T lymphocyte subsets. Behring Inst Mitt. 1993;92:44–50. [PubMed] [Google Scholar]
- 42.Hogg N, Berlin C. Structure and function of adhesion receptors in leukocyte trafficking. Immunol Today. 1995;16:327–330. doi: 10.1016/0167-5699(95)80147-2. [DOI] [PubMed] [Google Scholar]
- 43.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1 and p150/95 glycoprotein. Annu Rev Med. 1987;38:175–194. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 44.Kuijpers TW, Harlan JM. Monocyte-endothelial interactions: insights and questions. J Lab Clin Med. 1993;122:641–651. [PubMed] [Google Scholar]
- 45.Mizgerd JP, Kubo H, Kutkoski GJ, Bhagwan SD, Scharffetter-Kochanek K, Beaudet AL, Doerschuk CM. Neutrophil emigration in the skin, lungs, and peritoneum: different requirements for CD11/CD18 revealed by CD18-deficient mice. J Exp Med. 1997;186:1357–1364. doi: 10.1084/jem.186.8.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmits R, Kündig TM, Baker DM, Shumaker G, Simard JJL, Duncan G, Wakeham A, Shahinian A, van der Heiden A, Bachmann MF, et al. LFA-1–deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med. 1996;183:1415–1426. doi: 10.1084/jem.183.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dewerchin M, Van Nufelen A, Wallays G, Bouche A, Moons L, Carmeliet P, Mulligan RC, Collen D. Generation and characterization of urokinase receptor–deficient mice. J Clin Invest. 1996;97:870–878. doi: 10.1172/JCI118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirchheimer JC, Remold HG. Endogenous receptor-bound urokinase mediates tissue invasion of human monocytes. J Immunol. 1989;143:2634–2639. [PubMed] [Google Scholar]
- 49.Gyetko MR, Chen G-H, McDonald RA, Goodman R, Huffnagle GB, Wilkinson CC, Fuller JA, Toews GB. Urokinase is required for the pulmonary inflammatory response to Cryptococcus neoformans. . J Clin Invest. 1996;97:1818–1826. doi: 10.1172/JCI118611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin αvβ3 binding to vitronectin. Nature. 1996;383:441–443. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- 51.Deng G, Curriden SA, Wang S, Rosenberg S, Loskutoff DJ. Is plasminogen activator inhibitor-1 the molecular switch that governs urokinase receptor–mediated cell adhesion and release? . J Cell Biol. 1996;134:1563–1571. doi: 10.1083/jcb.134.6.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kjoller L, Kanse SM, Kierkegaard T, Rodenburg KW, Ronne E, Goodman SL, Preissner KT, Ossowski L, Andreasen PA. Plasminogen activator inhibitor-1 represses integrin- and vitronectin-mediated cell migration independently of its function as an inhibitor of plasminogen activation. Exp Cell Res. 1997;232:420–429. doi: 10.1006/excr.1997.3540. [DOI] [PubMed] [Google Scholar]
- 53.Pedersen TL, Yong K, Pedersen JO, Hansen NE, Dano K, Plesner T. Impaired migration in vitro of neutrophils from patients with paroxysmal nocturnal haemoglobinuria. Brit J Haematol. 1996;95:45–51. [PubMed] [Google Scholar]
- 54.Poston RN, Haskard DO, Coucher JR, Gall NP, Johnson-Tidey RR. Expression of intercellular adhesion molecule-1 in atherosclerotic plaques. Am J Pathol. 1992;140:665–673. [PMC free article] [PubMed] [Google Scholar]