Cooperative Roles of CTLA-4 and Regulatory T Cells in Tolerance to an Islet Cell Antigen (original) (raw)
Abstract
Adoptive transfer of ovalbumin (OVA)-specific T cells from the DO.11 TCR transgenic mouse on a Rag−/− background into mice expressing OVA in pancreatic islet cells induces acute insulitis and diabetes only if endogenous lymphocytes, including regulatory T cells, are removed. When wild-type OVA-specific/Rag−/− T cells, which are all CD25−, are transferred into islet antigen–expressing mice, peripheral immunization with OVA in adjuvant is needed to induce diabetes. In contrast, naive CTLA-4−/−/Rag−/− OVA-specific T cells (also CD25−) develop into Th1 effectors and induce disease upon recognition of the self-antigen alone. These results suggest that CTLA-4 functions to increase the activation threshold of autoreactive T cells, because in its absence self-antigen is sufficient to trigger autoimmunity without peripheral immunization. Further, CTLA-4 and regulatory T cells act cooperatively to maintain tolerance, indicating that the function of CTLA-4 is independent of regulatory cells, and deficiency of both is required to induce pathologic immune responses against the islet self-antigen.
Keywords: autoimmunity, tolerance, diabetes, T cell activation, interferon gamma
Introduction
Multiple mechanisms are known to induce tolerance in mature T cell populations in peripheral lymphoid tissues. These mechanisms include anergy, deletion, and suppression by regulatory T (Treg) cells (1–4). We and others have shown previously that the inhibitory T cell receptor, CTLA-4, plays an essential role in maintaining unresponsiveness to tolerogenic forms of foreign and self-antigens (5–7). However, it is still not clearly established if CTLA-4 limits the magnitude of initial T cell activation or inhibits continued T cell expansion and differentiation into effector cells. Many investigators have also demonstrated a critical role of Treg cells in maintaining tolerance to a variety of self-antigens, including islet antigens (8, 9), but again the mechanisms by which Treg cells inhibit responses of autoreactive lymphocytes are not well established.
Most autoimmune diseases show complex, multigenic patterns of susceptibility. The existence of multiple susceptibility genes suggests that several pathways of self-tolerance need to be disrupted in order to trigger pathologic autoreactivity. In this study, we have examined the consequence of deleting CTLA-4 and eliminating Treg cells on the development of diabetes in a transgenic mouse model. Our results show that in the absence of an exogenous immunogenic stimulus acute, severe diabetes develops only if both CTLA-4 and Treg cells are nonfunctional. The cooperative role of these two tolerogenic pathways suggests that they serve distinct functions, perhaps at different stages of the T cell response.
Materials and Methods
Mice.
DO11.10 TCR transgenic mice were purchased from Jackson Laboratory, and Rag2−/− mice were purchased from Taconic Laboratories. Rat insulin promoter (RIP)-mOVA/Balb/c mice (referred to as RIP-mOVA mice), expressing OVA under the control of the RIP, were provided by Dr. William Heath (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia). The CTLA-4−/−DO11.10 mouse has been described previously (10). RIP-mOVA mice, CTLA-4−/−DO.11 mice, and the DO11.10 TCR transgenic mice were bred onto a Rag2−/− background. DO.11 homozygous mice were bred with the RIP-mOVA mice to generate DO.11 × RIP-mOVA mice. All mice were bred onto the Balb/c background for at least nine generations. Mice were housed in the University of California, San Francisco animal facility according to institutional guidelines.
Flow Cytometry.
Antibodies for flow cytometry were KJ1-26-APC, CD4-PerCP, CD4-PeCy5.5 (Caltag Laboratories), CD62L (PE and FITC), CD25 (PE and FITC), and IFNγ-PE. All antibodies were purchased from BD Biosciences unless otherwise stated. For intracellular IFNγ stains, cells (5 × 106/ml) were cultured in 200 μl complete medium (RPMI 1640 supplemented with 1 mM glutamine, penicillin, streptomycin, nonessential amino acids, sodium pyruvate, HEPES [all from Life technologies], 10% FCS [Sigma-Aldrich], and 5 × 10−5 M β-mercaptoethanol) and stimulated with 1 μg/ml OVA peptide (residues 323–339) for a total of 6 h. Brefeldin A (10 μg/ml) (Epicenter) was added for the last 3 h of incubation. The cells were then surface stained with CD4-PerCP and the clonotypic antibody KJ1-26-APC, fixed in 1% paraformaldehyde, permeabilized with 0.5% saponin (Sigma-Aldrich), and stained for intracellular IFNγ and intracellular KJ1-26, since the TCR is internalized upon activation.
Adoptive Transfers and Immunization.
For T cell transfers, LNs (axillary, brachial, mesenteric, popliteal, and submandibular) and spleens were pooled, red cells lysed, and single cell suspensions made. The cells were stained with CD4-PerCP and KJ1-26-APC to determine the number of DO.11 cells. Indicated numbers of cells were injected i.v. through the tail vein. Where indicated, mice were immunized s.c. in the flank, 24 h after adoptive transfer with 200 μg OVA peptide emulsified in IFA (Difco). For transfers of CD25+ and CD25− DO.11 cells, pooled LN and spleen cells were stained with CD4-PeCy5.5, KJ1-26-APC, CD25-PE, and CD62L-FITC. Cells were purified using high speed sorting (MoFlo; Cytomation) to separate CD25+CD62Lhi DO.11 (Treg cells) and CD25−CD62Lhi DO.11 (responder) populations.
Monitoring for Diabetes.
Adoptively transferred mice were followed for the development of diabetes by biweekly monitoring of blood glucose levels using an Elite glucometer (Bayer Corp.).
Histology.
Mice were killed using CO2 narcosis, and the pancreas was removed aseptically. For hematoxylin and eosin staining, the tissues were fixed in 10% phosphate-buffered formalin. For immunohistochemistry, acetone-fixed frozen tissues were stained with KJ1-26-biotin (Caltag Laboratories) followed by alkaline phosphate-conjugated streptavidin (BD Biosciences) to detect DO.11 cells. Sections were observed under light microscopy. Severity of insulitis was measured by the extent of islet infiltration using the criteria: no infiltration, infiltration around the periphery, or invasive infiltration.
Statistics.
The statistical significance of differences between groups was determined using the Student's t test.
Results and Discussion
CTLA-4−/− DO.11 T Cells Induce Diabetes without Immunization.
We have previously shown that OVA-specific DO.11/Rag−/− cells induce acute diabetes only in Rag−/− RIP-mOVA recipients (not in Rag+/− littermates) and that peripheral (s.c.) immunization with OVA in adjuvant is required (unpublished data). In striking contrast, we show here that transfer of T cells from CTLA-4−/− DO.11/Rag−/− donors induced diabetes in RIP-mOVA/Rag−/− recipients in the absence of peripheral immunization (Fig. 1, A compared with B). Importantly, the CTLA-4−/− T cells were not diabetogenic in lymphocyte-sufficient RIP-mOVA recipients, suggesting that even CTLA-4−/− T cells can be controlled by Treg cells (reference 11 and Fig. 1 A).
Figure 1.
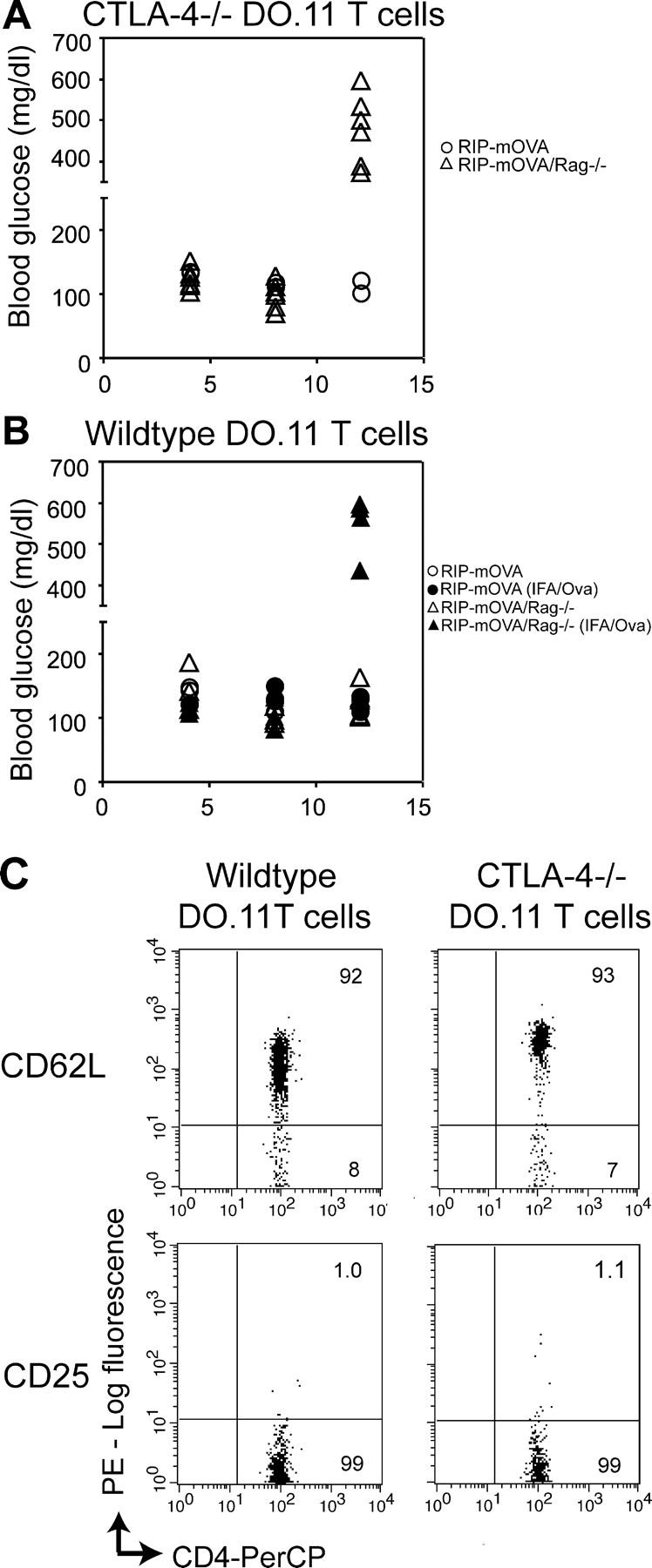
Naive CTLA-4−/− DO.11 T cells induce diabetes in a lymphocyte-deficient recipient, whereas naive wild-type DO.11 T cells require a priming signal. Naive CTLA-4−/− DO.11 (A) or wild-type DO.11 cells (B) (10 million per mouse) were transferred into either RIP-mOVA or RIP-mOVA/Rag−/− recipients, and some mice were immunized s.c. with 200 μg OVA peptide in IFA as indicated. The development of diabetes, measured by increase in blood glucose levels, is shown. The data represent the combined results of two independent experiments with a total of six RIP-mOVA/Rag−/− and three RIP-mOVA recipients per experimental group. In A, the blood glucose levels in RIP-mOVA versus RIP-mOVA/Rag−/− recipients were significantly different (P = 0.0002); in B, the blood glucose levels in the immunized RIP-mOVA/Rag−/− mice were different from unimmunized mice (P = 0.012) and from immunized RIP-mOVA mice (P = 0.0124). Note that the increased levels seen after transfer of wild-type DO.11 cells and immunization were not different from those after transfer of CTLA-4−/− DO.11 cells and no immunization (P = 0.376). (C) Wild-type and CTLA-4−/− DO.11 T cells are phenotypically naive; plots shown were gated on KJ1-26+ cells.
A possible explanation for the increased pathogenicity of the CTLA-4−/− T cells is that these cells are activated by endogenous antigens before adoptive transfer. However, as shown previously (5) both wild-type and CTLA-4−/− DO.11 T cells on a Rag−/− background express high levels of CD62L and undetectable levels of CD25 and are thus phenotypically and functionally naive (Fig. 1 C). It is also conceivable that CTLA-4 is involved in the function of Treg cells (11, 15), and therefore, deletion of CTLA-4 abrogates regulatory function in the population of transferred DO.11 cells. However, all the experiments have been done with DO.11 cells from DO.11/Rag−/− mice. In these “monoclonal mice,” there are no CD25+ cells (Fig. 1 C and reference 12) because there is no source of antigen that developing cells might encounter. Thus, the effect of eliminating CTLA-4 cannot be on regulatory cells and must be on effector T cell activation. Furthermore, these results show that in the absence of CTLA-4, a transgene-encoded self-antigen alone is sufficient to initiate T cell responses and cause disease in an animal lacking endogenous T cells.
Activation of Antigen-specific T Cells.
Histological examination of the pancreas from adoptive transfer recipients showed severe insulitis with massive infiltration of DO.11 cells only when CTLA-4−/− T cells were transferred into RIP-mOVA/Rag−/− mice (Fig. 2, E and F) or when wild-type DO.11 cells received a priming signal with OVA peptide and IFA (Fig. 2, C and D). When the transferred cells were wild-type DO.11 T cells, there was mild insulitis with infiltration predominantly around the periphery of the islets (Fig. 2, A and B).
Figure 2.
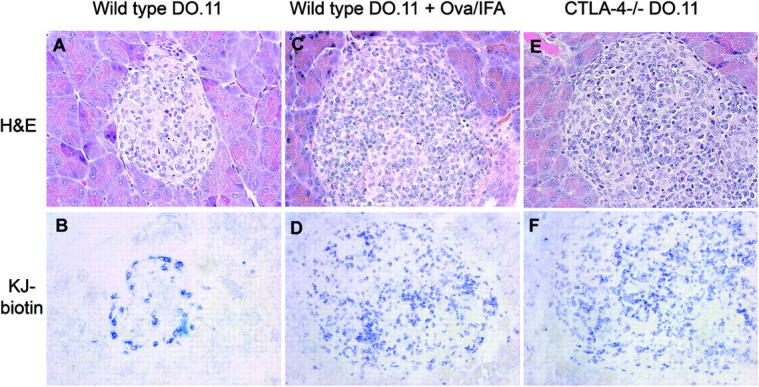
CTLA-4−/− DO.11 T cells cause severe insulitis in RIP-mOVA/Rag−/− mice. Naive wild-type or CTLA-4−/− DO.11 T cells (10 million per mouse) were transferred into RIP-mOVA/Rag−/− recipients and immunized with OVA peptide and IFA where indicated. The mice were killed 13 d after transfer, and the pancreas were either formalin fixed and stained with hematoxylin and eosin (left) or frozen, acetone fixed, and stained with KJ1-26 (right) and observed under 40× magnification. Wild-type DO.11 T cells (A and B); wild-type DO.11 T cells and immunized with OVA peptide + IFA (C and D); CTLA4−/− DO.11 T cells (E and F). Representative sections for each experimental group are shown.
We next examined the DO.11 cells in peripheral and pancreatic LNs by staining and flow cytometry. At day 3 posttransfer, wild-type DO.11 cells remained phenotypically naive in both the peripheral and pancreatic LNs if the mice were not immunized. After immunization, the wild-type DO.11 cells down-regulated CD62L and up-regulated CD25, both markers of activation (Fig. 3, A and B). Strikingly, the CTLA-4−/− DO.11 cells showed a similar pattern of activation, especially in the pancreatic LNs (where cells may encounter antigen), even in the absence of immunization (Fig. 3, A and B). At later time points, both wild-type and CTLA-4−/− DO.11 cells showed equal expansion in Rag−/− hosts (Fig. 4 A), both in the periphery and in the pancreatic LN, the site of antigenic stimulation. This expansion was likely due to homeostatic proliferation seen in lymphopenic recipient animals rather than stimulation by self-antigen. However, the CTLA-4−/− T cells showed a significantly higher frequency of IFNγ-producing cells in the pancreatic LN than did wild-type DO.11 cells (Fig. 4 B). Thus, CTLA-4 normally controls the development of self-antigen–reactive T cells into pathogenic effectors. These results, together with the development of diabetes as shown in Figs. 1, A and B, demonstrate that in order for antigen-specific T cells to proliferate in response to a self-antigen, differentiate into effector cells, infiltrate the antigen-expressing tissue, and induce acute disease, both CTLA-4 and Treg cells have to be nonfunctional.
Figure 3.

Transferred wild-type and CTLA-4−/−DO.11 T cells show similar activation profiles on encountering antigen. Naive CTLA-4−/− DO.11 or wild-type DO.11 cells (10 million per mouse) were transferred into RIP-mOVA/Rag−/− recipients, and some mice were immunized s.c. with 200 μg OVA peptide in IFA as indicated. Peripheral (PLN) and pancreatic (PANC) LNs were harvested 3 d later and cells were stained for CD4 and KJ1-26 and CD62L (A) and CD25 (B) expression. Plots shown are gated on KJ1-26+ cells. Numbers refer to the percentage of positive cells in the quadrant.
Figure 4.

T cell activation in the absence of CTLA-4 and endogenous lymphocytes. Naive wild-type or CTLA-4−/− DO.11 T cells (10 million per mouse) were transferred into either RIP-mOVA or RIP-mOVA/Rag−/− recipients. Mice were killed on day 13, and pancreatic and peripheral LN cells were stained for the presence of KJ1-26+CD4+ cells or restimulated with 1 μg/ml OVA peptide for 6 h, and stained for intracellular IFNγ. Shown are DO.11 cells as the percentage of total mononuclear cells recovered from peripheral and pancreatic LNs of RIP-mOVA/Rag−/− mice (A) or RIP-mOVA recipients (C). IFNγ-producing DO.11 cells as the percentage of total KJ+CD4+ cells in either pancreatic or peripheral LNs of RIP-mOVA/Rag−/− (B) or RIP-mOVA (D) recipients. The data represent pooled results of two independent experiments using a total of six RIP-mOVA/Rag−/− and three RIP-mOVA recipients per experimental group. In A, no difference was observed in numbers of wild-type and CTLA-4−/− DO.11 cells recovered from the peripheral (P = 0.09) or pancreatic LNs (P = 0.08). In B, CTLA-4−/− cells recovered from the pancreatic LN produced significantly more IFNγ that wild-type cells (P = 0.007).
The striking difference between naive wild-type and CTLA-4−/− cells in the induction of disease in a lymphopenic host led to the question: do CTLA-4 and Treg cells control distinct stages in antigen-specific T cell activation, expansion and/or differentiation? To address the influence of CTLA-4 in the presence of endogenous Treg cells, we examined the accumulation of wild-type or CTLA-4−/− DO.11 cells adoptively transferred into RIP-mOVA hosts. As is evident in Fig. 4 C, the presence of endogenous lymphocytes prevented expansion of both wild-type DO.11 and CTLA-4−/− DO.11 T cells, both in the periphery and at the site of antigenic stimulation, the pancreatic LN. In addition, the cells did not differentiate into effectors, indicated by the lack of IFNγ production (Fig. 4 D). Thus, both Treg cells and CTLA-4 control inappropriate T cell expansion and effector differentiation in response to a self-antigen, and it does not appear that they regulate different components of the T cell response, e.g., expansion and effector cell development.
Treg Cells Prevent Disease Induced by CTLA-4−/− DO.11 T Cells.
Balb/c animals are relatively resistant to autoimmune disease, possibly due to high activity of Treg cells (12). As demonstrated earlier (Figs. 1, A and B), we have never been able to break tolerance and induce diabetes in Balb/c animals that have intact lymphoid compartments. To confirm that acute diabetes required a deficiency of Treg cells, we asked if purified Treg cells could protect Rag-deficient animals from diabetes. To reconstitute Treg cells, we isolated OVA-specific CD25+ DO.11 T cells from DO.11 × RIP-mOVA mice, as described previously (13), and cotransferred these with CTLA-4−/− DO.11 T cells into RIP-mOVA Rag−/− recipients. As shown in Fig. 5 A, the CD25+ DO.11 T cells completely prevented disease induction by the CTLA-4−/− T cells, whereas cotransfer of CD25− DO.11 cells was not protective. These results are consistent with the view that RIP-mOVA/Rag−/− mice develop disease because they lack CD25+ Treg cells. Although it has been shown that cells transferred to lymphopenic animals are prone to induce autoimmune disease chiefly due to their homeostatic proliferation in this environment (14), we doubt that homeostatic proliferation alone is sufficient to induce disease in our model, since only cotransfer of CD25+ but not CD25− T cells had an inhibitory effect on the progression of diabetes. Furthermore, the recovery of DO.11 cells was not significantly different in the pancreatic and peripheral LNs of mice regardless of whether they received CD25− or CD25+ DO.11 cells (Fig. 5 B).
Figure 5.

Treg cells prevent induction of diabetes induced by CTLA-4−/− DO.11 T cells. (A) Naive CTLA-4−/− DO.11 cells (1.5 million) were transferred into RIP-mOVA/Rag−/− mice along with 0.75 million of either CD25+ or CD25− DO.11 T cells. The mice were followed for the development of diabetes. (B) DO.11 cells, shown as a percentage of total mononuclear cells, recovered from RIP-mOVA/Rag−/− mice 18 d posttransfer with 1.5 million CTLA-4−/− DO.11 cells and 0.75 million of either CD25+ or CD25− DO.11 T cells. The data represent pooled results of two independent experiments with three to four mice per experimental group. In A, blood glucose levels seen after transfer of CTLA-4−/− cells were significantly different from CTLA-4−/− + CD25+ cells (P < 0.05) but not from CTLA-4−/− + CD25− cells (P = 0.252).
The experiments in this work show that wild-type, antigen-specific T cells attack antigen-expressing islets and cause acute disease only if the T cells are first activated by intentional peripheral immunization with the same antigen in adjuvant. However, CTLA-4−/− T cells are pathogenic even without such immunization, implying that in the absence of CTLA-4, a self-antigen behaves like an immunogen. The pathogenicity of both immunized wild-type T cells and naive CTLA-4−/− T cells is prevented by the presence of endogenous Treg cells. Since both CTLA-4 and Treg cells must be absent in order to result in severe disease, these results suggest that the two mechanisms of tolerance do not serve the same role. Thus, although it has been suggested that CTLA-4 is involved in the suppressive function of Treg cells (11, 15), our results indicate that this cannot be the only function of CTLA-4.
A likely model for the cooperativity of CTLA-4 and Treg cells is that these inhibitory influences may act at different stages in the life of a T cell. A major function of CTLA-4 may be to increase the threshold for initial T cell activation (16), such that self-antigens by themselves are incapable of activating naive T cells. Thus, CTLA-4 would prevent normal DO.11 cells from being pathogenic unless its inhibitory effect was overcome by immunization with adjuvant. This role is consistent with the finding that wild-type T cells are not pathogenic unless they are first exposed to peripheral immunization with antigen and adjuvant, but CTLA-4−/− T cells are pathogenic after encounter with self-antigen alone. Treg cells may function later in T cell responses to block proliferation and differentiation at the site of self-antigen, regardless of the initiating trigger. This is why both activated wild-type T cells and naive CTLA-4−/− T cells are prevented from causing disease if Treg cells are present. It will be important to develop systems for addressing the possible sequential function of different tolerance mechanisms and to determine if genetic polymorphisms of CTLA-4 (17) need to be complemented by deficiencies of Treg cells in order to induce spontaneous autoimmune diseases in humans and experimental models.
Acknowledgments
We thank S. Jiang for help with cell sorting.
This work was supported by National Institutes of Health grant RO1 AI35297 to A.K. Abbas and grant K08 AI054366-01 to M.P. Eggena. V. Nagabhushanam is supported by the Juvenile Diabetes Research Foundation, and L.S.K Walker is supported by the Wellcome Trust.
M.P. Eggena, L.S.K. Walker, and V. Nagabhushanam contributed equally to the work.
L.S.K. Walker's present address is MRC Centre for Immune Regulation, University of Birmingham Medical School, Vincent Drive, Birmingham B15 2TT, England, UK.
Abbreviations used in this paper: RIP, rat insulin promoter; Treg cell, regulatory T cell.
References
- 1.Walker, L.S., and A.K. Abbas. 2002. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2:11–19. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz, R.H. 2003. T cell anergy. Annu. Rev. Immunol. 21:305–334. [DOI] [PubMed] [Google Scholar]
- 3.Arch, R.H., and C.B. Thompson. 1999. Lymphocyte survival—the struggle against death. Annu. Rev. Cell Dev. Biol. 15:113–140. [DOI] [PubMed] [Google Scholar]
- 4.Shevach, E.M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 5.Greenwald, R.J., V.A. Boussiotis, R.B. Lorsbach, A.K. Abbas, and A.H. Sharpe. 2001. CTLA-4 regulates induction of anergy in vivo. Immunity. 14:145–155. [DOI] [PubMed] [Google Scholar]
- 6.Walker, L.S., L.J. Ausubel, A. Chodos, N. Bekarian, and A.K. Abbas. 2002. CTLA-4 differentially regulates T cell responses to endogenous tissue protein versus exogenous immunogen. J. Immunol. 169:6202–6209. [DOI] [PubMed] [Google Scholar]
- 7.Eagar, T.N., N.J. Karandikar, J.A. Bluestone, and S.D. Miller. 2002. The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance. Eur. J. Immunol. 32:972–981. [DOI] [PubMed] [Google Scholar]
- 8.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+ CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 9.McHugh, R.S., E.M. Shevach, and A.M. Thornton. 2001. Control of organ-specific autoimmunity by immunoregulatory CD4(+)CD25(+) T cells. Microbes Infect. 3:919–927. [DOI] [PubMed] [Google Scholar]
- 10.Oosterwegel, M.A., D.A. Mandelbrot, S.D. Boyd, R.B. Lorsbach, D.Y. Jarrett, A.K. Abbas, and A.H. Sharpe. 1999. The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 163:2634–2639. [PubMed] [Google Scholar]
- 11.Read, S., V. Malmstrom, and F. Powrie. 2000. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itoh, M., T. Takahashi, N. Sakaguchi, Y. Kuniyasu, J. Shimizu, F. Otsuka, and S. Sakaguchi. 1999. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 162:5317–5326. [PubMed] [Google Scholar]
- 13.Walker, L.S., A. Chodos, M. Eggena, H. Dooms, and A.K. Abbas. 2003. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J. Exp. Med. 198:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gudmundsdottir, H., and L.A. Turka. 2001. A closer look at homeostatic proliferation of CD4+ T cells: costimulatory requirements and role in memory formation. J. Immunol. 167:3699–3707. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi, T., T. Tagami, S. Yamazaki, T. Uede, J. Shimizu, N. Sakaguchi, T.W. Mak, and S. Sakaguchi. 2000. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egen, J.G., and J.P. Allison. 2002. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 16:23–35. [DOI] [PubMed] [Google Scholar]
- 17.Ueda, H., J.M. Howson, L. Esposito, J. Heward, H. Snook, G. Chamberlain, D.B. Rainbow, K.M. Hunter, A.N. Smith, G. Di Genova, et al. 2003. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 423:506–511. [DOI] [PubMed] [Google Scholar]