Differential Requirement for TANK-binding Kinase-1 in Type I Interferon Responses to Toll-like Receptor Activation and Viral Infection (original) (raw)
Abstract
TANK-binding kinase-1 (TBK1) and the inducible IκB kinase (IKK-i) have been shown recently to activate interferon (IFN) regulatory factor-3 (IRF3), the primary transcription factor regulating induction of type I IFNs. Here, we have compared the role and specificity of TBK1 in the type I IFN response to lipopolysaccharide (LPS), polyI:C, and viral challenge by examining IRF3 nuclear translocation, signal transducer and activator of transcription 1 phosphorylation, and induction of IFN-regulated genes. The LPS and polyI:C-induced IFN responses were abolished and delayed, respectively, in macrophages from mice with a targeted disruption of the TBK1 gene. When challenged with Sendai virus, the IFN response was normal in TBK1−/− macrophages, but defective in TBK1−/− embryonic fibroblasts. Although both TBK1 and IKK-i are expressed in macrophages, only TBK1 but not IKK-i was detected in embryonic fibroblasts by Northern blotting analysis. Furthermore, the IFN response in TBK1−/− embryonic fibroblasts can be restored by reconstitution with wild-type IKK-i but not a mutant IKK-i lacking kinase activity. Thus, our studies suggest that TBK1 plays an important role in the Toll-like receptor–mediated IFN response and is redundant with IKK-i in the response of certain cell types to viral infection.
Keywords: LPS, innate immunity, interferon regulatory factor-3, viral infections, NF-κB
Introduction
In the evolutionary battle between viruses and their multicellular hosts, type I IFNs have emerged as critical mediators of host defense (1). Type I IFNs, including IFNα and IFNβ, are cytokines that are transcriptionally induced during viral infection and that signal through the type I IFN α/β receptor (IFNAR) to activate transcription of a large set of genes important in antiviral responses (2). IFNβ is first produced in response to the signaling cascade activated by innate immune viral detection and its transcription is controlled largely by the IFN regulatory factor-3 (IRF3) transcription factor (3–5).
IRF3 is localized in the cytoplasm in unstimulated cells and, upon stimulation, becomes activated by serine/threonine phosphorylation leading to nuclear translocation (4). IRF7, a close homologue of IRF3, is transcriptionally induced by IFNAR signaling and acts to promote further production of type I IFN (5–7). After production, IFNα/β binds to IFNAR in both an autocrine and paracrine manner and initiates activation of the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling pathway (2, 5). This activates the IFN-stimulated gene factor-3 transcription factor complex consisting of IRF-9, STAT1, and STAT2. IFN-stimulated gene factor-3 binds to IFN-stimulated response elements present in promoters of a large set of target genes important in antiviral responses (2, 5).
Toll-like receptors (TLRs) are a family of pattern recognition receptors that are known to recognize conserved microbial motifs and to subsequently induce signaling cascades that lead to proinflammatory cytokine production and maturation of antigen-presenting cells as well as enhancement of phagocytic activity of macrophages (8, 9). Several TLRs are able to recognize viral products including dsRNA (TLR3), the F protein of respiratory syncytial virus (TLR4), ssRNA and the antiviral imiquimod compounds (TLR7), and CpG DNA (TLR9; references 10–16).
Most TLR-mediated responses proceed through the Toll/IL-1 receptor domain–containing adaptor molecule MyD88 to the activation of NF-κB and MAP kinases (17). However, careful studies of MyD88-deficient mice revealed a MyD88-independent pathway that contributed to DC maturation and delayed NF-κB and MAP kinase activation (18, 19). Subsequently, TLR3 and TLR4, which recognize dsRNA and LPS respectively, were found to activate IRF3 and up-regulate a type I IFN–dependent gene program in a MyD88-independent manner and became characteristic of this pathway (20, 21). Thus, the signaling mediators leading to type I IFN production during TLR3/4 stimulation have been under heavy study. Recently, two additional adaptor molecules were identified as missing links between TLR3/4 and IRF3. Mice deficient in Toll/IL-1 receptor domain–containing adaptor-inducing IFN-β (TRIF/TICAM-1) were found to have defective type I IFN production and late NF-κB activation in response to stimulation of either TLR3 or TLR4, whereas mice deficient in TRIF-related adaptor molecule (TRAM/TICAM-2) were defective in these responses only during TLR4 stimulation (22–28).
TANK-binding kinase-1 (TBK1; T2K, NAK) and inducible IκB kinase (IKK-i; IKKɛ) were known previously as relatives of the IKKs that had an undefined role in NF-κB activation and might potentially act as IKK kinases (29). Recently, TBK1 and IKK-i were identified as having the ability to induce in vitro phosphorylation of IRF3 and IRF7 and to activate a luciferase reporter driven by the IFNβ promoter (30, 31). However, the specificity of TBK1 and IKK-i in type I IFN production by TLR3, TRL4, and viral stimulations remains to be elucidated.
In this paper, we further characterize the contribution of TBK1 in TLR3/4 signaling pathways and during viral infections in cells derived from TBK1−/− mice. We provide genetic evidence that TBK1 is required for IRF3 activation and production of type I IFN genes in response to TLR3 and TLR4 ligands, but not for NF-κB activation. Interestingly, during virus infection, a cell type–specific requirement for TBK1 was observed in which embryonic fibroblasts but not macrophages required TBK1. We suggest that differential expression of IKK-i may mediate this difference.
Materials and Methods
Mice and Cell Culture.
Generation of mice with a targeted disruption of the TBK1 gene have been described previously (32). TBK1−/− mice were generated by crossing mice heterozygous for a TBK1-deficient allele with TNFR1−/− mice (provided by T. Mak, University of Toronto, Toronto, Canada) for two generations to get TBK1−/−TNFR1−/− mice. BM–derived macrophages (BMMs) differentiated from marrow from 6–10-wk-old TBK1−/−TNFR1−/− mice and TBK1+/+TNFR1+/− or TBK1+/+ TNFR1−/− littermates were generated as described previously (33). In brief, BM cells were harvested from mice and allowed to differentiate for 7–8 d in 30% L929-conditioned media before assays were performed. TBK1 wild type and knockout immortalized murine embryonic fibroblasts (MEFs) have been described previously (32).
Virus Infections and Reagents.
BMMs were stimulated with LPS (Sigma-Aldrich) at 10 ng/ml and polyI:C (Amersham Biosciences) at 1 μg/ml. Sendai virus (SeV; Z strain) was a gift from D. Nayak (University of California Los Angeles, Los Angeles, CA; reference 34). Cells were infected at a multiplicity of infection of approximately five for the time points indicated.
Protein Extracts, Immunoblotting, and EMSA.
Cell lysates were fractionated into nuclear and cytoplasmic fractions as described previously (20). Nuclear fractions were probed with antibodies against IRF3 (Zymed Laboratories), p65, and USF2 (Santa Cruz Biotechnology, Inc.). Cytoplasmic fractions were probed with antibodies against phosphorylated STAT1 (pY701; Cell Signaling Technology) and STAT1 (Santa Cruz Biotechnology, Inc.). For EMSA, nuclear fractions were used in binding reactions with a γ[32P]-labeled NF-κB–specific oligonucleotide as described previously (35).
Vectors and Reconstituted Cell Lines.
Full length and kinase-inactive (KA; K38A) mutant forms of TBK1 and IKK-i were cloned from human expressed sequence tags (American Type Culture Collection) into the pEBB eukaryotic expression vector containing a puromycin-resistant gene. Vectors were transfected into TBK1−/− MEFs and stably expressing cell lines were selected in the presence of 2.5 μg/ml puromycin.
Quantitative Real-Time PCR and Northern Blot Analysis.
RNA was isolated using TRIzol (Invitrogen), and cDNA was synthesized using iScript (Bio-Rad Laboratories) according to the manufacturer's instructions. Quantitative RT-PCR (Q-PCR) analysis was performed using the iCycler thermocycler (Bio-Rad Laboratories) as described previously (20). Primer sequences for IFNβ, IP-10, IRF7, TNFα, IκBα, ICAM-1, and L32 have been described previously (20, 36). The following IL-15 primers were used: F, 5′-CACTTTTTAACTGAGGCTGGCATT-3′; and R, 5′-TCCAGTTGGCCTCTGTTTTAGG-3′. The following primers were used: IFNα5, F, 5′-TGACCTCAAAGCCTGTGTGATG-3′ and R, 5′-AAGTATTTCCTCACAGCCAGCAG-3′; and Mx1 primers, F, 5′-AAACCTGATCCGACTTCACTTCC-3′, and R, 5′-TGATCGTCTTCAAGGTTTC-CTTGT-3′. All gene expression data presented were normalized to L32 levels for each sample.
Northern blot analysis was performed using standard procedures. Probes for detection of TBK1 and IKK-i were PCR amplified from full-length human expressed sequence tags and labeled with α[32P].
Results
TBK1 Is Required for LPS-induced IRF3 Activation and Type I IFN Production.
The LPS-induced type I IFN response through TLR4 involves TRAM and TRIF-dependent, but MyD88-independent activation of IRF3 and transcriptional up-regulation of a subset of primary response genes including IFNβ. Subsequently, activation of the JAK–STAT pathway and up-regulation of a subset of secondary response genes occurs through auto/paracrine activity of type I IFNs (2, 20–28). To examine the role of TBK1 in LPS-induced activation of the type I IFN response, activation of IRF3 and STAT1 and induction of IFN-regulated genes was compared in TBK1+/+ and TBK1−/− BMMs at various time points after LPS stimulation. Although strong IRF3 nuclear translocation and STAT1 phosphorylation was observed in TBK1+/+ BMMs upon LPS stimulation, this response was either completely absent or greatly diminished in BMMs lacking TBK1 (Fig. 1, A and B). In response to LPS, TBK1−/− BMMs also failed to up-regulate transcription of IFNβ and IFN-mediated transcription of genes including IP-10, IFNα5, IRF7, IL-15, and Mx1 (Fig. 1 C). Because TBK1-deficient mice are embryonically lethal, TBK1−/− BMMs used here and in the subsequent experiments were derived from TBK1 and TNFR1 double knockout mice, which are viable. Although controls used in experiments shown in Figs. 1–4 are BMMs from TBK1+/+TNFR1+/− mice, similar results were obtained with BMMs from TBK1+/+TNFR1−/− mice (unpublished data). Thus, TBK1 is an essential and nonredundant component of LPS-induced IRF3 activation and type I IFN responses.
Figure 1.

TBK1 is required for LPS-mediated activation of type I IFN responses. TBK1+/+TNFR1+/− and TBK1−/−TNFR1−/− BMMs were stimulated with 10 ng/ml LPS for the indicated time points. (A) Nuclear fractions were probed for IRF3 and USF2 as a loading control for nuclear proteins. (B) Total cell extracts were probed for phospho-STAT1 and total STAT1. (C) Total RNA was extracted and analyzed by Q-PCR for expression of IFNβ, IP-10, IFNα5, IRF7, IL-15, and Mx1.
Figure 4.

TBK1-deficient BMMs have normal IFN responses to SeV. TBK1+/+TNFR1+/− and TBK1−/−TNFR1−/− BMMs were infected with SeV for the indicated time points. (A) Nuclear fractions were probed for IRF3 and USF2 as a loading control. (B) Total cell extracts were probed for phospho-STAT1 and total STAT1. (C) Total RNA was extracted and analyzed by Q-PCR for expression of IFNβ, IP-10, IFNα5, IRF7, IL-15, and Mx1.
TBK1 Is Required for dsRNA-mediated IRF3 Activation and Early But Not Late Induction of Type I IFN Responses.
The dsRNA analogue, polyI:C, can induce type I IFN responses through both TLR3-dependent and -independent mechanisms (10, 24, 37). To determine the requirement for TBK1 in type I IFN responses induced by dsRNA signaling pathways, the effect of polyI:C stimulation on TBK1−/− BMMs was analyzed. Similar to that seen with LPS stimulation, BMMs lacking TBK1 were unable to induce nuclear translocation of IRF3 in response to polyI:C (Fig. 2 A). Even during evaluation of an extended time course (up to 4 h after stimulation), IRF3 nuclear localization remained undetectable in TBK1−/− BMMs (unpublished data). However, unlike what was observed for LPS stimulation, polyI:C-induced STAT1 phosphorylation could be detected in TBK1−/− BMMs, albeit with delayed kinetics (Fig. 2 B). Gene expression analyses showed that IFNβ production was absent in TBK1−/− BMMs in response to polyI:C (Fig. 2 C). However, induction of secondary IFN-regulated genes, such as IP-10, IFNα5, IRF7, IL-15, and Mx1, were induced in a delayed fashion in TBK1−/− BMMs (Fig. 2 C and not depicted). Thus, in response to polyI:C, TBK1 is required for IRF3 activation and IFNβ production; however, there appears to be a TBK1-independent pathway that can up-regulate delayed IFNα and type I IFN–regulated gene production.
Figure 2.

TBK1-deficient BMMs have defective type I IFN responses to polyI:C. TBK1+/+TNFR1+/− and TBK1−/−TNFR1−/− BMMs were stimulated with 1 μg/ml polyI:C for the indicated time points. (A) Nuclear fractions were probed for IRF3 and USF2 as a loading control. (B) Total cell extracts were probed for phospho-STAT1 and total STAT1. (C) Total RNA was extracted and analyzed by Q-PCR for expression of IFNβ, IP-10, IFNα5, IRF7, IL-15, and Mx1.
Activation of NF-κB in Response to LPS and PolyI:C Does Not Require TBK1.
TBK1 is an IKK family member and was initially suggested to have a role in NF-κB activation (38, 39). To determine if TBK1 has a role in the NF-κB response to LPS or polyI:C, stimulated TBK1+/+ and TBK1−/− BMMs were assayed for activation of NF-κB using EMSA and by immunoblotting nuclear protein fractions for p65, the major NF-κB subunit. Neither assay indicated a defect in NF-κB activation in response to LPS or polyI:C in TBK1−/− BMMs (Fig. 3, A and B). Furthermore, Q-PCR analyses of the NF-κB–regulated genes ICAM-1 and IκBα revealed similar induction in TBK1+/+ and TBK1−/− BMMs after LPS or polyI:C stimulation (Fig. 3 C). Thus, TBK1 is not required for NF-κB activation in response to either LPS or polyI:C.
Figure 3.
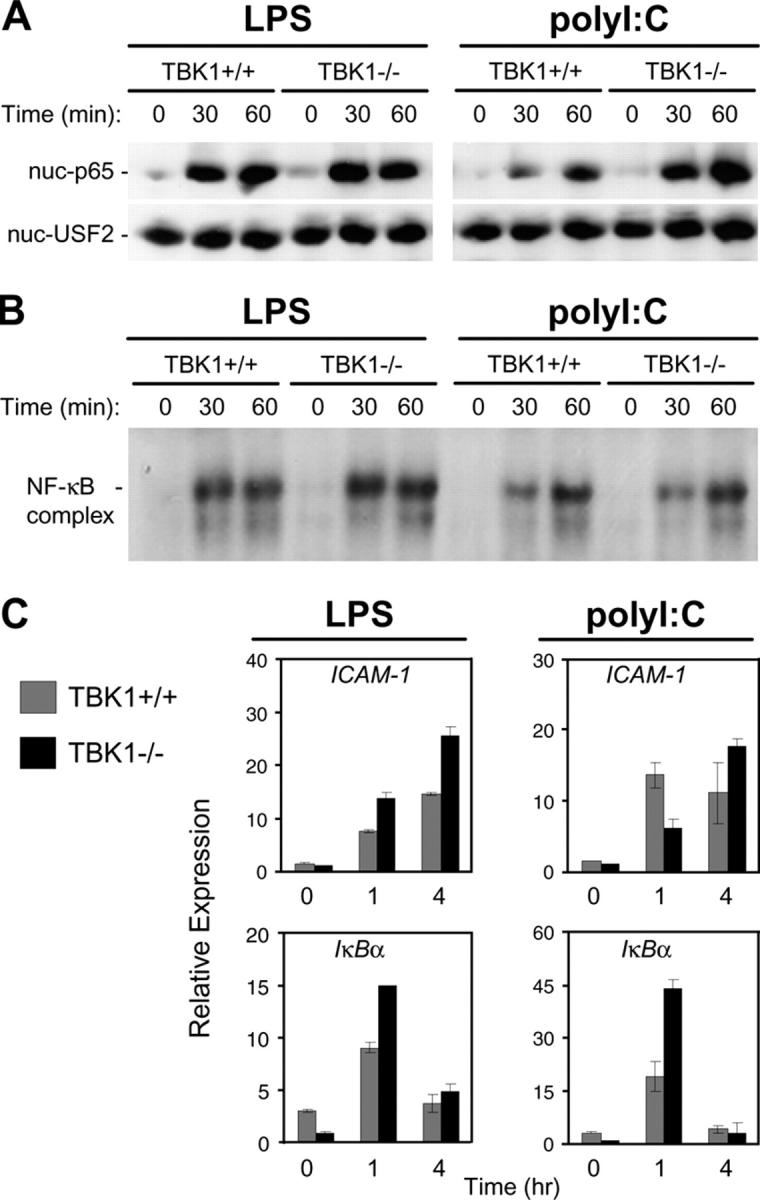
TBK1-deficient BMMs induce normal NF-κB responses to LPS and polyI:C. TBK1+/+TNFR1+/− and TBK1−/−TNFR1−/− BMMs were stimulated with 10 ng/ml LPS or 1 μg/ml polyI:C for the indicated time points. (A) Nuclear fractions were probed for p65 and USF2 as a loading control. (B) EMSA was performed by incubating nuclear extracts were with an NF-κB-specific oligonucleotide. (C) Total RNA was extracted and analyzed by Q-PCR for expression of ICAM1 and IκBα.
TBK1 Is Not Required for SeV-induced IRF3 Activation in BMMs.
Type I IFN production is a characteristic response of virally infected cells. TLR3 activation by viral dsRNA may mediate some aspects of viral recognition leading to type I IFN responses. However, it is unclear how different host cells detect various viruses and induce type I IFN responses during an actual infection. To determine the role for TBK1 in activation of the IFN response during an in vitro virus infection, we infected BMMs with SeV, a negative-stranded RNA virus. Surprisingly, in contrast to polyI:C stimulation, nuclear localization of IRF3 was normal in TBK1−/− BMMs during SeV infection (Fig. 4 A). Furthermore, although TBK1 mediated early induction of polyI:C-induced STAT1 phosphorylation, comparable kinetics and levels of STAT1 phosphorylation were observed between TBK1+/+ and TBK1−/− BMMs infected with SeV (Fig. 4 B). In accordance with normal STAT1 kinetics, production of IFNβ and the IFN-regulated genes IP-10, IFNα5, IRF7, IL-15, or Mx1 were comparable in TBK1+/+ and TBK1−/− BMMs during SeV infection (Fig. 4 C). Activation of NF-κB and NF-κB–regulated gene induction was normal in TBK1−/− BMMs (unpublished data). Thus, although TBK1 mediates IRF3 activation and IFNβ production as well as prompt induction of IFN-regulated genes in response to polyI:C, a TBK1-independent pathway exists for these responses during infection of BMMs with an RNA virus.
A Cell Type–dependent Requirement for TBK1 in Response to Virus Infection.
In addition to BMMs, we analyzed the requirement for TBK1 during SeV infection of several other cell types to determine if all cells can activate IFN responses independently of TBK1. Infection of thymocytes with SeV showed comparable kinetics and levels of STAT1 phosphorylation in TBK1+/+ and TBK1−/− thymocytes, indicating that TBK1 is not required for IFN responses in thymocytes (unpublished data).
We also examined the role for TBK1 in IRF3 activation during SeV infections in immortalized MEFs derived from wild-type and TBK1−/− littermates. In contrast with our observations in BMMs and thymocytes, IRF3 nuclear localization in response to SeV was absent in MEFs lacking TBK1 (Fig. 5 A). STAT1 phosphorylation was also largely reduced in TBK1−/− MEFs (Fig. 5 A). Furthermore, TBK1−/− MEFs failed to induce expression of IFNβ, IP-10, IRF7, or IL-15 in response to SeV infection (Fig. 5 B). However, NF-κB activation was unaffected as assessed by EMSA, p65 nuclear localization, and analysis of induction of IκBα and ICAM-1 genes (Fig. 5, C–E). Thus, we have observed a cell type–dependent requirement for TBK1 in the activation of IFN responses during infection with SeV, and have found a requirement for TBK1 in MEFs, but not in BMMs or thymocytes.
Figure 5.

TBK1-deficient MEF cells have impaired IFN responses to SeV. Wild-type and TBK1−/− MEF cells were infected with SeV for the indicated time points. (A) Nuclear fractions were probed for IRF3 and USF2 as a loading control. Total cell extracts were probed for phospho-STAT1 and total STAT1. (B) Total RNA was extracted and analyzed by Q-PCR for expression of IFNβ, IP-10, IRF7, and IL-15. (C) EMSA was performed by incubating nuclear extracts were with an NF-κB-specific oligonucleotide. (D) Nuclear fractions were probed for p65 and USF2. (E) Total RNA was extracted and analyzed by Q-PCR for expression of ICAM1 and IκBα.
Differential Expression of IKK-i May Be Responsible for Cell Type–dependent Requirements for TBK1 in Viral Activation of IFN Responses.
In addition to TBK1, IKK-i has also been identified as a putative IRF3 and IRF7 kinase (30, 31). IKK-i has been shown to be preferentially expressed in specific tissues, including spleen and peripheral blood lymphocytes, and expression can be induced in response to LPS and proinflammatory cytokines (40). We sought to determine if differential expression of IKK-i may mediate the discrepancy we observed when comparing the requirement for TBK1 in BMMs versus MEFs during SeV infection. Thus, we analyzed basal expression levels of IKK-i and TBK1 mRNA in BMMs and MEFs by Northern blot analysis. Similar expression levels of TBK1 were observed in BMMs and MEFs (Fig. 6 A). However, analysis of IKK-i mRNA levels in these cell types revealed strong expression in BMMs, whereas wild-type MEFs had barely detectable levels of IKK-i and these levels were even lower in TBK1−/− MEFs (Fig. 6 A).
Figure 6.
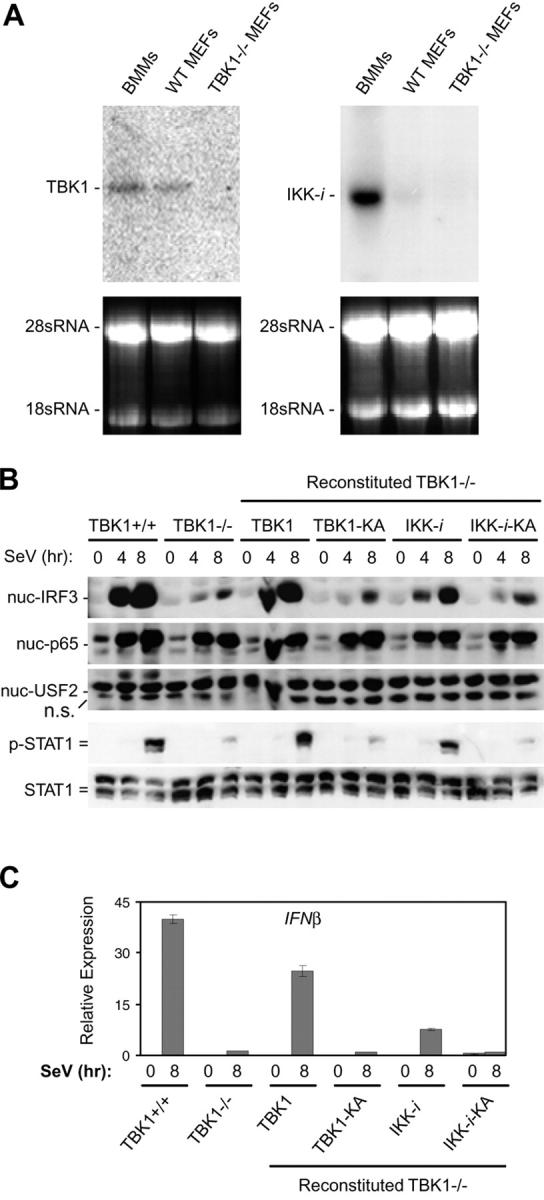
IKK-i is differentially expressed in BMMs and MEFs, and can rescue a TBK1 deficiency during SeV infection. (A) Total RNA extracted from BMMs, wild-type MEFs, and TBK1−/− MEFs were probed by Northern blot for expression of TBK1 and IKK-i. 28 and 18 sRNA bands are shown as loading controls. (B) Wild-type MEFs, TBK1−/− MEFs, and TBK1−/− MEFs reconstituted with wild-type TBK1 or IKK-i, or their kinase-inactive (KA) mutants were infected with SeV for the time points indicated. Nuclear fractions were probed for IRF3, p65, and USF2 as a loading control. Total cell extracts were probed for phospho-STAT1 and total STAT1. (C) Cells were infected with SeV as in B and total RNA was extracted and analyzed by Q-PCR for expression of IFNβ.
We determined if the introduction of IKK-i into TBK1−/− MEFs could rescue SeV-mediated IRF3 activation. TBK1−/− MEFs were stably transfected with vectors encoding either TBK1 or IKK-i or their respective KA versions, harboring a single K38A substitution shown to inactivate kinase activity (29, 38–40). Reconstituted cells were infected with SeV, and IRF3 nuclear localization was analyzed. TBK1−/− MEFs reconstituted with wild-type TBK1 but not KA TBK1 (TBK1-KA) were able to induce IRF3 nuclear localization, IFNβ production, and STAT1 phosphorylation similarly to wild-type MEFs (Fig. 6, B and C). In addition, TBK1−/− MEFs reconstituted with IKK-i could also rescue the deficiency of TBK1. This was dependent on the kinase activity of IKK-i as KA IKK-i (IKK-_i_–KA) could not rescue IRF3 activation, STAT1 phosphorylation, or IFNβ production (Fig. 6, B and C). Thus, IKK-i is able to mediate IRF3 activation in response to SeV in the absence of TBK1. Hence, our data indicate that IKK-i may be playing a redundant role with TBK1 during virus-induced IFN responses in cell types that express sufficient levels of IKK-i.
Discussion
Production of type I IFNs is critical to the host immune defense against viral infection and is a hallmark of virally infected cells. Induction of antiviral IFN responses can also occur as a result of TLR3/4 ligand stimulation. Although virtually all cell types can elicit an IFN response during viral infection, expression of TLRs is more restricted to immune cells and, thus, TLR-induced antiviral responses may be primarily important in these cell types. As the pathways involved in TLR3/4-induced type I IFN production are being carefully studied, an interesting question remains unanswered regarding the role of these pathways and signaling mediators during viral infections. In this work, we compare the role of the kinase TBK1 in IFN responses induced by LPS, polyI:C, and SeV. We have found a requirement for TBK1 in IRF3 activation and IFNβ production in response to LPS and polyI:C. However, during infection with SeV, we have found a cell type–dependent requirement for TBK1 for induction of IFN responses.
In response to LPS, BMMs derived from TBK1−/− mice were found to be defective in their ability to activate IRF3 and up-regulate IFNβ production. Furthermore, LPS induction of IFN-regulated secondary response genes was also TBK1 dependent. Thus, our data demonstrate that although IKK-i is coexpressed in BMMs, TBK1 plays a nonredundant role as the LPS-activated IRF3 kinase. Previous studies have also shown that cells from mice lacking TLR4, TRIF, or TRAM are completely defective in LPS-induced IRF3 activation and IFN production (21, 23, 24, 27). In addition, IRF3 is essential for LPS-mediated type I IFN responses (reference 41 and unpublished data). Therefore, our studies are consistent with a model of LPS-induced IFN induction proceeding through a linear signaling pathway dependent on TLR4, TRAM, TRIF, TBK1, and IRF3.
IRF3 activation and IFNβ production in response to polyI:C was also found to be dependent on TBK1. Previous studies have shown that TBK1 can interact with TRIF and phosphorylate IRF3 (30, 42). Therefore, it seems likely that TBK1 mediates dsRNA induction of type I IFN through the same pathway as TLR3, TRIF, and IRF3. However, although TBK1 seems to be required for all LPS-mediated IFN responses, polyI:C stimulation was able to induce TBK1-independent STAT1 phosphorylation and production of IFNα and IFN-regulated genes in a delayed but significant fashion. Studies in IRF3-deficient cells show a similar delay in STAT1 phosphorylation and induction of secondary IFN-regulated genes (reference 41 and unpublished data). Other studies have also indicated that alternative pathways exist for dsRNA induction of IFN responses. In cells lacking TLR3 or TRIF, dsRNA stimulation is still able to induce a delayed but significant amount of type I IFN and IFN-mediated gene induction, potentially mediated by protein kinase R recognition of intracellular dsRNA (10, 24, 43–45). Recently, a locus-mediating TRIF-independent up-regulation of costimulatory molecules was identified. Description of the gene mediating this phenotype may reveal a member of this alternative pathway (44). These studies highlight that host cells have evolved multiple mechanisms for detection of viral invasion. This phenomenon is particularly important because many viruses have the ability to inhibit various signaling mediators involved in activation of antiviral IFN responses.
During infection with SeV, we found a TBK1 deficiency in BMMs had minimal effects on IRF3 activation, IFNβ production, or production of IFN-regulated genes. However, all of these responses were dramatically abrogated during infection of TBK1−/− MEFs. The cell type–specific requirement for TBK1 during viral infection indicates that alternate mechanisms exist in BMMs but not in MEFs to phosphorylate IRF3. The related kinase IKK-i was expressed at much higher levels in BMMs as compared with MEFs. Reintroduction of either TBK1 or IKK-i but not their KA mutants into TBK1−/− MEFs could rescue activation of IRF3 and IFNβ production, indicating that IKK-i may be functionally redundant with TBK1 during viral infections. Studies in IKK-_i–_deficient and TBK1/IKK-i double knockout mice are needed to clarify the requirements for these kinases during viral infections.
TBK1 was initially identified as having a role in NF-κB activation, being able to induce NF-κB activation when overexpressed, as well as being able to in vitro phosphorylate IKKβ and Ser32 of IκBα (38, 39). However, in agreement with initial studies performed on TBK1−/− MEFs in which the process of activating NF-κB by TNF or IL-1 was not observably defective (32), our studies indicate that TBK1 is not required for NF-κB activation during either TLR3/4 stimulation or virus infection, as neither nuclear localization or DNA-binding ability were affected. This observation is consistent with previous biochemical studies indicating TLR-mediated MyD88-independent activation of NF-κB versus IRF3 responses diverge at TRIF (42).
Viruses are complex and diverse organisms and, thus, hosts have likely evolved multiple mechanisms for detecting viral invasion. Although we have characterized the role for TBK1 in response to LPS, polyI:C, and SeV infection, future studies are needed to address whether TBK1 plays a similar role during infections with different viruses, as well as to determine the role of IKK-i and other signaling mediators. Furthermore, the host receptors recognizing viral components have yet to be identified. Due to the differential requirement for TBK1 in various cell types, it will be interesting to explore the role for TBK1 in the whole animal response during in vivo infections to determine if cell types that elicit IFN production independently of TBK1 are sufficient for resistance to viral infection. Viruses can inhibit induction of type I IFNs in a variety of ways, many of which have not yet been elucidated, and further understanding of these pathways can help to better understand the mechanisms of virus-mediated inhibition of the IFN response (46).
Acknowledgments
We thank Dr. D. Nayak for providing us with Sendai virus. We give special thanks to Drs. G. Li and T.-T. Wu for guidance in virus infections.
A.K. Perry is supported by the Howard Hughes Medical Institute predoctoral fellowship (grant no. 59003787). J.B. Goodnough is supported by a UCLA Medical Scientist Training Program (GM08042). Part of this work was also supported by National Institutes of Health research grants RO1 CA87924, RO1 AI056154, and R37 AI47868.
Note added in proof. While this paper was prepared, a related paper was published (McWhirter, S.M., K.A. Fitzgerald, J. Rosains, D.C. Rowe, D.T. Golenbock, and T. Maniatis. 2004. Proc. Natl. Acad. Sci. USA. 101:233–238), indicating that TBK1 is required for type I IFN responses to viral infection in MEF cells.
Abbreviations used in this paper: BMM, BM-derived macrophage; IFNAR, type I IFN α/β receptor; IKK-i, inducible IκB kinase; IRF3, IFN regulatory factor-3; JAK, Janus kinase; KA, kinase-inactive; MEF, immortalized murine embryonic fibroblast; Q-PCR, quantitative RT-PCR; SeV, Sendai virus; STAT, signal transducer and activator of transcription; TBK1, TANK-binding kinase-1; TLR, Toll-like receptor; TRAM, TRIF-related adaptor molecule; TRIF, Toll/IL-1 receptor domain–containing adaptor-inducing IFN-β.
References
- 1.Muller, U., U. Steinhoff, L.F. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 2.Stark, G.R., I.M. Kerr, B.R. Williams, R.H. Silverman, and R.D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264. [DOI] [PubMed] [Google Scholar]
- 3.Juang, Y.T., W. Lowther, M. Kellum, W.C. Au, R. Lin, J. Hiscott, and P.M. Pitha. 1998. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc. Natl. Acad. Sci. USA. 95:9837–9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taniguchi, T., K. Ogasawara, A. Takaoka, and N. Tanaka. 2001. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19:623–655. [DOI] [PubMed] [Google Scholar]
- 5.Taniguchi, T., and A. Takaoka. 2002. The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr. Opin. Immunol. 14:111–116. [DOI] [PubMed] [Google Scholar]
- 6.Servant, M.J., B. Tenoever, and R. Lin. 2002. Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J. Interferon Cytokine Res. 22:49–58. [DOI] [PubMed] [Google Scholar]
- 7.Sato, M., N. Hata, M. Asagiri, T. Nakaya, T. Taniguchi, and N. Tanaka. 1998. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 441:106–110. [DOI] [PubMed] [Google Scholar]
- 8.Barton, G.M., and R. Medzhitov. 2003. Toll-like receptor signaling pathways. Science. 300:1524–1525. [DOI] [PubMed] [Google Scholar]
- 9.Doyle, S.E., R.M. O'Connell, G.A. Miranda, S.A. Vaidya, E.K. Chow, P.T. Liu, S. Suzuki, N. Suzuki, R.L. Modlin, W.C. Yeh, et al. 2004. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 11.Kurt-Jones, E.A., L. Popova, L. Kwinn, L.M. Haynes, L.P. Jones, R.A. Tripp, E.E. Walsh, M.W. Freeman, D.T. Golenbock, L.J. Anderson, and R.W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398–401. [DOI] [PubMed] [Google Scholar]
- 12.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 13.Diebold, S.S., T. Kaisho, H. Hemmi, S. Akira, and E.S.C. Reis. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 14.Hemmi, H., T. Kaisho, O. Takeuchi, S. Sato, H. Sanjo, K. Hoshino, T. Horiuchi, H. Tomizawa, K. Takeda, and S. Akira. 2002. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3:196–200. [DOI] [PubMed] [Google Scholar]
- 15.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745. [DOI] [PubMed] [Google Scholar]
- 16.Akira, S., and H. Hemmi. 2003. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 85:85–95. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi, O., and S. Akira. 2002. MyD88 as a bottle neck in Toll/IL-1 signaling. Curr. Top. Microbiol. Immunol. 270:155–167. [DOI] [PubMed] [Google Scholar]
- 18.Kaisho, T., O. Takeuchi, T. Kawai, K. Hoshino, and S. Akira. 2001. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J. Immunol. 166:5688–5694. [DOI] [PubMed] [Google Scholar]
- 19.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 11:115–122. [DOI] [PubMed] [Google Scholar]
- 20.Doyle, S., S. Vaidya, R. O'Connell, H. Dadgostar, P. Dempsey, T. Wu, G. Rao, R. Sun, M. Haberland, R. Modlin, and G. Cheng. 2002. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 17:251–263. [DOI] [PubMed] [Google Scholar]
- 21.Kawai, T., O. Takeuchi, T. Fujita, J. Inoue, P.F. Muhlradt, S. Sato, K. Hoshino, and S. Akira. 2001. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167:5887–5894. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, and S. Akira. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 169:6668–6672. [DOI] [PubMed] [Google Scholar]
- 23.Hoebe, K., X. Du, P. Georgel, E. Janssen, K. Tabeta, S.O. Kim, J. Goode, P. Lin, N. Mann, S. Mudd, et al. 2003. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 424:743–748. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 301:640–643. [DOI] [PubMed] [Google Scholar]
- 25.Oshiumi, H., M. Matsumoto, K. Funami, T. Akazawa, and T. Seya. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4:161–167. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgerald, K.A., D.C. Rowe, B.J. Barnes, D.R. Caffrey, A. Visintin, E. Latz, B. Monks, P.M. Pitha, and D.T. Golenbock. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 198:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto, M., S. Sato, H. Hemmi, S. Uematsu, K. Hoshino, T. Kaisho, O. Takeuchi, K. Takeda, and S. Akira. 2003. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 4:1144–1150. [DOI] [PubMed] [Google Scholar]
- 28.Oshiumi, H., M. Sasai, K. Shida, T. Fujita, M. Matsumoto, and T. Seya. 2003. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-beta. J. Biol. Chem. 278:49751–49762. [DOI] [PubMed] [Google Scholar]
- 29.Peters, R.T., and T. Maniatis. 2001. A new family of IKK-related kinases may function as I kappa B kinase kinases. Biochim. Biophys. Acta. 1471:M57–M62. [DOI] [PubMed] [Google Scholar]
- 30.Fitzgerald, K.A., S.M. McWhirter, K.L. Faia, D.C. Rowe, E. Latz, D.T. Golenbock, A.J. Coyle, S.M. Liao, and T. Maniatis. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. [DOI] [PubMed] [Google Scholar]
- 31.Sharma, S., B.R. tenOever, N. Grandvaux, G.P. Zhou, R. Lin, and J. Hiscott. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science. 300:1148–1151. [DOI] [PubMed] [Google Scholar]
- 32.Bonnard, M., C. Mirtsos, S. Suzuki, K. Graham, J. Huang, M. Ng, A. Itie, A. Wakeham, A. Shahinian, W.J. Henzel, et al. 2000. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19:4976–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chin, A.I., P.W. Dempsey, K. Bruhn, J.F. Miller, Y. Xu, and G. Cheng. 2002. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 416:190–194. [DOI] [PubMed] [Google Scholar]
- 34.Shioda, T., Y. Hidaka, T. Kanda, H. Shibuta, A. Nomoto, and K. Iwasaki. 1983. Sequence of 3,687 nucleotides from the 3′ end of Sendai virus genome RNA and the predicted amino acid sequences of viral NP, P and C proteins. Nucleic Acids Res. 11:7317–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liou, H.C., W.C. Sha, M.L. Scott, and D. Baltimore. 1994. Sequential induction of NF-kappa B/Rel family proteins during B-cell terminal differentiation. Mol. Cell. Biol. 14:5349–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle, S.E., R. O'Connell, S.A. Vaidya, E.K. Chow, K. Yee, and G. Cheng. 2003. Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4. J. Immunol. 170:3565–3571. [DOI] [PubMed] [Google Scholar]
- 37.Honda, K., S. Sakaguchi, C. Nakajima, A. Watanabe, H. Yanai, M. Matsumoto, T. Ohteki, T. Kaisho, A. Takaoka, S. Akira, et al. 2003. Selective contribution of IFN-{alpha}/{beta} signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA. 100:10872–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tojima, Y., A. Fujimoto, M. Delhase, Y. Chen, S. Hatakeyama, K. Nakayama, Y. Kaneko, Y. Nimura, N. Motoyama, K. Ikeda, et al. 2000. NAK is an IkappaB kinase-activating kinase. Nature. 404:778–782. [DOI] [PubMed] [Google Scholar]
- 39.Pomerantz, J.L., and D. Baltimore. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18:6694–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimada, T., T. Kawai, K. Takeda, M. Matsumoto, J. Inoue, Y. Tatsumi, A. Kanamaru, and S. Akira. 1999. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int. Immunol. 11:1357–1362. [DOI] [PubMed] [Google Scholar]
- 41.Sakaguchi, S., H. Negishi, M. Asagiri, C. Nakajima, T. Mizutani, A. Takaoka, K. Honda, and T. Taniguchi. 2003. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem. Biophys. Res. Commun. 306:860–866. [DOI] [PubMed] [Google Scholar]
- 42.Sato, S., M. Sugiyama, M. Yamamoto, Y. Watanabe, T. Kawai, K. Takeda, and S. Akira. 2003. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappaB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 171:4304–4310. [DOI] [PubMed] [Google Scholar]
- 43.Honda, K., S. Sakaguchi, C. Nakajima, A. Watanabe, H. Yanai, M. Matsumoto, T. Ohteki, T. Kaisho, A. Takaoka, S. Akira, et al. 2003. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA. 100:10872–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoebe, K., E.M. Janssen, S.O. Kim, L. Alexopoulou, R.A. Flavell, J. Han, and B. Beutler. 2003. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat. Immunol. 4:1223–1229. [DOI] [PubMed] [Google Scholar]
- 45.Diebold, S.S., M. Montoya, H. Unger, L. Alexopoulou, P. Roy, L.E. Haswell, A. Al-Shamkhani, R. Flavell, P. Borrow, and C. Reis e Sousa. 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 424:324–328. [DOI] [PubMed] [Google Scholar]
- 46.Katze, M.G., Y. He, and M. Gale, Jr. 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675–687. [DOI] [PubMed] [Google Scholar]