Repression of B7.2 on Self-reactive B Cells Is Essential to Prevent Proliferation and Allow Fas-mediated Deletion by CD4+ T Cells (original) (raw)
Abstract
Peripheral tolerance mechanisms normally prevent delivery of T cell help to anergic self-reactive B cells that accumulate in the T zones of spleen and lymph nodes. Chronic exposure to self-antigens desensitizes B cell antigen receptor (BCR) signaling on anergic B cells so that they are not stimulated into clonal expansion by CD4+ T cells but instead are eliminated by Fas (CD95)-induced apoptosis. Because a range of BCR-induced signals and responses are repressed in anergic B cells, it is not known which of these are critical to regulate for Fas-mediated peripheral tolerance. Display of the costimulatory molecule, B7.2 (CD86), represents a potentially important early response to acute BCR engagement that is poorly induced by antigen on anergic B cells. We show here that restoring B7.2 expression on tolerant B cells using a constitutively expressed B7.2 transgene is sufficient to prevent Fas-mediated deletion and to trigger extensive T cell–dependent clonal expansion and autoantibody secretion in the presence of specific T cells. Dysregulated expression of B7.2 on tolerant B cells caused a more extreme reversal of peripheral tolerance than that caused by defects in Fas or Fas ligand, and resulted in T cell–dependent clonal expansion and antibody secretion comparable in magnitude to that made by foreign antigen-specific B cells. These findings demonstrate that repression of B7.2 is critical to eliminate autoreactive B cells by Fas in B cell–T cell interactions. The possible role of B7.2 dysregulation in systemic autoimmune diseases is discussed.
Keywords: B7.2 (CD86), Fas (CD95), B cell antigen receptor, B cell, autoimmunity
CD4+ T cells, to initiate humoral immune responses against foreign antigens and simultaneously avoid autoimmunity, must selectively help foreign antigen–reactive but not self-reactive B cells. Clonal deletion of self-reactive T and B cells in the thymus and bone marrow provides only a partial bulwark against T cell–dependent autoantibody production, because a significant number of self-reactive T and B cells are exported to secondary lymphoid tissues. Once they reach the spleen and lymph nodes, interaction between self-reactive B and T cells must be controlled by peripheral tolerance mechanisms. Anergic B cells, recognizing model self-antigens or nuclear autoantigens that are prototypic targets in systemic lupus, concentrate as short-lived emigrants in the outer T zones adjacent to B cell–rich follicles in the spleen and lymph nodes (1–3). Paradoxically, this site appears to offer the greatest chance of interacting with Th cells. The Fas ligand (FasL)/Fas (CD95/APO-1)1 pathway plays a critical role in preventing delivery of help to this class of autoreactive B cells (4–8). During the early phases of anergic B cell–T cell interaction in vivo, CD40L on the T cell signals the B cell to elevate Fas expression and become sensitive to the apoptotic effects of FasL on the T cell (8). When the Fas gene is mutated, T cell–dependent activation of this class of self-reactive B cells cannot be aborted by this mechanism, and the anergic B cells gradually come to be triggered into high-rate autoantibody secretion (4, 6, 7) and to form large clones of autoantibody-producing cells in the T zones (5).
Signaling by the B cell antigen receptor (BCR) regulates whether B cells proliferate or undergo Fas (CD95)-induced apoptosis when they present antigen to CD4+ T cells in secondary lymphoid organs (6, 8–12). Acute BCR signaling during exposure to foreign antigens promotes clonal expansion and protects foreign-specific B cells against death triggered by FasL on the T cell. In anergic self-reactive B cells, BCR signaling is desensitized and qualitatively altered by chronic exposure to self-antigens, so that the nuclear factor of activated T cells (NFAT) and extracellular signal– regulatory kinase (ERK) pathways continue to be stimulated but the nuclear factor κB and c-Jun NH2-terminal kinase (JNK) signaling pathways are uncoupled from the BCR (12, 13). This modulation of BCR signaling allows the FasL/Fas pathway to selectively abort proliferation of self-reactive B cells, thus focusing T help onto foreign-specific B cells (6, 8). Because a range of signaling pathways and early responses to BCR signaling are repressed or absent in self-tolerant (anergic) B cells (12, 13), it is not known which must be regulated to allow Fas-mediated peripheral tolerance.
Surface display of the costimulatory molecule, B7.2 (CD86), is one of the early responses to acute BCR signaling that is repressed in self-tolerant B cells (2, 12, 14–18). Tolerant B cells present antigen to primary T cells effectively, but cannot stimulate cytokine secretion in vitro unless the poor display of B7.2 is compensated by adding a stimulatory antibody to CD28, a receptor for B7.2 that is displayed constitutively on T cells (15). CD28 transmits signals that synergize with the TCR for T cell activation and cytokine secretion (18). Antibody blocking and gene knockout experiments have established that B7.2 is important for initiating humoral immune responses to foreign antigens and for humoral autoimmunity (18–20). Although these experiments do not resolve whether B7.2 functions during activation of Th cells by dendritic cells, during T cell–B cell interactions, or both, they highlight the potential importance of repressing B7.2 on self-tolerant B cells.
To test the significance of the repressed B7.2 response to antigen on tolerant B cells, we describe here the effects of selectively restoring B7.2 expression on these cells using a constitutively expressed B7.2 transgene (21). Dysregulated expression of B7.2 on self-tolerant B cells did not interfere with desensitization of BCR signaling, but in the presence of autoantigen-specific T cells, it was sufficient to prevent their Fas-mediated deletion and to trigger extensive clonal expansion and autoantibody secretion. The reversal of peripheral tolerance induced by B7.2 dysregulation was much more extreme than the reversal caused by defects in Fas or FasL, and resulted in T cell–dependent clonal expansion and antibody secretion comparable in magnitude to that made by foreign antigen-specific B cells. These findings demonstrate that repression of B7.2 is critical to eliminate autoreactive B cells by Fas in B cell–T cell interactions in vivo. Based on these results, we suggest that failure to repress B7.2 on B cells may be equally or more important as an inherited or acquired component of systemic autoimmune diseases than defects in the Fas pathway itself.
Materials and Methods
Mice.
Mice carrying a transgene for B7.2 (line 7 in reference 21) were crossed with C57BL6/J hen egg lysozyme (HEL)/IgHEL double-transgenic mice (ML5/MD4-tg [22]) that had been previously crossed to B10.Br and selected for H-2k homozygosity. Blood from the resulting offspring was screened by FACS® to determine which combination of transgenes they carried. Because the MD4 IgHEL transgene and the H-2 complex are closely linked on chromosome 17 (∼1 cM apart), IgHEL-tg mice were H-2k/b heterozygotes. Wild-type and FasL-deficient non-tg or TCR-tg mice were bred as described (8). T cells heterozygous for the gld mutation of FasL were used because they develop in a nonautoimmune background but fail to eliminate B cells by Fas in the short-term adoptive transfer assays used here (6, 8). This failure is perhaps due to formation of trimers which consist of both mutant and wild-type FasL and have limited function. Recipient mice for adoptive transfers were non-tg or ML5 HEL-tg (C57BL6/J × B10.Br)F1 animals. Donor and recipient mice were between 6 and 20 wk of age. Sexes were matched as much as possible, although no difference was observed between male and female mice in the adoptive transfer assays described. Mice were MHC matched in all experiments.
Adoptive Transfers.
B220+ B cells and CD4+ T cells were purified as described previously (8). 1–3 × 106 B220+ B cells and 3–6 × 105 CD4+ T cells were mixed on ice and injected via the lateral tail vein into sublethally irradiated (750 cGy) recipient mice. 5 d after adoptive transfer, recipient mice were killed, and spleen cell suspensions were made by passing through a metal sieve. The cells were then counted by hemocytometer and suspended to appropriate concentrations for flow cytometric analysis or spot ELISA to determine the frequency of anti-HEL–secreting cells (8).
Flow Cytometry.
Cells were analyzed with a FACScan® (Becton Dickinson, Mountain View, CA) as described (6). The following antigens and antibodies were used: B220, RA3-6B2– FITC or –PE (Caltag Laboratories, Inc., South San Francisco, CA); IgMa, RS3.1-FITC; Fas, Jo2-PE (PharMingen, San Diego, CA); B7.2, GL1-PE (PharMingen); CD69, H1.2F3-FITC (PharMingen); HEL, HyHEL9-TRIcolor (custom conjugation; Caltag Laboratories, Inc.); Syndecan, 281.2-biotin followed by streptavidin-CyChrome (PharMingen); and OX40, OX40L–human Ig fusion protein or control human Ig followed by goat anti–human Ig-PE (gift of Dr. Wayne Godfrey, University of California at San Francisco, San Francisco, CA). Cell numbers for each experiment were determined by multiplying the frequency of B220+IgMa+ HEL-binding cells by the cell count as determined by hemocytometer.
Western Blots.
Western blots were performed as described (12).
In Vitro Cultures and Competitive Reverse Transcription PCR.
Cells were cultured at 37°C, 5% CO2 at 1 or 2 × 106 cells/ml together in 1 ml or 200 μl complete RPMI in 24- or 96-well round-bottomed plates (Corning Glass Works, Corning, NY), respectively. The anti-CD40 antibody, 1C10 (Southern Biotechnology Associates, Inc., Birmingham, AL), was used at 10 μg/ml, and the anti-Fas antibody, Jo2, was used at 100 ng/ml. Cells were harvested for FACS® analysis, or total RNA was prepared with Trizol (GIBCO BRL, Gaithersburg, MD) according to the manufacturer's instructions. Cell death was measured flow cytometrically by propidium iodide (PI) exclusion, and specific killing was determined by subtracting the percentage of PI+IgDa+ cells in the absence of Jo2 from the percentage of PI+IgDa+ cells in the presence of Jo2 and dividing by 100, minus the percentage of PI+IgDa+ cells in the absence of Jo2. Total RNA from each sample was reverse transcribed (Superscript II; GIBCO BRL) with oligo-dT (Pharmacia Biotech AB, Uppsala, Sweden) and subject to competitive reverse transcription (RT)-PCR as described (23). In brief, after reverse transcription, cDNA concentrations were normalized by titration samples against a known concentration of hypoxanthine phosphoribosyltransferase (HPRT) competitor plasmid. The normalized dilution of each sample cDNA was identified as that which gave equivalent HPRT PCR product to the competitor plasmid. The differences in the amount of HPRT PCR product generated for each sample were less than twofold. PCR for HPRT was repeated at the normalized sample cDNA dilutions, followed by PCR for IL-2, FasL, CD40L, and IL-4. The sample cDNA PCR product sizes for HPRT, IL-2, IL-4, FasL, and CD40L are 352, 247, 240, 350, and 443 bp, respectively. The competitor PCR product sizes for IL-2, IL-4, FasL, and CD40L are 320, 360, 510, and 603 bp, respectively. The pPQRS plasmid containing the competitors for HPRT, IL-2, and IL-4 was a gift of Sarah E. Townsend (John Curtin School of Medical Research). The competitor plasmid for FasL and CD40L was constructed as follows. An intron-spanning 350-bp fragment of FasL and a 443-bp fragment CD40L mRNA were PCR amplified from activated T cells (AGT GTG GCC CAT TTA ACA and ATG TTG ACA TAT AAA TGG TCA G for FasL, and GAA GCC AAC AGT AAT GCA and TGC CAG CAT CAG CCC TG for CD40L) and ligated to pBluescript (Stratagene Inc., La Jolla, CA) that had been cut with EcoRV (New England Biolabs, Inc., Beverly, MA) and incubated with dTTP (Pharmacia Biotech AB) plus Taq DNA polymerase (Promega Corp., Madison, WI) to generate pFasL and pCD40L. 160-bp fragments of the HEL gene were PCR amplified from ML5-tg mice by primers with 5′ NdeI and 5′ AvrII adapter sites for FasL and CD40L, respectively (GAA GAC ACA TAT GGA GCG TGA ACT GCG CGA AGA and GCG GTT CCA TAT GTC GGT ACC CTT GCA GCG GTT for FasL, and GCG CTG TCC TAG GGA GCG TGA ACT GCG CGA AGA and GCT GCA GCC TAG GTC GGT ACC CTT GCA GCG GTT for CD40L), and inserted into FasL and CD40L on pFasL and pCD40L in unique NdeI or AvrII sites, respectively, to generate pCFasL and pCCD40L.
Results
The B7.2 Transgene Does Not Interfere with B Cell Tolerance to HEL.
To study the functional significance of the failure of self-tolerant B cells to display B7.2, transgenic mice expressing high levels of B7.2 constitutively on B cells (line 7 in reference 21) were bred to mice carrying unlinked transgenes encoding both anti-HEL Ig (IgHEL) and HEL as a soluble serum protein (22). This cross produced mice with HEL-specific B cells that either had not previously been exposed to HEL antigen (naive) or had been chronically exposed to HEL (tolerant). In both types of progeny, half carried the B7.2 transgene (B7.2+ naive or tolerant) and half did not. In mice that inherited the B7.2 transgene, the constitutive level of B7.2 expressed on splenic B cells was comparable to that normally induced on naive HEL-specific B cells 6–20 h after acute exposure to HEL antigen (Fig. 1 a, and data not shown). Mice which express the B7.2 transgene alone have reduced and variable numbers of B cells through an antigen-nonspecific T cell–mediated mechanism (21). However, the additional presence of the HEL and IgHEL transgenes suppressed this phenotype. Thus, comparable numbers of B7.2+ or B7.2− tolerant B cells were present in the spleens of the two kinds of mice (3.44 ± 1.15 × 107 IgD+ cells for B7.2+/HEL/IgHEL mice compared with 2.33 ± 1.22 × 107 IgD+ cells for normal B7.2−/HEL/IgHEL mice). Also, unlike B7.2+ B cells in the absence of the IgHEL transgene, which are enlarged and activated (21), B7.2+ tolerant B cells are small and resting (Fig. 2 b, and data not shown).
Figure 1.


Antigen receptors are downregulated and desensitized normally on self-reactive B cells in B7.2-tg animals. (a) Spleen cells from IgHEL-tg mice (naive), HEL/IgHEL double-tg mice (tolerant), and HEL/IgHEL/B7.2 triple-tg mice (B7.2+ tolerant) were cultured in vitro with 50 ng/ml HEL for 17 h, and B cells were analyzed flow cytometrically for B7.2 expression (thick lines). Control cells were held on ice and analyzed in parallel (thin lines). Cells cultured in vitro for 17 h in media alone stained comparably to control cells held on ice (data not shown). Similar results were obtained upon stimulation with 500 ng/ml HEL. (b) Splenocytes from naive, tolerant, and B7.2+ tolerant mice were analyzed flow cytometrically for transgene-encoded surface IgMa and IgDa. (c) Splenocytes were stimulated and analyzed for CD69 expression as in a. Note that CD69 is induced to lower levels per cell on B7.2+ or B7.2− tolerant cells compared with naive cells. Similar results were obtained for both 50 and 500 ng/ml HEL. (d ) Naive, tolerant, and B7.2+ tolerant B cells were purified by selection on anti-B220 magnetic microbeads and stimulated for 3 min in vitro with 1 μg/ml HEL, and whole cell lysates were fractionated by SDS-PAGE and transferred to a nitrocellulose membrane. The Western blot was probed with antiphosphotyrosine, and then reprobed with a mixture of anti-Syk (gift of J. Roberts and A. Defranco, University of California at San Francisco) and anti-Lyn antibodies as a loading control. (e) Splenocytes from two control tolerant HEL/IgHEL mice and one B7.2+/HEL/IgHEL tolerant mouse were cultured in vitro for 0, 24, 48, or 72 h in 1C10 anti-CD40 antibody followed by an additional 5–6-h culture in the presence or absence of Jo2 anti-Fas antibody. Cell death was determined in triplicate cultures by flow cytometric analysis of uptake of the vital dye, PI, and positive staining for IgDa. Error bars indicate SD. Equivalent results were obtained with B220 in place of IgDa.
Figure 2.

B7.2 prevents FasL-mediated deletion and promotes proliferation of tolerant B cells upon interaction with antigen-specific T cells. (a) Purified B7.2+ or control tolerant B cells were adoptively transferred into irradiated non-tg recipients alone or with purified +/+ or gld/+ TCR-tg CD4+ T cells. 5 d after transfer, HEL-binding B cells were enumerated flow cytometrically in spleens from recipient mice. Means are shown for all groups. SD is indicated for groups of three mice; values for individual mice are shown for other groups. Horizontal line, Background staining in negative control animals injected with media only. (b–d ) The surviving transferred B cells had become activated as measured by cell size (b, Forward Scatter), recovery of surface IgM (c), and increased expression of Fas (d ).
B7.2+ tolerant B cells that had been chronically exposed to self-antigen in HEL/IgHEL-tg animals were tolerant to HEL and could be rendered sensitive to killing through Fas. Thus, the B7.2+ tolerant B cells selectively downregulated surface IgM but not IgD similarly to tolerant B cells lacking the B7.2 transgene (Fig. 1 b). The small difference in percentage of IgMhighIgDhigh cells between the B7.2+ and B7.2− tolerant splenocytes in Fig. 1 b was not reproducible, and represents natural variation of IgM expression on tolerant B cells presumably due to variation in blood HEL concentration. The mean ratio of IgMhighIgDhigh to IgMlowIgDhigh splenic B cells was found to be 18.6 ± 5.6 in IgHEL-tg mice, 0.5 ± 0.2 in HEL/IgHEL-tg mice, and 0.4 ± 0.2 in B7.2+ HEL/IgHEL-tg mice. Moreover, HEL antigen induced CD69 only to low levels (Fig. 1 c), and was unable to increase tyrosine phosphorylation of intracellular proteins such as syk, lyn, Igα, and Igβ (Fig. 1 d ) in tolerant B cells regardless of B7.2 expression. Importantly, tolerant and B7.2+ tolerant B cells were equally sensitive to Fas-mediated cytotoxicity in vitro after CD40 stimulation (Fig. 1 e).
B7.2 Expression on Tolerant B Cells Switches Their Fate from Clonal Elimination to Clonal Expansion.
The effect of B7.2 on antigen-specific B–T cell interactions was analyzed in vivo by purifying naive or HEL tolerant B cells either with or without the B7.2 transgene, mixing them with purified CD4+ T cells from 3A9 HEL-specific TCR-tg mice (15), and adoptively transferring the cell mixture into sublethally irradiated recipient mice. The fate of the B cells was then observed by flow cytometry to enumerate the number and phenotype of the transferred B cells and by spot ELISA to determine the number of B cells that had differentiated to secrete transgene-encoded anti-HEL antibody (Figs. 2 and 3). As reported previously, tolerant B cells lacking the B7.2 transgene were eliminated when adoptively transferred with wild-type (+/+) TCR-tg T cells (Fig. 2 a; references 6 and 8). This elimination was mediated by Fas, as FasL-deficient gld/+ TCR-tg T cells did not eliminate the tolerant B cells, but rather triggered blastogenesis and limited proliferation (Fig. 2 b; references 6 and 8). TCR gld/+ cells were used as described previously (6, 8), because in the short-term assay used here, they are as deficient in killing through Fas as gld/gld cells, but can be obtained from donors that do not have overt immunopathology. By contrast to the tolerant B cells lacking the B7.2 transgene, tolerant B cells that constitutively expressed high levels of B7.2 were not eliminated by antigen-specific T cells (Figs. 2 a and 3 a), and instead the population of transferred B7.2+ tolerant B cells enlarged (Fig. 2 b), upregulated IgM and Fas (Fig. 2, c and d ), and increased in number 20–100-fold (Fig. 2 a). Thus, although in the presence of antigen-specific gld/+ TCR-tg T cells, both B7.2+ and control tolerant B cells were protected from elimination and acquired similarly activated phenotypes, clonal expansion was much greater in the B7.2-tg tolerant B cells. Although observed in other experiments, the elimination of B7.2-tg B cells by antigen-nonspecific T cells (21) did not occur in the experiments presented here due to irradiation of recipient animals and the low number of adoptively transferred T cells.
Figure 3.
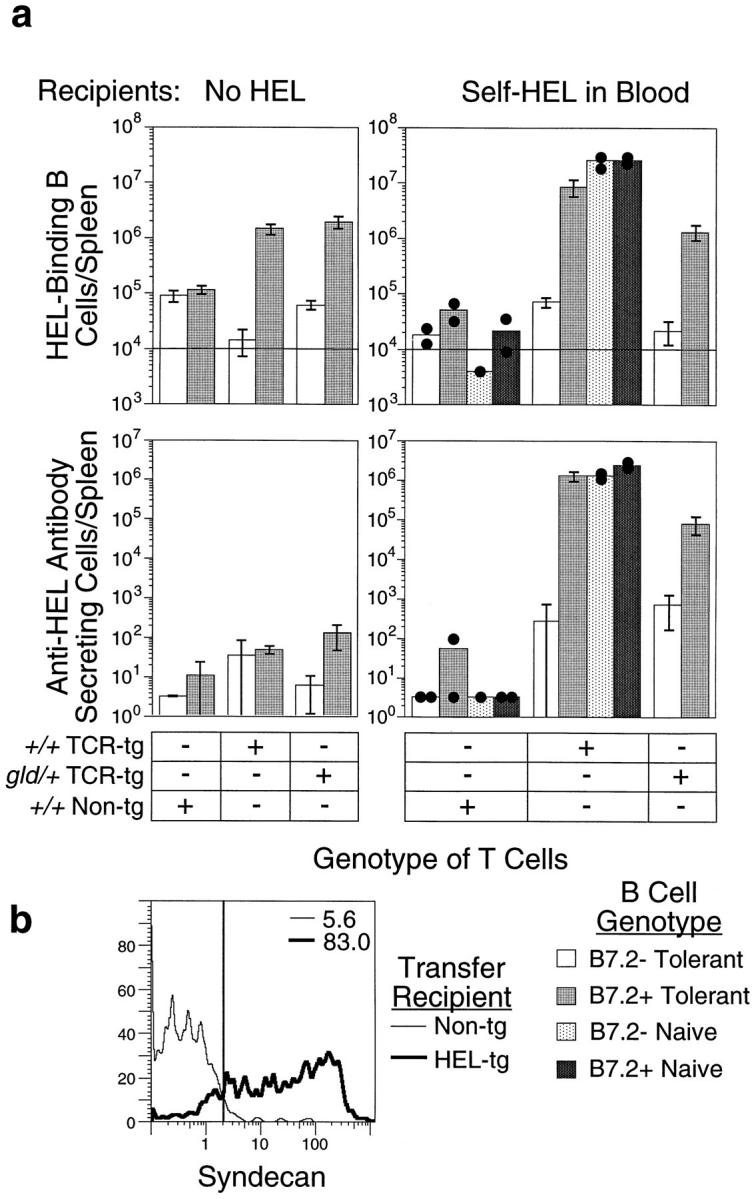
B7.2 provokes plasma cell differentiation and high-rate autoantibody secretion by tolerant B cells in the presence of antigen-specific T cells and continuous exposure to antigen. Purified naive or tolerant B cells with or without the B7.2 transgene were mixed with purified +/+ or gld/+ CD4+ non-tg or TCR-tg T cells and adoptively transferred into non-tg (No HEL) or HEL-tg (Self-HEL in Blood) recipients. 5 d later, spleens from recipient mice were analyzed flow cytometrically to enumerate the number and phenotype of transferred HEL-binding B cells and by spot ELISA to determine the number of cells secreting HEL-specific antibody in each recipient. (a) Numbers of HEL-binding B cells (top) and antibody-secreting cells (bottom) in the spleens of individual adoptive transfer recipients with (right) and without (left) self-HEL in the blood. Means are shown for all groups. SD is indicated for groups of three mice; values for individual mice are shown for other groups. Horizontal line, Background staining in negative control animals injected with RPMI only. (b) Expression of the plasma cell surface marker, Syndecan, on B7.2+ tolerant B cells 5 d after transfer with +/+ TCR-tg T cells into recipients with no circulating HEL (Non-tg; thin lines) or in recipients with HEL continuously present as a self-antigen in blood (HEL-tg; thick lines). The histograms show staining for Syndecan on gated IgMa+B220+ cells in the spleen.
B7.2 expression on tolerant B cells was sufficient to stimulate extensive B cell proliferation when they were transferred with antigen-specific T cells into non-tg recipients, but little or no secreted antibody was produced (compare Fig. 3 a, top left and bottom left, and few cells differentiating into Syndecan+ plasmablast cells in Fig. 3 b). To test if continuous exposure of the B and T cells to antigen was necessary to promote antibody production, B and T cells were adoptively transferred into HEL-tg recipients. As reported previously (6), antigen exposure in the absence of T cell help reduced B cell numbers to near background levels (Fig. 3 a, top right). However, in the continuous presence of antigen and antigen-specific T cells, proliferation of B7.2+ tolerant B cells was greatly increased, and most of the cells now differentiated into plasma cells, resulting in massive autoantibody secretion (Fig. 3, a and b, right). Under these conditions, only trace numbers of normal tolerant B cells were stimulated to proliferate and differentiate into autoantibody-secreting cells upon interaction with +/+ or gld/+ TCR-tg cells (Fig. 3 a, right). It is possible that the small number of normal tolerant B cells that successfully collaborate with T cells in this way represent cells at the upper end of the normal distribution of endogenous B7.2 on tolerant cells (Fig. 1 a), which is comparable to the B7.2 levels induced on naive cells or on B7.2+ transgene–expressing cells. The B7.2 transgene had little effect on proliferation and antibody production by naive B cells (Fig. 3 a). The number of antibody-forming cells generated by the B7.2+ tolerant B cell–T cell mixture was 2,000-fold higher than from tolerant B cells where B7.2 was repressed normally, and was comparable to the number produced by naive B cells under the same transfer conditions. Therefore, despite the fact that HEL induces a range of acute BCR signaling responses in the naive cells that are absent in tolerant B cells (12–14), restoring expression of B7.2 alone was sufficient to switch the fate of tolerant B cells from elimination to maximal clonal expansion and differentiation into plasmablasts.
Expression of B7.2 on Tolerant B Cells Alters the Pattern of Gene Induction in T Cells.
In contrast to naive B cells stimulated by foreign antigen, tolerant B cells have a selective costimulatory deficit for T cell cytokine production in vitro that can be reversed by antibody-mediated cross-linking of CD28 (15). To examine whether B7.2 expression on tolerant B cells altered cytokine gene expression in collaborating T cells, tolerant B cells with or without the B7.2 transgene were cultured in vitro with CD4+ T cells from non-tg or TCR-tg mice, and total RNA was prepared and converted to cDNA. The cDNAs were then normalized for HPRT concentration so as to produce less than twofold differences in HPRT PCR product when titrated against a known concentration of an HPRT competitor. After 16 h in vitro (Fig. 4 a), competitive PCR analysis showed that FasL mRNA was comparably induced in cultures with HEL-specific T cells (TCR) regardless of whether the tolerant B cells expressed B7.2 or not. By contrast, IL-2 mRNA was induced only in HEL-specific T cells cultured with B7.2+ tolerant B cells. Notably, message for IL-4, which has been previously been shown in vitro to rescue B cells from Fas-mediated apoptosis (11, 24–26), was detectable in cultures of HEL-specific T cells and B7.2+ tolerant B cells but not with tolerant B cells lacking the B7.2 transgene (Fig. 4 b). Additionally, the TNFR family member, OX40, was induced on T cells stimulated overnight in vitro with B7.2+ tolerant B cells but not with tolerant B cells without the B7.2 transgene. The selective induction of OX40 on T cells by B7.2+ tolerant B cells may be particularly relevant to autoimmunity, as OX40 has been implicated in the clonal expansion of B cell foci in the T zone (27), the primary site of clonal expansion and autoantibody production in MRL-lpr/lpr mice (5). Thus, selectively restoring B7.2 expression to tolerant B cells altered the cytokine gene induction profile in collaborating T cells to include a range of mitogenic and potentially antiapoptotic cytokines.
Figure 4.

Different cytokine genes are induced in the T cells as a result of B7.2 expression on tolerant B cells. (a and b) Purified B7.2+ or control tolerant B cells were cultured with equal numbers of purified CD4+ T cells from non-tg or 3A9 TCR-tg mice. Cells were harvested, and RNA was prepared after 16 (a) and 30 h (b). The RNA was converted to cDNA, and samples were normalized to each other for HPRT concentration by titrating each cDNA against a known concentration of competitor plasmid for HPRT by competitive PCR with HPRT-specific primers. When cDNAs were normalized so as to produce less than twofold differences in the amount of HPRT PCR product, they were analyzed by competitive PCR for transcripts encoding IL-2, FasL, CD40L, and IL-4. The PCR products from the exogenously added competitor plasmid (C ) and the sample cDNA (P) are indicated. The relative amounts of each RNA transcript in the original sample can be estimated by the strength of signal for each PCR product itself as well as by the reduction in intensity of the PCR product from the competitor plasmid (reference 23). Due to low RNA yield in samples with non-tg T cells and the low concentration of message, competitive PCR for IL-4 was conducted only from samples which included TCR-tg T cells. The competitive PCRs were repeated three times in each of two separate experiments with equivalent results to those shown. (c) Purified +/+ CD4+ T cells were cultured for 30 h without stimulus, in anti-CD3–coated wells, or with purified B7.2+ or control tolerant B cells. Cells were then analyzed flow cytometrically for OX40 by staining with OX40L-Fc (thick histograms) or a control Ig (thin histograms) followed by anti–human Ig conjugated to PE.
Discussion
The findings here demonstrate that selectively restoring B7.2 expression on otherwise self-tolerant B cells is sufficient to switch their fate from peripheral deletion to large-scale proliferation and autoantibody secretion during interactions with specific CD4+ T cells. Of the many early signals and responses to BCR engagement that are repressed or altered in tolerant B cells (12–14), the data here single out repression of the B7.2 response as being necessary to allow peripheral tolerance by FasL/Fas-mediated clonal abortion. The findings raise interesting questions about how B7.2 switches the outcome of interactions between tolerant B and T cells, and highlight the regulation of B7.2 in B cells as a key process that may be dysregulated by inherited or acquired triggers to systemic autoimmune disease.
Three possible mechanisms may in principle explain how dysregulated expression of B7.2 on tolerant B cells switches the outcome of interactions with autoantigen-specific T cells from deletion to autoimmunity. First, it is possible that self-reactive B cells maturing in a B7.2-tg mouse do not become functionally tolerant in the usual way. This possibility is ruled out by the data in Fig. 1, which show that the B7.2 transgene does not interfere with the BCR modulation and desensitization that characterizes HEL tolerant B cells, nor with the acquisition of sensitivity to Fas-induced apoptosis after activation by CD40 in vitro. The possibility that a minor subset of nontolerized B cells could be present in the B7.2+-tg mice and account for the large autoantibody response after transfer with T cells is excluded by the fact that the magnitude of the response is comparable to that of B7.2+ or B7.2− naive B cells, where all of the B cells are nontolerant (Fig. 3 a). The number of B cells transferred in these experiments is well below the saturating dose, so that even a 5–10% subset of nontolerant cells in the B7.2+ tolerant inoculum would be expected to mount a 10–20-fold reduced response.
Second, it is possible that B7.2 displayed on tolerant B cells engages CD28 on interacting T cells and transmits a retrograde signal into the B cell that blocks Fas-induced apoptosis and promotes proliferation and differentiation. The possibility of signal transduction by B7.2 has been raised previously, based on the presence of potential sites for protein kinase C phosphorylation (18). Indeed, an anti-B7.2 antibody has been reported to enhance B cell proliferation and differentiation (28). On the other hand, antibodies or soluble CD28-Fc or cytotoxic T lymphocyte–associated antigen 4 (CTLA4)-Fc ligands to B7.2 have not been observed to be mitogenic for B cells (18, 20), nor do they protect against Fas-mediated apoptosis (29).
The third interpretation for the effect of B7.2 is that it transmits a costimulus through CD28 in the T cell that alters the spectrum of cytokines/cell surface molecules made and displayed to the B cell, and these cytokines in turn promote B cell mitosis and block Fas-induced deletion. This conclusion is supported by the data in Fig. 4 and in the literature. Fig. 4 shows that dysregulated expression of B7.2 on tolerant B cells induces mRNAs for IL-2 and IL-4 in the T cells, and triggers surface expression of the TNFR homologue, OX40. IL-2 and IL-4 are potent mitogens for B cells (30), and OX40 is implicated in driving proliferation of B cells in the T zone by engaging OX40L on the surface of B cells (31). In addition to promoting mitogenesis, IL-4 has also been shown to inhibit the sensitization of CD40-activated B cells to Fas-induced apoptosis (11, 24–26). Indeed, HEL tolerant B cells are protected from Fas-induced apoptosis by IL-4 in vitro (26). Interestingly, transgenic mice that express IL-4 constitutively develop a systemic autoantibody syndrome reminiscent of MRL-lpr/ lpr mice, consistent with the notion that it blocks Fas-mediated peripheral tolerance (32). The failure of Fas-mediated B cell elimination caused by B7.2 was not simply due to proliferation outstripping FasL-induced death, because B7.2+ B cell proliferation was not increased further when the T cells were FasL deficient (Figs. 2 a and 3 a).
It is interesting that constitutive expression of B7.1 on B cells in transgenic mice prevents induction of an antibody response (33, 34), raising the possibility that B7.2 and B7.1 have distinct effects during T–B cell interactions. These effects could in principle be mediated through retrograde signaling via their distinct cytoplasmic regions or through their differential interactions with their receptors, CD28 and CTLA4 (18, 35). It is also intriguing that CD40 and IL-4 are also good inducers of B7.2 on B cells, and B7.2 responses to these stimuli are not depressed in tolerant B cells in vitro (12). CD40L from the T cells transmits signals that induce Fas on tolerant B cells during the sensitization phase preceding Fas-mediated deletion (8), so it might be expected that this signal would also upregulate B7.2 and block Fas-mediated deletion, as envisaged by Clark and Ledbetter (36). It is possible that B7.2 induction by CD40L during tolerant B cell–T cell interactions in vivo is either insufficient in amount or occurs too late to block deletion. Clearly, it will be important in the future to understand precisely how B7.2 and other surface molecules regulate both the induction and the delivery of Th cell molecules to B cells.
In addition to controlling B7.2 display, BCR signaling in response to antigen also regulates B cell–T cell interactions at several other levels. BCR signaling alters the migratory tropism of antigen-binding naive and tolerant B cells so that they accumulate preferentially in the T zone (1, 37). It is likely that this occurs by modulating the function of chemokine receptors such as Burkitt's lymphoma receptor 1 (BLR-1; reference 38). BCR signaling also induces production of chemokines for activated T cells such as macrophage-inflammatory protein (MIP)-1α and -1β (our unpublished observations). The combination of these effects appears likely to steer migration of antigen-binding B and T cells towards one another. BCR signals also promote cell cycle progression in their own right. These signaling responses are lost in tolerant B cells, and thus prevent activation of autoreactive B cells by T cell–independent antigens (12, 13). Finally, acute BCR signaling can block transmission of apoptotic signals from Fas through intrinsic pathways within the B cell (9–11, 24, 39, 40). This intrinsic effect of BCR signaling might work by altering expression of FLIP/Casper/FLAME/CASH/ I-FLICE (41–45), inhibitor of apoptosis proteins such as ILP (46) or Bcl-xL (47), and appears to be desensitized in tolerant B cells (8). It is conceivable that intrinsic regulation of the Fas pathway by the BCR acts in concert with the regulation of B7.2 and its effects as shown here, ensuring selective delivery of T cell help to the correct B cell targets in a variety of tissue locations and stages of the immune response.
The requirement for continued exposure to HEL antigen for differentiation of B7.2 tolerant B cells into plasma cells (Fig. 3) indicates that the BCR also regulates plasma cell differentiation independently of clonal expansion. There are several potential explanations for this finding. First, the altered signaling properties of desensitized BCRs on the B7.2+ tolerant B cell may not provide or may actively inhibit the necessary signals for differentiation of plasma cells (12, 13). However, as the B7.1 tolerant B cells become activated and proliferate, their receptors are upregulated, an indication that they are no longer desensitized. Additional antigen at this point might then provide the appropriate differentiation signals either within B cells or by providing additional costimulation to T cells. Second, the presence of antigen has been shown to alter patterns of B cell migration (1, 2, 48, 49). Therefore, antigen exposure may cause B or T cells to migrate to microenvironments conducive to B cell differentiation. Finally, tolerant B cells transferred into antigen-free recipient mice carry a finite supply of antigen and antigenic peptides with which to directly interact with antigen-specific T cells. Additional antigen may allow for stronger or repeated interactions of T cells with the B cells or other antigen-presenting cells.
The findings described here illuminate the importance of regulating B7.2 expression on B cells to ensure peripheral tolerance. Several studies have shown that systemic autoimmunity can be inhibited by blocking the interactions of B7.1 and B7.2 with their receptors, CD28 and CTLA4 (18, 20), but do not define which cells must express B7.1 or B7.2 for autoimmunity. The experiments described here demonstrate that in the presence of nontolerant T cells, the expression of B7.2 on self-reactive B cells is sufficient to cause systemic autoimmunity. B7.2 must be expressed on the autoreactive cells themselves, as the presence of neighboring cells which induce B7.2 normally does not cause the proliferation of tolerant B cells (12). The lack of a bystander effect is most likely due to directional display of cytokines and mitogenic factors (50). It is notable that dysregulated expression of B7.2 on self-reactive B cells leads to a much more extreme reversal of tolerance than that produced by simple Fas deficiency. Inherited Fas deficiency in C57BL/6 lpr/lpr mice allows self-reactive B cells to resist elimination by CD4+ T cells, but in this case they are stimulated to proliferate by the T cells to only a limited extent (references 6 and 8; Figs. 2 and 3). By contrast, the B7.2+ tolerant B cells undergo a very large clonal expansion. The strong effect of B7.2 on peripheral tolerance indicates that it will be important to define the biochemical pathway which normally regulates B7.2 expression on self-reactive B cells. Genetic mutations or polymorphisms in this pathway will be significant candidates for heritable predisposition to systemic lupus erythematosus, rheumatoid arthritis, and other human systemic autoimmune diseases.
Acknowledgments
This work was supported by grants from the National Institutes of Health, the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation (to B.C. Weintraub), and the Fonds de la Recherche en Santé du Québec (to S. Fournier). C.C. Goodnow was an investigator of the Howard Hughes Medical Institute.
Abbreviations used in this paper
BCR
B cell antigen receptor
CTLA4
cytotoxic lymphocyte–associated antigen 4
HEL
hen egg lysozyme
HPRT
hypoxanthine phosphoribosyltransferase
L
ligand
PI
propidium iodide
RT
reverse transcription
tg
transgenic
Footnotes
J.C. Rathmell's current address is Gwen Knapp Center for Lupus and Immunology Research, University of Chicago, 924 E. 57th St., R402, Chicago, IL 60637.
References
- 1.Cyster JG, Hartley SB, Goodnow CC. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. 1994;371:389–395. doi: 10.1038/371389a0. [DOI] [PubMed] [Google Scholar]
- 2.Cyster JG, Goodnow CC. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 1995;3:691–701. doi: 10.1016/1074-7613(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 3.Roark JH, Bui A, Nguyen KA, Mandik L, Erikson J. Persistence of functionally compromised anti-double-stranded DNA B cells in the periphery of non-autoimmune mice. Int Immunol. 1997;9:1615–1626. doi: 10.1093/intimm/9.11.1615. [DOI] [PubMed] [Google Scholar]
- 4.Rathmell JC, Goodnow CC. Effects of the lprmutation on elimination and inactivation of self-reactive B cells. J Immunol. 1994;153:2831–2842. [PubMed] [Google Scholar]
- 5.Jacobsen BA, Panka DJ, Nguyen KA, Erikson J, Abbas AK, Marshak-Rothstein A. Anatomy of autoantibody production: dominant localization of antibody-producing cells to T cell zones in Fas-deficient mice. Immunity. 1995;3:509–519. doi: 10.1016/1074-7613(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 6.Rathmell JC, Cooke MP, Ho WY, Grein J, Townsend SE, Davis MM, Goodnow CC. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+T cells. Nature. 1995;376:181–184. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- 7.Roark JH, Kuntz CL, Nguyen KA, Caton AJ, Erikson J. Breakdown of B cell tolerance in a mouse model of systemic lupus erythematosus. J Exp Med. 1995;181:1157–1167. doi: 10.1084/jem.181.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cells in vivo: dual roles for CD40- and Fas (CD95)-ligands modulated by the B cell antigen receptor. Cell. 1996;87:319–329. doi: 10.1016/s0092-8674(00)81349-5. [DOI] [PubMed] [Google Scholar]
- 9.Rothstein T, Wang J, Panka D, Foote L, Wang Z, Stanger B, Cui H, Ju S, Marshak-Rothstein A. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 1995;374:163–165. doi: 10.1038/374163a0. [DOI] [PubMed] [Google Scholar]
- 10.Lagresle C, Mondiere P, Bella C, Krammer PH, Defrance T. Concurrent engagement of CD40 and the antigen receptor protects naive and memory human B cells from APO-1/Fas–mediated apoptosis. J Exp Med. 1996;183:1377–1388. doi: 10.1084/jem.183.4.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Taniuchi I, Maekawa Y, Howard M, Cooper MD, Watanabe T. Expression and function of Fas antigen on activated murine B cells. Eur J Immunol. 1996;26:92–96. doi: 10.1002/eji.1830260114. [DOI] [PubMed] [Google Scholar]
- 12.Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, Howard M, Goodnow CC. Immunoglobulin signal transduction guides the specificity of B cell–T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. 1994;179:425–438. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, Lewis RS, Goodnow CC. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–428. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- 14.Eris JM, Basten A, Brink RA, Doherty K, Kehry MR, Hodgkin PD. Anergic self-reactive B cells present self antigen and respond normally to CD40-dependent T cell signals but are defective in antigen-receptor-mediated functions. Proc Natl Acad Sci USA. 1994;91:4392–4396. doi: 10.1073/pnas.91.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho WY, Cooke MP, Goodnow CC, Davis MM. Resting and anergic B cells are defective in CD28- dependent costimulation of naive CD4+T cells. J Exp Med. 1994;179:1539–1549. doi: 10.1084/jem.179.5.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenschow DJ, Sperling AI, Cooke MP, Freeman G, Rhee L, Decker DC, Gray G, Nadler LM, Goodnow CC, Bluestone JA. Differential upregulation of the B7-1 and B7-2 costimulatory molecules after Ig receptor engagement. J Immunol. 1994;153:1990–1997. [PubMed] [Google Scholar]
- 17.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 18.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 19.Borriello F, Sethna MP, Boyd SP, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- 20.Tivol EA, Scheitzer AN, Sharpe AH. Costimulation and autoimmunity. Curr Opin Immunol. 1996;8:822–830. doi: 10.1016/s0952-7915(96)80011-2. [DOI] [PubMed] [Google Scholar]
- 21.Fournier S, Rathmell JC, Goodnow CC, Allison JP. T cell-mediated elimination of B7.2 transgenic B cells. Immunity. 1997;6:327–339. doi: 10.1016/s1074-7613(00)80335-0. [DOI] [PubMed] [Google Scholar]
- 22.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 23.Reiner SL, Zheng S, Corry DB, Locksley RM. Constructing polycompetitor cDNAs for quantitative PCR. J Immunol Methods. 1993;165:37–46. doi: 10.1016/0022-1759(93)90104-f. [DOI] [PubMed] [Google Scholar]
- 24.Foote LC, Howard RG, Marshak-Rothstein A, Rothstein TL. IL-4 induces Fas resistance in B cells. J Immunol. 1996;157:2749–2753. [PubMed] [Google Scholar]
- 25.Nakanishi K, Matsui K, Kashiwamura S, Nishioka Y, Nomura J, Nishimura Y, Sakaguchi N, Yonehara S, Higashino K, Shinka S. IL-4 and anti-CD40 protect against Fas-mediated B cell apoptosis and induce B cell growth and differentiation. Int Immunol. 1996;8:791–798. doi: 10.1093/intimm/8.5.791. [DOI] [PubMed] [Google Scholar]
- 26.Foote LC, Marshak-Rothstein A, Rothstein TL. Tolerant B lymphocytes acquire resistance to Fas- mediated apoptosis after interleukin 4 but not after treatment with specific antigen unless a surface immunoglobulin threshold is exceeded. J Exp Med. 1998;187:847–853. doi: 10.1084/jem.187.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stüber E, Strober W. The T cell–B cell interaction via OX40-OX40L is necessary for the T cell–dependent humoral immune response. J Exp Med. 1996;183:979–989. doi: 10.1084/jem.183.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeannin P, Delneste Y, Lecoanet-Henchoz S, Gauchat J, Ellis J, Bonnefoy J. CD86 (B7-2) on human B cells: a functional role in proliferation and selective differentiation into IgE- and IgG4-producing cells. J Biol Chem. 1997;272:15613–15619. doi: 10.1074/jbc.272.25.15613. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Lenardo MJ. Essential lymphocyte function associated 1 (LFA-1): intercellular adhesion molecule interactions for T cell–mediated B cell apoptosis by Fas/ APO-1/CD95. J Exp Med. 1997;186:1171–1176. doi: 10.1084/jem.186.7.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 31.Stüber E, Neurath M, Calderhead D, Fell HP, Strober W. Cross-linking of OX40 ligand, a member of the TNF/NGF cytokine family, induces proliferation and differentiation in murine splenic B cells. Immunity. 1995;2:507–521. doi: 10.1016/1074-7613(95)90031-4. [DOI] [PubMed] [Google Scholar]
- 32.Erb KJ, Rüger B, von Brevern M, Ryffel B, Schimpl A, Rivett K. Constitutive expression of interleukin (IL)-4 in vivo causes autoimmune-type disorders in mice. J Exp Med. 1997;185:329–339. doi: 10.1084/jem.185.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sethna MP, van Parijs L, Sharpe AH, Abbas AK, Freeman GJ. A negative regulatory function of B7 revealed in B7-1 transgenic mice. Immunity. 1994;1:415–421. doi: 10.1016/1074-7613(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 34.Yuschenkoff VN, Sethna MP, Freeman GJ, Parker DC. Coexpression of B7-1 and antigen blocks tolerance induction to antigen presented by resting B cells. J Immunol. 1996;157:1987–1995. [PubMed] [Google Scholar]
- 35.Schweitzer AN, Borriello F, Wong RCK, Abbas AK, Sharpe AH. Role of costimulators in T cell differentiation: studies using antigen-presenting cells lacking expression of CD80 or CD86. J Immunol. 1997;158:2713–2722. [PubMed] [Google Scholar]
- 36.Clark EA, Ledbetter JA. How B and T cells talk to each other. Nature. 1994;367:425–428. doi: 10.1038/367425a0. [DOI] [PubMed] [Google Scholar]
- 37.Cyster JG. Signaling thresholds and interclonal competition in preimmune B-cell selection. Immunol Rev. 1997;156:87–101. doi: 10.1111/j.1600-065x.1997.tb00961.x. [DOI] [PubMed] [Google Scholar]
- 38.Forrester R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 39.Foote LC, Schneider TJ, Fischer GM, Wang JKM, Rasmussen B, Campbell KA, Lynch DH, Ju S, Marshak-Rothstein A, Rothstein TL. Intracellular signaling for inducible antigen receptor-mediated Fas resistance in B cells. J Immunol. 1996;157:1878–1885. [PubMed] [Google Scholar]
- 40.Choe J, Kim H, Zhang X, Armitage RJ, Choi YS. Cellular and molecular factors that regulate the differentiation and apoptosis of germinal center B cells. J Immunol. 1996;157:1006–1016. [PubMed] [Google Scholar]
- 41.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer J, Schröter M, Burns K, Mattmann C, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 42.Shu H, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6:751–763. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 43.Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Croce CM, Litwack G, Tomaselli KJ, Armstrong RC, Alnermi ES. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1- induced apoptosis. J Biol Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- 44.Goltsev YV, Kovalenko AV, Arnold E, Varfolomeev EE, Brodianskii VM, Wallach D. CASH, a novel caspase homologue with death effector domains. J Biol Chem. 1997;272:19641–19644. doi: 10.1074/jbc.272.32.19641. [DOI] [PubMed] [Google Scholar]
- 45.Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1 and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–17257. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 46.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 47.Schneider TJ, Grillot D, Foote LC, Nunez GE, Rothstein TL. Bcl-x protects primary B cells against Fas-mediated apoptosis. J Immunol. 1997;159:4834–4839. [PubMed] [Google Scholar]
- 48.Liu Y-J, Oldfield S, MacLennan ICM. Memory B cells in T cell-dependent antibody responses colonize the splenic marginal zones. Eur J Immunol. 1988;18:355–362. doi: 10.1002/eji.1830180306. [DOI] [PubMed] [Google Scholar]
- 49.Fulcher DA, Lyons AB, Korn SL, Cook MC, Koleda C, Parish C, Fazekas de St B, Groth, Basten A. The fate of self-reactive B cells depends primarily on the degree of antigen receptor engagement and availability of T cell help. J Exp Med. 1996;183:2313–2328. doi: 10.1084/jem.183.5.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kupfer H, Monks CR, Kupfer A. Small splenic B cells that bind to antigen-specific T helper (Th) cells and face the site of cytokine production in the Th cells selectively proliferate: immunofluorescence microscopic studies of Th–B antigen-presenting cell interactions. J Exp Med. 1994;179:1507–1515. doi: 10.1084/jem.179.5.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]