Crystal structure of hypothetical protein TTHB192 from Thermus thermophilus HB8 reveals a new protein family with an RNA recognition motif-like domain (original) (raw)
Abstract
We have determined the crystal structure of hypothetical protein TTHB192 from Thermus thermophilus HB8 at 1.9 Å resolution. This protein is a member of the Escherichia coli ygcH sequence family, which contains ∼15 sequence homologs of bacterial origin. These homologs have a high isoelectric point. The crystal structure reveals that TTHB192 consists of two independently folded domains, and that each domain exhibits a ferredoxin-like fold with a four-stranded antiparallel β-sheet packed on one side by α-helices. These two tandem domains face each other to generate a β-sheet platform. TTHB192 displays overall structural similarity to Sex-lethal protein and poly(A)-binding protein fragments. These proteins have RNA binding activity which is supported by a β-sheet platform formed by two tandem repeats of an RNA recognition motif domain with signature sequence motifs on the β-sheet surface. Although TTHB192 does not have the same signature sequence motif as the RNA recognition motif domain, the presence of an evolutionarily conserved basic patch on the β-sheet platform could be functionally relevant for nucleic acid-binding. This report shows that TTHB192 and its sequence homologs adopt an RNA recognition motif-like domain and provides the first testable functional hypothesis for this protein family.
Keywords: structural genomics, hypothetical protein, ygcH, ferredoxin-like fold, RNA recognition motif
The complete genome sequences of hundreds of organisms have been determined (Bernal et al. 2001). Comparative analysis of these diverse genome sequences accelerates the functional annotation of a gene. The primary approach in assigning a function to these new genes is to extrapolate the experimentally determined function of a protein from one organism to another on the basis of protein sequence similarity. The function of one-third to one-half of the predicted proteins, however, cannot be assigned by this sequence-based gene annotation (Galperin and Koonin 2004). Such functionally nonassigned proteins are classified as “hypothetical proteins.” Some hypothetical proteins are well conserved among many organisms and presumably perform a common biological role that is as yet undefined. The other hypothetical proteins are specific to only one organism where they exert a specialized role. Functional assignment of hypothetical proteins is, therefore, indispensable for understanding the fundamental or specific biological phenomena of diverse organisms. Because the three-dimensional structure of a protein is very important for the understanding of its function at the molecular level, structural analysis of hypothetical proteins can give valuable functional clues that may not be evident from sequence data alone (Yakunin et al. 2004).
An extremely thermophilic bacterium, Thermus thermophilus HB8, possesses the essential gene sets required for a free-living organism in spite of its small genome size (∼2 Mbp). In the genome of this organism, ∼40% of the predicted genes encode hypothetical proteins. As part of a structural and functional genomics project on T. thermophilus HB8 (http://www.thermus.org/), we selected hypothetical protein TTHB192 for functional annotation on the basis of its three-dimensional structure.
TTHB192 consists of 211 amino acid residues and is a member of the Escherichia coli ygcH sequence family, which consists of ∼15 sequence homologs of bacterial origin (Fig. 1A): The sequence identities range from 19% to 41%, with the PSI-BLAST E-values (Altschul et al. 1997) <10−15. All members of this family have high theoretical isoelectric points (p_I_s) of ∼10. There is no structural or functional information available for this protein family. In this study, we have solved the crystal structure of TTHB192 and thus present the first structure of this protein family. We show that this protein family adopts an RNA recognition motif (RRM)-like domain.
Figure 1.
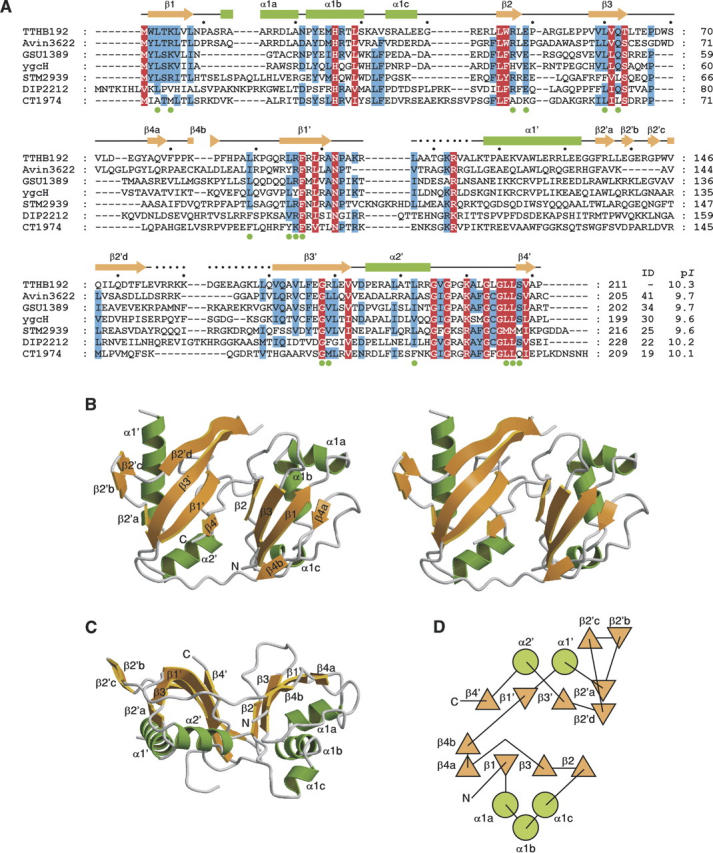
(A) Sequence alignment of TTHB192 and its sequence homologs. The homologs were identified by the second iteration cycle of PSI-BLAST sequence analysis (Altschul et al. 1997) and their sequences were aligned using Clustal W (Thompson et al. 1994). All homologs are annotated as hypothetical proteins from the following organisms: Avin3622, Azotobacter vinelandii; GSU1389, Geobacter sulfurreducens PCA; ygcH, Escherichia coli K12; STM2939, Salmonella typhimurium LT2; DIP2212, Corynebacterium diphtheriae NCTC 13129; CT1974, Chlorobium tepidum TLS. “ID” and “p_I_” represent percentage identity to TTHB192 and theoretical p_I_ of each homolog, respectively. Residues conserved in all sequences and in more than five of seven sequences are shown in white on red and shaded in blue, respectively. Of all these conserved residues, those that form the basic patch on the β-sheet platform of TTHB192 are indicated by green filled circles below the sequences. Secondary structural elements of TTHB192 are shown as green rectangles (α-helices) and orange arrows (β-strands). (B) Ribbon diagram of TTHB192 structure (stereo view). α-Helices are colored in green and labeled α1a–α2′; β-strands are orange and labeled β1–β4′ (′ denotes the C-terminal domain). The N and C termini are labeled. (C) Different view of TTHB192 structure. This figure was drawn after a 90° rotation of B around the horizontal axis. (D) Topological diagram of TTHB192. Circles represent α-helices, and triangles represent β-strands.
Results and Discussion
The crystal structure of TTHB192 was determined by the multiple wavelength anomalous dispersion (MAD) method and refined to 1.9 Å resolution (Table 1). The final model contains 188 of the 211 amino acids and 100 water molecules. Residues 108–116 and 156–169 are not included due to poor electron density. The crystal structure reveals that TTHB192 consists of two independently folded domains (Fig. 1B): the N-terminal domain (residues 1–85) and the C-terminal domain (residues 94–211). The two tandem domains face each other and generate a β-sheet platform (Fig. 1C). Each domain adopts an α + β structure consisting of a four-stranded antiparallel β-sheet packed on one side by α-helices (Fig. 1D). The four β-strands in each domain are topologically arranged in the order 2314. All structures containing this four-stranded β-sheet domain have essentially the same fold, known as the ferredoxin-like fold (Zhang and Kim 2000), and TTHB192 constitutes a member of the ferredoxin-like fold family in the Structural Classification of Proteins database (Murzin et al. 1995).
Table 1.
X-ray data collection and refinement statistics

To obtain clues as to the function of TTHB192, we searched for structural homologs in the Protein Data Bank (PDB) using the DALI server (Holm and Sander 1998). This search revealed that in spite of no detectable sequence homology, structurally similar proteins with a root-mean-square deviation (RMSD) of <3.5 Å for >60 paired Cα atoms commonly have a four-stranded β-sheet domain known as the ferredoxin-like fold. These proteins include elongation factor 1-β fragment (PDB code 1B64, _Z_-score = 5.4, RMSD = 2.6 Å), hypothetical protein HI0828 (PDB code 1MWQ, _Z_-score = 5.2, RMSD = 3.4 Å), Sex-lethal protein fragment (PDB code 1B7F, _Z_-score = 4.5, RMSD = 2.9 Å), splicing factor U2AF 65 kDa subunit fragment (PDB code 1O0P, _Z_-score = 4.3, RMSD = 3.1 Å), metallochaperone atx1 (PDB code 1CC8, _Z_-score = 4.3, RMSD = 2.8 Å) and poly(A)-binding protein fragment (PDB code 1CVJ, _Z_-score = 4.2, RMSD = 3.1 Å). The structures of the Sex-lethal protein (Sxl) fragment (Handa et al. 1999) and poly(A)-binding protein (PABP) fragment (Deo et al. 1999) demonstrate that each has two tandem repeats of the four-stranded β-sheet domain (Fig. 2A), whereas the other structural homologs comprise one copy of this domain. Visual inspection of these protein structures reveals that TTHB192 displays overall structural resemblance to the Sxl and PABP fragments, although the domain arrangement of TTHB192 is the other way around to those of the Sxl and PABP fragments (Figs. 1B, 2A).
Figure 2.

Comparison of TTHB192 and its structural homologs. Ribbon diagrams (A) and electrostatic potential surfaces (B) of the Sxl (left) and PABP (right) fragments. The orientations of each protein in B are similar to those in A. Secondary structural elements are colored and labeled according to Figure 1B. The single-stranded RNA bound to each protein is shown as a ball-and-stick model, and the 5′ and 3′ ends of the RNA are labeled. In A, the N and C termini of the protein are labeled. Electrostatic surface potentials are shown in blue and red for the positively and negatively charged regions, respectively (blue = +20 k_B_T, red = −20 k_B_T), where _k_B is the Boltzmann constant and T is the temperature. (C) Molecular surfaces of TTHB192, colored by electrostatic potential (left) and sequence conservation (right). The electrostatic surface potential is colored in the same way as in B. In the right panel, evolutionarily-conserved residues, which are shown in white on red and shaded in blue in Figure 1A, are colored in green. The orientations in C are similar to those in Figure 1B.
The four-stranded β-sheet domain in the Sxl and PABP fragments is well known as the RRM domain and is the most widely found RNA binding motif in all kingdoms of life (Burd and Dreyfuss 1994; Maris et al. 2005). The RRM domain has two conserved signature sequence motifs called RNP1 and RNP2 with the consensus sequences (Arg/Lys)-Gly-(Phe/Tyr)-(Gly/Ala)-(Phe/Tyr)-(Ile/Leu/Val)-X-(Phe/Tyr) and (Ile/Leu/Val)-(Phe/Tyr)-(Ile/Leu/Val)-X-Asn-Leu, respectively (where X is any amino acid, and an underlined amino acid has a solvent-accessible side chain for RNA recognition) (Burd and Dreyfuss 1994; Kielkopf et al. 2004; Maris et al. 2005). The signature motifs form the central two β-strands, with RNP1 in β3 and RNP2 in β1. RNA recognition is primarily mediated through hydrogen bonding and base stacking interactions with solvent-exposed charged and aromatic side chains within these motifs (Maris et al. 2005). RRM domains are often found as multiple copies within an RNA-binding protein (Maris et al. 2005). In Sxl and PABP fragments, the tandem RRM domains make a positively charged β-sheet surface (Fig. 2A,B), which is in good agreement with their relatively high theoretical p_I_s of between 7.0 and 9.7 (Kielkopf et al. 2004).
TTHB192 and its sequence homologs also have strongly high theoretical p_I_s of between 9.6 and 10.3 (Fig. 1A). The highly basic nature of these proteins suggests the affinity for nucleic acid. Consistent with the high p_I_, TTHB192 has large continuous basic patches on its surface. One of the basic patches is formed on the β-sheet platform (Fig. 2C, left), which spatially corresponds to the RNA binding sites of Sxl and PABP fragments. Furthermore, this patch is composed of the evolutionarily conserved residues (Fig. 2C, right). The central two β-strands for possible nucleic acid binding have the following consensus sequences (Fig. 1A): (Tyr/Trp)-Leu-(Ser/Thr)-(Arg/Lys)-(Leu/Val) for β1, (Ile/Leu/Val)-Leu-(Ile/Leu/Val)-Gln-(Ser/Thr) for β3, (Leu/Val)-(Arg/Lys)-Phe-X-(Ile/Leu/Val) for β1′ and Val-X-(Phe/Tyr)-X-Gly-(Leu/Val/Met)-Leu for β3′ (where the underlined amino acid has a solvent-accessible side chain). These consensus sequences are different from the RNP1 and RNP2 motifs. In spite of differences in the domain arrangement and the consensus sequence motifs between TTHB192 and RRM domains, the presence of an evolutionarily conserved basic patch on the β-sheet platform could be functionally relevant for nucleic acid-binding. We conclude that TTHB192 and E. coli ygcH sequence homologs adopt an RRM-like domain with a different signature motif and could function as nucleic acid-binding proteins. This functional clue could be valuable for understanding the results of transcriptome analysis of T. thermophilus HB8, which show that mRNA expression of TTHB192 increases in the early log phase and reaches its plateau in the late log phase (data not shown).
Clustered regularly interspaced short palindromic repeats (CRISPRs) are a family of repetitive DNA sequences found in bacteria and archaea (Jansen et al. 2002). Four CRISPR-associated protein families, which always appear near a repeat cluster, are suggested to be involved in DNA metabolism (Jansen et al. 2002). In addition, a number of uncharacterized protein families are found to be associated with CRISPR elements (Haft et al. 2005). E. coli ygcH is annotated as the CRISPR-associated protein, CT1974 family, and this gene is clustered with several CRISPR-associated genes (Haft et al. 2005). TTHB192 gene is also clustered with CRISPR-associated genes, and the gene arrangement is conserved in several bacteria including E. coli. The crystal structure reported here provides the first testable functional hypothesis (i.e., nucleic acid-binding) for the CRISPR-associated protein, CT1974 family.
Materials and methods
Protein expression and purification
The TTHB192 gene was amplified by polymerase chain reaction from T. thermophilus HB8 genome and subcloned into the NdeI and BamHI sites of pT7Blue (Novagen). The sequences of the forward and reverse primers were 5′-ATATcatatgTGGCTCACTAAGCTCGTGCT-3′ (NdeI site in lowercase) and 5′-ATATggatccTTATTAGGGGGCCACGGAAAGGAGGCCGAGGCC-3′ (BamHI site in lowercase), respectively. The plasmid was digested by NdeI and BamHI, and the resulting DNA fragment containing the TTHB192 gene was subcloned into the NdeI and BamHI sites of pET-11a (Novagen). E. coli BL21(DE3) cells were transformed by the expression plasmid and the transformants were grown at 37°C overnight in medium containing 1.0% polypepton, 0.5% yeast extract, 0.5% NaCl, and 50 μg/mL ampicillin (pH 7.0). The cells were lysed by sonication in 20 mM Tris-HCl (pH 8.0) containing 50 mM NaCl and 5 mM β-mercaptoethanol. The lysate was incubated at 70°C for 10 min and ultracentrifuged at 200,000_g_ at 4°C. TTHB192 in the clear supernatant was purified by successive chromatography steps on RESOURCE PHE, RESOURCE S, Bio-Scale CHT10-I, and HiLoad 16/60 Superdex 75-pg columns (all Amersham Biosciences except for Bio-Scale CHT10-I from Bio-Rad).
Crystallization and data collection
The crystals of TTHB192 were obtained by the hanging-drop vapor diffusion method at 20°C. A protein solution containing 9 mg/mL TTHB192, 20 mM Tris-HCl (pH 8.0) and 1 mM dithiothreitol was mixed (1:1) with a precipitant solution containing 20% polyethylene glycol monomethyl ether 2000 and 0.1 M N,_N_-bis(2-hydroxyethyl) glycine (pH 8.2). An iridium (Ir) derivative was made by soaking the native TTHB192 crystal overnight with 10 mM ammonium hexachloroiridate. For diffraction data collection, the native and Ir-derivative crystals were flash-frozen after soaking for ∼1 min in Paraton-N (Hampton Research). All data sets were collected under a liquid nitrogen stream (100 K) using the beamline BL26B2 at SPring-8 (Hyogo, Japan) (Ueno et al. 2004). The native and Ir-derivative MAD data sets were processed and scaled with the program packages HKL2000 (Otwinowski and Minor 1997) and CrystalClear (Rigaku MSC) (Pflugrath 1999), respectively (Table 1).
Structure determination
The positions of Ir-site and the initial phases were determined with the program SOLVE (Terwilliger and Berendzen 1999) using the MAD data sets. The initial model was built with the program ARP/wARP (Perrakis et al. 1999). The model was refined automatically against the native data set at 1.9 Å resolution using the program LAFIRE (Yao et al. 2006) with the program CNS (Brünger et al. 1998), followed by final manual fitting and checking using the program Xtalview/Xfit (McRee 1999). The final model was validated using the program PROCHECK of the CCP4 suite (Collaborative Computational Project, Number 4 1994). The refinement statistics are summarized in Table 1. Molecular graphic figures were drawn using the programs MolScript (Kraulis 1991) and Raster3D (Merritt and Bacon 1997). The topological diagram was generated by the TOPS server (Michalopoulos et al. 2004). The molecular surface was created with the program SPOCK (Christopher 1998). The atomic coordinates and structure factors have been deposited in the PDB (http://www.rcsb.org/) under accession code 1WJ9.
Acknowledgments
We thank Drs. R. Hirose and K. Hamada for assistance during the data collection at SPring-8 synchrotron radiation beam line, BL26B2. This work was supported in part by the RIKEN Structural Genomics/Proteomics Initiative and the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Reprint requests to: Seiki Kuramitsu, RIKEN SPring-8 Center, Harima Institute, 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5148, Japan; e-mail: kuramitu@spring8.or.jp; fax: +81-791-58-2892.
Abbreviations: p_I_, isoelectric point; RRM, RNA recognition motif; MAD, multiple wavelength anomalous dispersion; PDB, Protein Data Bank; RMSD, root-mean-square deviation; Sxl, Sex-lethal protein; PABP, poly(A)-binding protein; CRISPR, clustered regularly interspaced short palindromic repeat.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal A., Ear U., Kyrpides N. 2001. Genomes OnLine Database (GOLD): A monitor of genome projects world-wide Nucleic Acids Res. 29 126–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S.et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Burd C.G. and Dreyfuss G. 1994. Conserved structures and diversity of functions of RNA-binding proteins Science 265 615–621. [DOI] [PubMed] [Google Scholar]
- Christopher J.A. In SPOCK: The structural properties observation and calculation kit (program manual) . 1998. The Center for Macromolecular Design, Texas A&M University, College Station, TX.
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Deo R.C., Bonanno J.B., Sonenberg N., Burley S.K. 1999. Recognition of polyadenylate RNA by the poly(A)-binding protein Cell 98 835–845. [DOI] [PubMed] [Google Scholar]
- Galperin M.Y. and Koonin E.V. 2004. “Conserved hypothetical” proteins: Prioritization of targets for experimental study Nucleic Acids Res. 32 5452–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- e60Haft D.H., Selengut J., Mongodin E.F., Nelson K.E. 2005. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes PLoS Comput. Biol. 1. [DOI] [PMC free article] [PubMed]
- Handa N., Nureki O., Kurimoto K., Kim I., Sakamoto H., Shimura Y., Muto Y., Yokoyama S. 1999. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein Nature 398 579–585. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander C. 1998. Touring protein fold space with Dali/FSSP Nucleic Acids Res. 26 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R., Embden J.D., Gaastra W., Schouls L.M. 2002. Identification of genes that are associated with DNA repeats in prokaryotes Mol. Microbiol. 43 1565–1575. [DOI] [PubMed] [Google Scholar]
- Kielkopf C.L., Lucke S., Green M.R. 2004. U2AF homology motifs: Protein recognition in the RRM world Genes & Dev. 18 1513–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Maris C., Dominguez C., Allain F.H. 2005. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression FEBS J. 272 2118–2131. [DOI] [PubMed] [Google Scholar]
- McRee D.E. 1999. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density J. Struct. Biol. 125 156–165. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon D.J. 1997. Raster3D: Photorealistic molecular graphics Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Michalopoulos I., Torrance G.M., Gilbert D.R., Westhead D.R. 2004. TOPS: An enhanced database of protein structural topology Nucleic Acids Res. 32 D251–D254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A.G., Brenner S.E., Hubbard T., Chothia C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris R., Lamzin V.S. 1999. Automated protein model building combined with iterative structure refinement Nat. Struct. Biol. 6 458–463. [DOI] [PubMed] [Google Scholar]
- Pflugrath J.W. 1999. The finer things in X-ray diffraction data collection Acta Crystallogr. D Biol. Crystallogr. 55 1718–1725. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen J. 1999. Automated MAD and MIR structure solution Acta Crystallogr. D Biol. Crystallogr. 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno G., Hirose R., Ida K., Kumasaka T., Yamamoto M. 2004. Sample management system for a vast amount of frozen crystals at SPring-8 J. Appl. Crystallogr. 37 867–873. [Google Scholar]
- Yakunin A.F., Yee A.A., Savchenko A., Edwards A.M., Arrowsmith C.H. 2004. Structural proteomics: A tool for genome annotation Curr. Opin. Chem. Biol. 8 42–48. [DOI] [PubMed] [Google Scholar]
- Yao M., Zhou Y., Tanaka I. 2006. LAFIRE: Software for automating the refinement process of protein-structure analysis Acta Crystallogr. D Biol. Crystallogr. 62 189–196. [DOI] [PubMed] [Google Scholar]
- Zhang C. and Kim S.H. 2000. The anatomy of protein beta-sheet topology J. Mol. Biol. 299 1075–1089. [DOI] [PubMed] [Google Scholar]