Recombinant Virus Vaccination against “Self” Antigens Using Anchor-fixed Immunogens (original) (raw)
. Author manuscript; available in PMC: 2008 Feb 22.
Published in final edited form as: Cancer Res. 1999 Jun 1;59(11):2536–2540.
Abstract
To study the induction of anti-“self” CD8+ T-cell reactivity against the tumor antigen gp100, we used a mouse transgenic for a chimeric HLA-A*0201/H-2 Kb molecule (A2/Kb). We immunized the mice with a recombinant vaccinia virus encoding a form of gp100 that had been modified at position 210 (from a threonine to a methionine) to increase epitope binding to the restricting class I molecule. Immunogens containing the “anchor-fixed” modification elicited anti-self CD8+ T cells specific for the wild-type gp100209–217 peptide pulsed onto target cells. More important, these cells specifically recognized the naturally presented epitope on the surface of an A2/Kb-expressing murine melanoma, B16. These data indicate that anchor-fixing epitopes could enhance the function of recombinant virus-based immunogens.
Introduction
The recent cloning of TAAs2 recognized by T lymphocytes has led to the development of recombinant and synthetic anticancer vaccines; the first of these candidate vaccines are currently being tested in clinical trials (1-4). However, recent clinical trials using recombinant virus-based immunogens (such as vaccinia virus, fowlpox virus, or adenovirus) encoding tumor rejection antigens have not yet demonstrated consistent antitumor immune responses or tumor reduction (Ref. 5 and Rosenberg et al.).3 These negative results may be attributed, in part, to preexisting antivector antibodies that neutralize the viral vaccines (5). However, no CD8+ T-cell reactivity was induced in individuals who received the recombinant avipox virus, fowlpox, which has little-to-no antibody cross-reactivity with vaccinia virus (Rosenberg et al.3 and Ref. 6). Therefore, factors other than neutralizing antibodies may explain why it is difficult to induce antitumor immunity using recombinant viral vectors. Among the possible factors are the regulatory effects of central or peripheral tolerance on the T-cell repertoire (7). In addition, the TAA epitopes may not be efficiently presented to the immune system because of interdeterminant competition with peptides derived from the vector (8). In an attempt to augment the immunogenicity of TAA expressed by recombinant viral vaccines, we modified an anchor residue of a self-epitope within the full-length melanoma-associated antigen, gp100, to increase peptide binding to class I molecules without compromising the interaction of the MHC/peptide complex with the T-cell receptor (9).
Human gp100, a nonmutated “self”-antigen expressed by melanocytes, pigmented retinal cells, and most melanoma lesions, is not expressed in other normal tissues or nonmelanoma tumors (10, 11). Parkhurst et al. (12) have shown that the engineering of a key anchor residue at position 2 of the HLA-A*0201-restricted peptide gp100209–217 [threonine (T) to an optimal anchor methionine (M), gp100209–217(210M)] yields a peptide with a 9-fold better binding to HLA-A*0201 and an increased ability to sensitize PBL from HLA-A*0201-positive melanoma patients in vitro. Furthermore, when the modified peptide was administered with interleukin 2 as an adjuvant, over 40% of patients with metastatic melanoma experienced tumor regression (4). In this study, we show that the immunization of A2/Kb transgenic mice with recombinant viral vectors expressing “anchor-fixed” peptides, engineered to have enhanced binding to MHC, induced immune responses against a naturally processed, self-peptide of gp100. Thus, this approach may overcome the negative effects of both immune tolerance and intramolecular competition.
Materials and Methods
Animals and Cell Lines
The HLA-A*0201/Kb (A2/Kb) transgenic mice used in this study were a kind gift of L. Sherman (The Scripps Research Institute, La Jolla, CA) and have been described previously (13). These animals express a chimeric class I molecule composed of the _α_1 and _α_2 domains of the HLA-A*0201 class I antigen and the _α_3, cytoplasmic, and transmembrane domains of the mouse H-2Kb class I molecule. We transfected B16 with a plasmid expressing the A2/Kb molecule driven by the SV40 promoter (B16/A2/Kb) and selected this cell line in 1 _μ_g/ml puromycin (Life Technologies, Inc., Grand Island, NY). We maintained B16/A2/Kb, B16.WT, and the T2 cell line (HLA-A*0201+, TAP-deficient, T-B cell hybrid) in CM containing RPMI 1640, 10% heat-inactivated FCS (both obtained from Biofluids, Rockville, MD), 0.03% fresh l-glutamine, 100 _μ_g/ml streptomycin, 100 units/ml penicillin (all from NIH, Media Unit, Bethesda, MD) and 50 _μ_g/ml gentamicin sulfate.
Peptides
Each of the peptides used in this study was prepared under Good Manufacturing Practice (GMP) conditions by Multiple Peptide Systems (San Diego, CA): (a) gp100154–162, KTWGQYWQV; (b) gp100209–217, ITDQVPFSV; and (c) gp100209–217(210M), IMDQVPFSV. The identity of each of the peptides was confirmed by mass spectral analysis. The peptides were >98% pure as assessed by high pressure liquid chromatography.
Recombinant Virus Construction
Recombinant viruses were constructed by the insertion of foreign sequences into the genome of a plaque-purified isolate from the Wyeth strain of vaccinia virus as described previously (14). In each virus, the foreign gene is under the control of the vaccinia 40K (H5R) promoter (15). Each virus also contains the Escherichia coli lacZ gene under the control of the fowlpox virus C1 promoter used for the selection of recombinant virions (16). The full-length (FL) gp100 gene was altered by site-directed mutagenesis at codon 210, changing a threonine codon to methionine, and at codon 288, changing an alanine codon to valine. This modified gene was inserted into the _Hin_dIII M region of the vaccinia virus genome to produce rVV-_FL_gp100(210M). The native form of the human gp100 gene was inserted into the _Hin_dIII M region of the vaccinia genome to produce rVV-_FL_gp100. The gp100 minigenes were constructed using synthetic oligonucleotides encoding the leader sequence from the adenovirus type 5 E3/19K gene joined to gp100 codons 209 to 217, with either methionine or the native threonine at codon 210 (17). The minigenes were inserted into the _Hin_dIII J region of the vaccinia genome and were designated rVV-ESgp100209–217(210M) and rVV-ESgp100209–217, respectively. rVV expressing the full-length tyrosinase gene (rVV-Tyr) was generated in a similar manner.
51Chromium (51Cr) and Cytokine Release Assays
Secondary in vitro lymphocyte populations were generated by harvesting spleens of mice 3–6 weeks after immunization and culturing single-cell suspensions of splenocytes in T-25 flasks (Nunc, Roskilde, Denmark) at a density of 6 × 106 cells/ml with 2 _μ_g/ml antigenic peptide at a total volume of 10 ml of CM containing 0.1 mm nonessential amino acids, 1.0 mm sodium pyruvate (both from Biofluids), and 5 × 10−2 ME (Life Technologies, Inc., Rockville, MD) in the absence of interleukin 2. Splenocytes were harvested and washed in CM before testing in a 51Cr release or cytokine release assay. Six-h 51Cr release assays were performed as described previously (6). Briefly, 2 × 106 target cells were incubated in 0.2 ml of CM labeled with 200 _μ_Ci of Na51CrO4 for 90 min. Peptide-pulsed T2 cells were incubated for 1 h with 1 _μ_g/ml (approximately 1 _μ_m) antigenic peptide during labeling. Target cells were mixed with effector cells for 6 h at 37°C at 25:1 E:T ratio. The amount of 51Cr released was determined by gamma counting, and the percentage of specific lysis was calculated as follows:
Experimentalcpm−spontaneouscpmMaximalcpm−spontaneouscpm×100
For cytokine release assays, 105 effector cells were coincubated with 105 target cells in a total of 200 _μ_l for 18–24 h in a 96-well plate. At this time, 150 _μ_l of supernatant were harvested and tested for the presence of murine IFN-γ or GM-CSF by ELISA as described previously (18).
Results
Comparison of Human and Murine gp100 Epitope Sequences
To explore the relevance of immunizing A2/Kb transgenic mice with human gp100, we compared the human and murine sequences from two well-characterized HLA-A*0201-restricted epitopes from gp100 (19). Human gp100154–162 differs from the mouse sequence by one amino acid at the nonanchor position 5, where glutamine (KTWG_Q_YWQV) replaces lysine (KTWG_K_YWQV). In contrast, the sequence for gp100209–217 is identical in mouse and human gp100 (ITDQVPFSV). Therefore, for studies in the A2/Kb transgenic mouse model, we focused our efforts on the induction of self-reactive, gp100209–217-specific CD8+ T-lymphocyte reactivity.
No Induction of Self-Reactive CD8+ T Cells after Immunization with rVV Encoding Wild-Type Full-Length Human gp100 (FLgp100)
To study the induction of anti-gp100 immune responses in A2/Kb transgenic mice, we cultured splenocytes from mice immunized with rVV-_FL_gp100 with each of the human gp100 epitopes, gp100154–162 or gp100209–217. There were high levels of gp100154–162-specific lysis from rVV-_FL_gp100 cultures stimulated with gp100154–162 (Fig. 1). In contrast, we could not detect gp100209–217-reactive CD8+ T cells after stimulation of the rVV-_FL_gp100-immune splenocytes with the gp100209–217 peptide (Fig. 1). Furthermore, in vitro stimulation of rVV-_FL_gp100 immune splenocytes with an anchor-fixed modified peptide, gp100209–217(210M), did not lead to the induction of gp100209–217-specific lysis (Fig. 1). Thus, we could not detect specific reactivity against the self-epitope, gp100209–217, using the unmodified, full-length gp100, despite the ability of this recombinant viral vaccine to elicit powerful immunoreactivity against the “nonself,” gp100154–162 epitope.
Fig. 1.
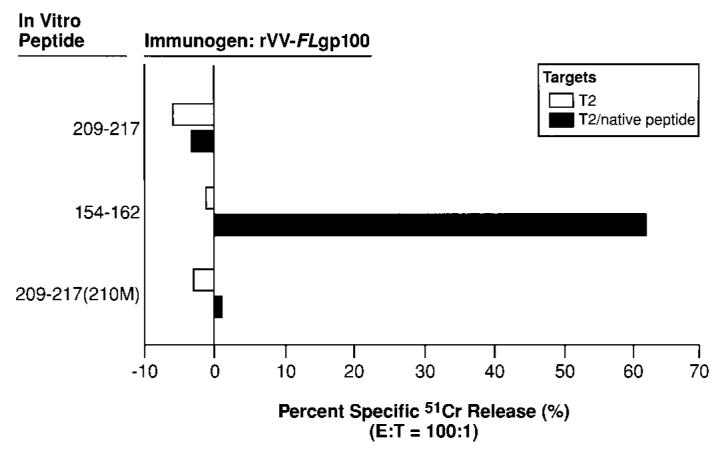
Immunization with recombinant poxviruses expressing gp100 induced CD8+ T-cell lytic responses against gp100154–162 but not against gp100209–217. A2/Kb transgenic mice were immunized once i.v. with 107 pfu of rVV-_FL_gp100. Three to 6 weeks later, pooled splenocytes (two mice per group) were harvested and stimulated for 6 days with 2 _μ_g of the indicated peptides per ml. Cultures were then assayed for specific lytic activity in a 51Cr-release assay against either T2 cells alone (□) or T2 cells pulsed with the relevant native peptides, gp100154–162 or gp100209–217 (■). A 100:1 E:T ratio is shown. This experiment was repeated two times with similar findings.
Enhanced Recognition of a Recombinant Vaccine Expressing an Anchor-fixed Modification of Full-Length gp100
To determine whether in vivo immunization with anchor-fixed immunogens could alter the weak immunogenicity of the native gp100209–217 epitope, we modified the gp100209–217 epitope in the full-length gp100 cDNA by site-directed mutagenesis at amino acid number 210 from a threonine to a methionine [rVV-_FL_gp100(210M)]. To test whether rVV-_FL_gp100 and rVV-_FL_gp100(210M) expressed the gp100209–217 and gp100209–217(210M) epitopes, respectively, we used them to infect an HLA-A*0201 positive, gp100 negative breast cancer cell line, MDA 231. The infected cells were then tested for T-cell recognition using two human HLA-A*0201-restricted T-cell clones designated Clone 70 and DW2C8—specific for gp100154–162 and gp100209–217, respectively. Clone 70 secreted background levels of IFN-γ in response to MDA 231 cells infected with a control rVV expressing human tyrosinase (Fig. 2). Clone 70 produced similar amounts of IFN-γ when it was cocultured with the tumor cells infected with rVV-_FL_gp100 compared with cells infected with rVV-_FL_gp100(210M), which suggested a similar level of infection by the two rVV. The gp100209–217-specific T-cell clone, DW2C8, secreted 15 times more IFN-γ when cocultured with the MDA 231 cells infected with the anchor-fixed rVV-_FL_gp100(210M) than with cells infected with rVV-_FL_gp100 (Fig. 2). These findings suggest that the anchor-fixed modification in gp100 results in enhanced presentation of the gp100209–217(210M) epitope compared with the gp100209–217 of native gp100.
Fig. 2.
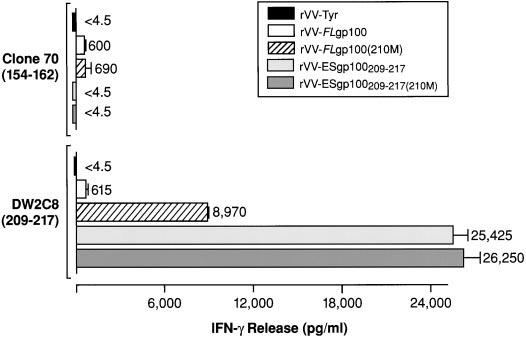
T-cell recognition of gp100-specific epitopes of rVV-infected HLA-A*0201+, gp100− breast cancer cell line. The MDA 231 HLA-A*0201+, gp100− breast cancer cell line was infected at an MOI of 10 for 1 h at 37°C with rVV-Tyrosinase (rVV-Tyr), rVV-_FL_gp100, rVV-_FL_gp100(210M), rVV-ESgp100209–217, or rVV-ESgp100209–217(210M). The infected MDA 231 cells were washed and plated at 105/well and coincubated with 2.5 × 104 human T-cell clones for 18 h. Clone 70 specifically recognizes gp100154–162, and clone DW2C8 recognizes either gp100209–217 or gp100209–217(210M). Supernatants were assayed for human IFN-γ release by ELISA.
Induction of Self-Reactive, gp100209–217-specific CD8+ T Cells Using a Recombinant Anchor-fixed Immunogen
To determine whether we could elicit antigen-specific reactivity against the “self” peptide, gp100209–217, using anchor-fixed modifications, we vaccinated A2/Kb Tg mice with either rVV-_FL_gp100 or rVV-_FL_gp100(210M); (Table 1A). Stimulation of rVV-_FL_gp100 immune splenocytes with gp100154–162 elicited specific IFN-γ release against T2 pulsed with gp100154–162 but not against media alone or T2 pulsed with gp100209–217 (Table 1A). We observed no gp100209–217-specific cytokine release from the rVV-_FL_gp100 immune splenocytes after restimulating with either the native gp100209–217 or the anchor-fixed gp100209–217(210M) peptides (Table 1A). The rVV-_FL_gp100(210M)-immune splenocytes—stimulated in vitro with the modified gp100209–217(210M) peptide—secreted high levels of IFN-γ when cocultured with T2 cells pulsed with the native gp100209–217 but not when T2 cells were pulsed with a control peptide, gp100154–162. Thus, to induce gp100209–217-specific, CD8+ T-cell-mediated cytokine release, it was necessary to immunize with the anchor-fixed modifications.
Table 1. Induction of gp100209–217-specific IFN-g release after immunization with rVV expressing the anchor-fixed modification.
A2/Kb transgenic mice were immunized one time i.v. with 1 × 107 pfu of either rVV-_FL_gp100 or rVV-_FL_gp100(210M) (Table 1A), rVVESgp100209–217 or rVVESgp100209–217(210M) (Table 1B). Three to 6 weeks later, splenocytes were harvested and stimulated two times in vitro with 2 _μ_g/ml of gp100154–162, gp100209–217, or gp100209–217(210M) as described in the “Materials and Methods” section. Seven days after the second stimulation, 105 effector cells were admixed with 105 target cells in each well of 96-well plates. Eighteen to 24 h later, supernatants were harvested and assayed by ELISA for the presence of murine IFN-γ.
| A. Full-length gp100 constructs | ||||||
|---|---|---|---|---|---|---|
| Immunogena:In vitro stimulator: | IFN-γ release (pg/ml) | |||||
| rVV-_FL_gp100 | rVV-_FL_gp100(210M) | |||||
| 154–162 | 209–217 | 209–217(210M) | 154–162 | 209–217 | 209–217(210M) | |
| Media | 1,117 | 1,057 | 999 | 1,743 | 497 | 681 |
| T2/154–162 | 25,412 | 1,476 | 1,228 | 23,097 | 611 | 662 |
| T2/209–217(210M) | 1,483 | 942 | 1,031 | 1,965 | 440 | 23,027 |
| T2/209–217 | 1,490 | 1,673 | 1,063 | 2,035 | 618 | 15,699 |
| B. ES-minigene constructs | ||||||
| Immunogen: In vitro stimulator: | IFN-γ release (pg/ml) | |||||
| rVV-ESgp100209–217 | rVV-ESgp100209–217(210M) | |||||
| 154–162 | 209–217 | 209–217(210M) | 154–162 | 209–217 | 209–217(210M) | |
| Media | 692 | 7,820 | 1,177 | 1,136 | 3,694 | 1,025 |
| T2 alone | 1,177 | 6,489 | 1,435 | 3,788 | 6,566 | 1,792 |
| T2/154–162 | 1,279 | 7,205 | 980 | 4,624 | 7,517 | 860 |
| T2/209–217(210M) | 1,540 | 9,409 | 2,334 | 6,650 | 8,533 | 35,902 |
| T2/209–217 | 1,223 | 6,181 | 1,792 | 6,524 | 6,817 | 33,116 |
We next investigated the possibility that the methionine substitution at position 210 of gp100 in rVV-_FL_gp100(210M) resulted in the formation of a new translation initiation site, overcoming the need to fully process the gp100. An eight-amino-acid-long peptide corresponding to gp100210–217(210M) was compared functionally with both the gp100209–217 and the gp100209–217(210M) 9-mers for recognition by specific T-cell clones. We found that the 8-mer peptide was significantly less well recognized than either the parental or the modified gp100210–217 peptide (data not shown), a finding inconsistent with the hypothesis that the truncated product was responsible for the increased immunogenicity of the modified gp100.
Enhanced Recognition of rVVs Expressing Minimal Determinant Epitopes of gp100
We have reported previously (17) that facilitating antigen processing enhances the immunogenicity of some antigens. In these cases, we used methods that eliminate the need for protease-mediated cleavage and for TAP-mediated transport by using minimal determinant epitopes preceded by endoplasmic reticulum insertion signal sequences (ES). Thus, this approach increases the amount of peptide complexed with MHC class I on the cell surface (17, 20). To determine whether this increased amount of peptide/ MHC complexes would break tolerance to the self-gp100209–217 epitope, we generated rVV constructs expressing either the native gp100209–217 (rVV-ESgp100209–217) or the anchor-fixed gp100209–217(210M) (rVV-ESgp100209–217(210M)) minimal determinant epitopes preceded by the ES leader sequence.
Next, we infected the MDA 231 HLA-A*0201+, gp100− cell line with each of the recombinant viruses and tested for recognition by the gp100209–217-specific, human T-cell clone DW2C8 to confirm that they expressed either the gp100209–217 or gp100209–217(210M) epitopes (Fig. 2). Large quantities of human IFN-γ were present after coincubation of the gp100209–217-specific T-cell clone (DW2C8) admixed with MDA 231 cells infected with either rVV-ESgp100209–217 or rVV-ESgp100209–217(210M). DW2C8 released 2–3 times more IFN-γ when target cells were infected with minigene-containing viruses than when they were infected with the rVV expressing the full-length, anchor-fixed gp100 construct, rVV-_FL_gp100(210M), and about 40 times as much as with the native, full-length gp100 construct, rVV-_FL_gp100. These data suggest that the use of minigene constructs enhances the level of peptide complexed with MHC class I for recognition by CD8+ T cells.
Induction of Self-Reactive, gp100209–217-specific CD8+ T Cells with rVV Expressing the Anchor-fixed Minimal Determinant
To determine whether we could induce native gp100209–217 CD8+ T-cell reactivity using vaccinia viruses encoding minimal determinant epitopes preceded by endoplasmic reticulum leader sequences, we vaccinated A2/Kb Tg mice with either rVV-ESgp100209–217 or rVV-ESgp100209–217(210M) (Table 1B). There was no gp100209–217-specific cytokine release from the rVV-ESgp100209–217 immune splenocyte cultures after restimulating with either the wild-type gp100209–217 or the anchor-fixed gp100209–217(210M) peptides (Table 1B). However, there was IFN-γ release from the rVV-ESgp100209–217(210M) splenocyte culture stimulated in vitro with the anchor-fixed gp100209–217(210M) peptide against T2 pulsed with either the wild-type or anchor-fixed peptide but not against T2 pulsed with gp100154–162. We, therefore, conclude that even with the enhanced presentation of the gp100209–217 using minigene constructs, we still needed the anchor-fixed modification to induce CD8+ T-cell immunoreactivity against the native gp100209–217 epitope.
CD8+ T-Cell Recognition of Naturally Processed and Presented gp100209–217 by Murine Melanoma, B16/A2/Kb
To determine whether rVV-ESgp100209–217(210M)-induced T cells recognize gp100209–217 naturally presented with MHC class I on the surface of tumor cells, we assayed splenocyte cultures derived from mice immunized with rVV-Tyrosinase, rVV-ESgp100209–217, or rVVESgp100209–217(210M) for the ability to recognize and lyse the murine melanoma, B16, transfected with the chimeric A2/Kb molecule (B16/ A2/Kb; Fig. 3A). Only the rVV-ESgp100209–217(210M)-immune splenocytes stimulated in vitro with gp100209–217(210M) peptide specifically lysed murine melanoma, B16/A2/Kb (Fig. 3A). The rVVESgp100209–217(210M)-induced splenocytes cultured in vitro with gp100209–217(210M) also specifically secreted both IFN-g and GMCSF in response to B16/A2/Kb (Fig. 3,B and C) but did not release IFN-γ or GM-CSF when cocultured with wild-type murine melanoma, B16.WT (HLA-A*0201 negative). This result indicated that the gp100209–217-specific reactivity was HLA-A*0201-restricted (Fig. 3, B and C). In addition, this gp100209–217(210M)-specific T-cell culture recognized as little as 10 ng/ml gp100209–217 peptide pulsed onto T2 cells, as assessed by the release of IFN_γ_, which indicated high avidity for wild-type peptide complexed with MHC (data not shown). Despite the induction of the antiself immunoreactivity, we did not observe autoimmune vitiligo in response to rVV-_FL_gp100 or rVV-_FL_gp100(210M) vaccination.
Fig. 3.
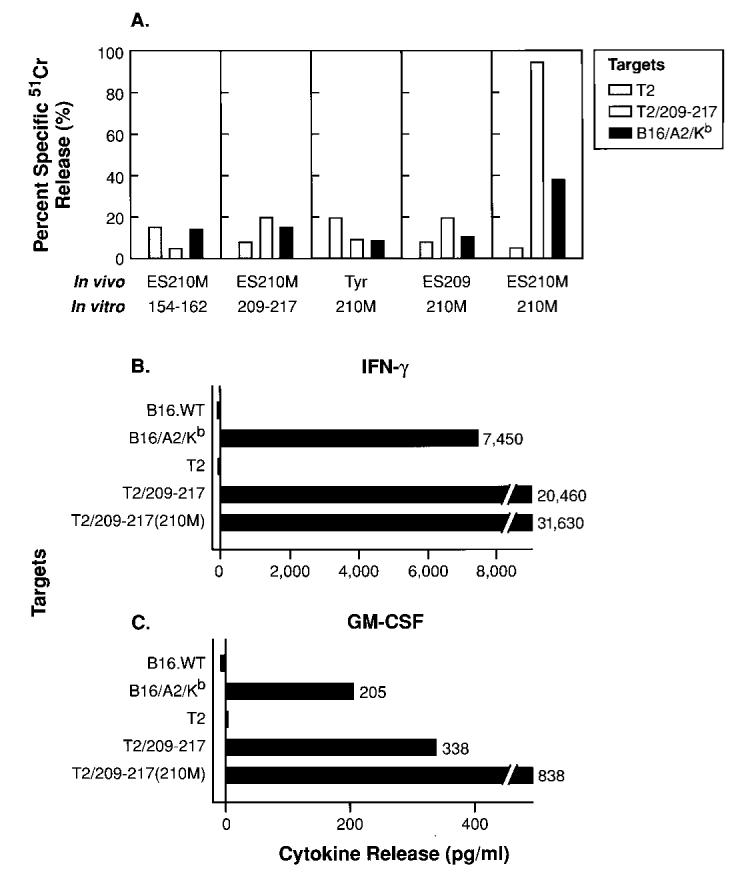
Immune recognition of the gp100209–217 epitope naturally processed and presented by B16/A2/Kb. In A, A2/Kb transgenic mice were immunized once i.v. with 107 pfu of rVV-Tyrosinase (Tyr), rVV-ESgp100209–217 (ES209), or rVV-ESgp100209–217(210M) (ES210M). Three to 6 weeks later, pooled splenocytes (two mice per group) were harvested and stimulated in vitro for 7 days with 2 _μ_g of gp100154–162 (154–162), gp100209–217 (209–217), or gp100210–217(210M) (210M) peptides per ml. Cultures were restimulated once with the same peptide. After 7 days in culture, splenocytes were assayed at a 25:1 E:T ratio for specific lytic activity in a 51Cr-release assay against T2 cells alone, T2 pulsed with the relevant gp100209–217, or a murine melanoma stably transfected with the chimeric A2/Kb molecule, B16/A2/Kb. In B and C, the rVV-ESgp100209–217(210M)-immune splenocyte culture stimulated in vitro with gp100209–217(210M) was further assayed for cytokine release in response to a panel of target cells. Specifically, 105 effector cells were admixed with 105 target cells in each well of 96-well plates. Eighteen to 24 h later, supernatants were harvested and assayed by ELISA for the presence of either murine IFN-γ (B) or murine GM-CSF (C).
Together, these results show that CD8+ T-cell reactivity against a self-epitope, gp100209–217, from melanoma antigen gp100, can be induced after in vivo immunization with rVV constructs when anchor-fixed modifications of the epitope are used.
Discussion
Peptides can be modified at the anchor residues in a way that increases their binding to class I molecules without compromising the interaction of this complex with the T-cell receptor. Furthermore, by stabilizing peptide/MHC complexes, specific CD8+ T-lymphocyte responses can be enhanced, and tolerance can be broken. In the clinic, immunization with viral vectors encoding full-length gp100 have not consistently elicited immunoreactivity against gp100209–217, an epitope with low binding affinity to HLA-A*0201 (Ref. 5 and Rosenberg et al.).3 gp100209–217 may compete for presentation by MHC class I molecules with other epitopes from gp100, or even more likely, with immunodominant epitopes derived from the recombinant virus (8). Alternatively, mechanisms of central or peripheral tolerance having negative regulatory effects on the T-cell repertoire may also explain the lack of immunoreactivity against the self-epitope, gp100209–217 (7). In this study, “anchor-fixing” the gp100209–217 epitope within the full-length gp100 molecule not only increased the amount of peptide presented complexed with MHC class I but also induced in vitro epitope-specific CD8+ T-cell reactivity (Fig. 2 and Table 1A). The gp100209–217-specific CD8+ T cells were not merely low-affinity CTLs that recognized exogenously loaded peptides at nonphysiologically high concentrations, but they also recognized the wild-type epitope naturally expressed by the murine melanoma, B16 transfected with A2/Kb.
Peptide elution studies that measured approximate numbers of peptides presented by cells that were infected with rVV that expressed different molecular forms of antigen showed an enhancement of peptide presentation in cells infected with rVV minigenes compared with rVV-expressing full-length proteins (20). Specifically, although only 30 peptides per cell were presented by cells infected with rVV encoding the full-length form of nucleoprotein from influenza virus, up to 55,000 copies of peptides were recovered from cells expressing nucleoprotein minigene products (20). Furthermore, we have previously demonstrated that facilitating antigen presentation by vaccinating with minigene constructs enhanced immunogenicity compared with immunizing with full-length antigens (17). rVV encoding the gp100209–217 minigene preceded by an endoplasmic reticulum insertion signal sequence expressed increased amounts of gp100209–217 compared with the rVV encoding the full-length gp100, as assessed by T-cell recognition (Fig. 2). Despite enhanced presentation, rVV-ESgp100209–217 failed to elicit CD8+ T cells reactive against the gp100209–217 native epitope. Only the rVV expressing the anchor-fixed minigene induced immunity against the wild-type gp100209–217. This result suggests that the stability of peptides associated with MHC, not simply the peptide levels, was critical to induce reactivity against the weak immunogen, gp100. One explanation for this may be that anchor-fixing increased the binding stability to MHC class I by potentially decreasing the rate of dissociation of the peptide from the MHC class I molecule (21).
The amount of peptide presented by the MDA 231 cells infected with rVV-ESgp100209–217 was apparently similar to those infected with rVV-ESgp100209–217(210M) (Fig. 2). We have repeated this experiment using cells infected at different MOI in an attempt to detect differences between the wild-type and anchor-fixed minigene forms of the immunogens. We, nevertheless, could not detect clear differences in the in vitro recognition of vaccinia infected target cells. The in vivo results, on the other hand, are highly reproducible. Only the anchor-fixed immunogen is effective at eliciting self-reactive T cells. What can account for this apparent discrepancy? It seems that the viruses produce high levels of these peptides even at lower MOI, and that the levels produced are not limiting for recognition by CD8+ T cells. This situation parallels the in vitro recognition assays using T2 cells pulsed with either 1 _μ_g/ml gp100209–217 peptide or 1 _μ_g/ml gp100209–217(210M) peptide. Both are recognized similarly, an observation likely due to the saturation of the MHC class I on the cell surface.
Several papers have also reported the use of anchor-fixing modifications to enhance cytotoxic T-cell responses against self-molecules (4, 18, 22). Recently, in a mouse model, we elicited CD8+ T cells against the murine homologue of gp100 using the human sequence gp10025–33, which was shown to bind to MHC H-2 Db 100-fold better than the murine gp10025–33 sequence. This fortuitous difference between species altered the predicted MHC binding rank from number 14 to number 1 (18). In addition, a peptide from the nonmutated murine tyrosinase-related protein-1 could be anchor-fixed, which enabled it to elicit tumor-reactive CD8+ T cells as well as tumor protection (22). In human clinical trials using peptides as immunogens, 10 (91%) of 11 of patients vaccinated with the gp100209–217(210M) peptide successfully induced CD8+ T cells reactive with native gp100209–217, whereas only 2 of 8 patients who received the gp100209–217 peptide exhibited gp100209–217-specific reactivity (4).
The similarity of these results to previous findings in melanoma patients documents the potential utility of the HLA/A2/Kb transgenic model system to predict the immunogenicity of other self-antigens (4). Peptides can also be engineered to optimize immunity by modifying secondary anchor residues, by removing amino acids that may have deleterious effects on MHC/peptide contacts or by altering T cell receptor contact residues (9). In addition, with the creation of peptide libraries, a new range of T-cell epitopes can be identified for testing in vivo to explore their relative immunogenicity (23). This study suggests that anchor-fixed modifications of peptides may be a key to inducing reactivity against nonmutated “self”-TAAs expressed by recombinant virus-based immunogens.
Acknowledgments
We thank W. Overwijk for helpful discussions. We also thank M. Blalock for assistance with the graphics and L. Gritz and G. Mazzara for provision of the viruses and critical review of this manuscript.
Footnotes
2
The abbreviations used are: TAA, tumor-associated antigen; rVV, recombinant vaccinia virus; A2/Kb, HLA-A*0201/H-2 Kb chimeric molecule; HLA, human leukocyte antigen; CM, complete media; GM-CSF, granulocyte macrophage colony-stimulating factor; MOI, multiplicity/multiplicities of infection; pfu, plaque-forming unit(s); _FL_gp100, full-length human gp100; TAP, transporter associated with antigen processing; ES, endoplasmic reticulum insertion signal sequence.
3
S. A. Rosenberg et al., unpublished data.
References
- 1.Hellstrom I, Hellstrom KE. Tumor vaccines—a reality at last? J. Immunother. 1998;21:119–126. [PubMed] [Google Scholar]
- 2.Mitchell MS. Immunotherapy of melanoma. J. Investig. Dermatol. Symp. Proc. 1996;1:215–218. [PubMed] [Google Scholar]
- 3.Fenton RG, Longo DL. Danger versus tolerance: paradigms for future studies of tumor-specific cytotoxic T lymphocytes. J. Natl. Cancer Inst. 1997;89:272–275. doi: 10.1093/jnci/89.4.272. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, Wunderlich JR, Parkhurst MR, Kawakami Y, Seipp CA, Einhorn JH, White DE. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA, Zhai Y, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Seipp CA, Einhorn JH, Roberts B, White DE. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J. Natl. Cancer Inst. 1998;90:1894–1900. doi: 10.1093/jnci/90.24.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irvine KR, Chamberlain RS, Shulman EP, Surman DR, Rosenberg SA, Restifo NP. Enhancing efficacy of recombinant anticancer vaccines with prime/boost regimens that use two different vectors. J. Natl. Cancer Inst. 1997;89:1595–1601. doi: 10.1093/jnci/89.21.1595. [DOI] [PubMed] [Google Scholar]
- 7.Benichou G, Takizawa PA, Ho PT, Killion CC, Olson CA, McMillan M, Sercarz EE. Immunogenicity and tolerogenicity of self-major histocompatibility complex peptides. J. Exp. Med. 1990;172:1341–1346. doi: 10.1084/jem.172.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng Y, Yewdell JW, Eisenlohr LC, Bennink JR. MHC affinity, peptide liberation, T cell repertoire, and immunodominance all contribute to the paucity of MHC class I-restricted peptides recognized by antiviral CTL. J. Immunol. 1997;158:1507–1515. [PubMed] [Google Scholar]
- 9.Berzofsky JA. Designing peptide vaccines to broaden recognition and enhance potency. Ann. NY. Acad. Sci. 1995;754:161–168. doi: 10.1111/j.1749-6632.1995.tb44449.x. [DOI] [PubMed] [Google Scholar]
- 10.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakker AB, Schreurs MW, de Boer AJ, Kawakami Y, Rosenberg SA, Adema GJ, Figdor CG. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J. Exp. Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, Kawakami Y. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J. Immunol. 1996;157:2539–2548. [PubMed] [Google Scholar]
- 13.Vitiello A, Marchesini D, Furze J, Sherman LA, Chesnut RW. Analysis of the HLA-restricted influenza-specific cytotoxic T lymphocyte response in transgenic mice carrying a chimeric human-mouse class I major histocompatibility complex. J. Exp. Med. 1991;173:1007–1015. doi: 10.1084/jem.173.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazzara GP, Destree A, Mahr A. Generation and analysis of vaccinia virus recombinants. Methods Enzymol. 1993;217:557–581. doi: 10.1016/0076-6879(93)17089-n. [DOI] [PubMed] [Google Scholar]
- 15.Gritz L, Destree A, Cormier N, Day E, Stallard V, Caiazzo T, Mazzara G, Panicali D. Generation of hybrid genes and proteins by vaccinia virus-mediated recombination: application to human immunodeficiency virus type 1 env. J. Virol. 1990;64:5948–5957. doi: 10.1128/jvi.64.12.5948-5957.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenkins S, Gritz L, Fedor CH, O'Neill EM, Cohen LK, Panicali DL. Formation of lentivirus particles by mammalian cells infected with recombinant fowlpox virus. AIDS Res. Hum. Retroviruses. 1991;7:991–998. doi: 10.1089/aid.1991.7.991. [DOI] [PubMed] [Google Scholar]
- 17.Restifo NP, Bacik I, Irvine KR, Yewdell JW, McCabe BJ, Anderson RW, Eisenlohr LC, Rosenberg SA, Bennink JR. Antigen processing in vivo and the elicitation of primary CTL responses. J. Immunol. 1995;154:4414–4422. [PMC free article] [PubMed] [Google Scholar]
- 18.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhai Y, Yang JC, Spiess P, Nishimura MI, Overwijk WW, Roberts B, Restifo NP, Rosenberg SA. Cloning and characterization of the genes encoding the murine homologues of the human melanoma antigens MART1 and gp100. J. Immunother. 1997;20:15–25. doi: 10.1097/00002371-199701000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anton LC, Yewdell JW, Bennink JR. MHC class I-associated peptides produced from endogenous gene products with vastly different efficiencies. J. Immunol. 1997;158:2535–2542. [PubMed] [Google Scholar]
- 21.Cerundolo V, Elliott T, Elvin J, Bastin J, Rammensee H-G, Townsend A. The binding affinity and dissociation rates of peptides for class I major histocompatibility complex molecules. Eur. J. Immunol. 1991;21:2069–2075. doi: 10.1002/eji.1830210915. [DOI] [PubMed] [Google Scholar]
- 22.Dyall R, Bowne WB, Weber LW, LeMaoult J, Szabo P, Moroi Y, Piskun G, Lewis JJ, Houghton AN, Nikolić-Žugić JN. Heteroclitic immunization induces tumor immunity. J. Exp. Med. 1998;188:1553–1561. doi: 10.1084/jem.188.9.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hemmer B, Vergelli M, Pinilla C, Houghten R, Martin R. Probing degeneracy in T-cell recognition using peptide combinatorial libraries. Immunol. Today. 1998;19:163–168. doi: 10.1016/s0167-5699(97)01217-6. [DOI] [PubMed] [Google Scholar]