Independent Introduction of Two Lactase-Persistence Alleles into Human Populations Reflects Different History of Adaptation to Milk Culture (original) (raw)
Abstract
The T−13910 variant located in the enhancer element of the lactase (LCT) gene correlates perfectly with lactase persistence (LP) in Eurasian populations whereas the variant is almost nonexistent among Sub-Saharan African populations, showing high prevalence of LP. Here, we report identification of two new mutations among Saudis, also known for the high prevalence of LP. We confirmed the absence of the European T−13910 and established two new mutations found as a compound allele: T/G−13915 within the −13910 enhancer region and a synonymous SNP in the exon 17 of the MCM6 gene T/C−3712, −3712 bp from the LCT gene. The compound allele is driven to a high prevalence among Middle East population(s). Our functional analyses in vitro showed that both SNPs of the compound allele, located 10 kb apart, are required for the enhancer effect, most probably mediated through the binding of the hepatic nuclear factor 1 α (HNF1α). High selection coefficient (s) ∼0.04 for LP phenotype was found for both T−13910 and the compound allele. The European T−13910 and the earlier identified East African G−13907 LP allele share the same ancestral background and most likely the same history, probably related to the same cattle domestication event. In contrast, the compound Arab allele shows a different, highly divergent ancestral haplotype, suggesting that these two major global LP alleles have arisen independently, the latter perhaps in response to camel milk consumption. These results support the convergent evolution of the LP in diverse populations, most probably reflecting different histories of adaptation to milk culture.
Introduction
The dairy culture was initiated some 10,000 years ago in the Middle East with the domestication of sheep, goat, and cattle.1–4 Lactase activity of intestinal cells, responsible for the digestion of the milk sugar, lactose, declines after weaning for most humans.5 However, in multiple global subpopulations, a genetic capacity of adult humans to digest milk sugar has evolved that results in the continuing expression of lactase by intestinal cells, a condition known as lactase persistence (LP) (MIM 223000).5 We and others have shown that a single allele, carrying the T−13910 variant 14 kb upstream of lactase (LCT) gene, fully correlates with LP in many global populations.5–7 Functional evidence for the C/T−13910 variant in the regulation of lactase activity in intestinal cells has also emerged, lending additional support for this variant being the true causative one.8–10 Functional studies in vitro have further shown that the LP trait-related T−13910 allele binds Oct-1 transcription factor more strongly than does the C−13910 allele. It has been further demonstrated that a wider DNA region encompassing the C/T−13910 variant contains an enhancer element with binding sites for several transcription factors such as Oct-1 and GATA-6 (region from −13909 to −13934), HNF4α and Fox/HNF3α (region −13857 to −13817), and Cdx-2 (region −14022 to −14032). All these factors probably contribute to the regulation of the lactase gene in intestinal cells.10 Furthermore, the expression of Oct-1 has been shown to drive the reporter gene expression from both T−13910 and C−13910 variant/LCT promoter constructs only when it is coexpressed with HNF1α, suggesting that the −13910 enhancer effect is mediated through HNF1α bound to the proximal promoter of the LCT gene.
Recently published data indicated the lack of the T−13910 variant among Sub-Saharan African populations, known to show high prevalence of LP.11 This finding suggested the presence of yet unidentified additional mutation(s) underlying LP globally. Recently, sequence analyses of the 3 kb region flanking the T/C−13910 have identified new regional mutations, seemingly associated with LP phenotype (G−13915, C−13913, G−13907, and C−14010) in various Middle East and African populations.12,13 The C−14010 allele was strongly associated with LP among Tanzanians and Kenyans whereas the G−13915 and G−13907 alleles were too rare to explain the LP prevalence observed in these populations. These data further underline the necessity to uncover additional LP variants in global populations.12,13
In the Middle East, a high frequency of the LP phenotype has been reported among a few pastoralist groups such as Saudi Arabians and Bedouins of Sinai,14–16 among which the Arabian camel (Dromedary camelous) has been the main domesticated animal used for milk.14,17 Here, we analyzed the population samples from Saudi Arabia and other Middle East populations to address the possibility of the presence of yet other unidentified LP variants among these populations that use camel milk. We applied a strategy of deep sequencing of the alleles, selected based on matched haplotypes of the critical DNA regions (47 kb) in two populations with opposite lactase-persistence/nonpersistence (LP/LNP) phenotypes, and we identified a novel compound allele underlying LP in these populations.
Material and Methods
Samples
A total of 124 samples representing five different geographical regions from Saudi Arabia were first genotyped for the C/T−13910 SNP and next sequenced for the 900 bp region flanking the C/T−13910. In addition, a global sample consisting of 143 samples from 12 different populations as well as all the samples of one region of Saudi Arabia were sequenced for the intron 13 (3218 bp) (between −11015 and −14234 bp from the first ATG of the LCT gene) where the C/T−13910 variant resides and intron 9 (1292 bp) of the MCM6 gene where the second variant, in almost complete LD in European LP alleles, G/A−22018, resides. To characterize the wider allelic background of LP alleles, we genotyped 19 biallelic markers over 2 Mb region flanking the LCT gene (see Figure S1 available online). The global sample was collected from different global populations and included CEPH samples of European origin, as well as samples from Saudi Arabians, Ob-Ugrics, Kalash, Baluchi, Iranians, Arabs, Moroccans, Saharawis, Fulani Sudanese, Gaali from Northern Sudan, and Mahas from Sudan.18 In addition, South Korean and Somalian samples, verified for LP by disaccharidase activities in intestinal biopsies, were included.
Genotyping
Genomic DNA was extracted from blood samples according to standard procedures.19 We genotyped 18 SNPs and 1 in/del covered 2 Mb around the LCT gene by using PCR minisequencing reactions in the global sample.20 For sequencing, the overlapping PCR products were amplified and carried out in a 50 μl volume with genomic DNA (100 ng), primers (20 ng each), dNTPs (200 μM), and 0.5 U of Taq polymerase (Dynazyme, Finnzymes) in a standard buffer. Purified PCR products (15–40 ng) were cycle sequenced with BigDye terminator chemistry (PE Biosystems). Data were analyzed with ABI Sequencing Analysis 3.3 (PE Biosystems) and Sequencher 4.1 (Gene Codes).
Analyzing the Identified Target Region by Haplotype-Matching Strategy
In order to identify the LP correlating variant(s) in the random population samples from Saudis, we have used the exclusion strategy for the target region by comparing the regional haplotype (in nonphenotyped Saudi Arabia samples) with the matched LNP haplotype (in phenotyped South Korean samples verified for LP by disaccharidase activities for intestinal biopsies) samples. The rationale of this strategy is that because we lack the phenotype data in Saudi samples, we cannot carry out a direct association study with the detected SNPs. Instead, we have used a reverse direction by excluding the entire target region, with similar haplotypes of phenotyped samples as a proxy for establishing the association with LP (Figure 1). We sequenced the 71 kb region between markers D2S3012 and D2S3016 (Figure 2). We amplified the region in overlapping PCR products and sequenced them from two samples from Saudis and two South Korean samples, selected for the similar haplotype but phenotyped to represent LNP phenotype. We constructed haplotypes for the selection of the matching samples for sequence analysis. We genotyped 9 biallelic markers in 25 Saudi samples and 23 South Korean samples and constructed the haplotypes with PHASE version 2.1.1. One single haplotype in the Saudi sample correlated with the LP prevalence in the population (56%) and was used as the “matching” criteria. We chose two samples homozygous for this allele among Saudi samples and two samples from LNP-phenotyped South Koreans with matching haplotypes. The following sequence analysis was expected to identify the polymorphisms that differentiate between these two haplotypes, whereas the variants nonrelevant for LP would be shared by these alleles with the similar haplotype background. We initially hypothesized that one major haplotype carrying the LP mutation(s) would be driven to high frequency among the Saudis by natural selection. Any variant that was present on the haplotype before initiation of the selection would have the same frequency as the target polymorphism, whereas any polymorphism accumulated later on (after the introduction of the LP mutation) in the Saudi lineage would have different levels of frequency, depending on the time scale from the most recent ancestor. In our case, any polymorphisms accumulated only in the Saudi lineage compared to the South Korean lineage would thus become a target for further analysis in this population.
Figure 1.
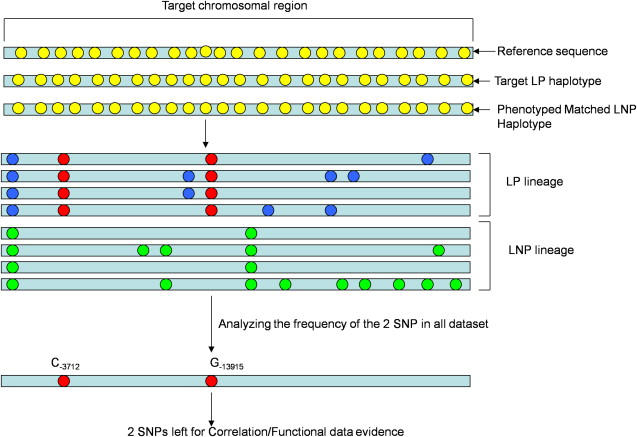
A Strategy to Identify the Causing Variant(s) from Random Population Samples through the Exclusion of the Target Region by Comparing the Haplotype of Nonphenotyped Individuals with a Matched Haplotype from Phenotyped Individuals as a Proxy for Establishing the Correlation
Small yellow circles represent the variants between the reference sequence in databases and the matched haplotypes. In our case, there were 71 such variants. Based on the matched haplotype approach, we reduced the number of potentially causative LP variants from 71 to 8 (represented by the blue and the red circles) showing an accumulation on the LP samples. The green circles represent the variants that originate on the LNP samples. Further genotyping of these variants in population samples showed that only two variants (red circles) stand out as candidates for LP.
Figure 2.
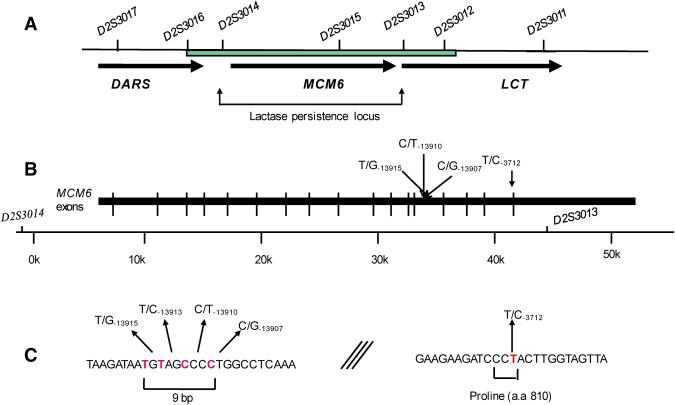
Map Showing the Sequenced Region and the LP-Related SNPs within Intron 13 and Exon 17 of the MCM6 Gene
(A) The physical map showing the restricted LP locus between markers D2S3013 and D2S3014 and the region that was sequenced between the markers D2S3012 and D2S3016 (marked as a green box).
(B) The physical map of the MCM6 gene showing the location of the SNPs identified within intron 13 and exon 17 of MCM6 gene.
(C) The sequence of the LP region indicating the European LP variant C/T−13910 and showing clustering of the LP mutations within a very short region and the Arab T/C−3712 variant in exon 17 of the MCM6 gene.
Plasmid Constructs and Transfection Assays
Human LCT promoters construct (pGL3 LPH1085−13910C)8 was used as template to generate mutations in a 455 bp region around position −13910. Mutation (G−13915) was introduced by polymerase chain reaction (PCR) mutagenesis by overlap extension32 with the pGL3 LPH1085−13910C plasmid as template. The mutated PCR fragments were cloned into SalI site of the pGL3 LPH1085, generating pGL3 LPH1085−13915G.8 In addition, PCR products from C−3712 and T−3712 regions (685 bp) were cloned in pCR2.1 TA (Invitrogen, Carlsbad, CA). The C−3712 and T−3712 fragments were cloned into the Kpn1 site in pGL3 LPH1085, producing pGL3 LPH1085−3712C and pGL3 LPH1085−3712T, respectively. The C−3712 and T−3712 were also inserted into pGL3 LPH1085−13915G and pGL3 LPH1085−13915T, generating the pGL3 LPH1085 −3712G −13915G plasmid and pGL3 LPH1085 −3712T −13915T, respectively. All pGL3 plasmids were verified by sequencing.
Caco2 cells were grown, and transfections were performed as previously described with or without HNF1α and Oct-1 expression plasmid.8,10 The cells were harvested and analyzed 2 days after transfection for luciferase and β-galactosidase activity with the Dual Light system (Applied Biosystems, Foster City, CA). The luciferase measurements were normalized and corrected for transfection efficiencies with the β-galactosidase activities.
Electrophoretic Mobility Shift Assay
Nuclear extracts were isolated from differentiated Caco2 cells.21 The following double-stranded oligonucleotides were used as probes and competitors for the EMSA: C−13910: upper strand, 5′-AAAGATAATGTAGCCCCTGGCCTCAA-3′, lower strand, 5′-ATTGAGGCCAGGGGCTACATTATCTT-3′; T−13910: upper strand, 5′-AAAGATAATGTAGTCCCTGGCCTCAA-3′, lower strand, 5′-ATTGAGGCCAGGGACTACATTATCTT-3′; G−13915: upper strand, 5′-AAAGATAAGGTAGCCCCTGGCCTCAA-3′, lower strand, 5′-ATTGAGGCCAGGGGCTACCTTATCTT-3′; G−13907: upper strand, 5′-AAAGA TAATGTAGCCCGTGGCCTCAA-3′, lower strand, 5′-ATTGAGGCCACGGGCTACATTATCTT-3′; C−3712 upper strand, 5′-AGATCCCCACTTGGTAGTTAACCCTAACTA-3′; C−3712 lower strand, 5′-ATAGTTAGGGTTAACTACCAAGTGGGGATC-3′; T−3712 upper strand, 5′-AGATCCCTACTTGGTAGTTAACCCTAACTA-3′; T−3712 lower strand, 5′- ATAGTTAGGGTTAACTACCAAGTAGGGATC-3′.
Electrophoretic mobility shift assays (EMSAs) were performed by adding approximately 5 μg of nuclear extract to 10 μl of gelshift buffer (25 mmol/L Tris [pH 7.8], 5 mmol/L MgCl2, 6 mmol/L KCl, 0.5 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L dithiothreitol, 0.5 mmol/L phenylmethylsulfonyl fluoride, 5% Ficoll, and 2.5% glycerol). Unspecific DNA binding was prevented by adding 1 μg dIdC (Boehringer Mannheim, Mannheim, Germany) and 2.5 pmol unrelated double-stranded oligonucleotide. After 20 min of preincubation on ice, 32P-labeled oligonucleotides (25 fmol) were added and incubated for another 20 min on ice. 2.5 pmol unlabeled double-stranded oligonucleotides were added to analyze for specificity of the generated DNA/protein complexes. The samples were separated by electrophoresis on a 5% nondenaturing polyacrylamide gel. After electrophoresis, the gels were dried and analyzed by autoradiography by phosphoimaging technology (Amersham Biosciences). HNF1α supershift analyses were performed by adding a polyclonal antibody against rat HNF1α in the preincubation step.
Estimation of the Age of Lactase-Persistent Alleles
We have estimated the most recent common ancestor (TMRCA) of the G−13915-C−3712 allele by three different methods. The first was LD decay method for marker D2S3014 showing the highest LD with the LP phenotype in our sample of Finnish families.5 The second was the average square distance method (ASD) with three markers (D2S3013, D2S3015, and D2S3016) showing less LD with the LP phenotype in the Finnish family data.7 In methods one and two, the analyses were performed on 88 random Saudi samples for the G−13915-C−3712 allele, with a rich algorithm and Y time program, respectively.22 Lastly, we applied a phylogeny-based method, with the first two methods as calibration points for the estimation of mutation rate in this method.23 The estimates were obtained with NETWORK4.1.1.2 program, by means of the measure rho statistic (ρ), the average number of mutations from the root (which was H1 in the networks), including the standard deviations (SD) and a generation time of 25 years in every population with the formula t = ρ/μ, where t is the time since the MRCA and μ is the mutation rate for the region per year. We used two different calibration points for estimating the mutation rate for the 31448 bp region. The first one is 4.54 × 10−8/bp/year, considered as the lower bound for mutation rate calibration. It is based on the LD decay for marker D2S3014 showing the highest LD with the LP phenotype in the Finn families.5 The second one is 2.59 × 10−8/bp/year, considered as the high bound for mutation rate calibration. It is based on the average square distance method (ASD) with three markers (D2S3013, D2S3015, and D2S3016) showing less LD with the LP phenotype in the Finn family data.7 In addition, we also used two different calibration points for estimating the mutation rate for the intron 13 (3218 bp) region analyzed in the network. The first calibration point is the same as the second one mentioned above; the second one is 1.04 × 10−9/bp/year, based on the age estimates acquired by ASD, with markers D2S3012 and D2S3014 in 88 random Saudi samples.33
Statistical Analyses
Haplotype estimations were made via hierarchical steps. First, the 31.4 kb region haplotype for every individual (8 SNPs and one indel) was determined with the program PHASE version 2.1.124,25 as a frame backbone for building the whole haplotype. Second, the haplotypes within the introns 13 and 9 of the MCM6 gene within the 30 kb region were resolved visually. Last, we fitted the data to a model that minimizes the number of implied historical crossovers in the population, and by using the program PHASE version 2.1.1,24,25 we obtained the full haplotype (more than 2 Mb for every individual for the extended haplotypes (Figure S1). The minimum number of mutations necessary to generate the observed haplotypes was inferred by median-joining (MJ) networks23 (implemented in the program NETWORK 4.1.1.2). The construction of the haplotype network was conducted first for the intron 13 region based on 26 variants detected from the sequence of intron 13 (3218 bp) in the global sample and for the ∼31 kb region with 47 variants (40 variants were detected from the sequencing of 5381 bp of introns 13 and 9 and exon 17 of the MCM6 gene in the global samples in addition to genotyping additional 6 SNPs and one indel polymorphisms over this region). We further typed additional 10 distant markers covered over 1.1 Mb of the LCT region to monitor the decay of the haplotypes.
The haplotype test of Hudson et al.26 was used on the Saudi population to address whether the variation detected within the region could be explained by neutral model. Data sets expected under the neutrality assumption and conditioned on the observed number of segregating sites were generated by a coalescent simulation program, ms.27 We assumed a recombination fraction of 1 cM = 1 Mb and chose the mutation rate so that after ascertainment the expected SNP density would match the observed density to some extent. We explored three demographic models. The first is a constant population with effective population size 104 for the last 5000 generations. The second is a recent expansion model in which the population was stable at 104 for 4000 generations and then began to expand 1000 generations ago to 107. The third assumes that a severe bottleneck 2000 generations ago happened, during which the size of the population was reduced from 104 to 103. This population was then recovered back to 104 in 500 generations and remained stable for another 500 generations, at which point the population began expanding 1000 generations ago to 107. The estimated p value from 1000 random coalescent simulations has been estimated. In addition, we used the long-range haplotypes tests: the extended haplotype homozygosity (EHH), defined as the probability that any two randomly chosen chromosomes carrying the core haplotype of interest are identical by descent,28 and the relative EHH (REHH), the factor by which EHH decays on the tested core haplotype compared to the manner in which it decays on all other core haplotypes combined. We selected a single SNP at a time as a core haplotype and calculated the EHH and REHH as described for all 19 SNPs analyzed with Sweep software program.28 Because there is no genotype data available for Saudis to assess the significance of the EHH and REHH values obtained, we evaluated the significance by using 1000 random coalescent simulations under the three demographic models described earlier. The haplotypes were placed into 20 bins based on their frequency, and p values were obtained by log-transforming the EHH and REHH in the bins to achieve normality, and we calculated the mean and standard deviation.
Then, we estimated the coefficient of selection, s, by applying a formula29 that relates the frequency in generation t + 1(pt+1) to the frequency in generation t(pt): In[p/q] + 1/q = In[p0/q0] + 1/q0 + st. In this formula, qt = 1 − pt, _w_11 is the relative fitness of individuals homozygous for the selected allele, _w_12 is the relative fitness of heterozygous individuals, and _w_22 is the relative fitness of individuals homozygous for the unselected allele. We assumed a dominant model for LP, i.e., _w_11 = _w_12 = 1 and _w_22 = 1 − s. We also assumed the initial frequency _p_0 to be 1/1000 (corresponding to a new mutation in a population with an effective size between 500; larger population sizes yield even higher coefficients of selection). Starting from that initial frequency, we calculated values of _w_22 that would yield a frequency of p = p = 0.57 after 4091 (2046–6136) years of selective pressure for the Saudi Arabian population (assuming 25 years/generation). Then we estimated the coefficient of selection associated with carrying at least one copy of the LP allele G−13915 and allele T−13910 in Moroccans, Saharawi, and Arabs based on the age of the alleles in these populations.
Results
Analysis of the Critical DNA Region among Middle East Populations
We first monitored for the presence of the European LP mutation among Saudi samples by genotyping the C/T−13910 variant in 124 samples from five different regions of Saudi Arabia, with a well-established high (>80%) prevalence of LP phenotype.14,30 Only one sample was heterozygous for the European C/T−13910 variant, indicating an almost total lack of the LP-related T−13910 allele. Thus it became obvious that the T−13910 allele does not explain the high prevalence of LP. We hypothesized that the presence of yet unidentified mutations would underlie LP in the Middle East.
A major caveat of our study is that we are lacking reliable LP/LNP phenotype data for Saudi samples. The low number of samples also prevented us from performing trivial association analysis with any nucleotide variant identified by sequencing. Instead, we adopted a haplotype-matching-based strategy to establish the critical LP variant(s). We first genotyped nine biallelic markers between markers D2S3013 and D2S3014 (Figure 2), as described earlier,18 to select for a closely matched haplotypes in two populations representing nonphenotyped Saudi samples and phenotyped South Korean samples verified for LNP. We hypothesized that the closely matched alleles would differentiate only in the critical SNPs, which could be identified by sequencing (Figure 1). The SNP haplotype analysis revealed the presence of one major haplotype that correlated perfectly with the prevalence of LP (56%) among the Saudis (Figure S1). We chose two homozygote samples for this haplotype from Saudis and two matched LNP phenotyped samples from South Koreans (C/C−13910 genotyped and verified by disaccharidase activities for intestinal biopsies) for regional resequencing (Figures 1 and 2; Figure S1). Our further analyses were then targeted only to the variants that differentiated Saudi from South Korean samples (Figure 1; Figure S1).
Sequence comparison of the critical 47 kb DNA region in the four samples from Saudis and Koreans to the reference human genome sequence revealed 71 variants (Table 1), 28 (∼40%) of them being absent in the GenBank data set (Table 1). Only 8 DNA variants differed between Korean and Saudi samples (present in the Saudi but not in the Korean lineage), three of them were heterozygous in only one sample (ss# 79088040, ss# 79088043, ss# 79088045), two were heterozygous in both samples (ss# 79088032, ss# 79088043), and the remaining three variants were homozygous in both Saudi samples (ss# 79088024, ss# 79088029, ss# 79088033) (Table 1). The further analysis of these variants in the regional Saudi samples revealed a low frequency not compatible with LP prevalence (Table 2) for all except two SNPs that remained as excellent candidates for the LP mutation (Table 2). These variants were T/G−13915 (ss# 79088033) and a newly described mutation, synonymous SNP in exon 17 of the MCM6 gene T/C−3712 (3712 bp 5′ from the ATG of LCT gene) (ss# 79088029) (Figure 2; Figure S1).
Table 1.
The SNPs Observed in the 47 kb Region Sequenced between Markers D2S3013 and D2S3014 in the Four Matched Haplotype Samples
| rs# or ss# | Positiona | Variation | S8 | S11 | K4 | K5 | Reference |
|---|---|---|---|---|---|---|---|
| rs10928552 | +1210 | C/T | TT | TT | TT | TT | CC |
| rs1185269 | +1193 | A/T | TT | TT | TT | TT | AA |
| rs11898981 | +1154 | T/C | CC | CC | CC | CC | TT |
| ss79088024 | +1150 | TA9/23 | TA9/9 | TA9/9 | TA23/23 | TA21/23 | TA22/19 |
| ss79088022 | +1062 | T/C | TT | TT | TT | TC | TT |
| ss79088025 | +1046 | A/G | GG | GG | GG | GG | AA |
| rs6730196 | +980 | C/A | AA | AA | AA | AA | CC |
| ss79088049 | +696 | G/G | CC | CC | CG | CC | CC |
| ss79088026 | −689 | A/G | GG | GG | GG | GG | AA |
| ss79088027 | −969 | T/C | CC | CC | CC | CC | TT |
| rs34496521 | −1651/2 | T11/T13 | T13/T13 | T13/T13 | T13/T13 | T13/T13 | T11/T11 |
| rs6742283 | −2131 | G/A | AA | AA | AA | AA | GG |
| ss79088028 | −3058 | A15/A14 | A14/A14 | A14/A14 | A14/A14 | A14/A14 | A15/A15 |
| ss79088029 | −3712 | T/C | CC | CC | TT | TT | TT |
| rs4988279 | −4170 | −/C | CC | CC | CC | CC | −/− |
| rs3820790 | −4482 | A/T | TT | TT | TT | TT | AA |
| ss79088030 | −5440 | G/A | AA | AA | G/A | AA | GG |
| rs3739020 | −7755 | T/G | GG | GG | GG | GG | TT |
| rs4988267 | −8170 | G/A | AA | AA | AA | AA | GG |
| ss79088031 | −10406 | T/C | TT | TT | T/C | T/C | TT |
| ss79088032 | −11000 | T/A | TA | TA | TT | TT | TT |
| rs4988252 | −11624 | T/G | GG | GG | GG | GG | TT |
| rs4988251 | −11677 | T/G | GG | GG | GG | GG | TT |
| rs2082730 | −12005 | C/A | AA | AA | AA | AA | CC |
| rs4988243 | −12967 | A/G | GG | GG | GG | AG | AA |
| rs6752360 | −13134 | T/A | AA | AA | AA | AA | TT |
| rs6752362 | −13135 | T/G | GG | GG | GG | GG | TT |
| rs4988239 | −13237 | −/GAGAG | GAGAG | GAGAG | GAGAG | GAGAG | −/− |
| rs4954492 | −13735 | G/T | TT | TT | TT | TT | GG |
| ss79088033 | −13915 | T/G | GG | GG | TT | TT | TT |
| ss79088034 | −16580 | A12/A13 | A13/A13 | A13/A13 | A13/A13 | A13/A13 | A12/A12 |
| ss79088035 | −17389 | GA4/− | −/− | −/− | GA4/A4 | ||
| ss79088035 | −17390 | del1340 −/− | +/+ | +/+ | del1340 −/− | del1340 −/− | +/+ |
| rs3213871 | −20486 | C/T | TT | TT | TT | TT | CC |
| rs309174 | −23747 | G/T | TT | TT | TT | TT | GG |
| ss79088036 | −24649 | A/T | AA | AA | AA | AT | AA |
| ss79088037 | −25095 | +/del30 | −/− | −/− | −/− | +/+ | |
| ss79088038 | −26591/5 | CA4/− | −/− | −/− | −/− | −/− | CAAAA |
| rs4988183 | −27310 | T/C | CC | CC | CC | CC | TT |
| rs309175 | −27369 | C/A | AA | AA | AA | AA | CC |
| rs309177 | −28089 | T/C | CC | CC | CC | CC | TT |
| ss79088039 | −30183 | T/C | CC | CC | CC | CC | TT |
| ss79088069 | −30948 | del 211 | −/− | −/− | −/− | −/− | +/+ |
| rs3769002 | −31268 | T/C | CC | CC | CC | CC | TT |
| rs309812 | −32250 | C/A | AA | AA | AA | AA | CC |
| rs218505 | −33447 | C/G | GG | GG | GG | GG | CC |
| rs4954513 | −33644 | G/A | AA | AA | AA | AA | GG |
| rs309127 | −34589 | T/C | CC | CC | CC | CC | TT |
| rs309129 | −35257 | A/G | GG | GG | GG | GG | AA |
| rs3835798 | −36670 | A7/6 | A6/6 | A6/6 | A6/6 | A6/6 | A7/7 |
| rs309131 | −36688 | T/C | CC | CC | CC | CC | TT |
| rs309132 | −38275 | C/G | GG | GG | GG | GG | CC |
| rs191079 | −39035 | T/C | CC | CC | CC | CC | TT |
| ss79088040 | −40046 | G/C | GG | GC | GG | GG | GG |
| ss79088041 | −40368 | C/G | CC | CG | CC | CC | CC |
| ss79088042 | −41588 | A/G | GG | GG | GG | GG | AA |
| rs13421746 | −41789 | C/A | AA | AA | AA | AA | CC |
| rs10539209 | −42078/9 | CT/− | −/− | −/− | −/− | −/− | CT |
| rs7608980 | −42173 | G/A | AA | AA | AA | AA | GG |
| rs16832138 | −42614 | G/A | AA | AA | AA | AA | GG |
| rs36097947 | −42816 | T21/23 | T23/23 | T23/23 | T23/23 | T23/23 | T21/21 |
| ss79088043 | −42888 | C/T | CC | CC | TT | TT | CC |
| ss79088044 | −43055 | G/A | GA | GA | GG | GG | GG |
| rs632632 | −43483 | T/C | CC | CC | CC | CC | TT |
| ss79088045 | −44514 | A/C | AA | AC | AA | AA | AA |
| D2S3014 | −44748 | AC22/19 | AC19/19 | AC19/19 | AC19/19 | AC19/19 | AC22/22 |
| ss79088046 | −45174 | G/− | −/− | −/− | −/− | −/− | G/G |
| ss79088047 | −45175 | C12/9 | C9/9 | C9/9 | C9/9 | C9/9 | C12/12 |
| ss79088048 | −45187 | (TC)4/4/− | TC4/4 | TC4/4 | TC4/4 | TC4/4 | −/− |
| rs666407 | −45505 | A/G | GG | GG | GG | GG | AA |
Table 2.
The Genotype Frequencies of the T/G−13915 and T/C−3712 SNPs in Middle East Populations
| Genotype T/G−13915 | Allele | Genotype T/C−3712 | Allele | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Population | n | TT | TG | GG | G−13915 (%) | TT | TC | CC | C−3712 (%) | % of LP Based on Allele Frequency |
| Saudi Arabians | ||||||||||
| Region 1 | 25 | 6 | 10 | 9 | 56 | 6 | 10 | 9 | 56 | 76 |
| Region 2 | 25 | 6 | 13 | 6 | 50 | 6 | 13 | 6 | 50 | 76 |
| Region 3 | 25 | 3 | 10 | 12 | 68 | 3 | 10 | 12 | 68 | 88 |
| Region 4 | 24 | 5 | 9 | 10 | 60 | 5 | 9 | 10 | 60 | 79.2 |
| Region 5 | 25 | 7 | 12 | 6 | 48 | 7 | 12 | 6 | 48 | 72 |
| Total | 124 | 27 | 54 | 43 | 57 | 27 | 54 | 43 | 57 | 78.2 |
| Jordanians | 56 | 50 | 6 | 0 | 5.4 | ND | ND | ND | ND | |
| Moroccans | 12 | 10 | 2 | 0 | 8.3 | 10 | 2 | 0 | 8.3 | 16.6 |
| Saharawi | 11 | 7 | 4 | 0 | 18.2 | 7 | 4 | 0 | 18.2 | 36.4 |
| Arabsa | 19 | 16 | 2 | 1 | 10.5 | 16 | 2 | 1 | 10.5 | 15.8 |
| Mahas, N. Sudan | 15 | 11 | 3 | 1 | 16.7 | 11 | 3 | 1 | 16.7 | 26.7 |
| Somaliansb | 62 | 60 | 2 | 0 | 1.6 | 60 | 2 | 0 | 1.6 |
Analysis of two critical SNPs in all 124 samples from Saudis samples shows a complete co-occurrence of C−3712 and G−13915 in a total of 140 chromosomes (Table 2). To establish the full allelic diversity for the critical DNA regions, we sequenced the 3218 bp region flanking the C/T−13910 and T/G−13915 and ∼900 bp flanking the T/C−3712 variant in all the Saudi samples (Table 2) as well as in our “global” sample set consisting of 143 DNA:s from 12 different populations (Figure S1; see Material and Methods).
The T/G−13915 variant was found at a high frequency ranging from 72% to 88% among Saudis (Table 2) and correlated well with the reported frequency of LP in these populations (Table 3). The −13915 variant is only 5 bp away from the European LP variant C/T−13910 and lies within an Oct-1 motif. Interestingly, another variant (C/G−13907) 3 bp downstream from the C/T−13910 was identified in two individuals among 124 samples tested (Table 3; Figure 2C). Our sequencing effort also identified a variant A/G−12962 in complete LD with T/G−13915. However, sequencing of the 143 global samples revealed the presence of this SNP in all populations at the frequency of 29% also among 14 South Korean samples (all verified for LP by disaccharidase activities of intestinal biopsies). Thus, we can exclude the SNP A/G−12962 as a causative LP variant in this population, and this SNP probably has simply been “hitchhiking” with T/G−13915 (Figure S1).
Table 3.
The Allele Frequencies of the SNPs Detected in Middle East Populations and Finns
| Allele Frequencies (±SD) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Population | n | C−3712 | T−13910 | G−13915 | G−13907 | C−13913 | Total | Reported Frequency of LP Based on LTTa |
| Saudi Arabia | 248 | 0.570 (0.031) | 0.004 (0.004) | 0.570 (0.031) | 0.008 (0.006) | 0 | 0.582 (0.031) | 0.592 (0.096) |
| Jordan | 112 | ND | 0.054 (0.021) | 0.054 (0.021) | 0 | 0.009 (0.009) | 0.117 (0.030) | 0.124 (0.044) |
| Mahas | 30 | 0.17 (0.069) | 0 | 0.17 (0.069) | 0 | 0 | 0.17 (0.069) | |
| Gaali | 20 | 0 | 0 | 0 | 0.05 (0.049) | 0 | 0.05 (0.049) | |
| Iran | 42 | 0 | 0.10 (0.03) | 0 | 0 | 0 | 0.10 (0.030) | 0.09 (0.045) |
| Morocco | 24 | 0.083 (0.056) | 0.21 (0.08) | 0.083 (0.056) | 0 | 0 | 0.294 (0.09) | |
| Saharawi | 22 | 0.182 (0.082) | 0.23 (0.09) | 0.182 (0.082) | 0 | 0 | 0.412 (0.10) | |
| Arabsb | 40 | 0.105 (0.048) | 0.13 (0.05) | 0.105 (0.05) | 0 | 0 | 0.235 (0.07) | |
| Finns | 1876 | 0 | 0.575 (0.011) | 0 | 0 | 0 | 0.575 (0.011) | 0.588 (0.023) |
We sequenced the identified DNA regions in 56 random Jordanian samples (i.e., another Middle East population) known to be of mixed origin.31 This analysis revealed—in addition to the presence of mutations C/T−13910 and T/G−13915 in six samples—a third variant, T/C−13913, only 3 bp apart from the C/T−13910 in one sample (Tables 2 and 3; Figure 2C). Interestingly, in our global samples, one tested and confirmed LP case from Denmark with German ancestors and genotype C/C−13910 was also found to be heterozygous for the SNP T/C−13913. A recent report has shown the presence of a new mutation G/A−13914 in another LP case from Germany, making the immediate vicinity of C/T−13910 region a potential region for the accumulation of the rare alleles associated with LP.32 Our sequence analysis of two African tribes in Northern Sudan, the Gaali and the Mahas, revealed the presence of the G−13915 allele (17%) among the Mahas (the tribes of proposed Arabic origin) and G−13907 allele (5%) among Gaali. In West Arabic populations of Northern Africa (Moroccans and Saharawis) and in East Arabic populations (Iraqis, Syrians, Lebanon's, and Palestine's), the sequence analysis revealed the presence of both T−13910 and G−13915 but at different frequencies, implying genetic heterogeneity behind LP in these populations.
Resequencing our 143 “global” samples from 12 different populations showed an almost total lack of the _C−3712_-G−13915 allele in the local neighboring populations of Arabs, such as Iranians. This would suggest that this compound allele would originate from the Arabian Peninsula (Table 2; Figure S1). The LP prevalence figures and the frequency of the compound allele correlate perfectly among Saudis (Table 3).5,14,31,33,34 Finally, sequence analysis of 62 individuals from East Africa (Somalia), phenotyped via disaccharidase activity measurements of intestinal biopsies, revealed the presence of _C−3712_-G−13915 in only two samples, both of them phenotyped as LP (Table 2).
Functional Analysis of the Identified Variants
To address the real functional significance of four LP-associated variants (T/C−3712, T/G−13915, C/G−13907, and T/C−13913), we performed electrophoretical mobility shift assay (EMSA) for the DNA segments, carrying these novel variants by using nuclear extracts from the intestinal cell line Caco2. We have previously shown10 that Oct-1 binds more strongly to T−13910 than C−13910 (Figure 3A, lane 1 and 2), correlating with the increased enhancer activity of the T−13910 variant. We also found a strong specific Oct-1 binding to the G−13907 probe similar to the T−13910 Oct-1 binding (Figure 3A, lane 5). On the contrary, the G−13915 bound more weakly to Oct-1 (Figure 3A, lane 3) than even the C−13910 (Figure 3A, lane 2), whereas the C−13913 (Figure 3A, lane 4) has the same affinity as C−13910. A strong specific binding was observed to both probes C−3712 and T−3712 (Figure 3B, lanes 1–4 and 6–9). A search of the TRANSFAC database version 10 for the flanking sequences of −3712 revealed a binding site for Hepatocyte Nuclear Factor 1 α (HNF1α). A supershift analysis with HNF1α antibodies revealed a supershift of both the C−3712 and the T−3712 complex, demonstrating the specific binding of HNF1α (Figure 3B, lanes 5 and 10). Although the result demonstrates a functional role of this SNP, we did not observe a differential binding activity between the C−3712 and T−3712 probes.
Figure 3.
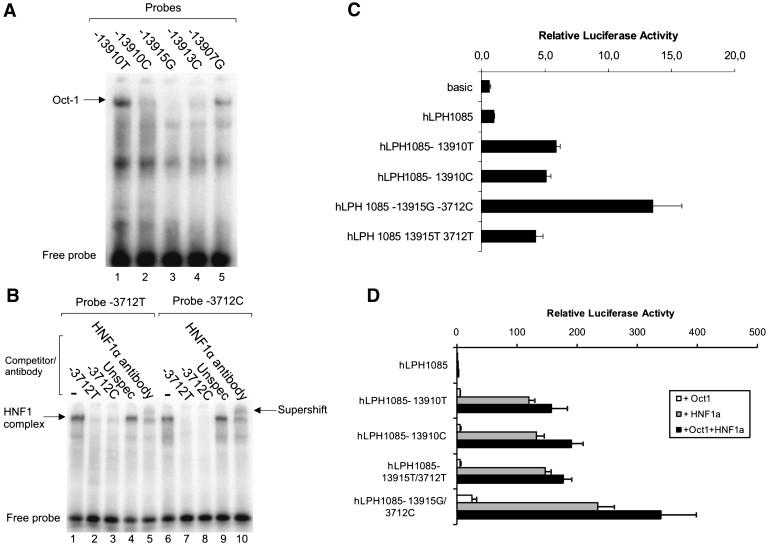
EMSA for Analyses of Protein/DNA Interactions and Relative Luciferase Activity of Intestinal Caco2 Cell Line Cotransfected with Lactase Promoter-Reporter Constructs Containing the Identified Variants
(A) Electrophoretical mobility shift assay (EMSA) for analyses of protein/DNA interactions of the identified variants (lanes 1–5) with double-stranded oligonucleotides primers and nuclear extracts from the intestinal cell line Caco2. The arrow indicates the Oct-1 complexes.
(B) The EMSA for the T/C−3712 showing the specific binding of HNF1α to both probes. The HNF1α supershift analyses were performed by adding a polyclonal antibody against rat HNF1α.
(C and D) Relative luciferase activity of intestinal Caco2 cell line cotransfected with lactase promoter-reporter constructs containing the 455 bp fragments of the regions flanking the T/G−13915 and 685 bp fragment of the region flanking T/C−3712.
(D) The G−13915-C−3712 and T−13915-T−3712 regions were analyzed together for the effect of overexpression of HNF1α and Oct-1 on the G−13915-C−3712 and T−13915-T−3712. The luciferase activity was corrected for transfection efficiency and normalized to the expression of pGL3-hLPH1085, n = 4.
To analyze further the functional significance of the Arabic LP allele(s) for the LCT promoter activity, constructs were designed to mimic the compound alleles carrying G−13915-C−3712 or T−13915-T−3712 variants and analyzed together in connection with the human LCT promoter. The result was striking differential reporter gene expression stimulation by the G−13915-C−3712 combination compared to the T−13915-T−3712 variant (Figure 3C). Because the T−13910 enhancer activity has previously been shown to be mediated through HNF1α and Oct-1 interaction,10 we examined the effect of overexpression of HNF1α and Oct-1 on the G−13915-C−3712 and T−13915-T−3712 constructs. The reporter gene expressions from both constructs were significantly increased by the HNF1α expression but not by the Oct-1 expression alone (Figure 3D). When both transcription factors were overexpressed, Oct-1 significantly increased the reporter gene expression. Similar results were obtained with the T−13910 and the C−13910 constructs (Figure 3D).10 The results indicate that Oct-1 and HNF1α are central regulators of the LCT promoter activity and that their binding is affected by the G−13915-C−3712 and T−13915-T−3712. The G−13915 was more transcriptionally active than C−3712, but the binding of HNF1α, one of the main LCT promoter regulators, to the T/C−3712 SNP indicates that this SNP in vivo might have the critical regulatory role.
Age Estimate for the Compound LP Allele
We tried to estimate the age of the most recent common ancestor (TMRCA) of the compound C−3712-G−13915 allele by different methods (Table 4 and Material and Methods). The age estimates for the C−3712-G−13915 allele was 4000 years (95% CI; 250–27,575) in Saudi Arabia, a population known to be the source of the migration into other Arab populations in the last several thousand years. This would provide additional support for the origin of the C−3712-G−13915 allele among Arabs (Table 4).
Table 4.
Age Estimates of the TMRCA for the Lactase Persistence G−13915
| Age Estimates | LD Decay | |||
|---|---|---|---|---|
| Population | ASDa (95% CI) | With Rho: Years (± SD) | D2S3012 (95% CI) | D2S3014 (95% CI) |
| Saudi Arabia | 4,000 (250–27,575) | 22,200 (21,500–23,000) | 1,500 (1,450–1,575) | |
| Based on 3218 bp sequence | 4,091 (± 2,045)b | |||
| 1,670 (± 835)c | ||||
| Based on genotyping 9 SNPs | 4,421 (± 2,211)b | |||
| 1,805 (± 903)c | ||||
| Non-Saudi | ||||
| Arab-Morocco-Saharawi | 1,275 (125–16,900) | 1,962 (± 981)b | 1,100 (825–1,400) | 15,975 (9,350–23,350) |
| 801 (± 400)c | ||||
| Arabs | 882 (± 441)b | |||
| 360 (± 180)c | ||||
| Morocco | 1,171 (± 585)b | |||
| 478 (± 239)c | ||||
| Saharawi | 3,375 (± 2,386)b | |||
| 1,378 (± 974)c |
Haplotype Analysis of the LP Alleles and Implication for Dairy Culture
We analyzed the broader LP background haplotype by genotyping total of 19 biallelic markers spanning more than 2 Mb DNA regions flanking the LCT gene in Saudis and Northern Europeans (Figures 4A and 4B). This haplotype analysis revealed that the C−3712 and G−13915 variants reside on the same allelic haplotype extending more than 1.3 Mb (844 kb 3′LCT, and 471 kb 5′ LCT gene) in the Saudi samples, quite analogous to the 2 Mb haplotype carrying the T−13910-G−22018 variants in European populations (Figures 4A and 4B). Furthermore, the analysis revealed a similar trend toward one intact haplotype carrying the C−3712-G−13915 variant allele in other Arab populations (Saharawi, Arabs, Moroccans, and Mahas) (Figure S1).
Figure 4.
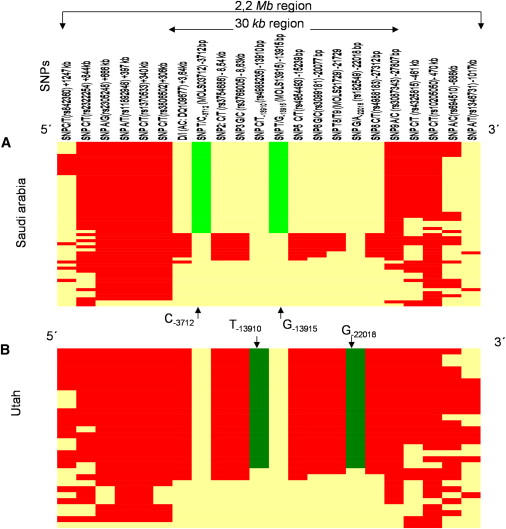
Long-Range Extended Haplotypes for the LP-Associated Alleles Constructed at Various Distances
Haplotypes are depicted from Saudi Arabia (A) and Utah (B) samples showing the core haplotype containing the European LP allele G−22018-T−13910 and the Arab LP allele C−3712-G−13915 at various distances. The core region containing G−22018-T−13910 and C−3712-G−13915 are shown in dark and light green, respectively, and the haplotypes are oriented from left to right. The derived allele at each SNP is shown in red while the ancestral allele is shown in yellow.
To compare the details of allelic backgrounds on which the C−3712-G−13915 and G−13907 variants have occurred versus the allelic background of the European LP variant, T−13910, we constructed the median joining (MJ) haplotype network. This network was based on 26 variants detected from the sequence analysis of intron 13 (3218 bp) of the MCM6 gene in the Saudi samples combined with 143 global samples from 12 worldwide populations (Figure 5A; Figure S1). The network analysis indicated that LP alleles carrying T−13910 and G−13907 variants share a common ancestral allelic background, here referred as H84, whereas the G−13915 LP allele has a completely different background allele, here referred as H107 (Figure 5A). To obtain a better resolution of the background alleles for these variants, we extended the DNA region used in network analysis to the wider ∼31 kb segment and constructed the network with a total of 47 biallelic variants. The analysis confirmed that the T−13910 and G−13907 alleles indeed reside on the same ancestral background allele, LNP H84, whereas the C−3712-G−13915 allele resides on a different ancestral background allele, LNP H107 (Figure 5B). We further monitored these haplotypes up to 1.1 Mb by typing additional distant markers. The allele H84 was almost intact up to 1.1 Mb, providing a probable genetic reflection to shared cattle domestication culture by Eurasians and Africans in the very recent history (i.e., within 10,000 years) (Figures 5C and 5D), most probably in the Middle East.
Figure 5.
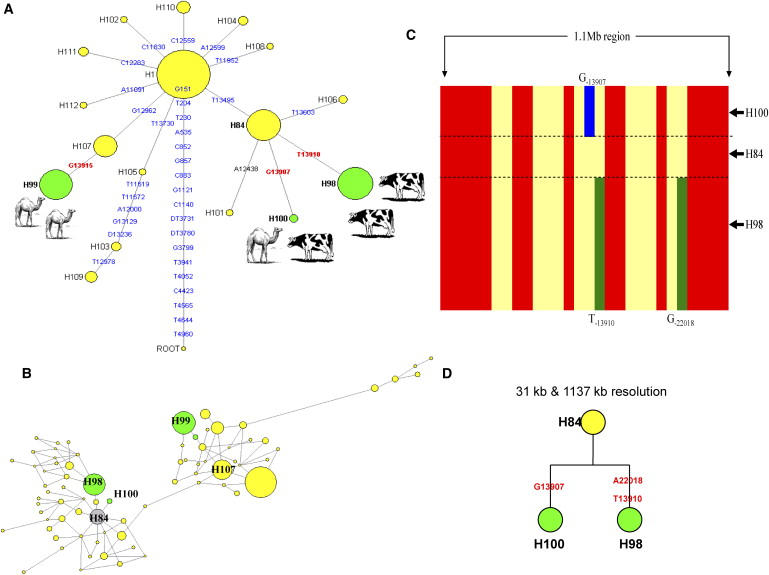
Haplotype Networks Showing the Relationships of the LP-Associated Alleles to Each Other in the Networks
(A) Haplotype network of intron 13 of MCM6 gene in the global population sample. The network was constructed with 26 SNPs (identified by sequence analysis of 170 global population samples) spanning 3218 bp of the intron 13 of the MCM6 gene. The network shows that the European LP European allele T−13910 (H98) and the African G−13907 allele (H100) have the same lactase nonpersistence (LNP) ancestral background allele (H84). The root was based on the chimpanzee sequence of intron 13 of MCM6 gene. Yellow circles represent LNP alleles whereas the green represent the LP alleles. Size of the circles is proportional to the frequency in our global samples. The G−13915 (H99) has a different background haplotype (H107) (potentially coevolved with the domestication of camels), whereas T−13910 (H98) and G−13907 (referred here as H100) share a same background haplotype (H105) (most likely coevolved with the domestication of cattle). The derived mutations are shown on the branches; the bold red mutations represent mutations associated with LP.
(B) Haplotype network consisting of 47 SNPs spanning 31 kb region between intron 1 of LCT gene and intron 7 of MCM6 gene in 170 global samples. The network obtained with data across this wider DNA region still indicates that H98 and H100 share a common ancestor background allele (H84, gray), whereas the LP H99 shows a different ancestral background allele (H107).
(C) Plot of the haplotypes H84, H98, and H100 spanning more than 1.1 Mb showing that the LNP H84 shares the same ancestral background haplotypes with LP H98 and H100 up to 1.1 Mb. The red indicates the derived allele; yellow indicates the ancestral allele at the SNP sites analyzed. Blue indicates the derived allele G−13907 at the SNP site; green indicates the derived alleles T−13910 and G−22018 at those SNP sites.
(D) Phylogenetic relationship between the haplotypes H98, H100, and H84 at 31 kb and 1137 kb resolution would provide genetic evidence that the Europeans and the Africans most probably have a shared a cattle domestication culture in very recent historical times.
Selection of the LCT Region
Because the LCT region shows most evidence of selection among human genome loci,6 we wanted to see whether any evidence could be obtained for selection for the compound LP allele. We consequently performed the haplotype test with ms simulation program to test whether this haplotype structure in Saudi Arabia would occur under neutrality.27 In Saudi samples, the haplotype analysis shows that out of 50 chromosomes carrying the C−3712-G−13915 allele, 12 chromosomes were identical over 2 Mb regions and 23 chromosomes were identical over 1.5 Mb regions (Figure 4A; Figure S1). Coalescent simulations were conditioned on the number of segregating sites observed in the data, and with three different demographic models: constant population model, recent expansion model, and a bottleneck model (see Material and Methods). The estimated p value was determined from 1000 random coalescent simulations, observing 24% haplotype H99 among 50 chromosomes under constant population model (p = 0.001), expansion model (p = 0.008), and bottleneck model (p = 0.009). This would indicate that the data set significantly deviates from neutrality and would indicate strong selection. Next we performed the long-range haplotype test (LRH) as described earlier.6 We used a single SNP at a time for the core marker, and both the extended haplotype homozygosity (EHH) and relative extended haplotype homozygosity (REHH) were computed for each core haplotype i and compared at increasing distances x from the core markers. In the Saudi samples (Figures 6A and 6C), the analysis showed that the REHH of the G−13915 core haplotypes was always significantly superior to the REHH of the T−13915 core haplotypes on both sides (REHH = 8.31 up to 857 Kb on proximal side for 3′ of the LCT gene, and REHH = 6.53 up to 673 Kb on distal side for 5′ of the LCT gene). Analogously, a similar situation holds for the Utah population regarding the European C/T−13910 variant (Figures 6B and 6D). To assess the significance of REHH values among Saudis, we applied the coalescent simulations under three different demographic models. These models were applied before giving the estimated p value from 1000 random coalescent simulations. The models used were constant population model (p = 0.000678 for the proximal side and p = 0.002249 for the distal side), expansion model (p = 0.000966 for the proximal side and p = 0.000656 for the distal side), and bottleneck model (p = 0.00119 for the proximal side and p = 0.000827 for the distal side) (Figures 6E and 6F). Again, similar results were obtained for the European C/T−13910 variant in Utah with the estimated p value under constant population model (p = 1.61 × 10−5 proximal side and p = 0.000515 distal side), expansion model (p = 6.72 × 10−5 for the proximal side and p = 0.00102 for the distal side), and bottleneck model (p = 5.15 × 10−5 for the proximal side and p = 0.000776 for the distal side) (Figures 6E and 6F).
Figure 6.
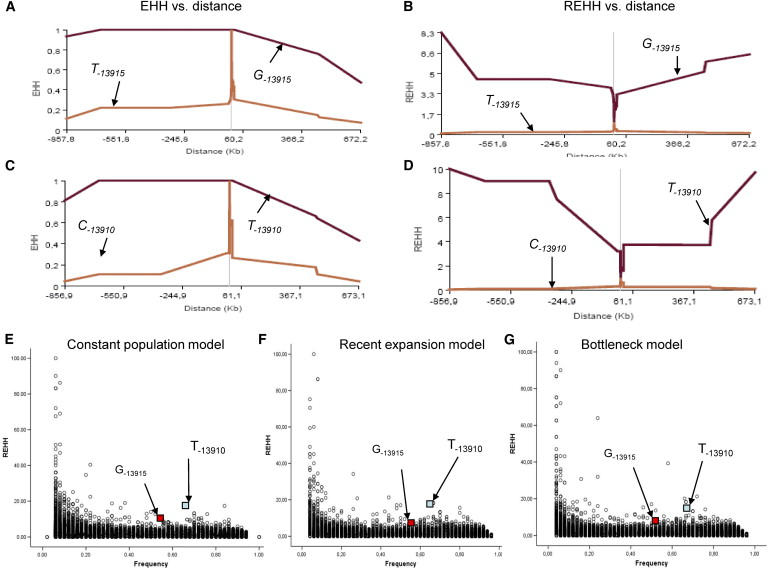
The Extended Haplotype Analyses for the LP-Associated Alleles
(A–E) The EHH and REHH analyses of the two alleles for the Arab LP/LPN variant T/G−13915 in the Saudi Arabian population (A and B) and European C/T−13910 variant in the Utah population (C and D) plotted against the distance in kb from the analyzed consecutive SNPs on both sides via Sweep software program.
(E–G) The REHH for G−13915 and T−13910 (small squares pointed by the arrows) are plotted against allele frequency in comparison with the coalescence simulation data of 1000 replica under constant population size (E), recent expansion (F), and bottleneck model (G), with recombination 1 cM = 1 Mb by ms program.
To estimate the selection coefficient, s, needed to increase the frequency of the C−3712-G−13915 allele in different populations, we applied the general selection formula.29 We assumed a dominant model for lactase persistence that is _w_11 = _w_12 = 1 and _w_22 = 1 – s and initial frequency _p_0 (0.001). We estimate the coefficient of selection associated with carrying at least one copy of the lactase-persistence allele G−13915 to be 0.0507 (0.0337–0.1007) for the Saudi population (Table 5). Estimates for C−3712-G−13915 and T−13910 alleles in Moroccans, Saharawis, and Arabs as well as Finns, Fulani, and individuals from Utah based on the age of the alleles in these populations revealed very strong selection pressure (s > 0.03) for the C−3712-G−13915 and T−13910 alleles (Table 5).
Table 5.
The Parameters Used for Estimation of the s Given the Age by Means of the General Selection Formula
| Populationa | Allele | Ages of the Allele (g ± SD)b | Current Allele Frequency (p)c | Selection Coefficient (s) |
|---|---|---|---|---|
| 1. Saudi Arabians | G−13915 | 163 (±81) | 0.57 | 0.0507 (0.0337–0.1007) |
| 2. Moroccans | G−13915 | 47 (±24) | 0.08 | 0.0968 (0.0650–0.1978) |
| T−13910 | 82 (±41) | 0.21 | 0.0713 (0.0479–0.1426) | |
| 3. Saharawi | G−13915 | 135 (±95) | 0.18 | 0.0415 (0.0244–0.1402) |
| T−13910 | 149 (±74) | 0.26 | 0.0417 (0.0277–0.0828) | |
| 4. Arabs | G−13915 | 35 (±17) | 0.10 | 0.1377 (0.0909–0.2677) |
| T−13910 | 39 (±28) | 0.10 | 0.1236 (0.0719–0.4381) | |
| 5. Utahans | T−13910 | 223 (±25) | 0.74 | 0.0484 (0.0437–0.0545) |
| 6. Fulani | T−13910 | 245 (±28) | 0.48 | 0.0318 (0.0286–0.0358) |
| 7. Finns, East | T−13910 | 217 (±34) | 0.54 | 0.0380 (0.0328–0.0450) |
| 8. Finns, West | T−13910 | 208 (±23) | 0.62 | 0.0434 (0.0391–0.0488) |
| 9. Basques | T−13910 | 208 (±23) | 0.66 | 0.0457 (0.0412–0.0514) |
Discussion
Haplotype-matching strategy has been suggested as a powerful approach in the initial identification of phenotype causing DNA variants because it reduces the number of variants subjected to follow-up analysis by at least a factor of 20.35 In a classical disease gene hunt, a well-established phenotype is an essential requirement to prove the genotype-phenotype association with the candidate variants. In the current study, the adopted haplotype-matching strategy provides unequivocal evidence that this strategy can be helpful even when no phenotype data exists. After screening the 71 kb critical region 5′ of the LCT gene of random population samples with only prevalence data for the LP trait, two sequence variants among the 71 identified variants remained as candidates for LP mutations among Saudi populations.
Based on the information of the prevalence of LP among Saudis,14 the role of natural selection that shaped the LCT region,6,7,36,37 and the availability of LNP phenotyped samples from another population, the matching haplotype strategy successfully identified the potential allele most probably harboring the LP mutation(s) among the Saudis and helped select the suitable DNA:s for sequencing.14 We were able to reduce the number of variants to be followed up by genotyping from 71 to 8. When tested in different populations, only two of them (T/C−3712, T/G−13915) remained as candidates for LP mutations in Saudi populations.
The critical functional role of the genomic DNA region encompassing the European C/T−13910 LP/LNP variant represents the obvious target for the search of additional LP variants in populations that demonstrate high LP but lack the C/T−13910 variant.8–10,12,13 By sequencing of this restricted region, two groups have independently shown recently the presence of new LP variants in the immediate vicinity of C/T−13910 in various population samples.12,13 In the first report, the sequence analysis of 700 bp around C/T−13910 identified three LP-associated variants, G−13915, C−13913, and G−13907, in 94 Sudanese samples phenotyped for LP.12 Although the G−13915 allele showed significant association with the LP status, it was not frequent enough to explain the entire LP observed in the Sudanese population. Our data agree with this finding in the analysis of the Northern Sudanese Tribe, Mahas, in which the G−13915 allele seem to explain almost 50% of the variation of the reported LP.30 In the second report, the sequence analysis of the 3314 bp region (encompassing the C/T−13910 in 69 LP and 40 LNP individuals from Sudan, Kenya, and Tanzania) revealed the presence of G/C−14010 correlating with LP, but their genotyping also revealed the presence of C/G−13907 and T/G−13915.13 These variants, however, failed to show significant association with LP because of the small number of the samples with G−13907, G−13915 alleles in their data set.13
Analysis of these SNPs in various Middle Eastern and African groups revealed that in most cases, the frequencies are significantly lower than the published prevalence values for LP. However, initial functional evidence and some evidence for selection supported the role of these alleles in relation to the LP.12,13 It should be noted that very restricted DNA regions, only 700 bp in the first report12 and 5 kb (introns 13 and 9 of the MCM6 gene)13 in the second report, were sequenced, and these SNPs collectively would explain a very small fraction of the phenotypic variations (20%) underlying LP in these populations. This reflects the high degree of heterogeneity of LP in Africa and implies that additional LP variants remain to be found. Unfortunately, it was not possible in this study to apply the haplotype-matching strategy for the other identified SNPs because of the low number of the samples harboring these SNPs.
The other, newly identified cSNP C/T−3712 of the compound LP allele is a synonymous SNP affecting the proline 810 codon of the MCM6 gene, the neighboring gene for LCT. The MCM6 protein is involved in the initiation of DNA replication. The protein is required to ensure that DNA replication occurs only once during the cell cycle.38 Although interesting, there is no evidence that the MCM6 gene per se would be important for the LP/LNP trait. Unlike the LCT, the expression of MCM6 is not restricted in its tissue distribution and does not show person-to-person variation on the level of expression in adult intestine.39 Further, there is no evidence for _cis_-acting regulators shared by these two genes.39 However, we did not analyze the functional consequences of this synonymous SNP here and this would certainly be warranted.
Our data from in vitro functional studies indicate that the binding of Oct-1 and HNF1α, the central regulators of the LCT promoter activity, is affected by the G−13915-C−3712 and T−13915-T−3712 variants. Our EMSA experiments showed that despite the G−13915 allele location within the motif Oct-1, which we have been shown previously to bind more strongly to the LP-associated T−13910 allele than the LNP-associated C−13910,10 the LP allele with G−13915 bound more weakly to Oct1 than even the LNP-associated C−13910 allele. Although this result was also observed in the recent report,12 our data found a strong specific Oct-1 binding to the G−13907 probe similar to the T−13910 Oct-1 binding, and also found that the C−13913 allele shows the same binding affinity as the LNP-associated C−13910 allele. These results would differ from the preliminary data by Ingram et al. with no binding to the C−13913 and only marginal to the G−13907.12 However, because no data was shown to support these results, it is possible that the evidence was not unequivocal.
Our result also showed that both the LP-associated C−3712 and LNP-associated T−3712 alleles bind strongly and specifically to the HNF1α but without a differential binding between the C−3712 and T−3712 alleles. Taken separately, the G−13915 allele shows no binding to the protein but significantly drives the expression of the reporter construct compared to the wild allele. Meanwhile, the C−3172 allele shows strong protein binding activity similar to the wild allele but with minimal drive of the reporter gene expression. G−13915 and C−3712 reside on the same allelic background only 10 kb apart, and when the compound allele was tested in vitro, we found a striking differential reporter gene expression stimulation that significantly increased by HNF1α expression but not by Oct-1 expression alone for the G−13915-C−3712 construct compared to the T−13915-T−3712 construct. When both transcription factors were overexpressed, Oct-1 significantly increased the reporter gene expression. Similar results were obtained with the T−13910 and the C−13910 constructs.10 This data would suggest that either the G−13915 is the critical variant underlying the LP and the C−3712 reflects the high regional LD (a situation resembling the combination of T−13910 and G−22018),5 or more likely, both SNPs are required in vivo for the enhancer effect through the binding of the hepatic nuclear factor 1 α (HNF1α). Given the complexity of the in vivo interactions, further analysis of these issues is needed to differentiate whether one or both of these variants represent the real culprit for LP.
The network analysis would indicate that the European T−13910 and the East African G−13907 LP alleles share a common ancestral allelic background (here labeled as H84) whereas the Arab G−13915 allele has a completely different background allele (labeled here as H107) (Figure 5A). This result would contradict the proposal by Tishkoff et al., who suggested different background haplotypes for G−13907 and T−13910 alleles.13 However, the recent report of Ingram et al. would support our interpretation; they also showed that the G−13907 allele most probably occurred on the so-called A haplotype background, the same haplotype that harbors the T−13910 allele (which we called here H98) (Figures 5A and 5B).12 Actually, the network analysis based on the variants over 98 kb presented by the Tishkoff et al. Figure 4 shows that the T−13910 and G−13907 alleles are very near to each other and probably share the same ancestral background allele.13 This result would justify the hypothesis that the European T−13910 and East African G−13907 LP alleles might have arisen because of a common domestication event of the cattle whereas the C−3712-G−13915 allele in Arabia most likely arose due to the separate domestication event of camels. This slightly far-reaching proposal is analogous to the previous interpretations: the presence of the LP T−13910 allele among three North African Berber populations (from Morocco and Algeria) has been taken as a genetic evidence for shared origin of the dairy culture between North African populations and Eurasians.40 Additional analyses of the East African samples could shed light on the origin of the G−13907 allele and its relationships to the domestication of milk-producing species within Africa. In our study, we did not detect the LP-associated allele, C−14010, found recently among Tanzanians and Kenyans, but reviewing the data from Tishkoff et al. would reveal that the C−14010 allele most likely originated on different background allele, most likely H1 in our data set (Figure 5A).13
Our age estimate of the G−13915 allele of ∼4095 (±2045) years in the Arabian Peninsula would suggest that the introduction of this LP variant might be associated with the domestication of the Arabian camel more than 6000 years ago.17 An analogous concept for the major European mutation was also supported by maximum likelihood analysis for the T−13910 allele, which likely arose after the domestication of cattle 5,000–10,000 years ago.6,7,18,41 Interestingly, similar age estimates were observed also for the LP mutation C−14010, detected in East African populations.13
Similar to the European LP variant, multiple tests for the selection applied to our data show that the LP haplotype structure among the Saudis significantly deviates from neutrality and would indicate strong selection. The estimates of the selective advantage of Saudi G−13915 allele would be in good agreement with the reported estimates for the selective advantage of LP6,13,42,43 and also very comparable for the protection provided by resistance to malaria in malaria-endemic regions. In these areas, the selective advantage in a region endemic for malaria has been estimated at 0.02–0.05 for G6PD deficiency and 0.05−0.18 for the sickle-cell trait.44
In summary, we show evidence of a new Arab mutation underlying LP among Saudis. We have also demonstrated that the European T−13910 LP and the Arab C−3712-G−13915 LP variants have emerged from different allelic backgrounds and driven to very high frequencies in different populations that presumably had different histories of animal domestication and dairy culture. We also provide functional data for the combined effect of two variants characterizing the Arab LP allele of the LCT gene.
Supplemental Data
One supplemental figure can be found with this article online at http://www.ajhg.org/cgi/content/full/82/1/57/DC1/.
Supplemental Data
Document S1. One Figure
Web Resources
The URLs for data presented herein are as follows:
- Arlequin program, http://lgb.unige.ch/arlequin/
- GenBank, http://www.ncbi.nlm.nih.gov/SNP/
- ms simulation program, http://home.uchicago.edu/∼rhudson1
- NETWORK 4.1.1.2 program, http://www.fluxus-engineering.com
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim (for Lactase, adult-type hypolactasia, Lactase persistence)
- PHASE2.1 program, http://stephenslab.uchicago.edu/software.html
- Sweep software program, http://www.broad.mit.edu/mpg/sweep/
- TRANSFAC data base version 10, http://www.biobase-international.com
- Ytime program, http://www.ucl.ac.uk/tcga/software/index.html
Accession Numbers
The GenBank accession number for the SNPs reported in this paper have been deposited in db SNP under ss# 79088022–79088069.
Acknowledgments
We are grateful to the participants for giving their samples to this study. We particularly thank Irma Järvelä and Antti Sajantila for their help in collecting the population samples and Sarah Tishkoff for fruitful discussions. Laura Palotie is appreciated for the revision of language. The Center of Excellence in Complex Disease Genetics of the Academy of Finland, the Biocentrum Helsinki Foundation, the Research and Science Foundation of Farmos, Helsinki, and the Finnish Culture Foundation are greatly acknowledged.
References
- 1.Loftus R.T., MacHugh D.E., Bradley D.G., Sharp P.M., Cunningham P. Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. USA. 1994;91:2757–2761. doi: 10.1073/pnas.91.7.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loftus R.T., Ertugrul O., Harba A.H., El-Barody M.A., MacHugh D.E., Park S.D., Bradley D.G. A microsatellite survey of cattle from a centre of origin: the Near East. Mol. Ecol. 1999;8:2015–2022. doi: 10.1046/j.1365-294x.1999.00805.x. [DOI] [PubMed] [Google Scholar]
- 3.Zeder M.A., Hesse B. The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago. Science. 2000;287:2254–2257. doi: 10.1126/science.287.5461.2254. [DOI] [PubMed] [Google Scholar]
- 4.Beja-Pereira A., Caramelli D., Lalueza-Fox C., Vernesi C., Ferrand N., Casoli A., Goyache F., Royo L.J., Conti S., Lari M. The origin of European cattle: evidence from modern and ancient DNA. Proc. Natl. Acad. Sci. USA. 2006;103:8113–8118. doi: 10.1073/pnas.0509210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enattah N.S., Sahi T., Savilahti E., Terwilliger J.D., Peltonen L., Jarvela I. Identification of a variant associated with adult-type hypolactasia. Nat. Genet. 2002;30:233–237. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 6.Bersaglieri T., Sabeti P.C., Patterson N., Vanderploeg T., Schaffner S.F., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coelho M., Luiselli D., Bertorelle G., Lopes A.I., Seixas S., Destro-Bisol G., Rocha J. Microsatellite variation and evolution of human lactase persistence. Hum. Genet. 2005;117:329–339. doi: 10.1007/s00439-005-1322-z. [DOI] [PubMed] [Google Scholar]
- 8.Troelsen J.T., Olsen J., Moller J., Sjostrom H. An upstream polymorphism associated with lactase persistence has increased enhancer activity. Gastroenterology. 2003;125:1686–1694. doi: 10.1053/j.gastro.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 9.Olds L.C., Sibley E. Lactase persistence DNA variant enhances lactase promoter activity in vitro: functional role as a cis regulatory element. Hum. Mol. Genet. 2003;12:2333–2340. doi: 10.1093/hmg/ddg244. [DOI] [PubMed] [Google Scholar]
- 10.Lewinsky R.H., Jensen T.G., Moller J., Stensballe A., Olsen J., Troelsen J.T. T-13910 DNA variant associated with lactase persistence interacts with Oct-1 and stimulates lactase promoter activity in vitro. Hum. Mol. Genet. 2005;14:3945–3953. doi: 10.1093/hmg/ddi418. [DOI] [PubMed] [Google Scholar]
- 11.Mulcare C.A., Weale M.E., Jones A.L., Connell B., Zeitlyn D., Tarekegn A., Swallow D.M., Bradman N., Thomas M.G. The T allele of a single-nucleotide polymorphism 13.9 kb upstream of the lactase gene (LCT) (C-13.9kbT) does not predict or cause the lactase-persistence phenotype in Africans. Am. J. Hum. Genet. 2004;74:1102–1110. doi: 10.1086/421050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingram C.J., Elamin M.F., Mulcare C.A., Weale M.E., Tarekegn A., Raga T.O., Bekele E., Elamin F.M., Thomas M.G., Bradman M., Swallow D.M. A novel polymorphism associated with lactose tolerance in Africa: multiple causes for lactase persistence? Hum. Genet. 2007;120:779–788. doi: 10.1007/s00439-006-0291-1. [DOI] [PubMed] [Google Scholar]
- 13.Tishkoff S.A., Reed F.A., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cook G.C., al-Torki M.T. High intestinal lactase concentrations in adult Arabs in Saudi Arabia. BMJ. 1975;3:135–136. doi: 10.1136/bmj.3.5976.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussein L., Flatz S.D., Kuhnau W., Flatz G. Distribution of human adult lactose phenotypes in Egypt. Hum. Hered. 1982;32:94–99. doi: 10.1159/000153266. [DOI] [PubMed] [Google Scholar]
- 16.Hussein L., Ezzilarab A. The frequency distribution of lactose malabsorption among adult populations from the eastern and western Egyptian deserts. Biochem. Genet. 1994;32:331–342. doi: 10.1007/BF02426895. [DOI] [PubMed] [Google Scholar]
- 17.Peters J. Tierarztl. Prax. Ausg. G Grosstiere Nutztiere. 1997;25:559–565. [PubMed] [Google Scholar]
- 18.Enattah N.S., Trudeau A., Pimenoff V., Maiuri L., Auricchio S., Greco L., Rossi M., Lentze M., Seo J.K., Rahgozer S. Evidence for still ongoing convergence evolution of the lactase persistence T-13910 alleles in humans. Am. J. Hum. Genet. 2007;81:615–625. doi: 10.1086/520705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vandenplas S., Wiid I., Grobler-Rabie A., Brebner K., Ricketts M., Wallis G., Bester A., Boyd C., Mathew C. Blot hybridisation analysis of genomic DNA. J. Med. Genet. 1984;21:164–172. doi: 10.1136/jmg.21.3.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Syvanen A.C., Sajantila A., Lukka M. Identification of individuals by analysis of biallelic DNA markers, using PCR and solid-phase minisequencing. Am. J. Hum. Genet. 1993;52:46–59. [PMC free article] [PubMed] [Google Scholar]
- 21.Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. John Wiley & Sons; New York: 2002. Current Protocols in Molecular Biology. [Google Scholar]
- 22.Behar D.M., Thomas M.G., Skorecki K., Hammer M.F., Bulygina E., Rosengarten D., Jones A.L., Held K., Moses V., Goldstein D. Multiple origins of Ashkenazi Levites: Y chromosome evidence for both Near Eastern and European ancestries. Am. J. Hum. Genet. 2003;73:768–779. doi: 10.1086/378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandelt H.J., Forster P., Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 24.Stephens M., Smith N.J., Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephens M., Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hudson R.R., Bailey K., Skarecky D., Kwiatowski J., Ayala F.J. Evidence for positive selection in the superoxide dismutase (Sod) region of Drosophila melanogaster. Genetics. 1994;136:1329–1340. doi: 10.1093/genetics/136.4.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudson R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- 28.Sabeti P.C., Reich D.E., Higgins J.M., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald G.J. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 29.Hartle D.L., Clark A.G. Third Edition. Sinauer; Sunderland, UK: 1997. Principles of Population Genetics. [Google Scholar]
- 30.Bloom G., Sherman P.W. Dairying barriers affect the distribution of lactose malabsorption. Evol. Hum. Behav. 2005;26:301.e1–301.e33. [Google Scholar]
- 31.Hijazi S.S., Abulaban A., Ammarin Z., Flatz G. Distribution of adult lactase phenotypes in Bedouins and in urban and agricultural populations of Jordan. Trop. Geogr. Med. 1983;35:157–161. [PubMed] [Google Scholar]
- 32.Tag C.G., Schifflers M.C., Mohnen M., Gressner A.M., Weiskirchen R.A. Novel proximal −13914G>A base replacement in the vicinity of the common-13910T/C lactase gene variation results in an atypical LightCycler melting curve in testing with the MutaREAL lactase test. Clin. Chem. 2007;53:146–148. doi: 10.1373/clinchem.2006.077529. [DOI] [PubMed] [Google Scholar]
- 33.Snook C.R., Mahmoud J.N., Chang W.P. Lactose tolerance in adult Jordanian Arabs. Trop. Geogr. Med. 1976;28:333–335. [PubMed] [Google Scholar]
- 34.Sahi T. Hypolactasia and lactase persistence. Historical review and the terminology. Scand. J. Gastroenterol. Suppl. 1994;202:1–6. doi: 10.3109/00365529409091739. [DOI] [PubMed] [Google Scholar]
- 35.Spencer D.H., Bubb K.L., Olson M.V. Detecting disease-causing mutations in the human genome by haplotype matching. Am. J. Hum. Genet. 2006;79:958–964. doi: 10.1086/508757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hollox E.J., Poulter M., Zvarik M., Ferak V., Krause A., Jenkins T., Saha N., Kozlov A.I., Swallow D.M. Lactase haplotype diversity in the Old World. Am. J. Hum. Genet. 2001;68:160–172. doi: 10.1086/316924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahi T. Genetics and epidemiology of adult-type hypolactasia. Scand. J. Gastroenterol. Suppl. 1994;202:7–20. doi: 10.3109/00365529409091740. [DOI] [PubMed] [Google Scholar]
- 38.Tsuruga H., Yabuta N., Hosoya S., Tamura K., Endo Y., Nojima H. HsMCM6: a new member of the human MCM/P1 family encodes a protein homologous to fission yeast Mis5. Genes Cells. 1997;2:381–399. doi: 10.1046/j.1365-2443.1997.1290327.x. [DOI] [PubMed] [Google Scholar]
- 39.Harvey C.B., Wang Y., Darmoul D., Phillips A., Mantei N., Swallow D.M. Characterisation of a human homologue of a yeast cell division cycle gene, MCM6, located adjacent to the 5′ end of the lactase gene on chromosome 2q21. FEBS Lett. 1996;398:135–140. doi: 10.1016/s0014-5793(96)01189-1. [DOI] [PubMed] [Google Scholar]
- 40.Myles S., Bouzekri N., Haverfield E., Cherkaoui M., Dugoujon J.M., Ward R. Genetic evidence in support of a shared Eurasian-North African dairying origin. Hum. Genet. 2005;117:34–42. doi: 10.1007/s00439-005-1266-3. [DOI] [PubMed] [Google Scholar]
- 41.Holden C., Mace R. Phylogenetic analysis of the evolution of lactose digestion in adults. Hum. Biol. 1997;69:605–628. [PubMed] [Google Scholar]
- 42.Aoki K. A stochastic model of gene-culture coevolution suggested by the “culture historical hypothesis” for the evolution of adult lactose absorption in humans. Proc. Natl. Acad. Sci. USA. 1986;83:2929–2933. doi: 10.1073/pnas.83.9.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoegerman S.F., Schenck R.A. Evolution of lactase persistence. Lancet. 1989;1:493. doi: 10.1016/s0140-6736(89)91388-3. [DOI] [PubMed] [Google Scholar]
- 44.Tishkoff S.A., Varkonyi R., Cahinhinan N., Abbes S., Argyropoulos G., Destro-Bisol G., Drousiotou A., Dangerfield B., Lefranc G., Loiselet J. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science. 2001;293:455–462. doi: 10.1126/science.1061573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. One Figure