Activity-dependent Synaptogenesis: Regulation by a CaM-kinase kinase/CaM-kinase I/βPIX Signaling Complex (original) (raw)
. Author manuscript; available in PMC: 2008 Apr 1.
Summary
Neuronal activity augments maturation of mushroom-shaped spines to form excitatory synapses, thereby strengthening synaptic transmission. We have delineated a Ca2+-signaling pathway downstream of the NMDA receptor that stimulates calmodulin-dependent kinase kinase (CaMKK) and CaMKI to promote formation of spines and synapses in hippocampal neurons. CaMKK and CaMKI form a multi-protein signaling complex with the guanine nucleotide exchange factor (GEF) βPIX and GIT1 that is localized in spines. CaMKI-mediated phosphorylation of Ser516 in βPIX enhances its GEF activity, resulting in activation of Rac1, an established enhancer of spinogenesis. Suppression of CaMKK or CaMKI by pharmacological inhibitors, dominant-negative (dn) constructs and siRNAs, as well as expression of the βPIX Ser516Ala mutant, decreases spine formation and mEPSC frequency. Constitutively-active Pak1, a downstream effector of Rac1, rescues spine inhibition by dnCaMKI or βPIX S516A. This activity-dependent signaling pathway can promote synapse formation during neuronal development and in structural plasticity.
Introduction
Dendritic spines are essential for proper functioning of the nervous system as they are the primary postsynaptic recipients of excitatory neurotransmission in the CNS (Carlisle and Kennedy, 2005; Hayashi and Majewska, 2005; Tada and Sheng, 2006). The role of spines in cognition is suggested by the fact that several forms of mental retardation (e.g., Down's, Rett, Fragile X and fetal alcohol syndromes) exhibit a reduction in spine density as well as a predominance of very long, thin filopodia at the expense of mature mushroom-shaped spines (Kaufmann and Moser, 2000; van Galen and Ramakers, 2005). Spine density and morphology are regulated developmentally as well as in response to synaptic plasticity. During synaptogenesis, dendritic filopodia are thought to represent initial dendritic projections which, if they contact an axon, can mature into mushroom-shaped spines that constitute functional synapses (Ethell and Pasquale, 2005; Matsuzaki et al., 2004; Ziv and Smith, 1996).
Spine morphology is very dynamic and modulated by several signaling pathways that regulate its actin-rich cytoskeleton (Bonhoeffer and Yuste, 2002; Calabrese et al., 2006; Dillon and Goda, 2005; Fischer et al., 1998; Hering and Sheng, 2001). One of the best characterized pathways for modulating actin dynamics involves the Rho family of small GTPases (G proteins), Rho, Rac and Cdc42 (Nakayama et al., 2000; Raftopoulou and Hall, 2004; Tada and Sheng, 2006). Intriguingly, seven genes that regulate Rho GTPase signaling were detected in a screen for genes that cause nonspecific X-linked mental retardation (Ramakers, 2002). There are several downstream effectors of these GTPases, including the WASP/WAVE family, Rho-associated kinase (ROCK) and p21-activated kinase Pak1, that modulate actin polymerization (Ethell and Pasquale, 2005; Tada and Sheng, 2006). The activation state of these small G proteins is determined by their GTP-loading dictated by exchange factors called guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) (Rossman et al., 2005). GEFs promote GTP-loading whereas GAPs inactivate G proteins by promoting hydrolysis of the bound GTP.
Two recent studies have demonstrated a role in spinogenesis for the GEF βPak-interacting exchange factor (βPIX) (Zhang et al., 2003; Zhang et al., 2005). βPIX contains an N-terminal SH3 domain, a Dbl motif containing Rac/Cdc42 GEF activity, a pleckstrin homology domain, and a GIT1-binding region near its C-terminus (Rosenberger and Kutsche, 2006). Subcellular localization of βPIX within dendritic spines to the postsynaptic density is mediated largely by scaffold proteins such as GIT1 (Ko et al., 2003; Zhang et al., 2003) and Shank (Park et al., 2003). Since βPIX is a Rac GEF and one of the major effectors of Rac is Pak1, which binds to the SH3 domain of βPIX, βPIX coordinates Rac-dependent activation of Pak1 (Mott et al., 2005). Rac activation has been demonstrated in spines (Zhang et al., 2005), and overexpression of dominant-negative Rac decreases the number of spines and synapses in cultured hippocampal neurons and slices (Nakayama et al., 2000; Penzes et al., 2003; Zhang et al., 2003). There are numerous downstream effectors of Pak1 that regulate actin dynamics (Bokoch, 2003).
Although signaling downstream of βPIX has received significant attention, little is known about upstream pathways that modulate the βPIX signal complex. A likely signaling molecule is intracellular Ca2+ which is regulated by neuronal activity and is known to modulate spine morphology and actin dynamics (Konur and Ghosh, 2005). Effects of Ca2+ on actin and spine morphology/motility are complex and determined by its mode of entry, concentration and temporal duration (Oertner and Matus, 2005). Ca2+ entry through NMDA receptors (NMDARs) stabilizes spine morphology (Ackermann and Matus, 2003) and may represent a mechanism for activity-dependent increases in spine number and volume (Maletic-Savatic et al., 1999). There are several mechanisms that might mediate these effects including Ca2+-regulated, actin-binding proteins such as gelsolin (Star et al., 2002) or profilin (Ackermann and Matus, 2003). A role for Ca2+/CaM-dependent protein kinases (CaMKs) has also been implicated through the use of the general CaMK inhibitor KN-62 that blocks activity- and NMDAR-dependent remodeling of dendritic spines (Matsuzaki et al., 2004). Using pharmacological inhihibitors, dominant-negative and constitutively-active constructs and siRNAs, we have identified a novel signaling pathway upstream of βPIX by which NMDAR activation during neuronal development or plasticity can modulate spinogenesis. CaMKK/CaMKI interacts with βPIX/GIT1 and mediates phosphorylation of Ser516 in βPIX to enhance Rac activity and promote formation/stabilization of mushroom-shaped spines.
RESULTS
Interaction of CaMKs with the βPIX/GIT1 complex
To determine if members of the CaMK cascade (i.e., CaMKK, CaMKI, CaMKIV) are constituents of multiprotein signaling complexes, we initiated a proteomics approach to identify potential interacting proteins. Members of the CaMK cascade containing a Flag-epitope were transfected into HEK293 cells, purified by immunoprecipitation, washed, and associated proteins identified by tandem mass spectrometry (Natsume et al., 2002). Several members of the CaMK cascade exhibited interactions with numerous proteins (data not shown), the most intriguing of which were protein 14-3-3 and βPIX, a Rac1/Cdc42 GEF. Identification of 14-3-3 was confirmatory as we previously demonstrated that 14-3-3 interacts with and inhibits PKA-phosphorylated CaMKK (Davare et al., 2004). However, βPIX was of special interest in the context of recently described roles of CaMKK and CaMKI in neuronal development of axons (Wayman et al., 2004) and dendrites (Takemoto-Kimura et al., 2007; Wayman et al., 2006). The neuronal actin cytoskeleton is regulated in numerous and complex ways by small G proteins such as Rac and Cdc42 (Nakayama et al., 2000; Raftopoulou and Hall, 2004; Tada and Sheng, 2006).
Next, we co-expressed in HEK293 cells Flag-tagged CaMKK (α and β isoforms), CaMKI (α,β,δ,γ isoforms) or CaMKIV with a signaling complex (βPIX, GIT1, Pak1) previously demonstrated to regulate the actin cytoskeleton (Mott et al., 2005; Rosenberger and Kutsche, 2006). Only βCaMKK exhibited robust interaction with all three members of the complex: βPIX, GIT1 and Pak1 (Fig. 1A). To confirm whether these interactions occur in brain, CaMKI, CaMKII, CaMKIV and CaMKK were immunoprecipitated from brain lysates of p6 rats, and the presence of co-precipitated βPIX and GIT1 was determined. The antibodies for immunoprecipitation of CaMKI and CaMKK were specific for the α and β isoforms, respectively - other isoform specific antibodies were not available. Again, βCaMKK exhibited significant co-precipitation of the multiple isoforms of brain βPIX as well as GIT1 (Fig. 1B), consistent with the results from the HEK293 cells. Since our subsequent studies were conducted using hippocampus, we also examined the presence of this βCaMKK/βPIX/GIT1 complex in hippocampal lysates from p12 rats (Fig. 1C). In this co-immunoprecipitation complex we were able to also detect CaMKI using both the αCaMKI antibody as well as a phospho-specific antibody against the CaMKK phosphorylation/activation site, Thr177 (Haribabu et al., 1995). To further verify the existence of CaMKI in this complex, brain lysates from postnatal day 0 to adult were immunoprecipitated with antibody to αCaMKI - western analysis revealed the co-immunoprecipiation of βPIX and GIT1 (Fig. 1D). These results demonstrate the formation of a signalsome containing βCaMKK, αCaMKI, βPIX and GIT1 in brain.
Figure 1.
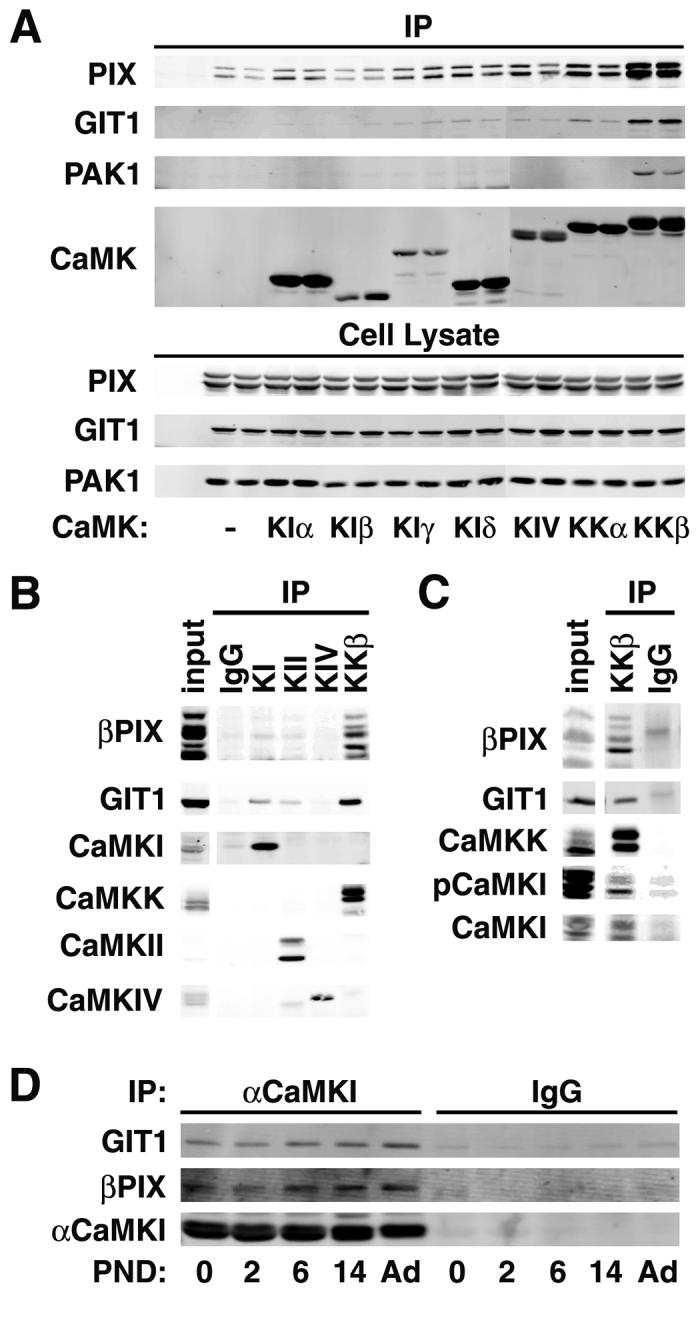
βCaMKK and αCaMKI interact with the βPIX/GIT1 complex. A. Co-immunoprecipitation from HEK293 cells. HEK293 cells were transfected with α (upper band) and βPIX (myc-tagged), GIT1 (EGFP fusion), Pak1 (Xpress-tagged) and the indicated FLAG-tagged CaMK. Duplicate cell lysates were immunoprecipated with FLAG antibody, washed, and western-blotted for the proteins indicated on the left. All of the transfected proteins were expressed to similar levels (see cell lysate) under all protocols. B. Co-immunoprecipitation from rat brain lysate. Brain lysates from P6 rat were immunoprecipitated with antibodies to αCaMKI, CaMKII, CaMKIV or βCaMKK, immunoprecipitates were washed and western-blotted for the proteins indicated on the left. C. Co-immunoprecipitation from rat hippocampus. Hippocampal lysates from P12 rat were immunoprecipitated with antibody to βCaMKK, washed and western-blotted for βPIX, GIT1, CaMKK, CaMKI, and phosphoCaMKI (pCaMKI). D. Co-precipitation of βPIX and GIT1 with αCaMKI. Brain lysate from the indicated postnatal days (PND) were immunoprecipitated with antibody to αCaMKI, washed and western blotted.
Since the proteomic and HEK293 cell experiments indicated direct binding of βPIX to βCaMKK, we mapped out their interaction domains. A kinase-dead (KD, K194E) mutant of βCaMKK bound βPIX similar to wild-type βCaMKK (Supplemental Fig. S1), and the catalytic domain (residues 137-519) did not interact (Fig. S2C), indicating that kinase activity is not required for this interaction. Binding of βPIX to βCaMKK was not affected by the absence or presence of Ca2+/CaM (Fig. S2B, last two lanes), suggesting that this complex may exist constitutively with non-activated CaMKK. Binding studies using fragments of βCaMKK or βPIX revealed that the N-terminus of βPIX (residues 1-97) interacted with the N-terminus of βCaMKK (residues 1-136) (Fig. S2). This region of βPIX contains the SH3 domain that binds multiple proteins including Pak1 (Mott et al., 2005), but a mutation of the SH3 domain (SHm, W43P, W44G), which obviated Pak1 binding, did not disrupt binding to βCaMKK (Fig. S1). The only known function of the N-terminus of CaMKK is to bind protein 14-3-3 (Davare et al., 2004). Thus, we explored whether binding of 14-3-3 to PKA-phosphorylated βCaMKK affected its interaction with βPIX, but binding of 14-3-3 had no effect (data not shown).
Phosphorylation of βPIX by CaMKK/CaMKI
Protein kinases are often co-localized with their substrates to confer subcellular specificity to signaling pathways (Pawson and Scott, 2005), so we explored whether βPIX might be a substrate for the βCaMKK/CaMKI pathway. Two sites in βPIX have been described: phosphorylation of Thr526 by Pak1 (Shin et al., 2002) and Ser516 and Thr526 by PKA (Chahdi et al., 2005). In vitro 32P-phosphorylation of wild-type βPIX and the T526A and S516A mutants was performed using αCaMKI activated by βCaMKK as well as by PKA and Pak1. All kinases gave equivalent total 32P-incorporation into wild-type βPIX (Fig. 2A). Phosphorylation by αCaMKI, but not PKA or Pak1, was obviated in the S516A mutant whereas Pak1 appeared to predominantly phosphorylate T526. To further examine phosphorylation of βPIX in situ, we generated an antibody specific for phosphorylation of Ser516. Wild-type and the S516A mutant of myc-βPIX were expressed in HEK293 cells which were subsequently stimulated with ionomycin, forskolin or the phorbol ester PMA to activate endogenous CaMKs, PKA and PKC, respectively. In the absence of stimulation, there was no immunoreactivity whereas ionomycin-treatment strongly enhanced S516 phosphorylation of the wild-type but not S516A mutant βPIX (Fig. S3, left panel), confirming that the antibody detects phospho-Ser516. Note that Ca2+-dependent phosphorylation of Ser516 was blocked by co-transfected dominant-negative members of the CaMK cascade (Fig. S3, right panel), strongly implicating endogenous CaMKK and CaMKI as the catalysts for βPIX Ser516 phosphorylation in HEK293 cells. Stimulation with forskolin or PMA did not increase Ser516 phosphorylation, consistent with our observation that PKA does not phosphorylate this site in vitro (Fig. 2A).
Figure 2.
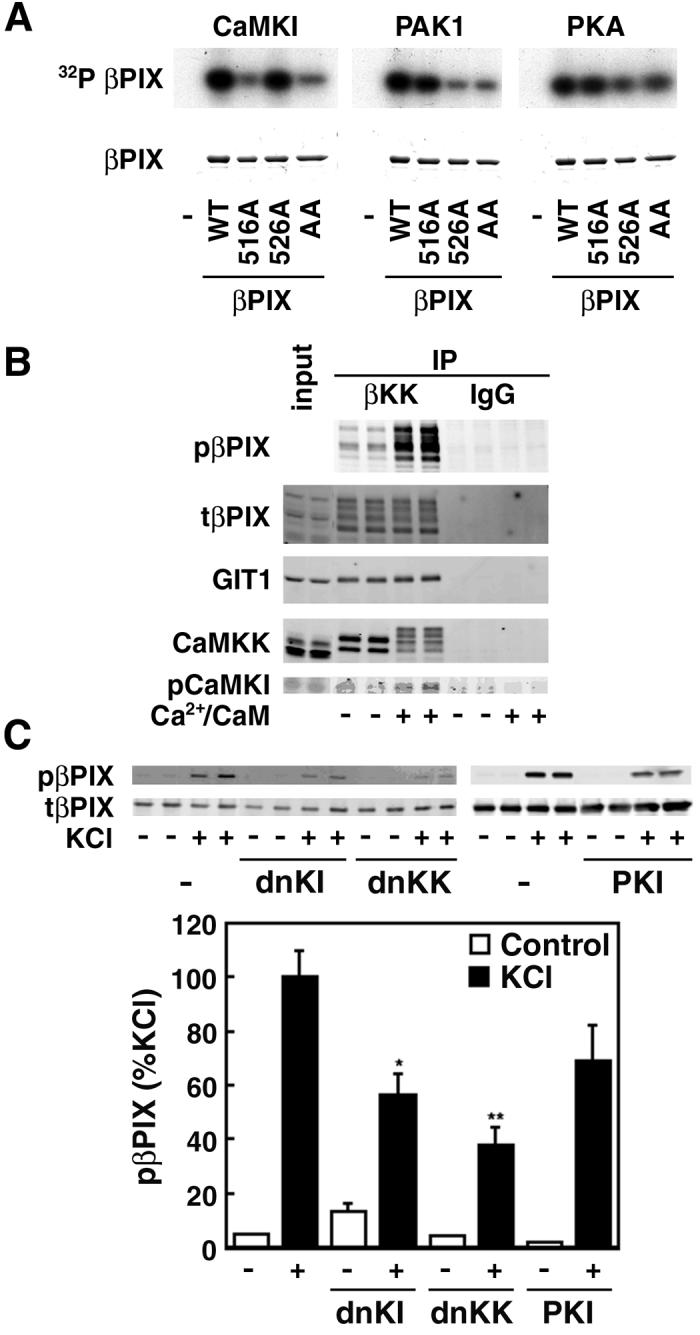
CaMKI phosphorylates Ser516 in βPIX. A. In vitro phosphorylation. Purified βPIX, wild-type (WT) or the indicated mutants (S516A, 516A; T526A, 526A; S516AT526A, AA), were subjected to in vitro phosphorylation using γ[32P]ATP and purified αCaMKI (activated by βCaMKK), PKA or Pak1. Reaction products were subjected to SDS/PAGE and autoradiography. B. Phosphorylation of βPIX Ser516 by CaMKI in the isolated multiprotein signaling complex. Brain homogenates from P6 rats were immunoprecipitated with anti-βCaMKK, washed, and incubated in the presence of Mg2+/ATP without or with Ca2+/CaM, subjected SDS/PAGE and western-blotted for phospho-βPIX, GIT1, CaMKK and phospho-CaMKI (pCaMKI). C. Phosphorylation in hippocampal neurons. Cultured hippocampal neurons (7DIV) were transfected with myc-βPIX and dnCaMKK, dnCaMKI or the PKA inhibitor protein PKI and treated with 0.5 μM tetrodotoxin in ACSF for 2 hrs to suppress neuronal activity. They were then incubated for 5 min with 56 mM KCl to activate Ca2+-dependent protein kinases and PKA. Cell lysates were immunoprecipitated with myc-antibody and western-blotted for phospho-S516 βPIX. *P<0.05, **P<0.001 compared to control.
To verify that phosphorylation of βPIX occurs within the βCaMKK/αCaMKI/βPIX/GIT1 signaling complex, brain lysates from p6 rats were immunoprecipitated with βCaMKK antibody to isolate this complex. The immunoprecipitate was then incubated with Mg2+/ATP with or without Ca2+/CaM and analyzed by western blot (Fig. 2B). There was robust Ca2+/CaM-dependent phosphorylation of Ser516 in βPIX within this complex. Note the mobility shift of CaMKK that occurs due to its autophosphorylation (Wayman et al., 1997) as well as an increase in pCaMKI in the complex due to its activation by CaMKK. When the complex was immunoprecipitated with an antibody to GIT1, a similar Ca2+/CaM-dependent in vitro phosphorylation of βPIX was observed (not shown).
Finally, we investigated Ca2+-dependent Ser516 βPIX phosphorylation in cultured hippocampal neurons. Neurons were transfected with myc-βPIX because we wanted to assess βPIX phosphorylation when endogenous kinases were suppressed by co-transfection with specific inhibitory constructs. Prior to elevation of intracellular Ca2+ by KCl depolarization, cells were treated for 2 hrs with 0.5 μM TTX to reduce basal phosphorylation due to the spontaneous neuronal activity in these cultures. KCl treatment gave a large increase in Ser516 phosphorylation (Fig. 2C), and this βPIX phosphorylation was significantly suppressed by co-transfection with either dnCaMKI or dnCaMKK. Elevated intracellular Ca2+ will also activate PKA through CaM-dependent adenylyl cyclase (Wong et al., 1999), and there may be some inhibition (P>0.05) by co-transfection with the PKA inhibitor PKI. We conclude that in hippocampal neurons CaMKK/CaMKI phosphorylated Ser516 in βPIX in response to robust Ca2+ elevation.
Regulation of βPIX GEF activity by CaMKI-mediated phosphorylation
Having demonstrated phosphorylation of Ser516 in βPIX by CaMKK/CaMKI in hippocampal neurons, the next question was whether this phosphorylation was regulatory. Of most interest would be if phosphorylation of βPIX enhanced activation of Rac and/or Cdc42 and their downstream targets leading to modulation of actin dynamics (Jaffe and Hall, 2005). HEK293 cells were transfected with wild-type or the S516A mutant of myc-βPIX without or with co-transfected caCaMKI. Cell lysates were mixed with GST fusions of either Rac1(T17N) or Cdc42(T17N). The T17N mutants bind GDP, but not GTP, thereby stabilizing the binding of Rac1 or Cdc42 to the GEF domain of βPIX (Feng et al., 2004). Binding of Rac1 and Cdc42 to βPIX were determined using GST pull-downs and then quantification of co-precipitated myc-βPIX. As shown in Fig. 3A, the presence of caCaMKI increased the interaction between βPIX and both Rac1 or Cdc42 by 3- to 4-fold. Most importantly, this enhanced interaction was obviated in the S516A mutant, indicating that phosphorylation of 516 in βPIX by CaMKI was likely responsible for this increased interaction.
Figure 3.
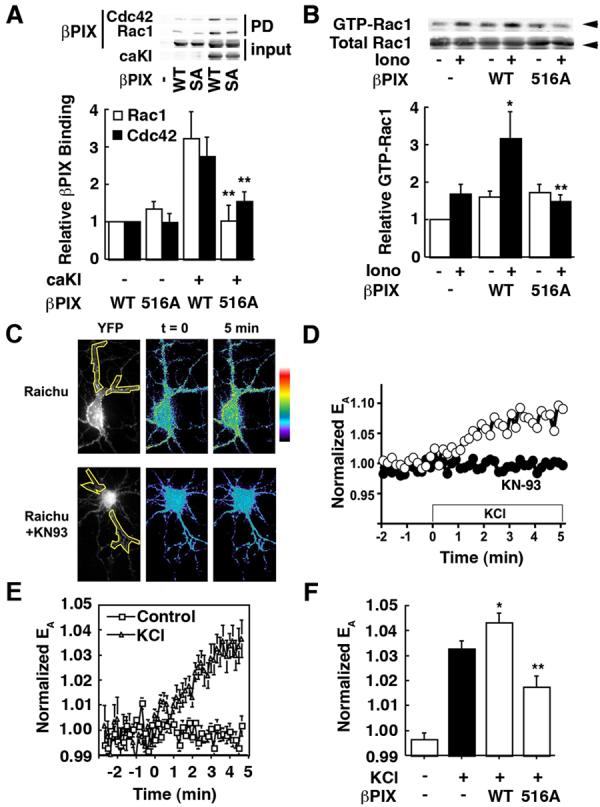
CaMKI-dependent phosphorylation of S516 stimulates βPIX GEF activity. A. Interaction of Rac/Cdc42 with βPIX. HEK293 cells were transfected with myc-βPIX (WT or S516A) and caCaMKI (caKI) as indicated. Cell lysates were mixed with GST-Rac1T17N or GST-Cdc42NT17N and the GST complex isolated by glutathionine-Sepharose. The myc-βPIX that co-purified with the GST-Rac1 or GST-Cdc42 was quantified by western blot. B. GEF activity of βPIX in HEK293 cells. HEK293 cells were transfected with βPIX (WT or S516A) and myc-Rac1 and treated for 3 min without or with 1 μM ionomycin. Cell lysates were mixed with GST fused to the CRIB domain of Pak1 (residues 67-150) which specifically binds to activated (i.e., GTP-loaded) Rac, and the GST complex was isolated by glutathione-Sepharose (Soderling et al., 2002). Bound Rac1 was quatified by western blotting. *P< 0.05 compared to control, **P< 0.05 compared to wild-type. C-E. Depolarization increases CaMK-dependent Rac GEF activity in hippocampal neurons. Hippocampal neurons, transfected with Raichu-Rac1, were live-imaged upon stimulation by 28 mM KCl without or with 1 hr pretreatment with the general CaMK inhibitor KN-93 (5 μM, panel C lower images, panel D solid circles). Normalized FRET efficiencies (EA, see Methods) or YFP signal (panel C, far left; region of proximal dendrites analyzed are outlined in yellow) are shown for representative cells (C, D) or multiple cells (E, n=15). See Fig. S4 for an enlargement of the horizonal dendrite in top of panel C. F. GEF activity of transfected βPIX in neurons. mRFP-βPIX WT (WT) or S516A mutant (516A) were co-transfected with Raichu-Rac1 (ratio 5:1) into hippocampal neurons. FRET efficiencies (EA) in proximal dendrites were averaged from 5 time points between 4-5 min. *P< 0.05 compared to Raichu alone, **P< 0.001 compared to wild-type (n=6-17).
Having demonstrated that CaMKI-mediated phosphorylation of Ser516 in βPIX enhanced its interaction with Rac and Cdc42, we next determined whether there was an effect on its GEF activity to stimulate GTP loading of Rac. The CRIB domain (residues 67-150) of Pak1, which specifically binds GTP-loaded Rac, fused to GST was used for a Pak pull-down assay. HEK293 cells were transfected with myc-Rac1 and wild-type or S516A βPIX. The cells were treated without or with ionomycin, and the GTP-loaded Rac that co-precipitated with GST-Pak1(67-150) was quantified. Transfected Rac1 exhibited some GTP loading that was elevated about 70% by ionomycin treatment (Fig. 3B), probably due to GEF activity of endogenous βPIX or Tiam (Fleming et al., 1999). Not surprisingly, transfection with wild-type βPIX enhanced basal GEF activity by about 70% that was further increased 2-fold in a Ca2+-dependent manner. These are the same conditions that enhanced phosphorylation of βPIX by CaMKK/CaMKI in HEK293 cells (Fig. S3). To ascertain whether the Ca2+-dependent increase in GEF activity of βPIX was due to phosphorylation of Ser516, we transfected the S516A mutant, and this construct did not exhibit Ca2+-dependent GEF activity (Fig. 3B).
Finally, Ca2+-dependent regulation of βPIX was assessed in dendrites of cultured hippocampal neurons by fluorescent resonance energy transfer (FRET) analysis of transfected Raichu-Rac. Raichu-Rac contains Rac and the CRIB domain of Pak1 flanked with YFP on the N-terminus and CFP on the C-terminus (Itoh et al., 2002). In the absence of GEF activity, GDP is bound to Rac so it does not bind the CRIB domain and there is no FRET between the YFP and CFP. Upon activation of a GEF to load GTP on Rac, Rac-GTP binds the CRIB domain, bringing YFP and CFP into close proximity to allow FRET. In Raichu-Rac tranfected hippocampal neurons, depolarization by KCl elicited an increase in dendritic Rac activation for at least 5 min (Figs. 3C-E, S4). The elevated Ca2+ presumably stimulated endogenous GEFs including βPIX, Tiam (Tolias et al., 2005) and perhaps others such as kalirin (Ma et al., 2003). This activation of Rac was mediated largely by CaM-dependent protein kinases since the KCl effect was completely obviated by the presence of the general CaM-kinase inhibitor KN-93 (Fig. 3C, D). We were unable to test effects of the CaMKK inhibitor STO-609 because of its photo-instability at the relevant wavelengths. Co-transfection of wild-type βPIX gave a modest but significant further increase in FRET whereas co-transfection with the S516A mutant acted in a dominant-interfering manner to partially suppress Ca2+-dependent Rac activation (Fig. 3F).
To summarize, this series of experiments in Fig. 3 demonstrate CaMKI-mediated phosphorylation of Ser516 in βPIX increased its interaction with Rac1/Cdc42 and enhanced its GEF activity towards Rac, a known modulator of the actin cytoskeleton.
Role of CaMKK/CaMKI-mediated phosphorylation of βPIX on synaptogenesis
Spine formation has been reported to be modulated by the βPIX/GIT1 complex (Zhang et al., 2003; Zhang et al., 2005), so we examined whether CaMKI-mediated phosphorylation of βPIX alters spinogenesis and synaptogenesis in DIV 7-12 cultured hippocampal neurons. This is the time period of developmental formation of spines (Fig. S5) and synapses - we are unable to reliably record AMPAR mEPSCs at DIV 7 (not shown) whereas DIV 12 neurons show abundant mEPSCs. Because our signaling pathway likely modulates the actin cytoskeleton in spines, we used mRFP-βactin, which has been used extensively (Brunig et al., 2004; Calabrese and Halpain, 2005; Fischer et al., 2000; Star et al., 2002), to quantify changes of spine density and morphology. β and γ actin are preferentially localized in dendritic spines (Kaech et al., 1997), the postsynaptic component of synapses. Expressed βactin is coincident with endogenous actin in spines and apparently present in trace amounts since rhodamine-phalloidin staining is not significantly increased in transfected vs non-transfected neurons (Kaech et al., 1997). Cultured hippocampal neurons were transfected with mRFP-βactin plus other plasmids on 7 DIV and fixed on 12-14 DIV. These neurons exhibit normal dendritic arborization with numerous dendritic protrusions (Fig. S6), and the density of mushroom-shaped spines and filopodia were quantified on 50 μm segments of three different dendrites for each cell. We identified spines by their mushroom-shaped morphology with intense puncta of mRFP-βactin at their tip (head) whereas filopodia were long, thin protrusions with low and constant mRFP-βactin expression throughout (Fig. 4A, left). When DIV 12 cultures tranfected with mRFP-βactin were also stained for the presynaptic marker synapsin 1 (Figs. 4A, S10), 97% of spines (n=616) showed apposing synapsin 1 staining whereas only 17% of filopodia (n=149) did. Thus, our operational definition identifies mushroom-shaped spines as morphological synapses, and we use the terms interchangeably. We also immunostained for pCaMKI which was distributed throughout the cell (excluding the nucleus, not shown) with apparent enrichment in spines (Fig. 4B, middle). Thus, spines in cultured neurons contain pCaMKI together with GIT1 and βPIX (Ko et al., 2003; Park et al., 2003; Zhang et al., 2003), consistent with a signaling complex. Furthermore, western blot analysis of purified rat postsynaptic densities (PSDs) revealed that α and βCaMKK and αCaMKI are present in the PSD of spines (Fig. S7).
Figure 4.
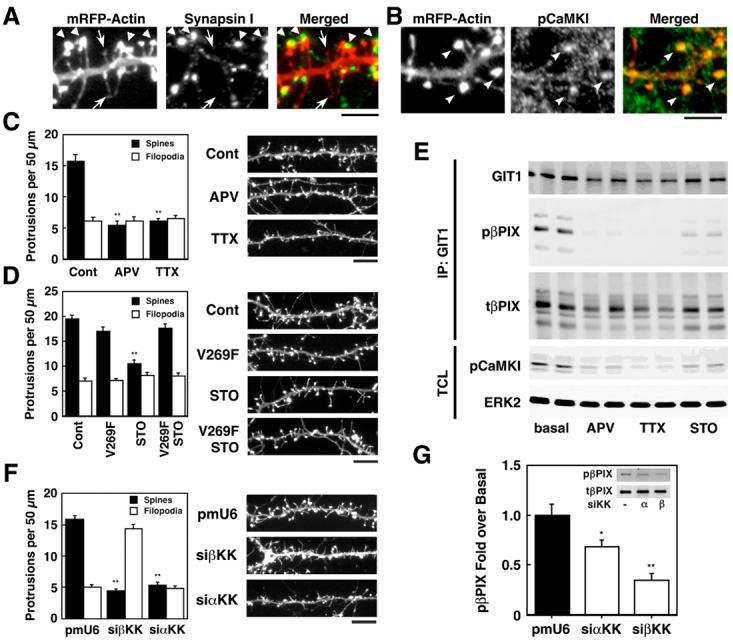
CaMKK modulates activity-dependent dendritic spine density and phosphorylation of βPIX. A, B. Cultured hippocampal neurons (7 DIV) were transfected with mRFP-βactin to visualize dendritic protrusions, fixed on DIV 14, and immunostained for the presynaptic marker synapsin 1 (A) or pCaMKI (B). A. The left panel shows mRFP-βactin; middle panel, synapsin 1; right panel, overlay. Spines (large arrows) were identified as mushroom-shaped projections with intense expression of mRFP-βactin at their tips (heads) whereas filopodia (small arrows) were thin with low expression of mRFP-βactin throughout. (Scale bar, 5 μm). B. The left panel shows mRFP-βactin; middle panel, phosphoCaMKI (pCaMKI); right panel, overlay. PhosphoCaMKI signals were identified in spines (arrow heads) (Scale bar, 5 μm). C,D,F. DIV 7 neurons were transfected with mRFP-βactin without or with the STO-insensitive mutant of βCaMKK (V269F) (D) or siRNAs specific for α or βCaMKK (F). Where indicated, neurons were treated for 4 days with 100 μM APV (block NMDAR), 1 μM TTX (inhibit neuronal activity) or 20 μM STO-609 (inhibit CaMKK). On 12 DIV (C, D) or 14 DIV (F) the neurons were fixed, dendrites were imaged and at least three different 50 μm sections per neuron (15-20 neurons per condition) were analyzed for protrusions. Representative dendrites are shown on the left, and summaries of the effects of transfections or pharmacological treatments on protrusion densities are shown on the right. **P<0.001, compared to control. Scale bar, 10 μm. E. On 12DIV, the neurons were lysed, immunoprecipitated with GIT1 antibody, western-blotted with GIT1, phospho-βPIX (pβPIX), βPIX (tβPIX), phosphoCaMKI (pCaMKI), and ERK2 (as loading control). G. DIV 7 neurons were transfected with myc-βPIX with or without siRNAs specific for α or β CaMKK. On 11 DIV, neurons were lysed, immunoprecipitated with myc antibody, western-blotted with phospho-βPIX (pβPIX) and myc (tβPIX). *P<0.05, **P<0.001 compared to control.
We first examined the role of spontaneous neuronal activity in spine formation. Figs. 4C and D demonstrate that about 2/3 of the dendritic projections fall into the mushroom-shaped spine/synapse category with 1/3 classified as filopodia. Treatment with the NMDAR antagonist APV or with the sodium channel blocker TTX dramatically decreased the density of spines with no effect on filopodia (Fig. 4C), demonstrating a strong positive role for afferent neuronal activity that is consistent with most (Collin et al., 1997; Kossel et al., 1997; Segal et al., 2003) but not all (McKinney et al., 1999) reports. Next, the potential role of CaMKK in spine formation was probed using STO-609, a pharmacological inhibitor of CaMKK whose specificity has been extensively characterized in vitro (Tokumitsu et al., 2002) and in cultured neurons (Schmitt et al., 2005; Wayman et al., 2004). Incubation of neurons with STO-609 gave a 50% suppression of spine formation, and this effect was reversed by transfection with a single-point mutant of βCaMKK (V269F) that is relatively insensitive to STO-609 inhibition (Tokumitsu et al., 2003) (Fig. 4D). This rescue experiment is very strong evidence that STO-609 effects were mediated by CaMKK.
Do the phosphorylation states of CaMKI and βPIX in the above experiments correlate with spine density? Lysates from cultured neurons treated with APV, TTX or STO-609 were incubated with antibody to GIT1 to immunoprecipitate the multiprotein complex that was then analyzed by western blots. In contrast to the experiment of Fig. 2C, endogenous neuronal activity was not suppressed. In the absence of treatments (i.e., basal), there was significant pCaMKI and pβPIX (Fig. 4E), consistent with a potential role of βPIX phosphorylation in spinogenesis. Incubation with APV, TTX and STO-609, all of which strongly suppressed spinogenesis, also decreased phosphorylation of both CaMKI and βPIX.
The role of CaMKK isoforms was further investigated using siRNAs to suppress expression of the endogenous kinases. Transfection of HEK293 cells with siRNAs for α or βCaMKK completely suppressed expression of their respective transfected CaMKK isoform (Fig. S8A). When cultured neurons were transfected with siRNA specific for either α or βCaMKK, both suppressed formation of spines (Fig. 4F) as well as phosphorylation of co-transfected myc-βPIX (Fig. 4G). Thus, reducing endogenous CaMKK isoforms with siRNAs was associated with suppression of both βPIX phosphorylation and formation of spines.
We next examined the roles of CaMKI, βPIX and Pak1 in regulating synaptogenesis. Figs. 5A-C demonstrate that dnCaMKI and the S516A mutant of βPIX blocked spine and synapse formation as did a mutant of βPIX (DHm) lacking a functional GEF domain. Importantly, the abilities of dnCaMKI and the S516A βPIX mutant to reduce spine number were rescued by co-expression of caPak1 (Fig. 5C), consistent with Pak1 being downstream of CaMKI and βPIX/Rac. We again used the siRNA approach to verify the results from overexpression of dnCaMKI and to investigate roles of specific CaMKI isoforms. The specificity of each siRNA for its corresponding CaMKI isoform has been established in HEK293 cells and used in cultured neurons (Wayman et al., 2006). The siRNA for αCaMKI phenocopied the effect of dnCaMKI, and the suppression of spine formation was rescued by expression of a human αCaMKI that was not susceptible to the siRNA (Fig. 5D). The efficacy of the siRNA for endogenous αCaMKI was verified by immunocytochemistry using an antibody that is specific for the α isoform. Neurons transfected with siRNA for αCaMKI showed little or no immunoreactivity compared to non-transfected neurons (Fig. S8B). On the other hand, siRNA for β (Fig. 5D) or δ (data not shown) CaMKI isoforms had little or no effect, indicating specificity among the various CaMKI isoforms. We were unable to test siRNA for γCaMKI because expression of this construct for 4 days was deleterious to the health of the cultured neurons. Moreover, siRNA for βPIX also blocked spine formation to the same extent as the siRNA for αCaMKI, and this was also rescued by expression of the siRNA-resistant human isoform of wild-type βPIX but not the S516A mutant (Fig. 5E). This result strongly supports the critical role of Ser516 in βPIX for its function in spinogenesis.
Figure 5.

Role of CaMKI and βPIX S516 phosphorylation on spinogenesis. A-C. Cultured hippocampal neurons (DIV 7) were transfected with mRFP-βactin plus either dnCaMKI or βPIX (WT, S516A or a mutant (DHm) devoid of GEF activity) ± caP ak1 as indicated. On DIV 14 neurons were fixed, stained for synapsin 1 and images were analyzed for density of spines and filopodia (A). Synapses (B) were identified as spines with adjacent staining of synapsin 1 (see Fig. S10). D, E. Neurons (DIV 7) were transfected with mRFP-βactin plus siRNAs for rat (Rn) α or βCaMKI (D) or βPIX (E) ± plasmids expressing human αCaMKI or human (Hs) βPIX (WT or S516A) as indicated. Spines/filopodia were analyzed as in Fig. 4 (**P<0.001, compared to control). HEK293 cells were transfected with Flag-tagged αCaMKI from rat (Rn) or human (Hs) plus siRNA for rat αCaMKI, lysed and immunoblotted for Flag and myosin heavy chain IIb (MHCIIb, loading control) (D, top panel). HEK293 cells transfected with Myc-tagged βPIX from rat or human or Myc-tagged rat αPIX plus siRNA for rat βPIX were immnoblotted for Myc and ERK2 (loading control), as indicated (E, top panel). F, G. Neurons (DIV 7) were co-transfected with mRFP-βactin plus other constructs as indicated, fixed on DIV14 and analyzed for spine head width (F) and length (G).
In addition to spine density, the diameter of the spine head and spine length are thought to be important parameters. Induction of LTP results in an increase in spine head size and a reduction in spine length (Yuste and Bonhoeffer, 2001). Therefore, we examined whether manipulation of the CaMKK/CaMKI/βPIX signaling complex would impact either spine head diameter or spine length. Expression of dnCaMKI or S516AβPIX or siRNA for αCaMKI or βPIX, all of which dramatically reduced spine density (Figs. 4, 5), had little effect on spine head width (Fig. 5F). However, disruption of this signaling pathway did significantly increase spine length (Fig. 5G).
Although transfected β-actin is present in low amounts compared to endogenous actin (Kaech et al., 1997), its expression alone might modulate spine density or morphology. To test this possibility, we quantified these parameters under our experimental conditions using transfected soluble tomato fluorescent protein (TFP). Spine density and spine head diameter in TFP-transfected neurons (Fig. S9) were not significantly different from cells expressing mRFP-β-actin (Figs. 4, 5). Spine length was not quantified with TFP because the intensity of TFP expression in the dendritic shaft artificially increased the apparent diameter of the dendrite (Fig. S9), making measurements of spine length inaccurate.
To further verify our conclusion from morphological evidence (Figs. 4A and S8) that most mushroom-shaped spines are functional synapses, we recorded miniEPSCs (mEPSCs) from 12 DIV cultured neurons. Figs. 6A-C show that transfection with either dnCaMKI or S516A βPIX gave a 60% reduction in the frequency of mEPSCs with little or no change (p>0.05) in amplitude. The resting membrane potential was not different between the various transfections (control, −57.7±1.3; dnCaMKI, −55.4±1.3; S516A βPIX, −56.7±1.5 (mV), n=8-12), demonstrating that expression of these constructs was not deleterious to the health of the neurons. The lack of effect of dnCaMKI and S516AβPIX expression on mEPSC amplitude is consistent with no effect on spine head diameter (Fig. 5F) since there appears to be a good correlation between the abundance of postsynaptic AMPARs and the size of dendritic spine heads (Matsuzaki et al., 2001). Moreover, mIPSC frequency and amplitude were not affected by transfection with either dnCaMKI or S516A βPIX (Figs. 6D-F), indicating specificity for excitatory neurotransmission. This is expected since most inhibitory synapses do not involve spines but occur directly on the dendrite.
Figure 6.

Effect of dnCaMKI and βPIX S516A mutant on mEPSCs and mIPSCs. Cultured neurons (DIV 7) were transfected with EYFP-γactin and either mRFP (controls), mRFP-dnCaMKI or mRFP-βPIX S516A mutant, and mEPSCs and mIPSCs were recorded on DIV 12. Representative traces of mEPSCs (A) or mIPSCs (D) recorded from control (Cont), dnCaMKI-expressing neuron (dnKI), or βPIX S516A-expressing neuron (516A). Frequencies (B, E) and amplitudes (C, F) of mEPSCs or mEPSCs. **P<0.005, compared to control. Note that suppression by dnCaMKI and βPIX S516A of mEPSCs, but not mIPSCs, correlates with their effects on synapse number from Fig. 5B. Scale bars: top, 20 pA and 250 ms; bottom, 100 pA and 250 ms.
Regulation of spinogenesis in organotypic hippocampal slices
To extend the above studies in cultured hippocampal neurons to a more physiological system, we chose cultured hippocampal slices. Organotypic slices have been used extensively to study hippocampal functions because they retain most of the cellular and morphological organization of the intact hippocampus (Bahr, 1995; Caeser and Aertsen, 1991). Slices from p7 pups were cultured for 2 days, subjected to biolistic transfection and then fixation on DIV 5. Transfected neurons were visualized with mRFP-βactin, and dendritic protrusions were analyzed by confocal microscopy. As in the cultured neurons, about 2/3 of projections were classified as spines (Fig. 7). Most importantly, spine formation was strongly suppressed by treatments with either APV, TTX, STO-609 or transfection with dnCaMKI or the S516A mutant of βPIX (Fig. 7). Thus, it appears that the same signaling pathway of activity-induced NMDAR-dependent activation of CaMKK/CaMKI phosphorylation of βPIX that regulates spinogenesis in cultured neurons is also operative in hippocampal slices.
Figure 7.
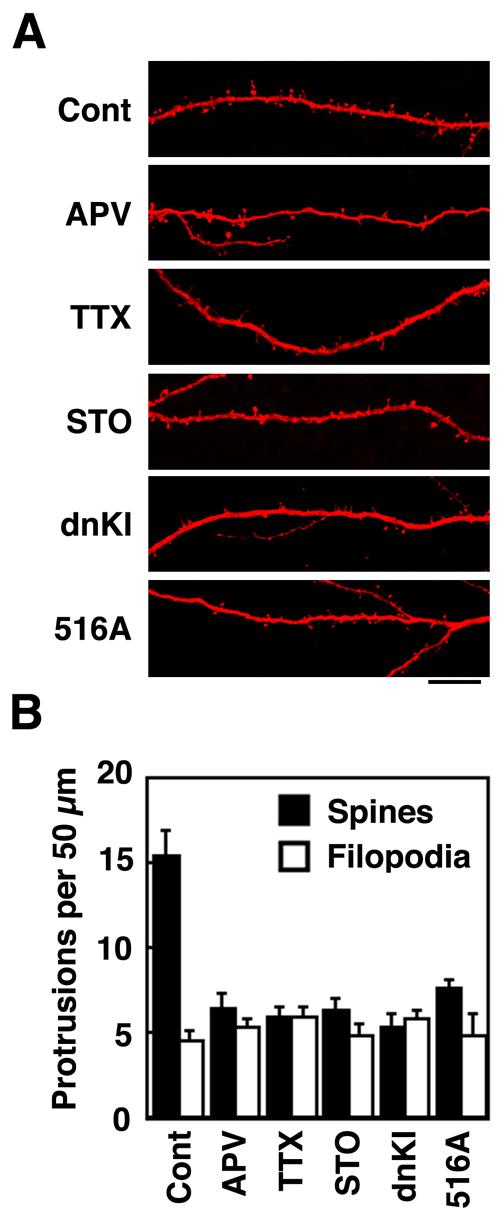
Regulation of spinogenesis in cultured hippocampal slices
Hippocampal slices from P7 rats were cultured for 2 days, subjected to biolistic transfection with mRFP-actin ± other plasmids as indicated. Neurons were treated without or with pharmacological reagents (100 μM APV, 1 μM TTX, or 20 μM ST O-609) as indicated. On DIV 5, dendritic protrusions were analyzed for spines and filopodia as in Fig. 5. A. Representative examples of apical CA1 dendrites from pyramidal neurons in hipocampal organotypic slice culture. Scale bar, 15 μm. B. Summary of the effects of transfections and pharmacological treatments on protrusion density.
Discussion
The present study delineates a signaling pathway triggered by afferent neuronal activity that couples stimulation of the NMDAR to activation of a signaling complex containing βCaMKK/CaMKI/βPIX that is localized in dendritic spines (Fig. 8). In this complex CaMKI phosphorylates Ser 516 in βPIX to stimulate its GEF activity, resulting in activation of Rac1 and Pak1 which are known to be essential for formation of spines in cultured hippocampal neurons (Zhang et al., 2003; Zhang et al., 2005). Although we chose to primarily investigate effects of βPIX phosphorylation on its Rac GEF activity, similar effects on Cdc42 activation appear to occur (Fig. 3A). The essential features of this signaling pathway that regulates activity-dependent spine and synapse formation were demonstrated in both cultured hippocampal neurons and organotypic slices. This constitutes the first report of a signaling complex involving the CaMKK/CaMKI cascade and identifies a precise mechanism for activation of Rac1 by neuronal activity. Consistent with this pathway, caPak1 was able to rescue spine formation that had been suppressed by dnCaMKI and S516A βPIX (Fig. 5C). There are several signaling pathways downstream of Pak1 (e.g., Lim-kinase (Meng et al., 2004; Schratt et al., 2006), myosin heavy chain IIb (Ryu et al., 2006)) that enhance spine maturation through regulation of the actin cytoskeleton (Hayashi et al., 2004; Luo et al., 1996; Tashiro and Yuste, 2004). Modulation of the βCaMKK/CaMKI pathway using pharmacological reagents, dominant-negative constructs as well as siRNAs had corresponding effects on spine formation. Mushroom-shaped spines, identified by intense puncta of βactin in their heads, showed a strong correlation (97%) with the presynaptic marker synapsin 1 (Fig. 4A) as well as co-localization with pCaMKI (Fig. 4B), indicating their presence in synapses. Thus, we conclude that this signaling pathway promotes not only spine formation but also synaptogenesis. Consistent with this conclusion, reductions in spine and synapse density (Fig. 5) correlated with reductions in mEPSC frequency with no effect on mIPSCs (Fig. 6), remarkably similar to the phenotype of cortical neurons from the mouse model of Rett Syndrome mental retardation (Dani et al., 2005).
Figure 8.
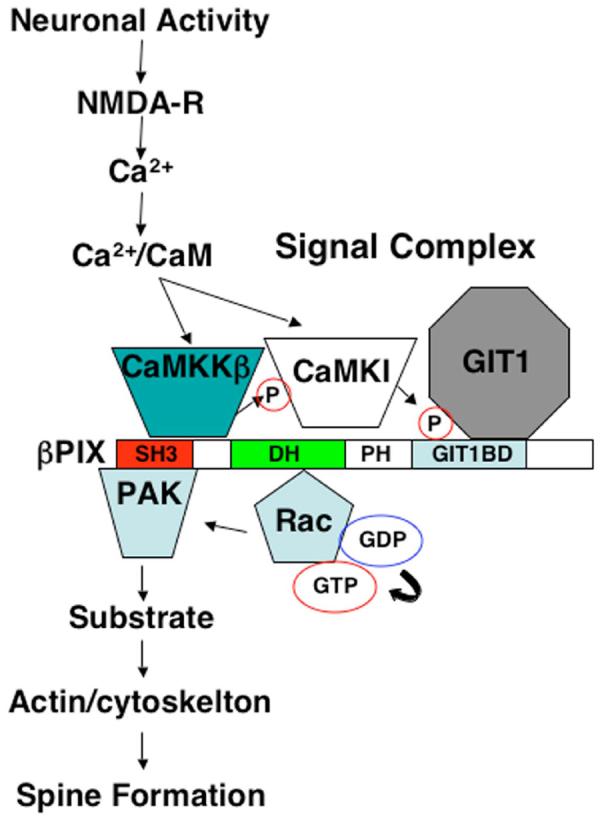
Model of CaMK/βPIX signaling pathway. Activity-dependent Ca2+ influx through the NMDA receptor of a spine activates the CaMKK/CaMKI/βPIX complex to promote CaMKI-mediated phosphorylation of S516 in βPIX with resultant activation of its GEF activity. The activated βPIX promotes GTP-loading of Rac (and/or Cdc42) to activate Pak and modulate actin dynamics and enhance spinogenesis and synapse formation.
The α, but not β or δ, isoform of CaMKI appeared to be responsible for spinogenesis through phosphorylation of βPIX; however, we could not exclude a potential role for γCaMKI since its siRNA was deleterious to the cultured neurons. Is βPIX the only target of αCaMKI that modulates spinogenesis? CaMKI can directly phosphorylate Ser19 in MLC in vitro and in transfected HeLa cells (Suizu et al., 2002), and MLC phosphorylation by Pak1 has been reported to enhance spine formation (Zhang et al., 2005). However, our data strongly support the conclusion that the major effect of endogenous βCaMKK/αCaMKI on spinogenesis is mediated largely through Ser516 phosphorylation in βPIX since the suppression of spine number and synapse formation by dnCaMKI or by siRNA were essentially identical to expression of the S516A βPIX or the βPIX mutant lacking GEF activity (Fig. 5A, B).
It is interesting that all the reagents that interfere with the CaMKK/CaMKI/βPIX pathway suppress formation of mushroom-shaped spines with little effect on filopodia number. The exception is the siRNA for βCaMKK that strongly decreased spine formation but also increased filopodia number (Fig. 4F). It seems likely that βCaMKK may be interfering with maturation of filopodia into mature spines, the major mechanism for spine formation and synaptogenesis (Matus, 2000; Ziv and Smith, 1996). This would be consistent with its co-localization with the βPIX/GIT1 complex that exists in spine heads (Zhang et al., 2003; Zhang et al., 2005). The siRNA for αCaMKK strongly reduced both spine and filopodia numbers. It is possible that αCaMKK may be regulating a different pathway that blocks formation or stabilization of filopodia that would not only reduce filopodia number but also spines because they mature from filopodia. Since αCaMKK only weakly interacts with βPIX, it is not clear how αCaMKK is exerting its effect. It may be targeting a different subcellular pool or isoform of CaMKI than the αCaMKI/βPIX complex, and mapping out this pathway will be a future endeavor. Interfering with the CaMK/βPIX pathway reduced spine/synapse density, increased the length of remaining spines but had no effect on spine head width. Physiological consequences of increasing spine length are not obvious, but it might further reduce the limited Ca2+ signaling from the spine head to the dendritic shaft (Sabatini et al., 2002). Such signaling in short spines has been proposed to be important in trafficking of AMPARs during certain forms of synaptic plasticity (Korkotian and Segal, 2006).
βPIX and its binding partner GIT1 are both multidomain proteins (Hoefen and Berk, 2006) that interact with numerous other proteins, such as hScrib (Audebert et al., 2004; Roche et al., 2002), piccolo (Kim et al., 2003), liprin-α (Kaufmann et al., 2002; Wyszynski et al., 1999), and Shank (Park et al., 2003), that are located both pre- and/or postsynaptically. Although it is likely that the βPIX/GIT1 complex may have presynaptic functions, the effects observed in this study are almost certainly postsynaptic. Our transfection efficiency of cultured neurons is very low (2-5%), so the probability that an axon from one transfected neuron forms a synapse with a spine from another transfected neuron is highly unlikely. Since βPIX regulates Rac, Cdc42 and Pak1 activation, postsynaptic effects on the actin cytoskeleton of filopodia and spines are likely involved (Calabrese et al., 2006). One reported mechanism is through Pak1-mediated phosphorylation of myosin II regulatory light chain (Zhang et al., 2005). Myosin IIB is present in the postsynaptic density of spines (Jordan et al., 2004; Peng et al., 2004) where it regulates spine morphology (Ryu et al., 2006).
It should be noted that expression of various constructs that inhibited either CaMKI or βPIX in our study gave about a 50% decrease in mushroom-shaped spines. However, suppression of by dominant-negative RacN17 produces an almost complete blockage of spinogenesis (Nakayama et al., 2000; Zhang and Macara, 2006; Zhang et al., 2003). This suggests the existence of mechanisms in addition to CaMKK/CaMKI/βPIX for activity-dependent regulation of Rac. Another Ca2+-dependent mechanism is the Rac GEF Tiam which also is involved in NMDAR-dependent regulation of spine formation (Tolias et al., 2005). Interference with endogenous Tiam using either siRNA or dnTiam (Tolias et al., 2005) or suppression of the Tiam-binding protein PAR-3 (Zhang and Macara, 2006) reduces spine density by 40-60%. NMDAR stimulation of Tiam GEF activity may be mediated by CaMKII-dependent phosphorylation of Tiam (Fleming et al., 1999), but this pathway could not be directly verified because the CaMKII regulatory phosphorylation site in Tiam has not been identified. Since we identified Ser516 in βPIX as the CaMKI phosphorylation site (Fig. 2A), we were able to demonstrate that this specific phosphorylation enhanced βPIX GEF activity (Fig. 3) and was critical in the regulation of spinogenesis (Fig. 5). Thus, multiple signaling pathways involving CaMKK/CaMKI/βPIX, Tiam and PAR3 all appear to modulate Rac-mediated spinogenesis. Since abnormalities in Rho-mediated spinogenesis are closely linked to several forms of mental retardation (Kaufmann and Moser, 2000; van Galen and Ramakers, 2005), it will be important to determine whether the CaMKK/CaMKI/βPIX pathway is perturbed in any of these neuropathologies.
The present work adds to the growing list of functions for the CaMKK/CaMKI pathway in neuronal development and plasticity by demonstrating its ability to activate the Rac GEF βPIX and thereby promote activity-dependent maturation of spines and synapses. Previously, we showed that CaMKK/CaMKI is essential for maintaining the morphology and motility of axonal growth cones and therefore basal axonal outgrowth (Wayman et al., 2004). Because CaMKK/CaMKI exerts Ca2+-dependent cross-talk with the MAP-kinase Erk (Schmitt et al., 2004), this signaling pathway accounts for Erk-mediated LTP (Schmitt et al., 2005) as well as for activity-dependent dendritic arborization (Wayman et al., 2006). These multiple functions, however, appear to be mediated by different CaMKI isoforms. For example, regulation of Erk activation and CREB-dependent dendritic arborization is mediated by the γ isoform of CaMKI whereas the current study implicated βCaMKK acting through αCaMKI. In summary, over the past couple years CaMKI has been transformed from an “orphan” kinase of unknown physiological function to a major player in neuronal development and plasticity.
METHODS (see Supplement Methods)
siRNA Constructs
siRNA constructs against the isoforms of CaMKI were previously described (Wayman et al., 2006). The siRNA constructs against αCaMKK, βCaMKK and βPIX were cloned into pmU6pro vector:αCaMKK, TTTGTGTTTGACCTCCTGAGAAATTGATATCCGTTTCTCAGGTCAAACATTTTTT; siβCaMKK, TTTGGGTCGAGAATTCAGTCAAACATTGATAGTCCGTGTTTGACTGAATTCTCGACCTTTTTT; siβPIX, TTTGTCAGCAACCGGCTCTTTGATTGATCCGTCAAAGAGCCGGTTGCTGATTTTTT.
Cell and organotypic culture and transfection
Medium-density (3.2 × 104 cells per square centimeter) hippocampal neurons were isolated from P0–2 Sprague Dawley rat as described (Wayman et al., 2006). Neurons were maintained in Neurobasal A media (Invitrogen, Gaithersburg,MD) supplemented with B27 (Invitrogen) and 0.5 mM L-glutamine with 5 μM cytosine-D-arabinofuranoside (AraC) added at 2 DIV. Neurons were cultured a further 5 days before transfection (LipofectAMINE 2000, Invitrogen) and/or treatment with various pharmacological reagents as specified. In each experiment we optimized DNA amounts, transfection reagent amounts, and transfection duration to minimize toxicity and maximize transfection efficiency. None of the transfections or drug treatments described in this work had noticeable effects on apoptosis as assessed by condensed nuclei using Hoechst staining. HEK293 from ATCC were cultured in Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum, penicillin/streptomycin, and L-glutamine at 37 °C in 5% CO2, 95% Air. HEK293 cells were transfected with Fugene6 (Roche, Mannheim, Germany) according to the manufacturer's protocols.
Organotypic hippocampal slices were prepared from P7 Sparague-Dawley rats as described previously (Wayman et al., 2006). To visualize transfected neurons and dendritic protrutions, slices were transfected with pCAGGS-mRFP1-βactin and pCAG-EGFP plus test plasmids using a Helios Gene Gun (BioRad) at DIV2. At 4 days post-transfection, slices were fixed, mounted, and imaged using a confocal microscope.
Quantification of spines
Medium density hippocampal neurons transfected on 7 DIV were fixed on either 12 DIV or 14 DIV with the PHEMS Fixative [4% paraformaldehyde in 60 mM PIPES, 25 mM Hepes (pH 7.4), 5 mM EGTA, 1 mM MgCl2 3% sucrose] for 20 min at room temperature. Fluorescent images were acquired using a cooled CCD camera (Hamamatsu Photonics, Shizuoka, Japan) attached to a Zeiss Axiplan2 (Carl Zeiss, Inc. NY, USA) inverted microscope with a 63X oil immersion lens. Morphometric measurements were performed using Openlab software (Improvision Inc. MA, USA). For protrusion density, the number of protrusions were counted from 15-30 neurons (∼200 μm total dendritic length per neuron). We identified spines by their mushroom-shaped morphology with intense puncta of mRFP-βactin at their tip (head) whereas filopodia were long, thin protrusions with low and constant mRFP-βactin expression throughout. Each experiment was repeated at least three times with independent neuronal preparations.
Immunocytochemistry
Hippocampal neuron were fixed in the PHEMS Fixative at 37 °C for 15 min; washed 3 times for 5 min in PBS permeabilized with 0.025% Triton X-100 in PBS for 5 min; washed 3 times for 5 min in blocking buffer [PBS containg 0.5% fish gelatin (Sigma), 0.05% Tween20]; stained with synapsin (1:2,000), or phospho-CaMKI (1:20), and Hoechst 33342 (1:10,000) in blocking buffer for 60 min at room temperature; after washed 3 times in blocking buffer, stained with appropriate secondary antibodies for 60 min. Coverslips were mounted on glass slides and analyzed by fluorescence microscopy.
Preparation of tissue and cell extracts
Soluble rat brain supernatants were prepared by dounce homogenization in lysis buffer (50 mM TrisHCl (pH8.5), 150 mM NaCl, 1% Deoxycholate, 10% Glycerol, 1 mM Na3VO4, 10 mM NaF, 1 mM β-glycerophosphate, 1 μM Microcystin-LR, complete tablet (Roche)) and centrifugation at 100 000 g for 30 min at 4°C. Soluble cell extracts were prepared from HEK293 cells or hippocampal neuron in lysis buffer [50 mM TrisHCl (pH8.0), 150 mM NaCl, 1% TritonX100, 10% Glycerol, 1 mM Na3VO4, 10 mM NaF, 1 mM β-glycerophosphate, 1 μM Microcystin-LR, 1 complete tablet (Roche)] and centrifugation at 10 000 g for 10 min at 4°C. Purified rat PSDs were a gift from Dr. Ayse Dosemeci (NIH).
Western blots and immunoprecipitation
Western blotting was performed as previously described (Davare et al., 2004) with quantification using the Odyssey near-infrared imaging platform system (LI-COR).
For imuunopreciptations, brain extracts were prepared as described above and subjected to immunoprecipitation for 2-4 hours using 4 μg of antibodies and 25 μl of protein G-conjugated agarose beads (Chemicon). Cell extracts were subjected to immunoprecipitation using 15 μl of the M2 anti-FLAG monoclonal antibody beads (Sigma) or 0.5 μg of anti-c-myc polyclonal antibodies immobilized on Sepharose beads, for 2-4 hours at 4 °C. Beads were washed once with 1 ml of lysis buffer, three times with 1 ml of wash buffer [20 mM TrisHCl (pH 8.0), 150 mM NaCl, 0.1% TritonX-100, 5% Glycerol]. Bound proteins were eluted with SDS/PAGE sample buffer, and subjected to immunoblotting as described above.
In vitro phosphorylation
Purified recombinant CaMKI (10 μM) was incubated with CaM-K K (0.5 μM) at 30 °C for 20 min in 50 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM DTT, 2 mM CaCl2, 20 μM CaM, and 200 μM of ATP. The reaction was initiated by the addition of ATP and terminated by a 10-fold dilution with ice-cold 50 mM HEPES (pH 7.5), 2 mg/ml bovine serum albumin, 10% ethylene glycol, and 2 mM EDTA. Purified GST-βPIX, wild-type or mutants (S516A, T526A or S516A/T526A) was incubated without or with activated CaMKI (100 nM), PKA (Davare et al., 2004) or Pak1 at 30 °C for 30 min in a solution containing 50 mM HEPES (pH 7.5), 10 mM MgCl2, 2.5 mM DTT, 1 mM CaCl2, 1.5 μM CaM, and 50 μM [γ32P]ATP (1,000 cpm/pmol). Ca2+/CaM was omitted for Pak1 phosphorylations. The reaction was terminated by the addition of SDS/PAGE sample buffer. The samples were subjected to SDS/PAGE followed by autoradiography.
Electrophysiology
Whole-cell voltage clamp recordings were performed on cultured hippocampal neurons using an Axopatch-200b amplifier (Molecular Devices). Cells were perfused (1 ml min−1) with perfusate containing (in mM): 140 NaCl, 2.5 KCl, 3 CaCl, 1 MgCl2, 5 HEPES and 25 D-glucose (pH = 7.3; 310 mosmol l−1). Miniature excitatory postsynaptic currents (mEPSCs) were isolated by adding picrotoxin (50 μM), strychnine (1 μM), and tetrodotoxin (0.5 μM) to the bath perfusate while 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM, Tocris) and D-(−)-2-amino-5-phosphonopentanoic acid (D-AP5, 50 μM, Tocris) were added to isolate miniature inhibitory postsynaptic currents (mIPSCs). Patch electrodes (4 – 6 MΩ) contained (in mM): 100 CsCH3O3S, 25 CsCl, 10 HEPES, 0.4 EGTA, 2 MgCl2, 4 Na2-ATP, 0.4 Na-GTP, and 10 phosphocreatine (pH = 7.3; 290-300 mosmol l−1). All experiments were carried out at room temperature (21 °C). Transfected cells were identified under epifluorescence. Only cells that had an input resistance >200 MΩ and resting membrane potentials <−50mV were considered for experiments. Access resistance (Ra) typically ranged between 10 - 15 MΩ and was compensated by ≥60%. Cells were rejected from analysis if Ra increased by more than 15%, or if the input resistance fell below 200 MΩ. Off-line analysis of mEPCSs was carried out using Clampfit v9.01.07 (Molecular Devices).
FRET Assay
Hippocampal neurons were transfected on DIV 4 with vectors encoding Raichu-rac (1011X) with or without wild-type or S516A βPIX mutant (1:5 ratio) and incubated 3 days at 37 °C. Cells were washed and preincubated in ASCF containing B27 supplement, FK506 (1 μM), cyclopsoprine A (1 μg/ml). Fluorescence from Raichu-rac was imaged and measured using an Axiovert 135TV microscope (Carl Zeiss Microimaging, Inc.) through an oil immersion objective (Plan-Apochromat 63x/1.4) equipped with cascade II:512 camera (Roper Bioscience) controlled by MetaMorph 6.3 (Universal Imaging Corp.) and a lambda 10-2 filter wheel (Sutter Instrument). The dual-emission images were obtained simultaneously through a dual-view module (Roper Bioscience) mounted with S470/30 and S535/30 emission filters and a 505 dcxr dichroic mirror (Chroma). Cells were illuminated with a 75-W Xenon lamp through D436/10 and 12.5% ND filter or HQ500/20 and 1.25% ND filter. The exposure time was 100 ms, and images were taken every 10 s. Sensitized FRET (Fsen) was calculated as reported elsewhere (van Rheenen et al., 2004). Briefly, after background subtraction, Fsen was calculated using fluorescent intensities from three images (Mdonor, MindirectAcceptor, MdirectAcceptor) using the following equation.
Fsen=(MindirectAcceptor−Mdonor∗β−MdirectAcceptor∗(γ−αβ))∕(1−βδ)
Apparent FRET efficiency (EA) was obtained by dividing Fsen by MdirectAcceptor. Note that we used ten times less illumination for MdirectAcceptor, which is compensated with a scale factor (S).
EA=S∗Fsen∕MdirectAcceptor
Constants for the equation were obtained by independent control experiments: α=5.6×10−5, β=0.628, γ=0.147, δ=3.7×10−4 and S=0.1, respectively. Cells were stimulated two minutes after recording. EA changes are shown after normalization. For each image set, the EA values before the stimulation were averaged and used as the reference value for normalization. Signals from 2-4 dendrite shafts were measured per cell and averaged values were used to represent single-cell responses.
Statistic
To determine whether significant differences existed among treatments, an analysis of variance was performed on the data with significance set at 0.05. To compare whether significant differences existed between two treatments, a Student's t test was performed on the data with significance set at 0.05. Significance levels (p value) are indicated in the figures.
Supplementary Material
01
Acknowledgements
We want to thank Drs. Gary Banker and Stefanie Kaech for advise and consultation throughout this study, Drs. Kohji Fukunaga, Hiroshi Tokumitsu, Michiyuki Matsuda, Ayse Dosemeci, Shelley Halpain and Gary Bokoch for reagents and Dr. John Scott for use of the imaging facility for the Raichu-Rac experiments. This work was supported by NIH grant NS027037 (TRS). TS was supported by the Human Frontier Science Program, DF from training grant DK007680, NH from NIH grant GM48231 (JD Scott) and TN was supported by the New Energy and Industrial Technology Development Organization of Japan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci. 2003;6:1194–1200. doi: 10.1038/nn1135. [DOI] [PubMed] [Google Scholar]
- Audebert S, Navarro C, Nourry C, Chasserot-Golaz S, Lecine P, Bellaiche Y, Dupont JL, Premont RT, Sempere C, Strub JM, et al. Mammalian Scribble forms a tight complex with the betaPIX exchange factor. Curr Biol. 2004;14:987–995. doi: 10.1016/j.cub.2004.05.051. [DOI] [PubMed] [Google Scholar]
- Bahr BA. Long-term hippocampal slices: a model system for investigating synaptic mechanisms and pathologic processes. J Neurosci Res. 1995;42:294–305. doi: 10.1002/jnr.490420303. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Yuste R. Spine motility. Phenomenology, mechanisms, and function. Neuron. 2002;35:1019–1027. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Brunig I, Kaech S, Brinkhaus H, Oertner TG, Matus A. Influx of extracellular calcium regulates actin-dependent morphological plasticity in dendritic spines. Neuropharmacology. 2004;47:669–676. doi: 10.1016/j.neuropharm.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Caeser M, Aertsen A. Morphological organization of rat hippocampal slice cultures. J Comp Neurol. 1991;307:87–106. doi: 10.1002/cne.903070109. [DOI] [PubMed] [Google Scholar]
- Calabrese B, Halpain S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron. 2005;48:77–90. doi: 10.1016/j.neuron.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Calabrese B, Wilson MS, Halpain S. Development and regulation of dendritic spine synapses. Physiology (Bethesda) 2006;21:38–47. doi: 10.1152/physiol.00042.2005. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Kennedy MB. Spine architecture and synaptic plasticity. Trends Neurosci. 2005;28:182–187. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Chahdi A, Miller B, Sorokin A. Endothelin 1 induces beta 1Pix translocation and Cdc42 activation via protein kinase A-dependent pathway. J Biol Chem. 2005;280:578–584. doi: 10.1074/jbc.M411130200. [DOI] [PubMed] [Google Scholar]
- Collin C, Miyaguchi K, Segal M. Dendritic spine density and LTP induction in cultured hippocampal slices. J Neurophysiol. 1997;77:1614–1623. doi: 10.1152/jn.1997.77.3.1614. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Saneyoshi T, Guire ES, Nygaard SC, Soderling TR. Inhibition of calcium/calmodulin-dependent protein kinase kinase by protein 14-3-3. J Biol Chem. 2004;279:52191–52199. doi: 10.1074/jbc.M409873200. [DOI] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Feng Q, Baird D, Cerione RA. Novel regulatory mechanisms for the Dbl family guanine nucleotide exchange factor Cool-2/alpha-Pix. Embo J. 2004;23:3492–3504. doi: 10.1038/sj.emboj.7600331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A. Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci. 2000;3:887–894. doi: 10.1038/78791. [DOI] [PubMed] [Google Scholar]
- Fleming IN, Elliott CM, Buchanan FG, Downes CP, Exton JH. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem. 1999;274:12753–12758. doi: 10.1074/jbc.274.18.12753. [DOI] [PubMed] [Google Scholar]
- Haribabu B, Hook SS, Selbert MA, Goldstein EG, Tomhave ED, Edelman AM, Snyderman R, Means AR. Human calcium-calmodulin dependent protein kinase I: cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin dependent protein kinase I kinase. Embo J. 1995;14:3679–3686. doi: 10.1002/j.1460-2075.1995.tb00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron. 2004;42:773–787. doi: 10.1016/j.neuron.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Majewska AK. Dendritic spine geometry: functional implication and regulation. Neuron. 2005;46:529–532. doi: 10.1016/j.neuron.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- Hoefen RJ, Berk BC. The multifunctional GIT family of proteins. J Cell Sci. 2006;119:1469–1475. doi: 10.1242/jcs.02925. [DOI] [PubMed] [Google Scholar]
- Itoh RE, Kurokawa K, Ohba Y, Yoshizaki H, Mochizuki N, Matsuda M. Activation of rac and cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol Cell Biol. 2002;22:6582–6591. doi: 10.1128/MCB.22.18.6582-6591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- Kaech S, Fischer M, Doll T, Matus A. Isoform specificity in the relationship of actin to dendritic spines. J Neurosci. 1997;17:9565–9572. doi: 10.1523/JNEUROSCI.17-24-09565.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann N, DeProto J, Ranjan R, Wan H, Van Vactor D. Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron. 2002;34:27–38. doi: 10.1016/s0896-6273(02)00643-8. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kim S, Ko J, Shin H, Lee JR, Lim C, Han JH, Altrock WD, Garner CC, Gundelfinger ED, Premont RT, et al. The GIT family of proteins forms multimers and associates with the presynaptic cytomatrix protein Piccolo. J Biol Chem. 2003;278:6291–6300. doi: 10.1074/jbc.M212287200. [DOI] [PubMed] [Google Scholar]
- Ko J, Kim S, Valtschanoff JG, Shin H, Lee JR, Sheng M, Premont RT, Weinberg RJ, Kim E. Interaction between liprin-alpha and GIT1 is required for AMPA receptor targeting. J Neurosci. 2003;23:1667–1677. doi: 10.1523/JNEUROSCI.23-05-01667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konur S, Ghosh A. Calcium signaling and the control of dendritic development. Neuron. 2005;46:401–405. doi: 10.1016/j.neuron.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Segal M. Morphological constraints on calcium dependent glutamate receptor trafficking into individual dendritic spine. Cell Calcium. 2006 doi: 10.1016/j.ceca.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Kossel AH, Williams CV, Schweizer M, Kater SB. Afferent innervation influences the development of dendritic branches and spines via both activity-dependent and non-activity-dependent mechanisms. J Neurosci. 1997;17:6314–6324. doi: 10.1523/JNEUROSCI.17-16-06314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Hensch TK, Ackerman L, Barbel S, Jan LY, Jan YN. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379:837–840. doi: 10.1038/379837a0. [DOI] [PubMed] [Google Scholar]
- Ma XM, Huang J, Wang Y, Eipper BA, Mains RE. Kalirin, a multifunctional Rho guanine nucleotide exchange factor, is necessary for maintenance of hippocampal pyramidal neuron dendrites and dendritic spines. J Neurosci. 2003;23:10593–10603. doi: 10.1523/JNEUROSCI.23-33-10593.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinor R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1505–1509. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Meng Y, Takahashi H, Meng J, Zhang Y, Lu G, Asrar S, Nakamura T, Jia Z. Regulation of ADF/cofilin phosphorylation and synaptic function by LIM-kinase. Neuropharmacology. 2004;47:746–754. doi: 10.1016/j.neuropharm.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Mott HR, Nietlispach D, Evetts KA, Owen D. Structural analysis of the SH3 domain of beta-PIX and its interaction with alpha-p21 activated kinase (PAK) Biochemistry. 2005;44:10977–10983. doi: 10.1021/bi050374a. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Yamauchi Y, Nakayama H, Shinkawa T, Yanagida M, Takahashi N, Isobe T. A direct nanoflow liquid chromatography-tandem mass spectrometry system for interaction proteomics. Anal Chem. 2002;74:4725–4733. doi: 10.1021/ac020018n. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Matus A. Calcium regulation of actin dynamics in dendritic spines. Cell Calcium. 2005;37:477–482. doi: 10.1016/j.ceca.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Park E, Na M, Choi J, Kim S, Lee JR, Yoon J, Park D, Sheng M, Kim E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem. 2003;278:19220–19229. doi: 10.1074/jbc.M301052200. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Protein phosphorylation in signaling--50 years and counting. Trends Biochem Sci. 2005;30:286–290. doi: 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- Penzes P, Beeser A,, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Roche JP, Packard MC, Moeckel-Cole S, Budnik V. Regulation of synaptic plasticity and synaptic vesicle dynamics by the PDZ protein Scribble. J Neurosci. 2002;22:6471–6479. doi: 10.1523/JNEUROSCI.22-15-06471.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger G, Kutsche K. AlphaPIX and betaPIX and their role in focal adhesion formation. Eur J Cell Biol. 2006;85:265–274. doi: 10.1016/j.ejcb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Ryu J, Liu L, Wong TP, Wu DC, Burette A, Weinberg R, Wang YT, Sheng M. A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron. 2006;49:175–182. doi: 10.1016/j.neuron.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Guire ES, Saneyoshi T, Soderling TR. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J Neurosci. 2005;25:1281–1290. doi: 10.1523/JNEUROSCI.4086-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JM, Wayman GA, Nozaki N, Soderling TR. Calcium activation of ERK mediated by calmodulin kinase I. J Biol Chem. 2004;279:24064–24072. doi: 10.1074/jbc.M401501200. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- Segal M, Greenberger V, Korkotian E. Formation of dendritic spines in cultured striatal neurons depends on excitatory afferent activity. Eur J Neurosci. 2003;17:2573–2585. doi: 10.1046/j.1460-9568.2003.02696.x. [DOI] [PubMed] [Google Scholar]
- Shin EY, Shin KS, Lee CS, Woo KN, Quan SH, Soung NK, Kim YG, Cha CI, Kim SR, Park D, et al. Phosphorylation of p85 beta PIX, a Rac/Cdc42-specific guanine nucleotide exchange factor, via the Ras/ERK/PAK2 pathway is required for basic fibroblast growth factor-induced neurite outgrowth. J Biol Chem. 2002;277:44417–44430. doi: 10.1074/jbc.M203754200. [DOI] [PubMed] [Google Scholar]
- Soderling SH, Binns KL, Wayman GA, Davee SM, Ong SH, Pawson T, Scott JD. The WRP component of the WAVE-1 complex attenuates Rac-mediated signalling. Nat Cell Biol. 2002;4:970–975. doi: 10.1038/ncb886. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Suizu F, Fukuta Y, Ueda K, Iwasaki T, Tokumitsu H, Hosoya H. Characterization of Ca2+/calmodulin-dependent protein kinase I as a myosin II regulatory light chain kinase in vitro and in vivo. Biochem J. 2002;367:335–345. doi: 10.1042/BJ20020536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Takemoto-Kimura S, Ageta-Ishihara N, Nonaka M, Adachi-Morishima A, Mano T, Okamura M, Fujii H, Fuse T,, Hoshino M, Suzuki S, et al. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIgamma. Neuron. 2007;54:755–770. doi: 10.1016/j.neuron.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Kobayashi R. A Single Amino Acid Difference between alpha and beta Ca2+/Calmodulin-dependent Protein Kinase Kinase Dictates Sensitivity to the Specific Inhibitor, STO-609. J Biol Chem. 2003;278:10908–10913. doi: 10.1074/jbc.M213183200. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- van Galen EJ, Ramakers GJ. Rho proteins, mental retardation and the neurobiological basis of intelligence. Prog Brain Res. 2005;147:295–317. doi: 10.1016/S0079-6123(04)47022-8. [DOI] [PubMed] [Google Scholar]
- van Rheenen J, Langeslag M, Jalink K. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J. 2004;86:2517–2529. doi: 10.1016/S0006-3495(04)74307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Tokumitsu H, Soderling TR. Inhibitory cross-talk by cAMP kinase on the calmodulin-dependent protein kinase cascade. J Biol Chem. 1997;272:16073–16076. doi: 10.1074/jbc.272.26.16073. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Valtschanoff JG, Naisbitt S, Dunah AW, Kim E, Standaert DG, Weinberg R, Sheng M. Association of AMPA receptors with a subset of glutamate receptor-interacting protein in vivo. J Neurosci. 1999;19:6528–6537. doi: 10.1523/JNEUROSCI.19-15-06528.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Horwitz AF. Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol. 2003;161:131–142. doi: 10.1083/jcb.200211002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. 2005;25:3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01