Pcons: A neural-network–based consensus predictor that improves fold recognition (original) (raw)
Abstract
During recent years many protein fold recognition methods have been developed, based on different algorithms and using various kinds of information. To examine the performance of these methods several evaluation experiments have been conducted. These include blind tests in CASP/CAFASP, large scale benchmarks, and long-term, continuous assessment with newly solved protein structures. These studies confirm the expectation that for different targets different methods produce the best predictions, and the final prediction accuracy could be improved if the available methods were combined in a perfect manner. In this article a neural-network-based consensus predictor, Pcons, is presented that attempts this task. Pcons attempts to select the best model out of those produced by six prediction servers, each using different methods. Pcons translates the confidence scores reported by each server into uniformly scaled values corresponding to the expected accuracy of each model. The translated scores as well as the similarity between models produced by different servers is used in the final selection. According to the analysis based on two unrelated sets of newly solved proteins, Pcons outperforms any single server by generating ∼8%–10% more correct predictions. Furthermore, the specificity of Pcons is significantly higher than for any individual server. From analyzing different input data to Pcons it can be shown that the improvement is mainly attributable to measurement of the similarity between the different models. Pcons is freely accessible for the academic community through the protein structure-prediction metaserver at http://bioinfo.pl/meta/.
Keywords: Fold recognition, threading, benchmark, web servers, LiveBench, CASP, CAFASP
As the genome projects proceed, we are presented with an exponentially increasing number of protein sequences, but with only a very limited knowledge of their structure or function. Because the experimental determination of protein structure or function is not a trivial task, the quickest way to gain some understanding of these proteins and their genes is by relating them to proteins or genes with known properties. Improving the algorithms that examine these relationships is a fundamental challenge in bioinformatics today. This work focuses on protein structure-prediction approaches, which are easier to quay and compare than function-prediction protocols.
There are many methods of database searches that have been developed to detect structurally related proteins based on single sequences (Needleman and Wunsch 1970; Smith and Waterman 1981), multiple sequence alignments or profiles (Gribskov et al. 1987; Altschul et al. 1997; Karplus et al. 1998; Rychlewski et al. 2000), and predicted (Fischer and Eisenberg 1996; Rice and Eisenberg 1997; Rost et al. 1997; Kelley et al. 2000) or experimentally determined (Jones et al. 1992) structures. Furthermore, some groups use other structural information from the template introducing special gap penalties in loop regions (Sanchez and Sali 1998), and other groups use special alignment techniques (Alexandrov and Luethy 1998). Several of these methods are available as web servers.
In several recent studies the ability of different methods to detect proteins that share the same fold has been studied (Abagyan and Batalov 1997; Di Francesco et al. 1997; Park et al. 1997, 1998; Brenner et al. 1998; Lindahl and Elofsson 2000). These studies have provided a rather clear picture with some important conclusions: (1) The common practice of describing similarity as the fraction of identical residues should be abandoned. (2) The exact choice of parameters such as gap penalties greatly affects the performance. (3) Methods using heuristic alignment techniques such as FASTA (Pearson and Lipman 1988; Pearson 1995) or BLAST2 (Altschul et al. 1997) do not perform as well as methods using optimal alignments. In many studies (Park et al. 1997, 1998; Salamov et al. 1999) it was also shown that the use of evolutionary information improves the detection rate, especially on the superfamily level (Lindahl and Elofsson 2000). Other studies revealed that the use of structural information also increases the ability to detect distantly related proteins (Lindahl and Elofsson 2000; Panchenko et al. 2000).
Lately, in addition to the evaluation of the ability of different methods to recognize the correct fold, some studies have also focused on the quality of the generated alignment (Domingues et al. 2000; Sauder et al. 2000; Bujnicki et al. 2001a; Elofsson 2001). An important conclusion from these studies is that for different targets the best predictions are often made by different methods. It is even quite common, for a single method and pairs of distantly related proteins, that the optimum choice of alignment parameters differs from case to case. Therefore, when evaluating the accuracy of a structure-prediction protocol in a large set, it is quite clear that its performance could be increased if different, best suited, approaches could be applied in appropriate cases.
In this study we have examined the possibility of combining several fold recognition methods to select the best prediction for each target. By making a consensus prediction, that is, selecting the most common model generated by all methods, we show that ∼8%–10% more correct predictions can be generated than by the best individual method used. The improvement is most significant for the difficult targets.
Some groups have used simple forms of consensus predictors, wherein several models are created for each sequence–template pair. The Inbgu method performs five alignments using combinations of single-sequence and profile data (Fischer 2000). The 3D-PSSM method performs three alignments for each sequence–template pair (Kelley et al. 2000). In both Inbgu and 3D-PSSM all alignments are made using predicted secondary-structure information for the query sequence and the experimentally determined secondary structure of the template protein. The alignments of Inbgu are made using either single-sequence or multiple-sequence information of the query and the template. In 3D-PSSM two alignments are made using the query sequence and two different template profiles, one derived from a superfamily-wide structural alignment. The third alignment uses the template sequence and a profile obtained from the query sequence. For each query–template pair these methods choose one alignment; 3D-PSSM chooses the highest scoring one, whereas Inbgu also takes the rank into account.
The server (Pcons) and the approach presented here differ significantly from earlier consensus predictors. A set of publicly available protein fold recognition web servers are used to produce input data. This way the consensus predictor can be improved if new structure-prediction servers become available or can automatically improve if the existing ones become more accurate. To efficiently combine the results of various methods all collected models are compared using a structural superposition algorithm. Pcons uses a set of neural networks to predict the quality and accuracy of all collected models, and if several servers predict one particular fold, Pcons assigns a high score to it. Pcons also differs from most earlier methods in the way that correct predictions are defined. The networks are trained to predict the quality of a model and not whether a correct fold is recognized. This might be advantageous as it is not trivial to uniquely define folds (Hadley and Jones 1999), and even if the correct fold is found, the alignment could potentially be wrong. Finally, the predicted model quality and the similarity to other models are used in the ultimate assessment and scoring of the evaluated model. A web server has been implemented providing access to this approach as a part of the protein structure-prediction metaserver at http://bionfo.pl/meta/
Results and Discussion
Pcons does not make any new predictions; instead, it uses predictions produced by six web servers. Pcons uses two types of information from the servers: the score and the overall fraction of other structurally similar models produced by other servers. Other types of input data, such as the length of the models, were also tested, but did not improve the performance and were therefore ignored. The similarity between two predictions is measured in two different ways, either by measuring the similarity of the models produced by the different servers or by measuring the similarity of the templates ideied by these servers. The reason for using both model and template similarities is that if the alignment is wrong the template similarity can still give information about similarities between predictions. For each pair of models or templates, a structural superposition is performed. Two models or templates are assumed to be similar if the structural alignment has a _P_-value <10−3 (Cristobal et al. 2001). Using different combinations of the similarity measures, five different networks were trained (see Table 1). For each fold recognition server a primary neural network layer was trained to predict the quality of a model based on the confidence score reported by the server and the fraction of other similar models. Finally, a secondary jury network was used to combine the results of the six primary networks for each individual server (see Fig. 1 ▶). It should be noted that the jury network is not necessary, but we found that including it increased the performance slightly. All comparisons were done using a fourfold cross-validation.
Table 1.
Description of the information used by the different neural networks used in this study
| Method | Scores | Model/ Template | All | First | Over cut-off | Similarity measure |
|---|---|---|---|---|---|---|
| NN-score | Yes | none | — | — | — | LGscore2 |
| NN-all | Yes | Both | Yes | — | — | LGscore2 |
| NN-combined | Yes | Both | Yes | Yes | Yes | LGscore2 |
| NN-noscore | No | Both | Yes | Yes | Yes | LGscore2 |
| NN-model | Yes | Model | Yes | Yes | Yes | LGscore |
Fig. 1.

Description of the NN-all neural networks used in this study. (Top panel) Generation of the inputs to NN-all. First, up to 60 models are collected from six different web servers. The structure of these models and the related templates are compared to the structure of all other models and templates. Three data points are fed into the network—the score, the fraction of similar models, and the fraction of similar templates. A separate network is trained for each server. For each model obtained from one server the log of LGscore2 is predicted by a first-layer neural network. The output from these networks is then fed into the jury network.
The first version of the network, NN-score, uses only the confidence scores reported by all servers, but NN-all in addition uses the fraction of all models and templates that are similar to the evaluated model. In the next version, NN-combined, we use two additional types of structural comparisons, the fraction of similar first-ranked models/templates and the fraction of similar models/templates that are above the proposed cutoff for each server. In another version, NN-noscore, we use the same information, but exclude the confidence scores. Finally, we created the NN-model network that can be used for fast comparison. Because of computational limitations, NN-model is the network used by the current Pcons implementation at http://bioinfo.pl/meta/.
Consensus predictions find more correct models for the difficult targets
This study is based on 125 different targets and 6 different servers producing up to 10 different models each. The LiveBench-1 experiment provided the input data (Bujnicki et al. 2001a). The target proteins were divided into 30 easy targets (EASY category) and the 95 remaining difficult targets (HARD category). The performance of several servers has been analyzed using this set of data in an earlier study (Bujnicki et al. 2001a).
In Table 2 the number of correct models from the individual servers and the consensus networks are shown. From Table 2 it is clear that the consensus networks detect more correct targets than any individual server. For the easy targets all the networks perform approximately as well as, but not better than, the best individual server. For the difficult targets, all the consensus methods that use structural comparisons detect significantly more targets (30) than any single server (21), whereas NN-score, that does not, shows no significant improvement.
Table 2.
Number of correct first ranked models by each server and network
| Method | Easy (30) | Hard (95) | All (125) |
|---|---|---|---|
| GenTHREADER | 23 | 13 | 36 |
| Sam-T98 | 22 | 16 | 38 |
| FFAS | 28 | 14 | 42 |
| Inbgu | 23 | 21 | 44 |
| 3D-PSSM | 22 | 21 | 43 |
| PDBBLAST | 28 | 10 | 38 |
| NN-score | 28 | 20 | 48 |
| NN-all | 27 | 29 | 56 |
| NN-combined | 28 | 30 | 58 |
| NN-noscore | 28 | 30 | 58 |
| NN-model | 28 | 29 | 57 |
It is not only important to idey a correct model, it is also important to be able to separate incorrect models from correct models. One way to study this is to sort the models according to the score and then plot the cumulative number of correct models versus incorrect models, as has been done in several earlier studies (Park et al. 1997, 1998). The consensus predictor NN-model finds ∼20% more correct models for any number of incorrect models than the best individual server (see Fig. 2 ▶). For clarity, we have only shown NN-model in this figure. However, the performance of the other networks is similar.
Fig. 2.

Cumulative plot of correct versus incorrect models. To make the curves easier to analyze they are smoothed by a running average. Correct and incorrect models are defined by using LGscore2 and a cutoff of 10−3. The _X_-axis reports the number of incorrect models according to Scop, and the _Y_-axis indicates the number of correct models.
There are four different networks that use structural information, NN-all, NN-combined, NN-noscore, and NN-model. From Table 2 it seems as if these four networks perform alike. However, from the correlations coefficients it can be seen that NN-model does not perform quite as well as the others (see Table 3). In Table 3, the correlation co efficients, the Matthews correlation coefficients (MCC), and the sensitivity/specificity values for all the consensus networks are shown. It can be seen that at a predicted LGscore of 10−3 ∼80% of the models are correct and up to 75% of all correct models are detected. At 10−5 the specificity is higher than 90%, but the sensitivity has dropped below 50%. Interestingly it seems as if NN-model has a slightly higher specificity but a lower sensitivity than the other networks.
Table 3.
Matthews Correlation Coefficients, specificity, sensitivity and correlation between the true and predicted LGscore2 coefficients of the different networks
| Method | Mc (10−3) | Spec/Sens (10−3) | Spec/Sens (10−5) | Correlation |
|---|---|---|---|---|
| NN-score | 0.54 | 0.80/0.46 | 0.91/0.36 | 0.61 |
| NN-all | 0.72 | 0.81/0.75 | 0.94/0.46 | 0.77 |
| NN-combined | 0.73 | 0.81/0.77 | 0.95/0.46 | 0.80 |
| NN-noscore | 0.74 | 0.84/0.75 | 0.95/0.48 | 0.81 |
| NN-model | 0.70 | 0.87/0.66 | 0.98/0.41 | 0.76 |
The good performance of NN-noscore was a surprise to us. We did not expect it to be possible to completely ignore the reported confidence score of the individual servers and still perform quite well. One possible explanation is that it is statistically unlikely that an incorrect top-ranking model would be similar to other top-ranking models. This would imply that any model that is similar to many other models is most likely correct.
The detailed analysis shows that the correlation coeffi cient and MCC for NN-score is substantially lower than for the networks that use structural comparisons. In Table 2, it is shown that NN-score does not detect more easy or difficult targets than the best servers. However, the overall performance is slightly better as different methods are better for easy or difficult targets. This indicates that the additional information extracted using the pairwise model or template comparisons is the main reason for the improvement achieved by Pcons.
Most selected models are the first-ranked models from Inbgu and 3D-PSSM
The neural network translated the confidence scores of the servers into values estimating the accuracy of the models. The final prediction choice is based on this translation, and the model with the highest estimated accuracy is selected. In Table 4, the origin of the models selected by the consensus predictor is shown. The most common predictions of the NN-score network originate from the Inbgu output, whereas for all other networks 3D-PSSM is the most frequent origin. Models with an origin in FFAS are quite common (15%–24%) as well, but GenTHREADER, Sam-T98, and PDBBLAST are less frequent. In contrast, the NN-noscore network selects the models with similar frequency from all servers, with a surprising exception in the case of GenTHREADER, which is chosen less frequently. As can be seen in Figure 2 ▶, GenTHREADER is one of the servers with the best ability to distinguish between the correct and incorrect models. On the other hand, the scoring function used by GenTHREADER makes it difficult for the neural network to distinguish between strong and very strong predictions. Therefore, in cases of trivial predictions other servers are favored. In addition, other benchmarks (Bujnicki et al. 2001a) have shown that even if the GenTHREADER prediction is correct, other servers frequently generate better models.
Table 4.
Illustration of which server the neural network prefers
| Method | GenTHREADER | Sam-T98 | FFAS | Inbgu | 3D-PSSM | PDBBLAST |
|---|---|---|---|---|---|---|
| NN-score | 6% | 11% | 15% | 40% | 19% | 9% |
| NN-all | 5% | 8% | 19% | 26% | 34% | 8% |
| NN-combined | 7% | 9% | 17% | 20% | 33% | 14% |
| NN-noscore | 4% | 14% | 23% | 19% | 25% | 16% |
| NN-model | 4% | 8% | 24% | 27% | 27% | 10% |
The model chosen by the consensus predictor is not always one of the models reported with rank one by the servers. Table 5 shows that a first-ranked model is chosen in 26% to 82% of the cases. When structural comparisons are included a significant number of higher ranked models are selected. The NN-model network selects more first-ranked models than the other networks, and the NN-combined network selects fewer than the NN-all network. This indicates that the more structural comparison terms are included the less important is the score.
Table 5.
The origin of the highest scoring model for each of the target sequences is analyzed
| Method | Rank 1 | Rank 2 | Rank 3 | Rank 4 to 10 |
|---|---|---|---|---|
| NN-score | 82% | 10% | 3% | 5% |
| NN-all | 48% | 12% | 7% | 33% |
| NN-combined | 42% | 11% | 8% | 39% |
| NN-noscore | 26% | 15% | 10% | 50% |
| NN-model | 57% | 14% | 5% | 25% |
The performance of Pcons is sustained on additional test sets
In addition to the jackknifed tests on the LiveBench-1 targets, two additional test sets were used: CASP4 and the LiveBench-2 set (Bujnicki et al. 2001b). In the CASP4 set as well as for some of the LiveBench targets we had no predictions from Sam-T98, as it has been replaced by Sam-T99. To compensate for this we retrained the networks without Sam-T98 on the LiveBench-1 targets. In these studies NN-model was used, because it is implemented as a part of the metaserver (http://bioinfo.pl/meta/).
In the automatic evaluation of the CAFASP2 results another method, MaxSub (Siew et al. 2000) was used to evaluate the targets. Using this evaluation Pcons did not perform significantly better than the best individual server. Pcons predicted 6 (out of 26) models correctly, with a total MaxSub score of 11.1. FFAS also predicted 6 models correctly with a total MaxSub score of 11.6. However, it should be noted that for 18 of the 26 targets none of the individual methods made a correct first-ranked prediction, that is, there are only 8 targets where the consensus predictor could possibly make a correct prediction. The main reason for Pcons not to perform better than FFAS is mainly attributable to one case (T0110), where the consensus prediction made a suboptimal choice by selecting the first model from Inbgu instead of a more accurate model from 3D-PSSM or FFAS, which was the second choice of the consensus predictor.
The LiveBench-2 targets were obtained in a similar way as for LiveBench-1 but during the next period of nine months. The consensus predictor performed better than any individual server, predicting the correct structure for 33% of the hard targets. The second best performance was obtained by 3D-PSSM, which predicted 30% of those targets correctly. More important, the specificity of Pcons is significantly better than for any of the individual servers on this test set; for instance, 82 correct models were found before the fifth incorrect model, whereas the second best server (mGenTHREADER) detected 54 (Bujnicki et al. 2001b). LiveBench is continuously measuring the performance of different web servers, and all data are available at http://bioinfo.pl/livebench/. It should be noted that additional servers could be included in future versions of the consensus predictor Pcons as a few new servers performed quite well on LiveBench-2. The second generation of Pcons based on 3D-PSSM, fugue, mGenTHREADER, FFAS, PDBBLAST, Inbgu, and Sam-T99 performs significantly better than the first generation (see http://bioinfo.pl/livebench/3/).
Conclusion
From this study it is evident that a combination of several servers improves the performance of a fold recognition protocol. More correct models are ideied, and the specificity is higher. We believe that there are three possible explanations for the improvements obtained with Pcons. (1) The structural similarity between top hits (models) used here is not sufficiently used by earlier fold recognition servers. From earlier CASP experiments, it has been a common practice among prediction teams to take into account several models obtained with any method, and select the most common fold among the top predictions. Pcons follows this strategy in an automated manner, and to our knowledge this has not been implemented in any single fold recognition method yet. As NN-score does not perform significantly better than the best individual servers, this is obviously the most important contribution. We believe it is likely that the results from individual servers can be improved by using a scoring dependent on the score for all sequence–template pairs of the same fold. (2) The consensus predictor uses multiple alignments for each target template pair. By using several alignments it is possible that one of them captures the important features of the correct structure. For instance, in some cases it might be better to use predicted secondary structures and in some not. As Pcons can choose models made by different techniques for different targets it might be able to use the best predictions of each server. An indication that this is true is that NN-score performs slightly better than the best server for all targets. (3) Pcons normalizes the scores so that they relate better to the quality of the model. Pcons is trained to predict the quality of the models and not simply idey if the correct fold is found or not. The advantage of this can be seen when the output from GenTHREADER is analyzed. GenTHREADER uses a network that is trained to separate correct and incorrect hits. This results in the confidence score of GenTHREADER being only marginally higher for an absolutely correct model than for a borderline case. Because Pcons is trained to predict the quality of the model its output would differ a lot between these two models.
Materials and methods
Test and training data
The LiveBench-1 set
This study is based on 125 different structure-prediction targets and 6 different servers producing up to 10 different models each. The input data come from the LiveBench program, which resulted in 125 targets submitted in the period between October 29, 1999 and April 6, 2000. The target proteins were divided into 30 easy targets (EASY category) and 95 difficult targets (HARD category). Per definition, easy targets were correctly predicted by PDBBLAST (Bujnicki et al. 2001a) with an expectation (E) value <10−5. This performance of the different servers using this set of data has been analyzed carefully in an earlier study (Bujnicki et al. 2001a).
Additional test sets
For additional tests we have used two other sets of targets. First, we have used the 26 hard targets that were evaluated in the fold recognition part of CAFASP2 (Siew et al. 2000). The easy targets were ignored as almost all the servers predicted these correctly. CAFASP is an evaluation of automatic servers that runs in parallel with CASP (Moult et al. 1999). Second, we have used a new LiveBench test set created between April 13, 2000 and December 31, 2000, termed LiveBench-2. This set contained 203 targets (Bujnicki et al. 2001b).
Measure of model/template similarity
To measure the similarity between models we used the LGscore algorithms (Cristobal et al. 2001). The LGscore is based on a method recently introduced by Levitt and Gerstein (1998) to calculate the significance of the similarity between two structures after a structural superposition. This measure is based on the following score:
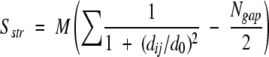
where M = 20, d ij is the distance between residues i and _j, d_0 = 5 L. and N gap is the number of gaps in the alignment.
To calculate the statistical significance of this score, Levitt and Gerstein (1998) used a set of structural alignments of unrelated proteins to calculate a distribution of _S_str dependent on the alignment length l. From this distribution a _P_-value dependent on _S_str and l was calculated.
Often, even though the fold is correctly predicted, the alignment is suboptimal. It is possible to ignore this problem if the model is superimposed with the correct structure before the evaluation. After the superposition, the structurally aligned residues are considered equivalent. From these equivalences it is possible to detect the most significant subset using the same algorithms as described above. In this study the superposition was made using a modified version of the algorithm used by Levitt and Gerstein (1998). After superposition, the most significant subset was found as for LGscore. This measure, termed LGscore2, is used to idey correct models in this study. In the NN-model as well as in the web server the alignment-dependent LGscore algorithm was used as it is much faster.
Evaluation of model quality
We used LGscore2 as the measure to evaluate model accuracy as has been done earlier in CAFASP and LiveBench (Bujnicki et al. 2001a). LGscore2 measures the quality of a model by finding the most significantly similar segment common to the model and the correct structure. In earlier studies we have shown that LGscore2 correlates quite well with the manual assessment by Murzin on CASP3 data (Cristobal et al. 2001). The correlation is also strong with several other measures used to evaluate model quality, such as GDT (Zemla et al. 1999), MaxSub (Siew et al. 2000), and sf4 (Lackner et al. 1999). None of these measures is ideal; however, it is our belief that the correlation is strong enough that it does not matter much which measure is used as a target function for the networks. An indication of this can be seen in the results from LiveBench-2, where Pcons performs equally well using MaxSub, LGscore, or LGscore2 for the model evaluation.
Model and template comparisons
The basis for the consensus predictor is six publicly available web servers—PDBBLAST, FFAS, Inbgu, GenTHREADER, Sam-T98, and 3D-PSSM. For each of the servers the top 10 hits were converted to pdb models, that is, for each query sequence up to 60 different models were created and the structures of the template proteins used were recorded. All models for the same target were compared with each other using the LGscore2 algorithm (Bujnicki et al. 2001a; Cristobal et al. 2001; Elofsson 2001). The templates were also compared to each other using LGscore2. If two protein structures (either models or templates) had an LGscore2 of 10−3 or better, they were considered similar.
For each model i, three variables were calculated. First, the fraction of other models exhibiting significant similarity according to LGscore2, that is:
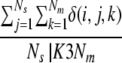
where _N_s is the number of servers, _N_m the number of models from a particular server, and δ(i,j,k) a function that is 1 if model i is similar to the model of rank k from server j and 0 otherwise.
Second, the fraction of models similar to this model and with a significant method score:
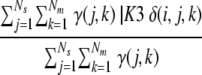
where γ(j,k) is a function that is 1 if the model with rank k from server j is higher than a cutoff (see Table 6) and 0 otherwise.
Table 6.
Description of the different servers that are the basis for the neural network used in this study and the proposed cut-off used
| Method | Cut-off |
|---|---|
| PDBBLAST | <0.1 |
| FFAS | >8 |
| 3D-PSSM | <0.37 |
| GenTHREADER | >0.7 |
| Sam-T98 | >20 |
| Inbgu | >12 |
Finally, the fraction of first-ranked models was measured as:
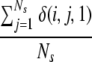
Three additional variables were calculated in the same way for the templates. These six variables, together with the scores taken from the individual methods, were fed into the network (see Fig. 1 ▶). Different combinations of these similarity measures were examined as described in Table 1.
In NN-model the similarities were measured differently. First, all template comparisons were ignored. Second, the LGscore algorithm was used for the model comparisons. This results in a method that is significantly faster than the other methods as no structural superpositions have to be made.
Neural networks
For the neural network implementations we used Netlab, a neural network package for Matlab (Bishop 1995; Nabney and Bishop 1995; Netlab: Netlab neural network software. http://www.ncrg.aston.ac.uk/netlab/). A linear activation function was chosen because it did not carry restrictions on the range of the output. The training was carried out using error back-propagation with a sum of square error function. The optimal gradient algorithm was used for training.
The neural network is built in two layers (see Fig. 1 ▶). The first layer consists of only one network for each method. The output from this neural network is fed into a final neural network and is thereby normalized. For a given output, the second-layer network will obviously receive input from one of the top networks. The second network is not really necessary as all the first-layer networks already are trained to predict the LGscore2. However, we have used it because it slightly improved the performance. We also tried to add additional information into this network, such as a threading score, but that did not increase the performance significantly.
The minimization of the error function (training) should be done with the optimal number of hidden nodes and training cycles to avoid overtraining and to minimize the training time. Method-NNs (first layer) were trained in 150–500 cycles with seven or nine hidden nodes, and the final-NN (second layer) was trained in 200 cycles with five hidden nodes. The magnitude of the error sum in the test and training set is monitored in each cycle of the training (Emanuelsson et al. 1999). The ultimate number of cycles is determined, when the error sum for the test set stops decreasing and starts to increase. It should be noted that both the number of hidden nodes and the number of training cycles are decided at one time before the rest of the experiment is carried out.
A key point for the performance of Pcons was to decide the parameter to which the networks were trained. We found that if the networks were trained to predict log(LGscore2), the results were significantly better than if trained on LGscore2 directly.
The neural network was trained using a fourfold cross-validation. For the additional test sets (CAFASP2 and LiveBench-2) the whole LiveBench-1 data set was used to train the network.
Acknowledgments
This work was supported by grants from the Swedish Natural Sciences Research Council, the Swedish Research Council for Engineering Sciences (TFR), and the Strategic Research Foundation (SSF). We are also grateful for comments made by Daniel Fischer, Bob MacCallum, and Gunnar von Heijne. It should also be noted that because the success of the consensus predictions would not be possible without the existence of the other servers, we are very grateful for their existence.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.08501.
References
- Abagyan, R.A. and Batalov, S. 1997. Do aligned sequences share the same fold? J. Mol. Biol. 273 355–368. [DOI] [PubMed] [Google Scholar]
- Alexandrov, N.N. and Luethy, R. 1998. Alignment algorithm for homology modeling and threading. Protein Sci. 7 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, Christopher M. 1995. Neural networks for pattern recognition. Oxford University Press, Oxford, UK.
- Brenner, S.E., Chothia, C., and Hubbard, T.J. 1998. Assessing sequence comparison methods with reliable structurally ideied distant evolutionary relationships. Proc. Natl. Acad. Sci. 95 6073–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujnicki, J.M., Elofsson, A., Fischer, D., and Rychlewski, L. 2001a. LiveBench-1: Continuous benchmarking of protein structure prediction servers. Protein Sci. 10 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2001b. Livebench-2: Large-scale automated evaluation of protein structure prediction servers. Protein Sci. (in press). [DOI] [PubMed]
- Cristobal, S., Zemla, A., Fischer, D., Rychlewski, L., and Elofsson, A. 2001. A study of quality measures for protein threading models. BMC Bioinformatics 2 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Francesco, V., Geetha, V., Garnier, J., and Munson, P.J. 1997. Fold recognition using predicted secondary structure sequences and hidden Markov models of protein folds. Proteins Suppl 1: 123–128. [DOI] [PubMed] [Google Scholar]
- Domingues, F.S., Lackner, P., Andreeva, A., and Sippl, M.J. 2000. Structure-based evaluation of sequence comparison and fold recognition alignment accuracy. J. Mol. Biol. 297 1003–1013. [DOI] [PubMed] [Google Scholar]
- Elofsson, A. 2001. A study on how to best align protein sequences. Proteins (in press).
- Emanuelsson, O., Nielsen, H., and von Heijne, G. 1999. ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci. 8 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, D. 2000. Hybrid fold recognition: Combining sequence derived properties with evolutionary information. In Pacific symposium on biocomputing (eds. R.B. Altman et al.), vol. 5, pp. 116–127. World Scieic. [PubMed]
- Fischer, D. and Eisenberg, D. 1996. Protein fold recognition using sequence-derived predictions. Protein Sci. 5 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribskov, M., McLachlan, A.D., and Eisenberg, D. 1987. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. 84 4355–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley, C. and Jones, D.T. 1999. A systematic comparison of protein structure classifications: SCOP, CATH and FSSP. Structure Fold Des. 7 1099–1112. [DOI] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. A new approach to protein fold recognition. Nature 358 86–89. [DOI] [PubMed] [Google Scholar]
- Karplus, K., Barrett, C., and Hughey R. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics 14 846–856. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Lackner, P., Koppensteiner, W.A., Domingues, F.S., and Sippl, M.J. 1999. Automated large scale evaluation of protein structure predictions. Proteins 37 (S3): 7–14. [DOI] [PubMed] [Google Scholar]
- Lindahl, E. and Elofsson, A. 2000. Ideication of related proteins on family, superfamily and fold level. J. Mol. Biol. 295 613–625. [DOI] [PubMed] [Google Scholar]
- Moult, J., Hubbard, T., Fidelis, K., and Pedersen, J.T. 1999. Critical assessment of methods of protein structure prediction (CASP): Round III. Proteins 37 (S3): 2–6. [PubMed] [Google Scholar]
- Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48 443–453. [DOI] [PubMed] [Google Scholar]
- Panchenko, A.R., Marchler-Bauer, A., and Bryant, S.H. 2000. Combination of threading potentials and sequence profiles improves fold recognition. J. Mol. Biol. 296 1319–1331. [DOI] [PubMed] [Google Scholar]
- Park, J., Teichmann, S.A., Hubbard, T., and Chothia, C. 1997. Intermediate sequences increase the detection of homology between sequences. J. Mol. Biol. 273 349–354. [DOI] [PubMed] [Google Scholar]
- Park, J., Karplus, K., Barrett, C., Hughey, R., Haussler, D., Hubbard, T., and Chothia, C. 1998. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 284 1201–1210. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. 1995. Comparison of methods for searching protein sequence databases. Protein Sci. 4 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, D.W. and Eisenberg, D. 1997. A 3D-1D substitution matrix for protein fold recognition that includes predicted secondary structure of the sequence. J. Mol. Biol. 267 1026–1038. [DOI] [PubMed] [Google Scholar]
- Rost, B., Schneider, R., and Sander, C. 1997. Protein fold recognition by prediction-based threading. J. Mol. Biol. 270 471–480. [DOI] [PubMed] [Google Scholar]
- Rychlewski, L., Jaroszewski, L., Li, W., and Godzik, A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 9 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamov, A.A., Suwa, M., Orengo, C.A., and Swindells, M.B. 1999. Genome analysis: Assigning protein coding regions to three-dimensional structures. Protein Sci. 8 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, R. and Sali, A. 1998. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. 95 13597–135602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauder, J.M., Arthur, J.W., and Dunbrack, R.L., Jr. 2000. Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins 40 6–22. [DOI] [PubMed] [Google Scholar]
- Siew, N., Elofsson, A., Rychlewski, L., and Fischer, D. 2000. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 16 776–785. [DOI] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Ideication of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Zemla, A., Venclovas, C., Moult, J., and Fidelis, K. 1999. Processing and analysis of CASP3 protein structure predictions. Proteins Suppl 3: 22–29. [DOI] [PubMed] [Google Scholar]