Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation (original) (raw)
Abstract
The proteasome is responsible for degradation of substrates of the ubiquitin pathway. 20S proteasomes are cylindrical particles with subunits arranged in a stack of four heptameric rings. The outer rings are composed of α subunits, and the inner rings are composed of β subunits. A well-characterized archaeal proteasome has a single type of each subunit, and the N-terminal threonine of the β subunit is the active-site nucleophile. Yeast proteasomes have seven different β subunits and exhibit several distinct peptidase activities, which were proposed to derive from disparate active sites. We show that mutating the N-terminal threonine in the yeast Pup1 β subunit eliminates cleavage after basic residues in peptide substrates, and mutating the corresponding threonine of Pre3 prevents cleavage after acidic residues. Surprisingly, neither mutation has a strong effect on cell growth, and they have at most minor effects on ubiquitin-dependent proteolysis. We show that Pup1 interacts with Pup3 in each β subunit ring. Our data reveal that different proteasome active sites contribute very differently to protein breakdown in vivo, that contacts between particular subunits in each β subunit ring are critical for active-site formation, and that active sites in archaea and different eukaryotes are highly similar.
The 20S proteasome is an abundant intracellular protease that has been implicated in many biological regulatory mechanisms. Association of 20S proteasomes with a 19S regulatory complex creates the 26S proteasome, which degrades proteins in an ATP-dependent and ubiquitin-dependent manner (1). The proteasome also participates in the degradation of misfolded or damaged proteins and in the generation of peptides for the major histocompatibility complex class I antigen presentation pathway in mammals (1, 2).
In the archaeon Thermoplasma acidophilum, a 20S proteasome has been described that has only two kinds of subunits, α and β, with seven α subunits in each of the outer two rings and seven β subunits in each inner ring (2). All eukaryotic 20S proteasome subunits are related in sequence to the archaeal subunits. In yeast, there are 14 genes encoding 20S proteasome subunits, and proteasomes form a uniform population in which each particle has the same 14 different subunits (3). The Thermoplasma proteasome has a single type of active site, whereas the eukaryotic particle has at least three (2). The archaeal enzyme efficiently cleaves model peptides only after hydrophobic residues (a “chymotrypsin-like” activity); the eukaryotic protease can, in addition, cleave peptides after basic (“trypsin-like” activity) and acidic [“peptidylglutamyl peptide hydrolyzing” (PGPH) activity] residues.
Solution of the Thermoplasma proteasome crystal structure revealed that the active sites are in the β subunits within a central chamber of the particle (4). Each β subunit is synthesized with a short N-terminal propeptide; the hydroxyl of the N-terminal Thr residue of the processed β subunit is responsible for nucleophilic attack on substrates (4, 5). Recently, it has been shown that the yeast Saccharomyces cerevisiae 20S proteasome is likely to use the same catalytic mechanism (6). Consistent with this, when mammalian proteasomes are treated with lactacystin, an irreversible proteasome inhibitor, one of the two rapidly modified sites in the β subunit X/MB1 is the N-terminal Thr (7). Interestingly, only three of the seven different β type subunits in a given eukaryotic 20S particle appear to have all the residues required for formation of such an N-terminal active site (5).
Earlier work from this laboratory (3) led to a model in which we proposed that interactions between specific subunits across the central β subunit rings were critical for establishment of individual proteasome active sites. This model received strong support from both the Thermoplasma proteasome-inhibitor structure (4) and recent experiments in yeast (6). However, it seems likely that specific interactions between heterologous β subunits within each β subunit ring will also be important for active site formation (8). Dick et al. (9) identified a specific Cys residue in the C10 β subunit of the bovine proteasome whose modification by _N_-ethylmaleimide (NEM) specifically inhibited trypsin-like activity; the residue in the archaeal enzyme that apparently corresponds to this Cys is at the interface between subunits within the β subunit ring (8). The C10 subunit does not have the N-terminal Thr residue shown to be required for catalysis in the Thermoplasma proteasome, so C10 modification by NEM may interfere with substrate binding or cleavage in a neighboring subunit in the same ring.
Here we show that the yeast proteasome is also specifically impaired in trypsin-like activity when treated with NEM. The yeast subunit most closely related to C10 is Pup3, which has the Cys corresponding to the critical NEM-modified C10 Cys. Our mutagenesis data indicate that Pup1 supplies the active site Thr nucleophile for the trypsin-like sites. By pseudoreversion analysis, we show that Pup1 is in direct contact with Pup3 in the β subunit ring. Disruption of this contact results in proteasomes with reduced trypsin-like activity, supporting the idea that intersubunit contacts in the β rings are important for formation of specific active sites. Unexpectedly, loss of the trypsin-like sites results in only minor defects in intracellular proteolysis and cell growth. Moreover, mutation of the putative active site Thr of Pre3 specifically blocks PGPH activity and has no obvious effect on growth. This contrasts with the major contribution of the chymotrypsin-like active sites to ubiquitin-dependent proteolysis and growth (6).
MATERIALS AND METHODS
Yeast and Bacterial Media and Methods.
Yeast rich (YPD) and minimal (SD) plates were prepared as described (10), and standard methods were used for genetic manipulation of yeast. Escherichia coli strains used were MC1061 and JM101, and standard methods were employed for recombinant DNA work.
Construction of New Proteasome Alleles.
A plasmid carrying the genomic PUP1 DNA sequence, pPHB2, was provided by T. Fox (11); the 2.3-kb _Xba_I fragment was excised from pPHB2 and subcloned into pRS316 (12). The PUP3 gene was isolated from a PCR with yeast genomic DNA, and a 1.6-kb _Eco_RI fragment bearing the PUP3 gene was subcloned into pRS316. Mutant alleles of PUP1 and PUP3 were generated by a two-step PCR procedure, starting with plasmid templates encoding the relevant wild-type genes. _Eco_RI-cut DNA fragments carrying the mutant alleles were cloned into low- and high-copy plasmids (13). An analogous approach was used to make pre3-T20A. Peptide-tagged derivatives of Pup1 proteins were made as follows. A triplicated hemagglutinin (HA) epitope-encoding DNA fragment was amplified by PCR from a plasmid template (14) with primers that also added _Xho_I sites. PCR products were digested with _Xho_I and inserted into the unique _Xho_I site in YCplac22PUP1 or YCplac22pup1-T30A. The proteins encoded by these plasmids have a 41-residue in-frame insertion.
Generation of Yeast Strains.
For making a strain with a chromosomal disruption of PUP1, a plasmid bearing the pup1::LEU2 allele (11) was cut with _Xba_I, and the digested DNA was transformed into MHY606 diploid cells. The pRS316PUP1 plasmid was introduced into one of the resulting heterozygous pup1::LEU2 disruptants, and haploid pup1::LEU2 (pRS316PUP1) segregants were isolated by tetrad dissection (PUP1, like all the β subunit genes, is essential for viability) (Table 1). To make a null allele of PUP3, the HIS3 gene was amplified with primers with 5′ sequence segments matching the beginning and end, respectively, of the PUP3 open reading frame (15); the amplified fragment was transformed into MHY606. To obtain haploid cells with a chromosomal _pup3-Δ_2::HIS3 allele, the heterozygous diploid was transformed with pRS316PUP3, cells were sporulated, and tetrads were dissected. To make MHY998, MHY991 cells were transformed with _Xho_I, _Hpa_I-cut pLH7 (obtained from F. Cross) and Leu− His+ transformants were identified. The leu2::HIS3 allele from pLH7 converted the pup1::LEU2 allele in MHY991 to pup1::leu2::HIS3. MHY1004, a strain in which the chromosomal copies of both PUP1 and PUP3 were inactivated, was obtained from a cross between MHY996 and MHY998.
Table 1.
Yeast strains
| Strain | Genotype |
|---|---|
| MHY50010 | MATa _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 |
| MHY50110 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 |
| MHY6063 | MHY500 × MHY501 |
| MHY9526 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 doa3-Δ1::HIS3 [YEpDOA3LS][YCpUbDOA3ΔLS] |
| MHY9736 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 doa3-Δ1::HIS3 [YEpDOA3LS][YCpUbdoa3ΔLS-T76A] |
| MHY991 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::LEU2 [pRS316PUP1] |
| MHY996 | MATa _his3-_Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup3-Δ_2::HIS3 [pRS316PUP3] |
| MHY998 | _MAT_α _his3-Δ_200 leu2-3,112::HIS3 ura3-52 lys2-801 trp1-1 pup1::leu2::HIS3 [pRS316PUP1] |
| MHY1004 | MATa _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::leu2::_HIS3 pup3-Δ_2::HIS3 [pRS316PUP1][pRS316PUP3] |
| MHY1066 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::LEU2 [YCplac22PUP1] |
| MHY1071 | MATa _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::leu2::_HIS3 pup3-Δ_2::HIS3 [YCplac22pup1-K58E][YEplac181pup3-E151K] |
| MHY1072 | MATa _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::leu2::_HIS3 pup3-Δ_2::HIS3 [YCplac22PUP1][YEplac181PUP3] |
| MHY1073 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::LEU2 [YCplac22pup1-T30A] |
| MHY1079 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::LEU2 [YCplac22HA-PUP1] |
| MHY1144 | _MAT_α _his3-Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pup1::LEU2 [YCplac22HApup1-T30A] |
| MHY1156 | _MAT_α _his3-_Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre3-Δ_2::HIS3 [YCplac22PRE3] |
| MHY1157 | _MAT_α _his3-_Δ_200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre3-Δ_2::HIS3 [YCplac22pre3-T20A] |
Plasmids carrying various pup1, pup3, or pre3 alleles were introduced into the appropriate yeast strains, and transformants were then plated on 5-fluoroorotic acid, which is toxic to _URA3_-expressing cells, to identify cells that had lost the _URA3_-bearing pRS316PUP1 and/or pRS316PUP3 plasmids or pRS316PRE3 plasmid.
Pulse–Chase and Immunoblot Analyses.
Pulse–chase analysis was carried out as described (10). Immunoprecipitations were done with anti-β-galactosidase antibodies (Cappel) or an anti-α2 antibody (10). Immunoblot analyses were performed as described (3). For detection of HA-tagged proteins, the 12CA5 monoclonal antibody (Babco, Richmond, CA) was used, followed by ECL detection (Amersham). The 20S proteasome antigens were detected with purified antibodies against yeast 20S proteasomes (a gift of K. Tanaka, University of Tokyo) followed by 125I-labeled protein A (ICN) and quantitated with a Molecular Dynamics PhosphorImager.
Protein Fractionation on Glycerol Density Gradients and Peptide Hydrolysis Assays.
Yeast extracts were prepared and glycerol gradient fractionation was performed essentially as in ref. 6, except that cells were grown in YPD, and lysis buffer contained 20 mM glucose and hexokinase (2.6 units/ml) to deplete ATP. Protein concentrations were determined by the Bradford method. Peptide hydrolysis by purified yeast 20S proteasomes was measured as described (3). _N_-CBZ-Gly-Gly-Leu-_p_-nitroanilide was used for testing chymotrypsin-like, _N_-CBZ-Leu-Leu-Glu-β-naphthylamide was used for PGPH, and _N_-CBZ-Ala-Arg-Arg-4-methoxy-β-naphthylamide was used for trypsin-like activities (where CBZ is carboxyl benzyl). The reactions were carried out at 30°C for 1 h with 50 μl of gradient fraction or 2 μg of purified enzyme.
RESULTS
Thr-30 of Pup1 and Thr-20 of Pre3 Are Essential for Specific Peptidase Activities.
Two yeast β-type subunits, Doa3 and Pre3, were shown to be synthesized with N-terminal propeptides (3), and a third, Pup1, was predicted to be processed by a closely related mechanism (3, 5). The N-terminal residues of the mature forms of these subunits are Thr residues and were predicted to provide the catalytic centers for the three major peptidase activities of the yeast proteasome (8). This prediction was fulfilled for Doa3, which bears the N-terminal Thr required for chymotrypsin-like activity (6).
Mutation of the predicted Pre3 active site Thr to Ala specifically eliminated PGPH activity (Fig. 1A). We then asked whether Pup1, the remaining β subunit with a predicted N-terminal Thr nucleophile, might be responsible for proteasomal trypsin-like activity. As shown in Fig. 1A, trypsin-like activity was reduced to background levels in pup1-T30A proteasomes, and chymotrypsin-like and PGPH activities were both ≈50% higher than in wild-type fractions. Quantitative immunoblot analysis using 125I-labeled protein A and anti-20S proteasome antibodies revealed that proteasome levels were 40–50% higher in the peak gradient fractions from pup1 extracts relative to wild-type or pre3 extracts despite similar total protein levels (data not shown). This could account for the ≈50% increase in chymotrypsin-like and PGPH activities seen in the pup1-T30A mutant.
Figure 1.
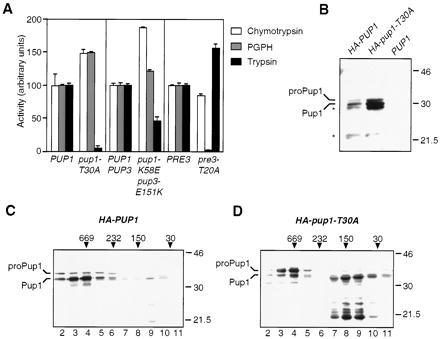
Effect of mutating the putative active-site Thr residues in Pup1 and Pre3 on proteasome function. (A) Peptidase activities of glycerol-gradient-purified proteasomes. Values are the mean of three measurements; error bars indicate standard deviations. (B) Anti-HA immunoblot analysis of spheroplast extracts of cells expressing HA-tagged proteins. Cells expressing untagged Pup1 were used as a control. The expected sizes for HA-proPup1 and HA-Pup1 are 33 kDa and 30 kDa, respectively, consistent with the sizes observed in this 10% gel. In higher percentage gels (e.g., C and D), the mobilities of the Pup1 derivatives suggested apparent masses slightly higher than predicted; the reason for this behavior is unknown. Asterisks mark apparent proteolytic fragments of HA-tagged proteins. Whether the protein that comigrates with the Pup1-sized species in the mutant is correctly processed is unknown (see D). Size standards are marked (in kDa). (C) Anti-HA immunoblot analysis of glycerol gradient-fractionated extract from HA-PUP1 cells. Positions of size standards in the gradient are indicated (in kDa) above the gel. Twelve 1-ml fractions were collected; the 20S proteasome activity peak is in fraction 4. (D) Same as C except the extract was derived from HA-pup1-T30A cells. The majority of HA-reactive protein migrated below the size of the full-length protein in fractions 7–10, and these species were also seen in HA-PUP1 extracts; they appear to be proteolytic fragments derived from full-length Pup1. It is not known if any of this proteolysis results from the HA insertion. The Pup1 in these fractions may be in proteasome assembly intermediates.
Effect of pup1-T30A on Propeptide Processing.
By analogy to the analysis of Doa3 (6), the pup1-T30A protein was expected to be defective for autocatalytic propeptide processing. To test this, we constructed a PUP1 derivative encoding a protein with a triplicated HA-epitope inserted into a region of Pup1 expected to face the proteasome exterior, based on the Thermoplasma proteasome structure. The HA-PUP1 allele rescued growth of a _pup1_Δ strain, although the cells grew slightly slower than wild-type cells; therefore, the HA-Pup1 protein was functional. An allele encoding a mutant pup1-T30A protein with the same HA tag resulted in slightly poorer growth than seen for HA-PUP1 cells.
Extracts were prepared from HA-PUP1 and HA-pup1-T30A cells and examined directly or fractionated on 10–40% glycerol gradients followed by immunoblot analyses using an anti-HA antibody (Fig. 1 B–D). A species whose size matched that of the predicted full-length HA-Pup1 protein was detected (Fig. 1B), but the bulk of HA-Pup1 appeared as a smaller species whose mass was consistent with that predicted for the protein if it lacked the 29-residue propeptide. Mutation of Thr-30 to Ala inhibited this processing (Fig. 1D). However, failure to remove the Pup1 propeptide did not prevent incorporation of the subunit into 20S particles; small amounts of unprocessed subunit were also observed in 20S particles from HA-PUP1 cells. A significant fraction of Pup1 from wild-type and especially mutant cells was not incorporated into 20S particles. (Much of this unincorporated protein appeared to have been proteolyzed during spheroplasting and fractionation to species smaller than correctly processed Pup1.) There was also a marked increase in total Pup1 levels when the processing site was mutated.
Effect of pup1-T30A and pre3-T20A on in Vivo Proteolysis.
Surprisingly, the pup1-T30A and pre3-T20A mutations caused little or no impairment of ubiquitin-dependent protein degradation in vivo despite the inactivation of the trypsin-like and PGPH sites, respectively. Degradation of the naturally short-lived MATα2 repressor (10) in pup1-T30A cells, for example, was inhibited by <15%, a difference that was within experimental error (Fig. 2A), and a similar lack of inhibition was seen with the α2 derivative _Deg1_α2-β-galactosidase (_Deg1_α2-βgal) (Fig. 2C). Leu-βgal, an artificial substrate of the N-end rule pathway (16), was degraded slightly slower than in wild-type cells (Fig. 2B), but another test substrate, ubiquitin-Pro-βgal, was not significantly affected (data not shown). Degradation of α2 occurred at wild-type rates in the pre3-T20A strain (Fig. 2D). For comparison, inactivation of the chymotrypsin-like sites in the Doa3 subunits caused a major reduction (≈6-fold) in MATα2 turnover (6).
Figure 2.

Ubiquitin-dependent proteolysis in pup1-T30A, pup1-K58E pup3-E151K, and pre3-T20A cells. (A) MATα2 degradation in pup1-T30A cells at 30°C. The half-life for α2 was ≈4 min in both wild-type (MHY1066) and mutant (MHY1073) cells. (B) Leu-βgal degradation in pup1-T30A cells. (C) _Deg1_α2-βgal degradation in pup1-T30A and pup1-K58E pup3-E151K cells. (D) MATα2 degradation in pre3-T20A cells. The half-life for α2 was slightly under 5 min in both wild-type (MHY1156) and mutant (MHY1157) cells.
Correspondingly mild effects on the growth and stress resistance of pup1-T30A cells were observed (Fig. 3), and no growth defects were seen for pre3-T20A cells under these same conditions (data not shown). This too contrasts with the severely impaired growth properties of doa3 mutants lacking chymotrypsin-like activity. Unexpectedly, the pup1-T30A mutant was more resistant to certain stresses. The ability to form colonies on 30 μM CdCl2 increased by at least 10-fold relative to otherwise isogenic wild-type cells (Fig. 3D). A small but reproducible increase in growth at 37.5°C was also detected with the mutant (Fig. 3B).
Figure 3.

Phenotypes of proteasome mutants. Exponentially growing cultures (in YPD) were plated in 1:10 serial dilutions on the indicated medium and incubated for 2 days (A) or 3 days (B–D). The yeast strains used were (from the top) MHY952, MHY973, MHY1066, MHY1073, MHY1072, and MHY1071. In strains MHY952 and MHY973, the Doa3 propeptide is expressed separately from the mature domain to circumvent the near-lethality associated with a failure to process proDoa3 with a T76A mutation (6). The MHY952 and MHY1073 strains both show an increase in Cd resistance.
Pup1 and Pup3 Are Neighbors in the β Subunit Rings.
As noted earlier, NEM modification of a specific Cys residue in the bovine homolog of the Pup3 β subunit, C10, specifically inhibits the trypsin-like activity of the 20S proteasome (9) even though C10/Pup3 is not predicted to contain a protease catalytic site. This NEM modification could be blocked by preincubation with leupeptin, a competitive inhibitor of the trypsin-like activity (9, 17). We first determined whether yeast 20S proteasomes were also inhibited by NEM. Treatment of purified proteasomes with 5 mM NEM almost completely eliminated the trypsin-like activity, but PGPH and chymotrypsin-like activities were unaffected (Fig. 4B). The Cys in C10 whose modification by NEM correlates with loss of trypsin-like activity in bovine proteasomes is conserved in yeast (Fig. 4A), suggesting that modification of this Cys in Pup3 is responsible for inactivation of the trypsin-like sites in the yeast enzyme.
Figure 4.

Putative Pup1–Pup3 interaction sites and effect of NEM on peptidase activities of purified proteasomes. (A) Pup1 and Pup3 β subunits in the regions surrounding the residues involved in the putative salt bridge (double arrow) and the region of Pup3 with the conserved Cys residue (arrowhead) whose modification by NEM in bovine C10 inhibits trypsin-like activity. Secondary structures expected based on the Thermoplasma structure (5) are also shown: s, β-strand; h, helix. (B) Inhibition by NEM of peptidase activities of purified yeast 20S proteasomes. Values are the mean of three measurements. Purified proteasomes were incubated with 5 mM NEM for 20 min at 30°C.
Because the catalytic residues for the trypsin-like sites reside in the Pup1 subunits (Fig. 1A), the inhibition of trypsin-like activity by NEM would be predicted to be due to perturbation of the active-site pocket in Pup1. The residue in the Thermoplasma proteasome that apparently corresponds to the NEM-modified Cys of C10 is at the interface between subunits within each β subunit ring. Therefore, we guessed that Pup1 and Pup3 might be direct neighbors. Previous pseudoreversion studies in yeast that were based on the Thermoplasma proteasome structure were used to demonstrate a direct interaction of Doa3 with Pre1 between the different β subunit rings that is critical for formation of the chymotrypsin-like sites (6). We used an analogous strategy to test for an interaction between Pup1 and Pup3. A salt bridge is observed between Lys-29 of each Thermoplasma β subunit with Glu-139 in each neighboring subunit in the same ring. Sequence alignments with eukaryotic β subunits suggested that this interaction might be conserved in a subset of proteasome pairings. For the putative Pup1-Pup3 contact, the salt bridge was predicted to be formed between Lys-58 of Pup1 (numbering refers to the unprocessed subunit) and Glu-151 of Pup3 (Fig. 4A). We reasoned that if this electrostatic interaction existed, a Lys → Glu mutation in Pup1 or a Glu → Lys mutation in Pup3 might be strongly deleterious, but proteasome function might be at least partially restored in a strain carrying both mutations.
Low- or high-copy plasmids carrying the pup1-K58E and pup3-E151K alleles were transformed into yeast strains bearing chromosomal deletions of either or both PUP1 and PUP3, with the corresponding wild-type genes present on plasmids that also carried the URA3 gene. Transformants were then plated on 5-fluoroorotic acid, a compound toxic to cells expressing URA3, to determine if colonies were able to form in the absence of the wild-type proteasome gene(s). Mutant pup1-K58E cells grew at wild-type rates at 30°C (Fig. 5) and grew essentially like wild-type cells at high temperature (37°C), low temperature (14°C), or in the presence of canavanine at 1 μg/ml. No defect in MATα2 degradation was seen, and glycerol gradient analysis revealed only a minor defect in trypsin-like activity (data not shown).
Figure 5.

Structure-based pseudoreversion analysis of the interaction between Pup1 and Pup3. MHY1004 cells transformed with the indicated plasmid-borne alleles were streaked on 5-fluoroorotic acid plates and grown at 30°C.
In contrast, the pup3-E151K allele did not allow colony formation even when expressed from a high-copy plasmid (Fig. 5). Strikingly, the pup1-K58E allele in either a low- or high-copy-number plasmid rescued the lethality of the pup3-E151K mutation, although growth was slower than that of wild-type cells (Figs. 3A and 5). This suppression was only seen when the pup3-E151K allele was on a high-copy-number plasmid, suggesting that the mutant protein was expressed poorly or did not fold or assemble into the proteasome as efficiently as its wild-type counterpart. To test whether suppression by pup1-K58E was allele-specific, we combined it with the pup3-G148L allele. The pup1-K58E pup3-G148L mutant failed to form colonies (Fig. 5); this synthetic lethality contrasted with the suppression of lethality seen with the pup1-K58E pup3-E151K double mutant. Because these pseudoreversion tests were structure-based, we conclude that Pup1 and Pup3 are neighbors within each central ring of the 20S proteasome.
Disruption of the Pup1–Pup3 Interface Inhibits Trypsin-Like Activity.
The slow growth of the pup1-K58E pup3-E151K strain suggested that the proteasomes in these cells were functionally perturbed. When glycerol-gradient-purified proteasomes from the mutant were examined, trypsin-like activity was slightly less than half that of wild-type particles (Fig. 1A). Chymotrypsin-like activity was consistently 50–80% above wild-type levels, which, as with the pup1-T30A fractionations, was likely due to proteasome levels in the peak gradient fractions being ≈50% above wild-type amounts based on quantitative immunoblots (data not shown). Although total PGPH activity was 20–30% above wild-type, this was less than the increase in either total proteasome levels or chymotrypsin-like activity, suggesting a slight defect in the specific activity of the PGPH sites in the mutant. The pup1-K58E pup3-E151K growth defect correlated with a defect in proteolysis. Degradation of the _Deg1_α2-βgal test substrate was markedly inhibited, which contrasted with the lack of a measurable defect in _Deg1_α2-βgal degradation in pup1-T30A cells (Fig. 2C).
DISCUSSION
We have shown here that the Pup1 subunit of the yeast proteasome provides the N-terminal Thr necessary for cleavage after basic residues in peptide substrates (trypsin-like activity) and Pre3 provides the Thr for cleavage after acidic residues (PGPH activity). Unlike loss of chymotrypsin-like activity by mutation of Doa3, loss of trypsin-like or PGPH activity is not associated with substantially decreased rates of ubiquitin-dependent protein degradation. Formation of the trypsin-like active sites depends on proper interaction between Pup1 and Pup3 in the same β subunit ring: NEM modification of a Pup3 residue at this interface abrogates trypsin-like activity, and perturbation of a salt bridge between these two subunits also inhibits the activity. These data provide insight into the subunit interactions important for formation of proteasome active sites and the different roles of these sites in intracellular proteolysis in vivo.
Proteasome Subunit Interactions and Active-Site Formation.
As noted earlier, there were reasons to suspect that the surfaces between β subunits within each central ring of the proteasome might help form the active sites. This has now been verified for the trypsin-like sites, which are specifically disrupted by perturbation of the Pup1–Pup3 interface. In this case, the residues directly involved in catalysis are all likely to be in Pup1, based on the archaeal proteasome structure and mutagenesis studies, but residues from Pup3 in the region facing Pup1 within the ring may help form the substrate binding pocket. Such a contribution would also be consistent with the severe effect on trypsin-like activity when a Pup3 Cys predicted to project toward the Pup1 active site is modified by NEM (Fig. 4). It follows from these findings and previous data (6) that each eukaryotic proteasome active site depends on a network of interactions between β subunits both within and between rings. This helps explain why proteolytic activity has never been observed with proteasome subparticles or individual subunits (18). It also suggests that proteasome cleavage specificity will depend strongly on subunits neighboring those bearing the residues contributing directly to catalysis.
We have now used a site-specific pseudoreversion strategy to map specific contacts between two pairs of subunits in the yeast proteasome. Previously, second-site suppressors were designed to relieve a predicted steric clash between bulky side chains of two subunits in mutant proteasomes (6). Here we manipulated charge–charge interactions to probe subunit associations. In each example, we not only established nearest neighbors but also ascertained the importance of distinct kinds of molecular interactions. The structure-based suppressor results, combined with the NEM inhibition data, also provide a compelling argument for close structural conservation among archaeal, yeast, and mammalian 20S proteasomes.
Proteasome Active Sites and Ubiquitin-Dependent Protein Degradation.
From the data in this paper and our earlier work (6), each of the three canonical peptidase activities associated with eukaryotic 20S proteasomes can now be assigned to particular N-terminal Thr-bearing β subunits. Mutation of the chymotrypsin-like sites in Doa3 correlates with a strong drop in ubiquitin-dependent proteolysis in vivo and with a significant growth defect (ref. 6 and Fig. 3). In contrast, elimination of either the trypsin-like sites by mutation of Pup1 or the PGPH sites by mutation of Pre3 results in only a minor defect in growth and minimal effects on ubiquitin-dependent proteolysis (Fig. 2).
The three kinds of eukaryotic proteasome active site might have been expected to make comparable contributions to protein breakdown except for potential differences in cleavage site preferences within substrates. As discussed above, however, the three known active centers of the yeast proteasome contribute very differently to at least the initial cleavages of protein substrates in vivo. Why might this be so? One possible explanation is that the chymotrypsin-like sites simply have the highest specific activity toward protein substrates. We also do not know how a substrate polypeptide chain is transported into or accommodated in the central proteasome chamber during degradation; it is possible that the chain is channeled preferentially to the chymotrypsin-like sites during processive protein breakdown.
These considerations notwithstanding, the chymotrypsin-like sites cannot be the only catalytic centers that contribute to ubiquitin-dependent proteolysis. When these sites are inactivated, the endogenous substrate MATα2 is still degraded with a half-life of ≈25 min (6), which is a smaller defect than observed with partial loss-of-function mutations in other proteasome components (19). The identification of a yeast mutant, pup1-K58E pup3-E151K, that has a strong defect in protein degradation yet high levels of chymotrypsin-like activity indicates that the latter activity is not sufficient for rapid ubiquitin-dependent protein breakdown. Degradation of proteins may be more sensitive than cleavage of short peptides to structural alterations in the proteasome.
There have been many in vitro studies on the ability of 20S proteasomes to cleave model peptide and protein substrates and on inhibition or activation of substrate hydrolysis by a variety of agents (ref. 18 and references therein). Attempts to link particular active sites defined with model peptides with the degradation of full-length proteins have yielded somewhat confusing results. For instance, with bovine proteasomes, dichloroisocoumarin (DCI) irreversibily inhibits the three peptidase activities discussed here, but this treatment actually stimulates cleavage of β-casein and certain model peptides (20). DCI treatment of Thermoplasma proteasomes, however, inhibits β-casein proteolysis (21). No in vitro studies have yet been done to determine the relative contribution of different active sites to degradation of polyubiquitinated substrates.
Regulation of Proteasome Levels in Vivo. Elimination of the trypsin-like activity of the proteasome by the pup1-T30A mutation results in a major increase in cellular Pup1 levels (Fig.
1 B–D) and ≈50% higher chymotrypsin-like and PGPH activities (Fig. 1A), with the latter increases correlating with a comparable increase in proteasome levels. These observations suggest a potential regulatory mechanism(s) that acts to increase proteasome levels when trypsin-like activity is inhibited (see below). In contrast, mutation of the PGPH sites (Fig. 1A) causes an increase in trypsin-like but not chymotrypsin-like activity and no change in proteasome levels, suggesting a specific influence of the Pre3 active sites on the Pup1 sites.
In the pup1-T30A mutant, protein degradation is only weakly inhibited and growth under most conditions is just slightly slower than that of wild-type cells (Figs. 2 and 3). Unexpectedly, growth of pup1-T30A cells is strongly enhanced relative to wild-type on plates containing Cd and slightly enhanced at high temperature (Fig. 3). The relationship between Cd resistance and ubiquitin-dependent proteolysis is not understood, but other mutations in the ubiquitin-proteasome pathway decrease resistance to the heavy metal (Fig. 3 and ref. 22). It is thought that Cd increases oxidative damage to proteins, which may make them substrates for the ubiquitin pathway. Why then does the pup1-T30A mutation enhance resistance to Cd and heat? We suggest that although the proteasome is essential for stress tolerance, a weakly compromised enzyme may lead to accumulation of low levels of abnormal proteins and, thereby, induction of stress proteins (23) and enhanced resistance to certain stresses. Treatment of mammalian cells with proteasome inhibitors at levels that are not strongly toxic has been recently shown to induce stress protein synthesis and thermotolerance (24). Interestingly, inverted repeats of the pentanucleotide sequence required for heat shock induction (25) are located 47 bp upstream of the PUP1 start codon, suggesting a possible feedback mechanism wherein an increase in proteasome substrates in the cell enhances expression of proteasomes themselves.
Acknowledgments
We thank P. Chen for the purified 20S proteasomes used for the NEM studies and for construction of the pre3-T20A allele, S.-J. Li for the immunoblot in Fig. 1B, T. Fox for the PUP1 and pup1::LEU2 clones, F. Cross for pLH7, and A. Amerik and P. Johnson for comments on the manuscript. This work was supported by National Institutes of Health Grant GM-46904. C.S.A. is a Howard Hughes Predoctoral Fellow.
ABBREVIATIONS
PGPH
peptidylglutamyl peptide hydrolyzing
NEM
_N_-ethylmaleimide
HA
hemagglutinin
βgal
β-galactosidase
Note Added in Proof
While this paper was under review, a crystal structure of the yeast 20S proteasome was reported (26). The subunit interactions demonstrated in the present work and previously (6) were confirmed. However, the authors concluded that both the trypsin-like and the chymotrypsin-like activities derive from the Doa3 subunit. This is clearly incorrect, based on the data in Fig. 1A and our earlier study (6).
References
- 1.Hochstrasser M. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 2.Rubin D M, Finley D. Curr Biol. 1995;5:854–858. doi: 10.1016/s0960-9822(95)00172-2. [DOI] [PubMed] [Google Scholar]
- 3.Chen P, Hochstrasser M. EMBO J. 1995;14:2620–2630. doi: 10.1002/j.1460-2075.1995.tb07260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 5.Seemüller E, Lupas A, Stock D, Löwe J, Huber R, Baumeister W. Science. 1995;268:579–582. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- 6.Chen P, Hochstrasser M. Cell. 1996;86:961–972. doi: 10.1016/s0092-8674(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 7.Fenteany G, Standaert R F, Lane W S, Choi S, Corey E J, Schreiber S L. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 8.Hochstrasser M, Papa F R, Chen P, Swaminathan S, Johnson P, Stillman L, Amerik A, Li S-J. Cold Spring Harbor Symp Quant Biol. 1995;60:503–513. doi: 10.1101/sqb.1995.060.01.054. [DOI] [PubMed] [Google Scholar]
- 9.Dick L R, Moomaw C R, Pramanik B C, DeMartino G N, Slaughter C A. Biochemistry. 1992;31:7247–7355. doi: 10.1021/bi00147a020. [DOI] [PubMed] [Google Scholar]
- 10.Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Cell. 1993;74:357–369. doi: 10.1016/0092-8674(93)90426-q. [DOI] [PubMed] [Google Scholar]
- 11.Haffter P, Fox T D. Nucleic Acids Res. 1991;19:5075. doi: 10.1093/nar/19.18.5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gietz R D, Sugino A. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 14.Tyers M, Tokiwa G, Nash R, Futcher B. EMBO J. 1992;11:1773–1784. doi: 10.1002/j.1460-2075.1992.tb05229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachmair A, Finley D, Varshavsky A. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 17.Savory P J, Rivett A J. Biochem J. 1993;289:45–48. doi: 10.1042/bj2890045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orlowski M, Cardozo C, Michaud C. Biochemistry. 1993;32:1563–1572. doi: 10.1021/bi00057a022. [DOI] [PubMed] [Google Scholar]
- 19.DeMarini D J, Papa F R, Swaminathan S, Ursic D, Rasmussen T P, Culbertson M R, Hochstrasser M. Mol Cell Biol. 1995;15:6311–6321. doi: 10.1128/mcb.15.11.6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pereira M E, Nguyen T, Wagner B J, Margolis J W, Yu B, Wilk S. J Biol Chem. 1992;267:7949–7955. [PubMed] [Google Scholar]
- 21.Akopian T N, Kisselev A F, Goldberg A L. J Biol Chem. 1997;272:1791–1798. doi: 10.1074/jbc.272.3.1791. [DOI] [PubMed] [Google Scholar]
- 22.Jungmann J, Reins H-A, Schobert C, Jentsch S. Nature (London) 1993;361:369–371. doi: 10.1038/361369a0. [DOI] [PubMed] [Google Scholar]
- 23.Ananthan J, Goldberg A L, Voellmy R. Science. 1986;232:522–524. doi: 10.1126/science.3083508. [DOI] [PubMed] [Google Scholar]
- 24.Bush K T, Goldberg A L, Nigam S K. J Biol Chem. 1997;272:9086–9092. doi: 10.1074/jbc.272.14.9086. [DOI] [PubMed] [Google Scholar]
- 25.Perisic O, Xiao H, Lis J T. Cell. 1989;59:797–806. doi: 10.1016/0092-8674(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 26.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik H D, Huber R. Nature (London) 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]