Interaction of measles virus glycoproteins with the surface of uninfected peripheral blood lymphocytes induces immunosuppression in vitro (original) (raw)
Abstract
A marked suppression of immune function has long been recognized as a major cause of the high morbidity and mortality rate associated with acute measles. As a hallmark of measles virus (MV)-induced immunosuppression, peripheral blood lymphocytes (PBLs) isolated from patients exhibit a significantly reduced capacity to proliferate in response to mitogens, allogens, or recall antigens. In an in vitro system we show that proliferation of naive PBLs [responder cells (RCs)] in response to a variety of stimuli was significantly impaired after cocultivation with MV-infected, UV-irradiated autologous PBLs [presenter cells (PCs)]. We further observed that a 50% reduction in proliferation of RCs could still be observed when the ratio of PC to RC was 1:100. The effect was completely abolished after physical separation of the two populations, which suggests that soluble factors were not involved. Proliferative inhibition of the RCs was observed after short cocultivation with MV-infected cells, which indicates that surface contact between one or more viral proteins and the RC population was required. We identified that the complex of both MV glycoproteins, F and H, is critically involved in triggering MV-induced suppression of mitogen-dependent proliferation, since the effect was not observed (i) using a recombinant MV in which F and H were replaced with vesicular stomatitis virus G or (ii) when either of these proteins was expressed alone. Coexpression of F and H, however, lead to a significant proliferative inhibition in the RC population. Our data indicate that a small number of MV-infected PBLs can induce a general nonresponsiveness in uninfected PBLs by surface contact, which may, in turn, account for the general suppression of immune responses observed in patients with acute measles.
Keywords: lymphoproliferation, morbillivirus
Measles virus (MV) is a major cause of child mortality in the Third World. The morbidity and mortality associated with the disease are mainly indirect effects caused by an MV-induced suppression of immune responses that lasts for weeks to months after acute measles (1). Patients typically reveal a pronounced lymphopenia that affects all lymphocyte subpopulations, but not the CD4/CD8 ratio (2), increased susceptibility to opportunistic infections, and strongly reduced or absent immune responses toward recall antigens (for review, see refs. 3 and 4). Peripheral blood mononuclear cells (PBMCs) isolated from patients generally fail to proliferate in response to allogens, mitogens, and recall antigens (5), which suggests a central block in lymphocyte activation. PBMCs are the prime target cells for viral replication during acute measles, and both lymphocytic and monocytic cells appear to be infected (6, 7). In cell culture, these subpopulations are also susceptible to infection, and the production of infectious virus is dependent on cellular activation (8). Both wild-type and vaccine MV strains are able to impair immune functions and this has mainly been investigated in cultured MV-infected PBMCs (4). Lymphocytic functions acquired prior to MV infection have generally been found to be unaffected, in contrast to those still to differentiate (9, 10). As observed in vivo, inhibition of proliferative responses to mitogens and recall antigens and strongly reduced cytotoxic T lymphocyte activities induced in mixed lymphocyte reactions were described (9, 11–13). The induction of activation markers such as interleukin 2 (IL-2)-receptors was not impaired in infected PBMC cultures and similar amounts of IL-2 were released as in mitogen-stimulated uninfected cultures (11, 12, 14). Mechanisms underlying functional impairments in MV-infected PBMC cultures remain largely undefined. It appears, however, that a large majority of these cells are arrested in the G1 phase of the cell cycle (12,15).
As the number of MV-infected PBMCs is usually low during acute measles, it is likely that the suppression of lymphocyte activation occurs independently of direct infection of target cells (4). Irradiated MV-infected PBMCs cause a significant inhibition of T-cell proliferation when added to fresh, autologous PBMCs (16). These authors reported that the effect was sensitive to the addition of MV-specific antibodies and was probably due to a soluble factor not identical to interferon or prostaglandin E. Using a similar experimental approach, we confirm that a very limited number of infected, UV-irradiated peripheral blood lymphocytes (PBLs) is sufficient to induce a strong proliferative inhibition of naive PBLs in response to a variety of stimuli. In contrast to the previous study, we found that a brief contact between the surface of the responder cells (RCs) and MV fusion (F) and hemagglutinin (H) glycoproteins coexpressed on the presenter cells (PCs) was necessary and sufficient for this effect. We also show that the membrane signal may not require the major MV receptor (CD46) on the target cells.
MATERIALS AND METHODS
Cell Lines and Viruses.
293 (human embryonic kidney), HeLa, and Vero cells were maintained in MEM containing 5% fetal calf serum (FCS). Human lymphoid (Jurkat: CD4+ T cells, BJAB: lymphoblastoid B cells) and monocytic {U-937, U-937-X [U-937 subclone deficient in CD46 expression (17)], HL60} and NCI H460 (human lung fibroblasts) were maintained in RPMI 1640 medium containing 10% FCS. MV vaccine strain Edmonston-B (ED) and vesicular stomatitis virus (VSV) (Indiana serotype) were grown and propagated in Vero cells; MV wild-type strains WTF and DLB (18) were propagated in BJAB cells. The recombinant virus, MGV, was created by replacing both the MV-F and MV-H coding regions with the reading frame encoding the VSV-G protein in the cloned MV-ED cDNA p(+)MV to yield p(+)MGV. The regions flanking the inserted gene were sequenced (the generation of the recombinant MV, as well as the biochemical characterization of the MGV are described in detail by Spielhofer and Naim, unpublished). Infectious virus was rescued after transfection of the recombinant cDNA along with pEMC-La into the helper cell line 293–3-46, which stably expresses MV-N and -P proteins and the T7 RNA polymerase as described (19). MGV stocks were amplified in Vero cells, titers were determined both by plaque titrations and indirect immunofluorescence. Final titers of 1 × 106 plaque-forming units/ml were reached. The ability of MGV to infect and replicate in PBLs was controlled by immunofluorescence and subsequent analysis using FACScan. 293-F and 293-H cell lines were generated basically as described using pCGΔ2F and pCGH for transformation of 293 cells (19, 20)
Cell Isolation and Culture.
PBMCs were isolated by Ficoll/Paque (Pharmacia) density gradient centrifugation of heparinized blood obtained from healthy adult donors. Monocytes were separated from PBLs by plastic adherence. PBLs and monocytes were cultured in RPMI 1640 medium/10% FCS. The purity of the monocytic and lymphocytic fractions was determined using specific antibodies (CD14, CD19, CD3). Rat PBLs were obtained after heart puncture by Percoll gradient centrifugation followed by removal of monocytes by adherence. Spleen cell suspensions from both BALB/c and C3H mice were prepared following standard procedures.
Immunohistochemistry.
Cell surface staining was performed using monoclonal antibodies directed against cell type specific markers (CD14, CD19, CD3), virus-specific proteins (MV-F, MV-H), or a polyclonal VSV-G-specific serum. Internal MV proteins were stained using a monoclonal MV-N-specific antibody. Stainings were analyzed by FACScan.
In Vitro Proliferation Assay.
PCs were generated by infecting freshly isolated PBLs in the presence of phytohemagglutinin (PHA) (2.5 μg/ml) or primary macrophages (stimulated with 10 μg/ml lipopolysaccharide) with MV-ED, MV-WTF, MV-DLB, MGV [each with a multiplicity of infection (MOI) of 1] or mock (with supernatant of Vero cells) for 48 hr. Expression of viral antigens was determined before UV irradiation of the PCs (0.25 J/cm2 in a biolinker). Proliferative inactivation was controlled after treatment by incorporation of [3H]thymidine. For 293 transfectants, 0.5 J/cm2 were required for complete inactivation. RCs were seeded in the presence of PHA (2.5 μg/ml) or concanavalin A (Con A) (5 μg/ml) into a 96-cluster plate with a density of 1 × 105 (human PBLs), 5 × 105 (rat PBLs and mouse spleen cells), and 5 × 104 (cell lines) in a volume of 100 μl per well. The PCs were added at the concentrations indicated in a volume of 100 μl per well, were incubated for 72 hr, and then labeled for 16 hr with [3H]thymidine (0.5 μCi/ml; 1 Ci = 37 GBq). Assays were performed in triplicate, harvested, and the incorporation rates of the label were determined using a β-plate reader.
RESULTS
Inhibition of Lymphocyte Proliferation in Vitro.
To induce immunosuppression, a presenter cell population was generated by infection of freshly isolated PBLs. These cells were first stimulated with PHA (2.5 μg/ml) and then infected with MV (MOI 1) (MV-PBLs or PCs) until 70–80% of the cells expressed MV antigens as determined by FACScan (Fig.1A). Cells (MV-PBLs, as well as a mock-infected control PBL culture) were UV-irradiated and mixed in decreasing proportions with autologous naive PBLs (RCs), which were simultaneously stimulated with PHA for 72 hr. Subsequently, cells were [3H]thymidine-labeled for a further 16 hr (yielding a total assay time of 88 hr) to determine the proliferative responses of the RCs. The rate of proliferative inhibition of the RCs caused by the cocultivation with the infected PCs was defined in relation to the positive control (mock-infected PCs). The inhibition of the proliferative response of the RC population was almost complete after cocultivation with MV-PBLs at a 1:1 ratio, with a 50% reduction observed at a ratio of 100:1, and a still significant inhibition of 10% at a ratio of 1000:1 RCs/PCs (Fig. 1B). For these and further experiments, proliferative inhibition rates below 10% were considered not to be significant. Because no differences were observed between vaccine- and wild-type strains in their ability to inhibit lymphoproliferation (representatively shown for MV-ED and MV-WTF, Fig.1B), the MV-ED strain was used for further experiments. In addition, after incubation with PCs the RCs also failed to proliferate in response to anti-CD3 stimulation and in mixed lymphocyte reactions (not shown).
Figure 1.
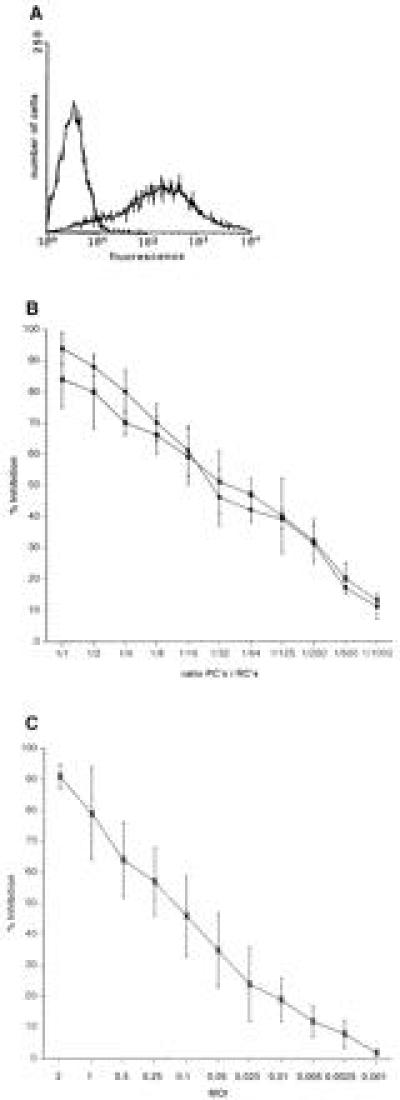
Induction of proliferative inhibition of PBLs after MV infection or cocultivation with MV-infected PBLs. (A) PHA-stimulated human PBLs were infected with MV-ED (MOI of 1) for 48 hr and stained for the expression of MV-H protein (MV-PBLs). The negative control was performed using a coronavirus-specific antibody. (B) MV-PBLs or mock-infected PBLs, respectively, were UV-irradiated and added in decreasing proportions as indicated in the axis legend (PCs) together with PHA (2.5 μg/ml) to naive autologous PBLs (RCs). Proliferative activity of the RCs was determined as described after a 72-hr incubation period followed by a further 16-hr labeling time. Each value represents the result of three independent experiments, and each was performed with triplicate assays. Open circles indicate the values obtained with MV-ED-infected PCs, solid circles indicate those with MV-WTF-infected PCs. (C) PBLs were infected with MV-ED at the MOIs indicated in the axis legend, stimulated with PHA (2.5 μg/ml), and their proliferative activity was determined after 72 hr by a labeling with [3H]thymidine for 16 hr. Proliferative inhibition is indicated in relation to the proliferation of uninfected, PHA-stimulated PBLs.
Proliferative inhibition of PBLs after MV-infection is well documented. Therefore, to evaluate direct effects of MV infection on the RCs, the amount of infectious MV present after UV-irradiation of the MV-PBLs and the number of infected RCs were determined. We found that the residual infectivity was less than 0.01 plaque-forming units/105 PCs; a dose that inhibits the proliferative response of PBLs infected directly maximally by 25% (Fig. 1C). This suggested that MV-infected PBLs were able to impair activation dependent proliferation of PBLs independently of infection of the RCs. In support of this interpretation, various amounts of MV (ranging from 5 × 105 to 1 × 104 plaque-forming units, corresponding to MOIs from 5 to 0.1/105 RCs, Fig.2, lanes 1–5) were UV-inactivated (UV-MV) and added to the RCs. Similar to MV-PBLs, proliferation of the RCs was markedly inhibited in a dose dependent manner after application of UV-MV, but not UV-VSV (grown and isolated under identical conditions to MV on Vero cells) (Fig. 2, lane 6). This indicated that inhibition was independent of viral replication. Moreover, a contribution of cellular proteins incorporated into the virion envelope from the surface of the PCs appeared unlikely as only UV-MV, but not UV-VSV (also grown on Vero cells) induced proliferative inhibition in the absence of PCs.
Figure 2.
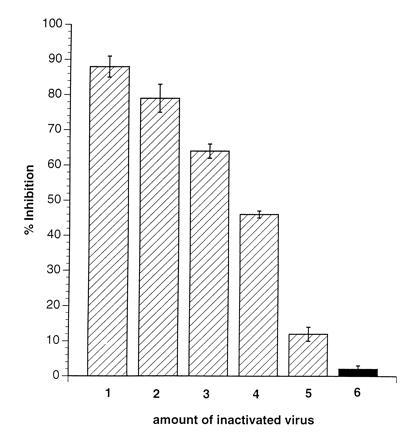
Inactivated MV, but not VSV, induces inhibition of mitogen-dependent proliferation of naive PBLs. RCs (naive PBLs) were incubated (in the presence of PHA) with amounts of MV-ED stock virus corresponding to MOIs of 5 (lane 1), 3 (lane 2), 2 (lane 3), 1 (lane 4), and 0.1 (lane 5) or VSV corresponding to an MOI of 3 (lane 6), which were previously inactivated by UV-irradiation (5 J/cm2). Proliferative inhibition was determined in relation to PHA-stimulated untreated controls after 72 hr and a 16-hr labeling period.
MV-Induced Proliferative Inhibition Is Cell Type but Not Species Specific.
UV-irradiated PCs efficiently impaired the mitogen-driven proliferative response of both autologous and allogeneic human PBLs in a dose dependent manner indicating that the phenomenon was not major histocompatibility complex-restricted (Table1; representative proliferative inhibition values determined at a ratio of 10:1 RCs/PC are indicated). Surprisingly, the mitogen-activated proliferation of PBLs isolated from Lewis rats and that of primary spleen cells from both C3H and BALB/c mice were also inhibited after cocultivation with human MV-PBLs. As mouse PBLs are not permissive for MV, the latter finding lends further support to our interpretation that the effect is independent of MV replication. Moreover, the effect is not confined to mitogen-dependent proliferation of primary lymphoid cells. The proliferation rates of human cell lines such as Jurkat (a CD4+ T-cell line), BJAB (B cells), HL60, and U-937 (both monocytic cell lines) were significantly reduced after cocultivation with MV-PBLs. No such effects were observed using cell lines of nonlymphoid origin such as HeLa, NCI H460, and 293. Most interestingly, values obtained for proliferative inhibition of U-937 and U-937-X cells [not expressing the major MV protein receptor CD46 (21, 22)] did not differ.
Table 1.
Various cell types are responsive to the induction of proliferative inhibition by MV-infected PCs
| Species | Cells | Proliferative inhibition, %* | |
|---|---|---|---|
| Human | PBLs† | Autologous | 74 ± 2 |
| Allogeneic | 71 ± 4 | ||
| Cell lines‡ | U-937 | 58 ± 6 | |
| U-937-X | 52 ± 4 | ||
| HL60 | 61 ± 8 | ||
| BJAB | 40 ± 13 | ||
| Jurkat | 65 ± 5 | ||
| HeLa, 293, | <10 | ||
| NCI H460 | |||
| Rat | PBLs§ | Lewis | 58 ± 3 |
| Mouse | Spleen cells§ | BALB/c | 70 ± 3 |
| C3H | 66 ± 7 |
Surface Contact Rather than Soluble Factors Induce Proliferative Inhibition of RCs.
Introduction of a 0.2-μm membrane filter to separate the RCs from either the UV-irradiated MV-PBLs (not shown) or from a mixture of MV-PBLs and RCs, completely prevented the induction of mitogen-unresponsiveness of the RCs (Fig. 3, lanes 1, 3, and 5). This indicated that neither PCs nor RCs after coculture with PCs released inhibitory factors that could account for the proliferative inhibition observed in nonseparated control cultures (Fig. 3, lanes 2, 4, and 6). This supported the requirement for a direct contact between RCs and MV constituents (Fig. 2). To define the kinetics for the induction of unresponsiveness in the RCs, MV-infected primary macrophages (MV-M) were used as PCs. We first demonstrated that these cells, when infected with MV, could functionally replace the infected PBLs as PCs (not shown). The UV-irradiated MV-M were separated from the RCs by anti-CD14 selection after a 10, 30, and 60 min coculture. After magnetic cell sorting separation of the MV-Ms, the RC population was negative for CD14 expression indicating that the removal of the PCs was complete (not shown). The proliferative response of the RCs contacted by the MV-Ms determined 72 hr later revealed that a contact of 10 min was sufficient to reduce mitogen-dependent proliferation of RCs by 22% (Fig. 4, lane 1), with a 65% inhibition being induced after a 1-hr contact period (Fig.4, lane 3).
Figure 3.
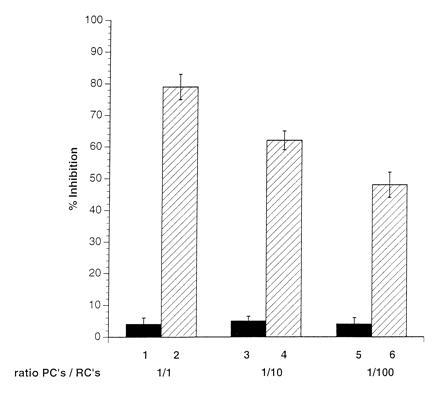
Physical separation of RCs from PCs abolishes the induction of proliferative inhibition. RCs were seeded into clusters of a 96-well plate and separated by filters with a pore size of 0.2 μm from a mixture of PCs/RCs (1:1, lane 1; 1:10, lane 3; 1:100, lane 5) in the presence of PHA. Filters were removed from the wells after 72 hr and the proliferative activity of the seeded RCs was determined after a further 16 hr of labeling. Control values indicate the results obtained in the standard proliferation assay using PC/RC ratios of 1:1 (lane 2), 1:10 (lane 4), and 1:100 (lane 6) in the absence of the filter membranes.
Figure 4.
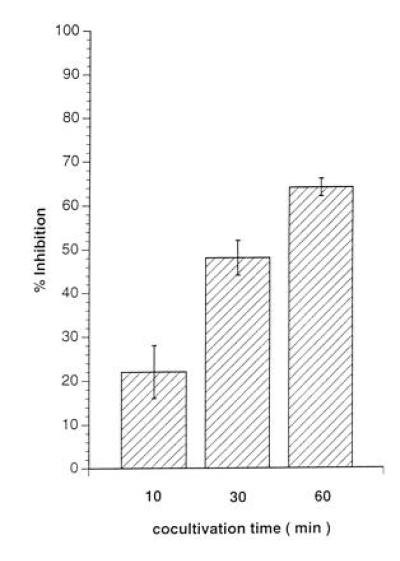
Short contact with PCs leads to proliferative inhibition of RCs. Primary macrophages were separated by adhesion from PBMCs, infected with MV (MOI 3 for 2 days until they were 80% positive for MV antigens) (MV-M) and UV-irradiated. For each value, 1 × 106 MV-M were mixed with 2 × 106 RCs in the presence of PHA and the PCs were separated by anti-CD14 magnetic cell sorting selection after 10 min (lane 1), 30 min (lane 2), or 60 min (lane 3) from the RCs. RCs were further cultivated in the absence of PCs for 72 hr and subsequently labeled for 16 hr.
The Expression of both MV Glycoproteins Is Necessary and Required for the Induction of Proliferative Inhibition.
To identify the viral constituents responsible for proliferative inhibition of RCs, we used a recombinant MV where the MV glycoproteins (fusion protein F and hemagglutinin H) had been replaced by VSV-G protein (MGV). The ability of the recombinant virus to infect PBLs and to replicate was first confirmed by the expression of internal MV antigens and VSV G protein and titration of supernatants from infected PBLs (not shown). Levels of proliferative inhibition obtained after cocultivation of RCs with MGV-PBLs were insignificant even when added at a 1:1 ratio [Fig.5, lane 1 compared with the MV-PBL control culture (lanes 2)] indicating that the inhibitory effect observed with MV-PBLs was mainly due to an interaction of the MV glycoproteins with the surface of the RCs. To determine the contribution of these two proteins in the inhibition process, we expressed MV F or H in transient expression systems. Because expression levels after transient transfection of pCG-F and pCG-H (20) were low and recombinant vaccinia viruses expressing MV-F and MV-H proteins could not be used in our system, because VV-T7 infection caused proliferative inhibition (not shown), 293 cells stably expressing either MV-F or MV-H proteins to high levels (Fig. 6A, 293-F and Fig.6B, 293-H) were used as PCs. Whereas UV-irradiated, MV-infected 293 cells were highly inhibitory (Fig. 6D, lane 10), 293-T7 (stably expressing T7 polymerase, lane 1), 293-F (lane 3), or 293-H cells (lane 6) did not exert any negative effect on the proliferation of the RCs at any concentration added. No effect was observed even after increasing the amount of the individual proteins expressed by transient supertransfections (Fig. 6D, 293-F plus pCG-F, lane 5; 293-H plus pCG-H, lane 7), indicating that neither protein alone could be the effector molecule. However, after transient transfection of pCG-H, leading to the expression of the F/H complex in about 30% of the cell population, 293-F cells were able to inhibit proliferation to 43% (Fig. 6D, lane 4). Proliferative inhibition induced by 293-H cells supertransfected with pCG-F was less pronounced (Fig. 6D, lane 8), which was probably due to the lower expression levels of the transiently transfected F protein. In contrast, 293-F and 293-H cells did not induce the effect when added together (Fig. 6D, lane 9). Taken together, these data clearly indicate that coexpression of both MV-F and MV-H proteins is required and sufficient to induce a state of nonresponsiveness to a variety of stimuli in cells of the lymphoid lineage.
Figure 5.
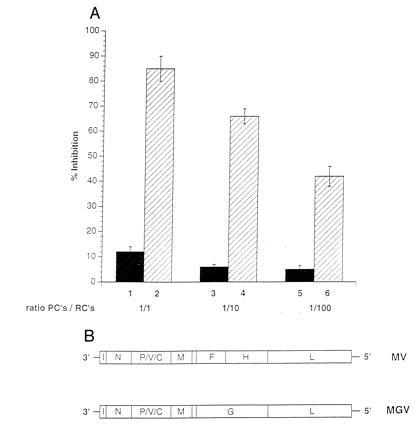
The MV glycoproteins are required for induction on nonresponsiveness of PBLs. PCs were infected with either MV-ED (lanes 2, 4, and 6) or MGV (schematically shown in B) (lanes 1, 3, and 5), UV-irradiated, and cocultivated at a PC/RC ratio of 1:1 (lanes 1 and 2), 1:10 (lanes 3 and 4), or 1:100 (lanes 5 and 6) for a total of 96 hr (72 hr plus 16 hr labeling), and proliferative inhibition was determined in relation to the control culture with mock-infected PCs. Infection levels in both PC populations were confirmed prior to the cocultivation by FACScan using MV-N- and MV-H-specific antibodies (for MV-ED) and MV-N and VSV-G-specific antibodies (for MGV).
Figure 6.
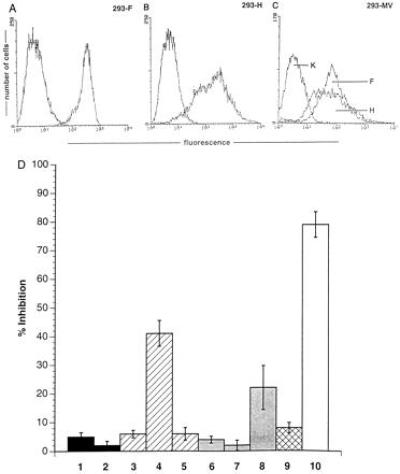
Both MV-F and MV-H proteins are required for the induction of nonresponsiveness of PBLs. (A) The expression levels of MV-F in 293-F cells, of MV-H in 293-H cells (B), and MV-F and MV-H in 293 cells productively infected with MV (C) were determined using specific antibodies. The negative controls were performed using a coronavirus-specific antibody. (D) Standard proliferation assays were performed using PBLs as RCs and 293-T7 (lane 1), 293-T7 supertransfected with pCG-H (lane 2), 293-F (lane 3), 293-F supertransfected with pCG-H (lane 4), 293-F supertransfected with pCG-F (lane 5), 293-H (lane 6), 293-H supertransfected with pCG-H (lane 7), 293-H cells supertransfected with pCG-F (lane 8), a 1:1 mixture of 293-F and 293-H (lane 9), or 293-MV infected (lane 10) cells, each at a PC/RC ratio of 1:2. Each value represents three independent experiments each performed as triplicate assay.
DISCUSSION
In view of the low number of MV-infected PBMCs detected in the course of an acute infection, the degree of suppression of immune reactivity during acute measles and for weeks afterwards has led to the suggestion that in a way similar to HIV infections, indirect mechanisms may be involved. We now provide evidence that a small number of MV-infected cells expressing the viral glycoproteins at their surface is able to induce a state of unresponsiveness to proliferation-activating signals in primary cells of the lymphoid and monocytic lineage in vitro. Moreover, proliferation of both lymphocytic and monocytic cells, but not of cells of other origin, was significantly impaired after cocultivation with MV-PBLs.
A number of investigations into MV-induced proliferative inhibition of PBLs have been carried out in vitro. However, most of these analyses have been performed using bulk cultures of PBLs infected in vitro and the effects observed have not been analyzed in the separate populations of MV-infected PBLs, and those that have remained uninfected and might have been contacted by infected cells. Thus, these previous data are not directly comparable with those obtained in our study. We also found that MV-infected PBLs reveal a markedly impaired ability to proliferate in response to mitogen and in mixed lymphocyte reactions (9, 11–14), with the extent of inhibition being directly related to the MOI used (13, 16) (Fig. 1C).
Sanchez-Lanier and coworkers previously described proliferative inhibition of uninfected PBMCs after cocultivation with MV-PBLs, which was prevented in the presence of MV antibodies (16). Their study suggested that soluble factors inhibitory to proliferation and not identical to interferon or prostaglandin E were involved. In contrast, physical separation of MV-PBLs and RCs by a permeable membrane completely abolished the induction of unresponsiveness in our experiments suggesting that soluble factors were not essential. The size exclusion range of the membrane filter used for separation strongly argues against the possibility that transfer of a soluble factor would have been prevented. Moreover, transfer of supernatants obtained from either PCs or from RCs after cocultivation with PCs (Fig.3) never induced proliferative inhibition of RCs (not shown). We cannot, however, rule out that depending on the experimental conditions, soluble factors that act over a short distance in the microenvironment surrounding the interacting cell surfaces may support contact-mediated inhibition. Our data clearly suggest that the expression of both viral glycoproteins (most likely as a complex, as colocalization of both proteins on the cell surface was required) is necessary and sufficient to induce unresponsiveness after a short contact and, thus, support our findings that complete, UV-inactivated MV particles were also able to induce the effect. In contrast to our findings, Sanchez-Lanier and colleagues (16) did not observe proliferative inhibition of RCs with inactivated MV. It is most likely that differences in the inactivation procedure (which was X-ray irradiation in their study) may have lead to functional impairment of the viral glycoprotein complex.
Our data reveal that coexpression of MV-F and MV-H is necessary and sufficient to induce unresponsiveness of naive PBLs to mitogen stimulation (Fig. 6D, lane 4). Inhibition induced with MV-infected 293 cells as PCs, however, was more pronounced (Fig.6D, lane 10), indicating that the signal transmitted was probably dependent on the amount of F/H complexes presented. The expression levels for the individual F or H proteins may be higher on 293-F or 293-H cells compared with 293-MV cells. However, the relative expression frequency of the F/H complex was higher in infected cells than in 293-F cells supertransfected with pCG-H. In addition to the number of F/H complexes, cellular surface protein expressed by the PCs may also contribute. Upregulation of lymphocyte function-associated antigen-1, as documented after MV-infection of MV-infected U-937 cells_in vitro_ (23), may strengthen the adhesion of PCs to the RCs in vivo, thus stabilizing the interaction between the F/H complex and the receptor(s) on the RCs. Unlike the viral constituents, the ligand(s) on the RC’s surface is undefined. Two proteins have been identified within the major MV receptor complex, CD46 and moesin (21, 22, 24, 25), and CD46 has already been successfully linked to contact-mediated signaling (26). It appears, however, that signals evoked by MV-H and CD46 contact leading to the down-regulation of CD46 from the cell surface may be different from those inducing unresponsiveness. First, down-regulation of CD46 is observed only with certain MV strains and not, for example, with the MV wild-type strain WTF (18, 27), which is highly potent in inducing proliferative inhibition (Fig. 1B). Second, proliferative inhibition was also induced in primary mouse lymphoid cells (Table 1), which only express a functional homologue of CD46 that does not serve as an MV receptor (28). Moreover, proliferation of cell lines such as HeLa, 293, and NCI H460, which clearly express CD46, was not significantly impaired after cocultivation with MV-PBLs (Table 1), whereas U-937 were sensitive to the effect independent of CD46 expression (U-937 versus U-937-X cells). Third, crosslinking CD46 on the surface of the RCs had no detectable impact on proliferative activity of these cells (not shown). Although moesin is widely expressed and highly conserved throughout all species analyzed so far, its role as a potential receptor for the induction of unresponsiveness is unclear, since some moesin-expressing cell lines, such as 293, HeLa, and NCI H460, fail to respond (Table 1).
Most data in this study were obtained using freshly isolated PBLs, and proliferative responses have mainly been analyzed after T-cell specific stimulation by, for example, PHA, Con A, anti-CD3, and mixed lymphocyte reaction. We have, however, no direct evidence for T-cell receptor involvement in the inhibitory effect because the proliferation of both CD4+ and CD8+ cells after mitogen stimulation (not shown), as well as that of TCR-negative cells such as U-937, HL-60, and BJAB (Table 1) is impaired. Moreover, the induction of proliferative inhibition was also observed using UV-MV, which lacks major histocompatibility complex molecules (Fig. 2).
It will be a high priority to determine the state of the RCs after PC contact. There was no evidence for a loss of viable cells in the course of our experiments and the RCs still appeared viable prior to harvesting as determined by trypan blue exclusion staining (not shown). One of the classical hallmarks of anergy is the rescue of proliferative activity by exogenously added IL-2. This has been attempted without success in MV-infected PBL cultures (14). Moreover, both levels of IL-2R expression and IL-2 released were not found to be affected in these cultures (12–14). As found in preliminary experiments, exogenous application of IL-2 did not restore mitogen-dependent proliferative activity of the RCs (not shown), which indicates that even if it occurs, MV-induced anergy may not be of the classical type. Apoptosis has been shown in a monocytic cell line 4 days after infection with low doses of MV and, more recently, in experimentally infected severe-combined immunodeficient-hu mice (29, 30), but has not been investigated in infected PBL cultures. Thus, future experiments in our system will have to determine whether the RCs are not in an anergic state, but rather are undergoing apoptosis or are primed to do so as has been observed for PBLs isolated from patients with HIV (reviewed in ref. 31).
Acknowledgments
We thank J. Schneider-Schaulies, S. Niewisk, C. Jassoy, A. Schimpl, and B. Askonas for helpful discussions; S. Mazgereanu for providing the rat PBLs; I. Johnston for critically reading the manuscript; M. Bayer for excellent technical assistance; and the Robert Pfleger Stiftung, the Deutsche Forschungsgemeinschaft, and the Schweizerische Nationalfond for financial support.
Footnotes
Abbreviations: MV, measles virus: MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cells; PBL, peripheral blood lymphocytes; RC, responder cells; PC, presenter cells; PHA, phytohemagglutinin; Con A, concanavalin A; VSV, vesicular stomatitis virus; IL-2, interleukin 2.
References
- 1.Clements C J, Cutts F. Curr Top Microbiol Immunol. 1995;191:13–34. doi: 10.1007/978-3-642-78621-1_2. [DOI] [PubMed] [Google Scholar]
- 2.Arneborn P, Biberfeld G. Infect Immun. 1983;39:29–37. doi: 10.1128/iai.39.1.29-37.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casali P, Nakamura M, McChesney M B. In: Virus-Induced Immunosuppression. Spector S, Bendinelli M, Friedman H, editors. New York: Plenum; 1989. pp. 345–373. [Google Scholar]
- 4.Borrow P, Oldstone M B A. Curr Top Microbiol Immunol. 1995;191:85–99. doi: 10.1007/978-3-642-78621-1_6. [DOI] [PubMed] [Google Scholar]
- 5.Smithwick E M, Berkovich S. In: Cellular Recognition. Smith R T, Good R A, editors. New York: Appleton Century Crafts; 1969. p. 131. [Google Scholar]
- 6.Whittle H C, Dossetor J, Oduloju A, Bryceson A, Greenwood B M. J Clin Invest. 1978;62:678–684. doi: 10.1172/JCI109175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esolen L M, Ward B J, Moench T R, Griffin D E. J Infect Dis. 1993;168:47–52. doi: 10.1093/infdis/168.1.47. [DOI] [PubMed] [Google Scholar]
- 8.Sullivan J L, Barry D W, Lucas S J, Albrecht P. J Exp Med. 1975;142:773–784. doi: 10.1084/jem.142.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galama J M D, Ubels-Postma J, Vos A, Lucas C J. Cell Immunol. 1980;50:405–415. doi: 10.1016/0008-8749(80)90294-4. [DOI] [PubMed] [Google Scholar]
- 10.Casali P, Rice G P A, Oldstone M B A. J Exp Med. 1984;159:1322–1337. doi: 10.1084/jem.159.5.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McChesney M B, Fujinami R S, Lampert P W, Oldstone M B A. J Exp Med. 1986;163:1331–1336. [PMC free article] [PubMed] [Google Scholar]
- 12.McChesney M B, Altman A, Oldstone M B A. J Immunol. 1988;140:1269–1273. [PubMed] [Google Scholar]
- 13.Yanagi Y, Cubitt B A, Oldstone M B A. Virology. 1992;187:280–289. doi: 10.1016/0042-6822(92)90316-h. [DOI] [PubMed] [Google Scholar]
- 14.Borysiewicz L K, Casali P, Rogers B, Morris S, Sissons J G P. Clin Exp Immunol. 1985;59:29–36. [PMC free article] [PubMed] [Google Scholar]
- 15.McChesney M B, Kehrl J H, Valsamakis A, Fauci S, Oldstone M B A. J Virol. 1987;61:3441–3447. doi: 10.1128/jvi.61.11.3441-3447.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez-Lanier M, Guerlin P, McLaren L C, Bankhurst A D. Cell Immunol. 1988;116:367–381. doi: 10.1016/0008-8749(88)90238-9. [DOI] [PubMed] [Google Scholar]
- 17.Schnorr J J, Dunster L M, Nanan R, Schneider-Schaulies J, Schneider-Schaulies S, ter Meulen V. Eur J Immunol. 1995;25:976–984. doi: 10.1002/eji.1830250418. [DOI] [PubMed] [Google Scholar]
- 18.Schneider-Schaulies J, Schnorr J J, Brinckmann U, Dunster L M, Baczko K, Schneider-Schaulies S, ter Meulen V. Proc Natl Acad Sci USA. 1995;92:3943–3947. doi: 10.1073/pnas.92.9.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dötsch C, Christiansen G, Billeter M A. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cathomen T, Buchholz C J, Spielhofer P, Cattaneo R. Virology. 1995;214:628–632. doi: 10.1006/viro.1995.0075. [DOI] [PubMed] [Google Scholar]
- 21.Dörig R E, Marcil A, Chopra A, Richardson C D. Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 22.Naniche D, Varior-Krishnan G, Cervoni F, Wild T F, Rossi B, Rabourdin-Combe C, Gerlier D. J Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Attibele N, Wyde P R, Trial J, Smole S C, Smith C W, Rossen R D. J Virol. 1993;67:1075–1079. doi: 10.1128/jvi.67.2.1075-1079.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunster L M, Schneider-Schaulies J, Löffler S, Lankes W, Schwarz-Albiez R, Lottspeich F, ter Meulen V. Virology. 1994;198:265–274. doi: 10.1006/viro.1994.1029. [DOI] [PubMed] [Google Scholar]
- 25.Schneider-Schaulies J, Dunster L M, Schwarz-Albiez R, Krohne G, ter Meulen V. J Virol. 1995;69:2248–2256. doi: 10.1128/jvi.69.4.2248-2256.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider-Schaulies J, Schnorr J J, Schlender J, Dunster L M, Schneider-Schaulies S, ter Meulen V. J Virol. 1996;70:255–263. doi: 10.1128/jvi.70.1.255-263.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider-Schaulies J, Dunster L M, Liebert U G, Kobune F, Rima B K, ter Meulen V. J Virol. 1995;69:7257–7259. doi: 10.1128/jvi.69.11.7257-7259.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunster L M, Schneider-Schaulies J, Dehoff M H, Holers V M, Schwartz-Albiez R, ter Meulen V. J Gen Virol. 1995;76:2085–2089. doi: 10.1099/0022-1317-76-8-2085. [DOI] [PubMed] [Google Scholar]
- 29.Esolen L M, Park S W, Hardwick M, Griffin D E. J Virol. 1995;69:3955–3958. doi: 10.1128/jvi.69.6.3955-3958.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Auwaerter P G, Kaneshima H, McCune J M, Wiegand G, Griffin D E. J Virol. 1996;70:3734–3740. doi: 10.1128/jvi.70.6.3734-3740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oyaizu N, Pahwa S. J Clin Immunol. 1995;15:217–231. doi: 10.1007/BF01540879. [DOI] [PubMed] [Google Scholar]