Synapses Between Corticotropin-Releasing Factor-Containing Axon Terminals and Dopaminergic Neurons in the Ventral Tegmental Area Are Predominantly Glutamatergic (original) (raw)
. Author manuscript; available in PMC: 2008 Jun 26.
Published in final edited form as: J Comp Neurol. 2008 Feb 1;506(4):616–626. doi: 10.1002/cne.21576
Abstract
Interactions between stress and the mesocorticolimbic dopamine (DA) system have been suggested from behavioral and electrophysiological studies. Because corticotropin-releasing factor (CRF) plays a role in stress responses, we investigated possible interactions between neurons containing CRF and those producing DA in the ventral tegmental area (VTA). We first investigated the cellular distribution of CRF in the VTA by immunolabeling VTA sections with anti-CRF antibodies and analyzing these sections by electron microscopy. We found CRF immunoreactivity present mostly in axon terminals establishing either symmetric or asymmetric synapses with VTA dendrites. We established that nearly all CRF asymmetric synapses are glutamatergic, insofar as the CRF-immunolabeled axon terminals in these synapses coexpressed the vesicular glutamate transporter 2, and that the majority of CRF symmetric synapses are GABAergic, insofar as the CRF-immunolabeled axon terminals in these synapses coexpressed glutamic acid decarboxylase, findings that are of functional importance. We then looked for synaptic interactions between CRF- and DA-containing neurons, by using antibodies against CRF and tyrosine hydroxylase (TH; a marker for DA neurons). We found that most synapses between CRF-immunoreactive axon terminals and TH neurons are asymmetric (in the majority likely to be glutamatergic) and suggest that glutamatergic neurons containing CRF may be part of the neuronal circuitry that mediates stress responses involving the mesocorticolimbic DA system. The presence of CRF synapses in the VTA offers a mechanism for interactions between the stress-associated neuropeptide CRF and the mesocorticolimbic DA system.
Indexing terms: dopamine, stress, reward, mesolimbic dopamine system
Corticotropin-releasing factor (CRF) is a 41-amino-acid neuropeptide isolated initially from ovine hypothalamus (Vale et al., 1981) and subsequently shown to be synthesized in numerous regions throughout the brain (Sawchenko et al., 1993; Swanson et al., 1983). CRF plays an important role in hypothalamic and extrahypothalamic responses to stress (Bale and Vale, 2004; Dunn and Berridge, 1990; Koob and Heinrichs, 1999; Vale et al., 1981) by interacting with different neuronal pathways, such as the hypothalamic-pituitary-adrenal (HPA) circuit (Bale and Vale, 2004; Dunn and Berridge, 1990; Vale et al., 1981) and the mesocorticolimbic dopamine (DA) system (Deutch and Roth, 1990; Kalivas et al., 1987).
The mesocorticolimbic DA system, which consists of DA-producing neurons concentrated in the ventral tegmental area (VTA) and DA axons targeting prefrontal cortex and limbic structures, plays a role in reward and motivation (Robinson and Berridge, 1993; Wise, 2002, 2004), including the rewarding effects of several drugs of abuse (Wise and Bozarth, 1987). Interaction between the stress and the mesocorticolimbic DA system is suggested from studies showing that mild foot shock induces metabolic activation of DA innervations in the prefrontal cortex (Thierry et al., 1976), whereas more severe stressors produce concurrent metabolic activation of DA terminals in the nucleus accumbens (Deutch and Roth, 1990). Functional interactions between stress and the mesocorticolimbic system have been further supported from microdialysis studies showing that different types of stressors induce release of DA in the prefrontal cortex (Abercrombie et al., 1989), in the shell of the nucleus accumbens (Kalivas and Duffy, 1995) and in the basolateral amygdala (Inglis and Moghaddam, 1999).
Earlier studies have suggested the involvement of CRF in mediating stress effects on the VTA. For instance, an increase in spontaneous motor activity is observed after injections of CRF into the VTA (Kalivas et al., 1987), indicating that cellular compartments within the VTA are responsive to CRF. The existence of a releasable pool of endogenous CRF within the VTA was recently revealed by microdialysis studies showing that foot shock induces release of CRF in the VTA (Wang et al., 2005). As opposed to the results obtained from these in vivo studies, analysis of post-mortem brain tissue indicated that CRF concentrations in the VTA did not change after stress exposure (Deutch et al., 1987). Electrophysiological studies demonstrate that, when CRF is applied to preparations of mouse midbrain slices, it potentiates synaptic transmission by N-methyl-D-aspartate (NMDA) receptors in VTA DAergic neurons (Ungless et al., 2003) and excites the majority of VTA DAergic neurons in rat midbrain slices (Korotkova et al., 2006). Together these observations indicate that DAergic neurotransmission in the VTA is affected by exogenous CRF. Hence, the VTA is a likely site for synaptic interactions between CRF and DAergic neurons.
The cellular nature of VTA endogenous CRF and the neuronal connectivity of CRF cells in the VTA are unknown. In this study, we used immunohistochemical electron microscopy to investigate the cellular distribution of endogenous CRF in the VTA and to determine the types of neuronal connections that these cells make in the VTA.
MATERIALS AND METHODS
Perfusions
In total, 24 male Sprague-Dawley rats (180–200 g) were used for these studies. Rats were singly housed on a 12-hour light schedule in a temperature-controlled (20°C) animal room and given access to standard rat chow and water ad libitum. All procedures were approved by the local Animal Care and Use Committee. Each animal was deeply anesthetized with chloral hydrate (300 mg/kg) and perfused transcardiacally through the left ventricle with 10 ml of 0.9% saline containing 1,000 U/ml heparin, 75 ml fixative containing 3.75% acrolein and 2% paraformaldehyde (PF), and finally 300 ml of 2% PF in 0.1 M phosphate buffer, pH 7.4 (PB). Each brain was removed and kept in the last fixative solution for 2 hours. Vibratome coronal sections (50 μm) were obtained from each brain, collected in PB, equilibrated in a solution containing 25% sucrose and 10% glycerol in PB, and frozen in liquid nitrogen. The detection of antigens was comparable using brain sections of animals perfused with different fixatives (4% PF with 0.15% glutaraldehyde and 15% picric acid, 4% PF with 0.15% glutaraldehyde, 2% PF with 0.1 M sodium metaperiodate and 4% PF with 0.1 M L-lysine). The best ultra-structural preservation was obtained with the fixative solution containing acrolein.
Immunohistochemistry
Primary antibodies
Five different commercially available primary antibodies were used in this study. The mouse monoclonal antityrosine hydroxylase (TH; Chemicon, Temecula, CA; code MAB318) antibody was raised against purified TH from the PC 12 pheochromocytoma cell line; this antibody recognizes recombinant TH by Western blot analysis (Wolf and Kapatos, 1989). We previously showed that the anti-TH antibody labels a single protein band in the molecular weight range of 56–60 kDA on Western blots prepared from VTA protein homogenates (Shepard et al., 2006). In addition, we had used this anti-TH antibody to demonstrate that midbrain unilateral lesion with 6-hydroxydopamine (6-OHDA) results in lack of TH immunolabeling in the lesioned site, whereas it is preserved in the contralateral unlesioned midrain (Sarabi et al., 2001). The mouse monoclonal antiglutamic acid decarboxylase (GAD; Chemicon; code MAB351) was raised against purified rat brain GAD (Chang and Gottlieb, 1988). The anti-GAD antibody recognizes the lower molecular weight component of both GAD65 and GAD67 isoforms present in the brain (Chang and Gottlieb, 1988). The polyclonal antivesicular glutamate transporter 1 (vGluT1; Chemicon; code AB5905) antibody was raised in guinea pig against the synthetic peptide GATHSTVQPPRPPP-PVRDY of the rat vGluT1. Similarly, the polyclonal anti-vesicular glutamate transporter 2 (vGluT2; Chemicon; code AB5907) antibody was raised in guinea pig against the synthetic peptide VQESAQDAYSYKDRDDYS of the rat vGluT2. The anti-vGluT1 and anti-vGluT2 antibodies recognize on Western blots the corresponding vGluT1 and vGluT2 proteins obtained by in vitro transcription-translation. As showed above, the antigenic peptides used to generate the anti-vGluT1 and anti-vGluT2 antibodies share homology in only two amino acids (DY). The lack of cross-reactivity between the anti-vGluT1 and the anti-vGluT2 antibodies was demonstrated by dot-blot analysis. A row of 1 μl serial dilutions of the vGluT1 antigenic peptide (1 to 0.001 μg) and a row of serial dilutions of the vGluT2 antigenic peptide (1 to 0.001 μg) were spotted onto two separated sheets of nitrocellulose paper. One sheet of nitrocellulose paper was probed with the anti-vGluT1 (1:4,000) antibody and the other with the anti-vGluT2 antibody (1:4,000), as previously detailed (Morales et al., 1996). On this type of test, the anti-vGluT1 antibody recognizes as little as 0.010 μg of the vGluT1 antigenic peptide but does not detect the vGluT2. Similarly, the anti-vGluT2 antibody recognizes as little as 0.010 μg of the vGluT2 antigenic peptide but does not detect the vGluT1. Two anti-CRF antibodies were used in this study, a polyclonal antibody provided by Dr. Wyle Vale (code PBL rC70; Salk Institute, La Jolla, CA) and a commercial antibody (Peninsula, San Carlos, CA; code T-4037). Both antibodies were raised in rabbit; the antibodies were generated against the human/rat (h/r)CRF peptide (SEEPPI SLDLTFHLLREVLEMARAEQLAQQAHSNRKLMEII-NH2), conjugated to human α-globulins (for PBL rC70 antibody) or keyhole limpet hemocyanin (for T-4037 antibody). By dot-blot analysis both anti-CRF antibodies recognize h/rCRF (code C-3042) but do not label rat urocortin I (code U-6631), mouse urocortin II (code U-9507), or human urocortin III (code U-1008). The h/r CRF and urocortins (Sigma, St. Louis, MO) were used at concentrations of 1–0.001 μg. In addition, control experiments for specificity of immunostaining obtained with anti-CRF, vGluT1, and vGluT2 antibodies included preadsorption of primary antibodies with antigens, as previously detailed (Morales et al., 1996). Vibratome brain sections from animals perfused with 2% PF containing 3.75% acrolein were used for these control experiments. Primary antibody preadsorption prevented immunostaining (for further information on antibodies see Table 1).
TABLE 1.
Primary Antibodies Used in This Study
| Antibody against | Source | Code | Host | Characterization |
|---|---|---|---|---|
| Corticotropin-releasing factor (CRF) | Peninsula | T-4037 | Rabbit | Immunolabel was eliminated by preadsorption of this antibody with synthetic CRF (Sigma C-3042; this study)The antibody does not cross-react with urocortins (I, II, or III; this study) |
| Corticotropin-releasing factor (CRF) | Dr. Wyle Vale | PBLrC70 | Rabbit | Immunolabel was eliminated by preadsorption of this antibody with synthetic CRF (Sigma C-3042; this study)The antibody does not cross-react with urocortins (I, II, or III; this study) |
| Tyrosine hydroxilase (TH) | Chemicon | MAB318 | Mouse | Western blot analysis demonstrated that this antibody recognizes the brain TH (Shepard et al., 2006)Lack of TH immunolabeling in 6-OHDA-lesioned midbrain but not in unlesioned midrain (Sarabi et al., 2001) |
| Glutamic acid decarboxylase (GAD) | Chemicon | MAB351 | Mouse | Western blot analysis demonstrated that this antibody recognizes the lower molecular weight components of GAD65 and GAD67 (Chang and Gottieb, 1988) |
| Vesicular glutamate transporter 1 (vGluT1) | Chemicon | AB5905 | Guinea pig | Immunolabel was eliminated by preadsorption of this antibody with the antigenic peptide (Chemicon AG208; this study)This antibody does not detect vGluT2 (this study) |
| Vesicular glutamate transporter 2 (vGluT2) | Chemicon | AB5907 | Guinea pig | Immunolabel was eliminated by preadsorption of this antibody with antigenic peptide (Chemicon AG209; this study)This antibody does not detect vGluT1 (this study) |
Single immunolabeling (immunoperoxidase protocol)
Vibratome tissue sections were incubated in 1% sodium borohydride in PB for 30 minutes to inactivate free aldehyde groups, rinsed in PB, and then incubated in blocking solution [4% bovine serum albumin (BSA) in PB] for 1 hour at room temperature. The sections were then incubated with a rabbit anti-CRF antibody (PBL rC70) at 1:4,000 dilution or anti-CRF antibody (T-4037) at a 1:4,000 dilution in the blocking solution for 48 hours at 4°C. Detection of primary antibodies was achieved by using a conventional avidin-biotin immunoperoxidase protocol (ABC Vectastain Elite Kit; Vector, Burlingame, CA) as follows: sections were incubated for 1 hour with an anti-rabbit biotinylated secondary antibody at 1:100 dilution in blocking solution. Sections were rinsed and then incubated for 2 hours in avidin-biotin peroxidase complex (ABC; 1:200 dilution) in PB. Peroxidase activity was detected by placing the sections in a solution containing 22 μg/ml diaminobenzidine (DAB; Electron Microscopy Sciences, Fort Washington, PA) and 0.003% hydrogen peroxide (H2O2) in PB for 10 minutes. Peroxidase reaction was stopped by rinsing sections in PB. Results reported in this study were performed with the T-4037 anti-CRF antibody and confirmed with the PBL rC70 anti-CRF antibody.
Double immunolabeling
Double immunolabeling was used to identify structures containing CRF (T-4037) in relation to those having the following cellular markers: vGluT1, vGluT2, GAD, or TH. CRF was detected by using the immunoperoxidase protocol, and immunogold-silver labeling was applied to detect vGluT1, vGluT2, GAD, or TH. In brief, vibratome sections were treated with 1% sodium borohydride dissolved in PB for 30 minutes, rinsed in PB, and incubated in a blocking solution containing 10% normal goat serum (NGS), 0.1% cold water fish gelatin, and 4% BSA in PB for 1 hour at room temperature. Sections were incubated in a cocktail of two different primary antibodies (CRF with vGluT1; CRF with vGluT2; CRF with GAD; or CRF with TH) for 48 hours at 4°C. Rabbit anti-CRF antibody was used at 1:4,000 dilution and mouse anti-TH antibody at 1:1,000 dilution. Guinea pig anti-vGluT1 and guinea pig anti-vGluT2 antibodies were used at 1:6,000 dilution. The mouse anti-GAD antibody was used at 1:200 dilution. After incubation with primary antibodies, sections were rinsed in PB and incubated overnight at 4°C in a cocktail of the two corresponding secondary antibodies: biotinylated goat anti-rabbit antibody (1:100 dilution) for detection of CRF-primary antibody complex and ultrasmall gold conjugated goat anti-guinea pig (1:100; Nanoprobes, Yaphank, NY) antibody for detection of vGluT1 or vGluT2-primary antibody complexes. Ultrasmall gold-conjugated goat anti-mouse antibody (1:100; Amershan, Arlington Heghts, IL) was used to detect anti-GAD and anti-TH primary antibodies. After several rinses in PB, sections were incubated in ABC (1:200) overnight at 4°C, rinsed, and then fixed with 0.5% glutaraldehyde in PB for 10 minutes. To remove phosphate ions before silver enhancement, sections were rinsed with 0.2 M sodium acetate buffer (SAB), pH 7.4. Silver intensification of gold particles was done for 10–15 minutes using a silver enhancement kit (Amershan and Nanoprobes); the reaction was stopped by transferring sections to SAB. To visualize CRF immunolabel, sections were incubated with 22 μg/ml DAB and 0.003% H2O2 in PB for 10 minutes.
Electron microscopy
Selected single- or double-immunolabeled sections of the VTA were fixed with 1% osmium tetroxide in 0.1 M PB for 1 hour and dehydrated through a series of graded alcohols (including 60 minutes in 70% alcohol containing 1% uranyl acetate), then with propylene oxide. Afterward, they were flat-embedded in Durcupan ACM epoxy resin (Electron Microscopy Sciences). Embedded sections were polymerized at 60°C for 2 days. Ultrathin sections (70 nm in thickness) were cut from the outer surface of the tissue with an ultramicrotome (Leica Microsystems, Wetzlar, Germany) using a diamond knife (Diatome, Fort Washington, PA). The sections were collected onto Formvar-coated single-slot grids and counterstained with Reynolds lead citrate (Reynolds, 1963). Sections were examined with a Tecnai G–12 transmission electron microscope (Fei Company, Hillsboro, OR). Digital electron micrographs were obtained with a digital micrograph 3.4 camera (Gatan, Inc., Pleasanton, CA) and adjusted to matching contrast and brightness in Adobe Photoshop (Adobe Systems, Mountain View, CA).
Ultrastructural analysis
Coronal vibratome sections of VTA corresponding to plates 71–79 (bregma −4.56 mm to −5.52 mm) of the rat brain atlas of Paxinos and Watson (2005) were used in this study. Brain sections from four rats were used for the ultrastructural analysis. At least four to six grids containing ultrathin sections were collected from four to six plastic-embedded sections of the VTA from the four rats. Differences among the means of the four animals were not significant. Data are presented as total number of synapses counted for the four animals. Synaptic contacts were classified according to their morphology and immunolabel, counted, and photographed at a magnification of ×6,800–13,000. When the identification of a synaptic contact was doubtful, contiguous sections were studied to determine the type of synapse as well as to confirm a positive immunolabeling pattern. The morphological criterion used for identification and classification of cellular components observed in these thin sections were as previously described (Peters et al., 1991). Briefly, neuronal cell bodies were identified by the presence of a large and round nucleus as well as rough endoplasmic reticulum and Golgi apparatus. Large and medium-sized dendrites were identified by the presence of endoplasmic reticulum, mitochondria, and microtubules. Axon terminals were characterized by the presence of synaptic vesicles and mitochondria, whereas axon preterminals and unmyelinated axons are smaller in diameter and occasionally contain a small number of synaptic vesicles. Asymmetric synapses were characterized by the presence of thick postsynaptic densities, whereas symmetric synapses have thin postsynaptic densities.
RESULTS
CRF immunolabeled axon terminals make both symmetric and asymmetric synapses on VTA neurons
CRF immunoreactivity was mostly found in axon terminals (Fig. 1A,B) and unmyelinated axons and occasionally in dendrites (data not shown). CRF-immunoreactive (-IR) axon terminals were observed making synaptic contacts mostly with small and medium-sized dendrites (Fig. 1C–E) and less frequently with neuronal cell bodies (not shown). CRF-IR axon terminals were found to establish asymmetric (Gray’s type I, characteristic of excitatory synapses) as well as symmetric (Gray’s type II, characteristic of inhibitory synapses) synapses on VTA neurons. CRF-IR axon terminals making asymmetric synapses had thick and electron-dense postsynaptic densities on the target dendrites (Fig. 1C,D). In contrast, CRF-IR axon terminals making symmetric synapses had thin postsynaptic densities (Fig. 1E,F).
Fig. 1.
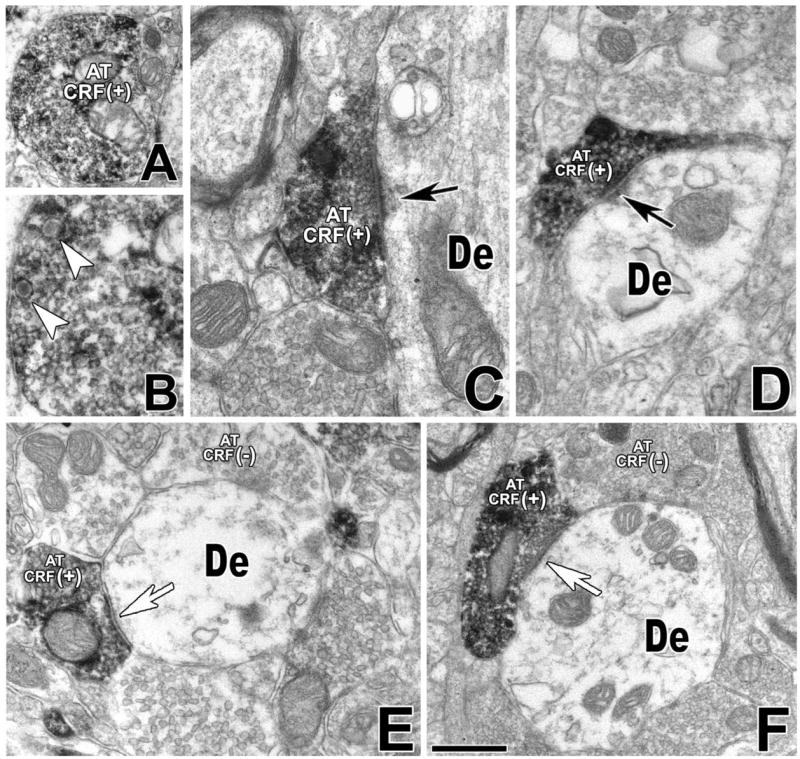
Detection of CRF-IR axon terminals in the VTA. Electron micrographs showing CRF immunoperoxidase labeling in the VTA. A: CRF-immunolabel is seen as black, diffuse deposits in an axon terminal. B: The same CRF-IR axon terminal at higher magnification shows dense-core vesicles (arrowheads). C: CRF-IR axon terminal (AT CRF+) forming an asymmetric synapse (black arrow) with an unlabeled dendrite (De). D: CRF-IR axon terminal (AT CRF+) makes an asymmetric synapse (black arrow) with a dendrite lacking CRF immunolabeling (De). E: CRF-IR axon terminal (AT CRF+) establishes a symmetric synapse (white arrow) with a dendrite lacking CRF immunolabeling (De), and the same dendrite receives synaptic contacts from axon terminals lacking CRF immunostaining (AT CRF−). F: A CRF-IR axon terminal (AT CRF+) makes a symmetric synapse (white arrow) with an unlabeled dendrite (De). The same dendrite makes synapses with other axon terminals lacking CRF immunolabeling (AT CRF−). Scale bar = 0.4 μm in F (applies to A,F); 0.18 μm for B; 0.25 μm for C,E; 0.3 μm for D.
From quantitative analysis, we concluded that CRF-IR axon terminals more frequently established asymmetric (60%) than symmetric synapses (40%; Table 2). Most CRF-IR axon terminals making asymmetric synapses were observed on dendrites (98% synapses of all CRF-IR asymmetric synapses, 277 synapses from a total of 282) and rarely on cell bodies (2% synapses of all CRF-IR asymmetric synapses, five synapses from a total of 282). Similarly, the majority of CRF-IR axon terminals making symmetric synapses were also observed on dendrites (72% of all CRF-IR symmetric synapses, 136 synapses from a total of 188) and less frequently on cell bodies (27% of all CRF-IR symmetric synapses, 52 synapses from a total of 188).
TABLE 2.
Frequency of CRF-IR Axon Terminals Making Asymmetric or Symmetric Synapses on Cell Bodies or Dendrites in the VTA
| Total number of counted synapses (n = 470) 100% | ||
|---|---|---|
| Postsynaptic targets of CR-IR axon terminals | Asymmetric synapses (n = 282) 60% | Symmetric synapses (n = 188) 40% |
| Cell bodies [total % of synapses on cell bodies (12%)] | 1% (n = 5) | 11% (n = 52) |
| Dendrites [total % of synapses on dendrites (88%)] | 59% (n = 277) | 29% (n = 136) |
CRF-IR axon terminals establishing asymmetric synapses preferentially coexpress vGluT2
Glutamate is transported into synaptic vesicles at presynaptic terminals by vesicular glutamate transporters (vGluTs). Three isoforms of vGluTs (1, 2, and 3) are found in the brain. Whereas vGluT1 and vGluT2 are restricted to known glutamatergic neurons, vGluT3 is present in GABAergic neurons, cholinergic interneurons, monoamine neurons, and glial cells (Fremeau et al., 2002; Gras et al., 2002). Therefore, the presence of vGluT1 and vGluT2 has become a reliable molecular phenotypic marker for the identification of glutamatergic neurons. To determine the probable glutamatergic nature of CRF-IR axon terminals forming asymmetric synapses, we analyzed the expression of vGluT1 and vGluT2 in these axon terminals. Although we often detected vGluT2 immunostaining in axon terminals that contained CRF immunolabeling (Fig. 2A–D), we rarely found vGluT1 immunoreactivity in CRF-IR axon terminals (Fig. 2E). Consistent with this observation, quantitative analysis of VTA sections doubly labeled with antibodies against CRF and VGluT2 showed that 96% of the CRF-IR axon terminals making asymmetric synapses (86 synapses from a total of 90) contained vGluT2 immunoreactivity, whereas only 4% (four synapses from a total of 90) lacked VGluT2 labeling. vGluT2-IR axon terminals containing or lacking CRF were observed making asymmetric synapses with the same dendrite (Fig. 2B). We often found two axon terminals, each coexpressing CRF and VGluT2 immunolabeling, establishing asymmetric synapses with the same dendrite (Fig. 2C), but we also observed a CRF-IR axon terminal containing vGluT2 labeling forming asymmetric synapses with two different dendrites (Fig. 2D).
Fig. 2.

CRF-IR axon terminals making asymmetric synapses in the VTA contain mostly vGluT2-immunolabel and rarely vGluT1-immunolabel. Electron micrographs showing CRF immunoperoxidase labeling (electron-dense, diffuse deposits) and vGluT2 or vGluT1 immunogold-silver labeling (highly electron-dense deposits). A: An axon terminal containing both CRF immunolabel and vGluT2 immunolabel (AT CRF+/vGluT2+) makes an asymmetric synapse (black arrow) with a dendrite lacking immunolabeling (De); note that this dendrite forms additional synapses with unlabeled axon terminals. B: An axon terminal containing both CRF immunolabel and vGluT2 immunolabel (AT CRF+/vGluT2+) forms an asymmetric synapse with an unlabeled dendrite (De). Note that the same dendrite makes another asymmetric contact with an axon terminal containing vGluT2 immunolabel but lacking CRF immunolabel (AT CRF−/vGluT2+). C: Two axon terminals containing both CRF immunolabel and vGluT2 immunolabel (AT CRF+/vGluT2+) form asymmetric synapses (black arrows) with the same dendrite lacking immunolabeling (De). D: An axon terminal containing both CRF immunolabel and vGluT2 immunolabel (AT CRF+/vGluT2+) establishes asymmetric synapses (black arrows) with two unlabeled dendrites (De). E: An axon terminal containing both CRF immunolabel and vGluT1 immunolabel (AT CRF+/vGluT1+) forms an asymmetric synapse (black arrow) with an unlabeled dendrite (De). Scale bar = 0.5 μm in C (applies to A,C); 0.35 μm for B; 0.40 μm for D,E.
Most CRF-IR axon terminals establishing symmetric synapses coexpress GAD immunoreactivity
To determine the possible GABAergic nature of CRF-IR axon terminals making symmetric synapses, we looked for expression of GAD in these axon terminals. Double-labeled CRF/GAD-IR axon terminals were observed to form synapses with GAD-IR cell bodies (Fig. 3A) and dendrites that either contained or lacked GAD immunoreactivity (Fig. 3B). We also found CRF-IR axon terminals lacking GAD that make asymmetric synapses with GAD-IR dendrites (Fig. 3C). Quantitative analysis of VTA sections doubly labeled with antibodies against CRF and GAD showed that 76% of the CRF-IR axon terminals making symmetric synapses (53 synapses from a total of 70) contained GAD immunoreactivity, whereas 24% (17 synapses from a total of 70) lacked it.
Fig. 3.
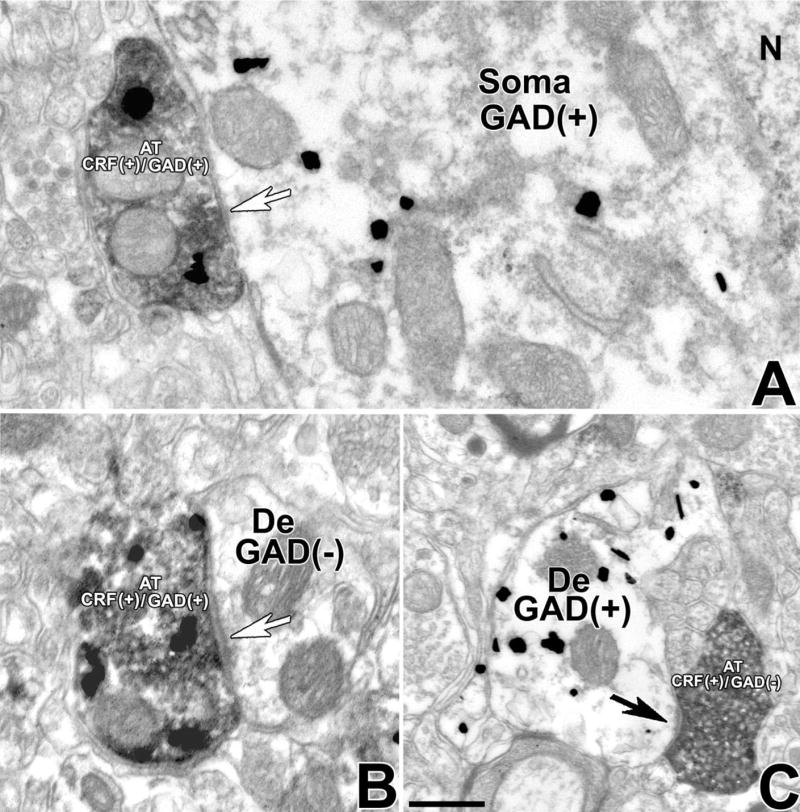
CRF-IR axon terminals making symmetric synapses in the VTA contain GAD. Electron micrographs showing CRF immunoperoxidase labeling (electron-dense, diffuse deposits) and GAD immunogold-silver labeling (highly electron-dense deposits). A: An axon terminal containing both CRF immunolabel and GAD immunolabel (AT CRF+/GAD+) establishes a symmetric synapse (white arrow) with a GAD-IR neuronal cell body (soma GAD+). N, nucleus. B: An axon terminal containing both CRF immunolabel and GAD immunolabel (AT CRF+/GAD+) forms a symmetric synapse (white arrow) with an unlabeled dendrite (De). C: An axon terminal containing CRF immunolabel lacking GAD immunolabel (AT CRF+/GAD−) makes an asymmetric synapse (black arrow) with a GAD-IR dendrite (De GAD+). Scale bar = 0.3 μm.
CRF-IR axon terminals make synapses on neurons that either contain or lack TH immunolabel
VTA sections were simultaneously incubated with antibodies against CRF and TH to detect anatomical interactions between neuronal profiles containing CRF immunostaining and those containing TH immunostaining. Ultrastructural analysis of CRF/TH-IR material further confirmed the presence of CRF-IR axons and axon terminals and TH-IR cell bodies and dendrites (Fig. 4A–C). CRF-IR axon terminals were found to establish asymmetric as well as symmetric synapses with either dendrites that contained (Fig. 4A,B) or lacked (Fig. 4C) TH immunostaining. Quantitative analysis of all CRF axon terminals making recognizable synaptic contacts indicated that 42% of them contacted TH-IR neurons and 58% contacted neurons lacking detectable levels of TH immunolabeling (Table 3).
Fig. 4.
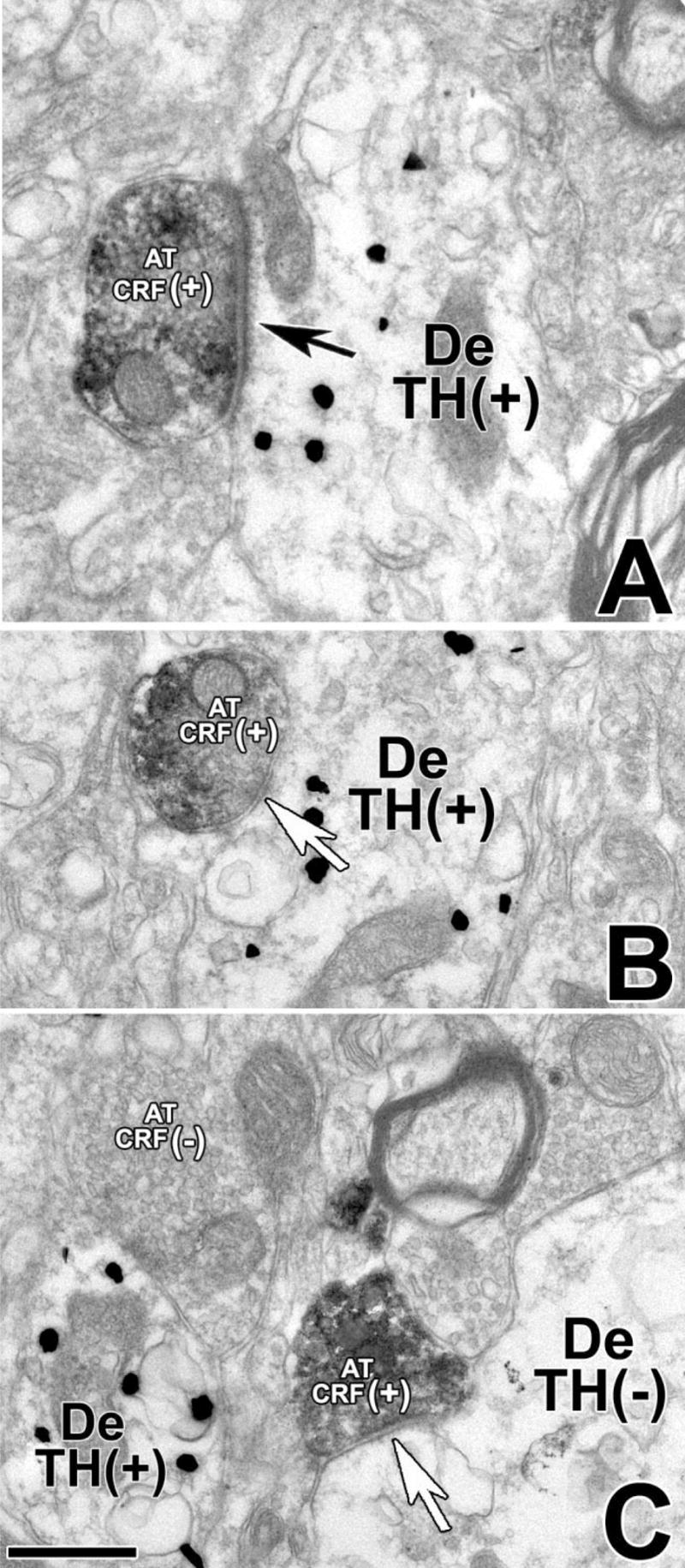
CRF-IR axon terminals make asymmetric or symmetric synapses with TH-IR dendrites in the VTA. Electron micrographs showing CRF immunoperoxidase labeling (electron-dense, diffuse deposits) and TH immunogold-silver labeling (highly electron-dense deposits). A: A CRF-IR axon terminal (AT CRF+) establishes an asymmetric synapse (black arrow) with a TH-immunolabeled dendrite (De TH+). B: A CRF-IR axon terminal (AT CRF+) establishes a symmetric synapse (white arrow) with a TH-IR dendrite (De TH+). C: A CRF-IR axon terminal (AT CRF+) makes a symmetric synapse (white arrow) on a dendrite lacking TH immunolabel (De TH−). Scale bar = 0.4 μm.
TABLE 3.
Frequency of CRF-IR Axon Terminals Making Asymmetric or Symmetric Synapses on Cell Bodies or Dendrites Containing (+) or Lacking (−) TH Immunoreactivity in the VTA
| Total number of counted synapses (n = 350) 100% | ||
|---|---|---|
| Postsynaptic targets of CR-IR axon terminals | Asymmetric synapses (n = 210) 60% | Symmetric synapses (n = 140) 40% |
| Total % of synapses on TH+neurons (42%) | ||
| Cell bodies TH+ | 2% (n = 7) | 1% (n = 3) |
| Dendrites TH+ | 33% (n = 116) | 6% (n = 22) |
| Total % of synapses on TH− neurons (58%) | ||
| Cell bodies TH− | 1% (n = 3) | 14% (n = 49) |
| Dendrites TH− | 24% (n = 84) | 19% (n = 66) |
CRF-IR axon terminals mostly establish asymmetric synapses on TH-IR dendrites
Further analysis of synapses between CRF-IR axon terminals and TH-IR neurons demonstrated that CRF-IR axon terminals mostly target TH-IR dendrites (93% of all CRF-TH synapses; Table 4) and rarely TH-IR cell bodies (7% of all CRF-TH synapses; Table 4). Whereas CRF-IR terminals were found to make asymmetric (Fig. 4A) as well as symmetric synapses on TH-IR neurons (Fig. 4B), CRF-IR axon terminals largely make asymmetric synapses on TH-IR neurons (83% of all counted CRF synapses on TH-IR neurons; Table 4) and much less frequently make symmetric synapses on these neurons (17% of all counted CRF synapses on TH-IR neurons; Table 4).
TABLE 4.
Frequency of CRF-IR Axon Terminals Making Asymmetric or Symmetric Synapses on TH-IR Cell Bodies or Dendrites in the VTA
| Total number of counted synapses (n = 148) 100% | ||
|---|---|---|
| Postsynaptic targets of CRF-IR axon terminals | Asymmetric synapses (n = 123) 83% | Symmetric synapses (n = 25) 17% |
| TH-IR cell bodies [total % of synapses on TH+ cell bodies (7%)] | 5% (n = 7) | 2% (n = 3) |
| TH-IR dendrites [total % of synapses on TH+ dendrites (93%)] | 78% (n = 116) | 15% (n = 22) |
Some CRF-IR axon terminals establish symmetric synapses and some establish asymmetric synapses on neurons lacking detectable levels of TH
As seen with TH-IR neurons, CRF-IR axon terminals mostly target dendrites (75% of all CRF-IR synapses on neurons lacking TH-IR; Table 5) and less often cell bodies (25% of all CRF-IR synapses on neurons lacking TH immunolabel; Table 5). Some CRF-IR terminals were found to make asymmetric and others to make symmetric synapses on neurons lacking TH label (Fig. 4C). The distribution of these synapses was more even (43% asymmetric and 57% symmetric; Table 5) than those found on TH-IR neurons (83% of all CRF-IR synapses were found to be asymmetric; Table 4).
TABLE 5.
Frequency of CRF-IR Axon Terminals Making Asymmetric or Symmetric Synapses on Cell Bodies or Dendrites Lacking TH-IR in the VTA
| Total number of counted synapses (n = 202) 100% | ||
|---|---|---|
| Postsynaptic targets of CR-IR axon terminals | Asymmetric synapses (n = 87) 43% | Symmetric synapses (n = 115) 57% |
| Cell bodies lacking TH-IR [total % of synapses on TH+ cell bodies (25%)] | 1% (n = 3) | 24% (n = 49) |
| Dendrites lacking TH-IR [total % of synapses on TH+ cell bodies (75%)] | 42% (n = 84) | 33% (n = 66) |
DISCUSSION
CRF in the VTA was found to be present mostly in axons and axon terminals but rarely in dendrites. The synaptic release of CRF from axon terminals in the VTA is likely to play a role in stress responses, in that Wang et al. (2005) demonstrated that foot shock-induced elevation of CRF in VTA is blocked by VTA infusion of the sodium channel blocker tetrodotoxin. We found that CRF-IR axon terminals in VTA make symmetric and asymmetric synapses largely with dendrites. We established that nearly all of the CRF-IR axon terminals making asymmetric synapses are glutamatergic and that the majority of those forming symmetric synapses are GABAergic, findings of functional importance. Because cholinergic neurons can also establish asymmetric synapses and monoaminergic neurons make symmetric synapses, we cannot rule out here the possibility that some CRF-IR axon terminals making either asymmetric or symmetric synapses may express neurotransmitters other than glutamate or GABA. CRF-IR axon terminals contact both DAergic and non-DAergic dendrites. However, we surprisingly found that most CRF-IR axon terminals contacting DAergic neurons form asymmetric synapses. The presence of CRF synapses in the VTA offers a mechanism for interactions between the stress-associated neurohormone CRF and the mesocorticolimbic DA system.
The VTA receives inputs from two types of CRF-containing neurons: CRF-glutamatergic and CRF-GABAergic
We concluded, based on morphological analysis, that about 60% of all CRF synapses in VTA are asymmetric (Gray’s type I), giving the characteristic thick postsynaptic density. The remaining 40% of the CRF synapses were identified as symmetric synapses (Gray’s type II), with a thin postsynaptic density. CRF-IR axon terminals making asymmetric or symmetric synapses had also been observed in the perilocus coeruleus area (Valentino et al., 2001). We found that, in asymmetric synapses, most of the CRF-IR axon terminals coexpress vGluT2 but rarely VGluT1. These results provide evidence supporting the notion that the vast majority of CRF asymmetric synapses are glutamatergic in the VTA.
GABA is an inhibitory neurotransmitter synthesized from glutamate by the enzyme GAD; GABA and GAD are considered dependable markers for the identification of GABAergic neurons. We found GAD coexpression with CRF in more than 60% of terminals forming symmetric contacts. These results provide evidence that CRF symmetric synapses are mostly GABAergic in the VTA. It is not clear whether the lack of GAD signal in 40% of the CRF-IR axon terminals making symmetric synapses was due to the lack of sensitivity of the procedure used or alternatively whether CRF-IR axon terminals making symmetric synapses may express neurotransmitters other than GABA.
The presence in VTA of some axon terminals coexpressing CRF and VGluT2 and others coexpressing CRF and GAD suggests that these CRF terminals come from at least two different types of neurons, CRF-glutamatergic and CRF-GABAergic. Based on review of the published literature, it is not possible to determine the probable source of CRF cells projecting to the VTA. Our preliminary in situ hybridization and tracing studies indicate that CRF/GABAergic neurons from the central amygdala, prefrontal cortex (PFC), or hypothalamus are unlikely to be the source of CRF innervating the VTA (unpublished results). We found CRF neurons coexpressing VGluT2 in the bed nucleus of the stria terminalis and the brainstem; some of these neurons appear to project to the VTA (unpublished results).
Stress and release of DA within the mesocorticolimbic system
It is not clear how CRF participates in the well-documented stress-induced release of DA in brain areas such as the medial PFC (Abercrombie et al., 1989), the shell of the nucleus accumbens (Kalivas and Duffy, 1995), and the basolateral amygdala (Inglis and Moghaddam, 1999). Previous studies showed that peripheral or intra-ventricular application of CRF causes release of DA, norephinephine, and most of their catabolites in the medial prefrontal cortex and medial hypothalamus (Lavicky and Dunn, 1993). Based on our results showing that mostly CRF/glutamatergic axon terminals target VTA DAergic neurons, it would appear that CRF/glutamatergic neurons play a role in the neuronal circuitry that mediates stress responses involving the DA mesocorticolimbic system. We propose that stress causes activation of CRF-glutamatergic neurons innervating the VTA, thus inducing local release of CRF, glutamate, or both and producing activation of VTA DAergic neurons. In support of this proposal, it was previously documented that footshock induces release of CRF in the VTA (Wang et al., 2005) and that CRF added to rat midbrain slice preparation directly excites the majority of VTA DAergic neurons (Korotkova et al., 2006). In addition, several studies indicate that glutamatergic projections to VTA, acting via both NMDA and non-NMDA-glutamate receptors, play a role in the stress-induced increase of DA release in the PFC (Enrico et al., 1998; Kalivas et al., 1989). For instance, application of the N-methyl-D-aspartate (NMDA) receptor antagonist 3-[(±)-2-carboxypiperazine-4-yl] propyl-1-phosphonic acid into the VTA prevents the foot shock stress-induced increase in PFC DA metabolites (Kalivas et al., 1989), whereas handling-stress-induced release of prefrontal DA is suppressed by infusion into the VTA of NMDA and non-NMDA-glutamate receptor antagonists (Enrico et al., 1998). Interactions between CRF and VTA DAergic neurons in mediating DA-stress-related responses is supported by observations that VTA DAergic neurons are contacted by CRF/glutamatergic neurons, that CRF directly excites VTA DAergic neurons, that stress induces release of CRF in the VTA, and that VTA glutamatergic neurotransmission mediates release of cortical DA.
Behaviors associated with intake of drugs of abuse are affected by stress. For instance, studies on drug self-administration in laboratory animals have shown that both physical and psychological stressors facilitate the acquisition of drug self-administration (Piazza and Le Moal, 1998). In addition, single or repeated exposure to stressful stimuli can increase the motor stimulant actions to subsequent amphetamine, cocaine, or morphine exposure (Kalivas and Stewart, 1991). Furthermore, stressors facilitate the reinstatement of drug taking even after prolonged periods of forced abstinence (Le et al., 2000). Although the neuronal pathways, neurons, and neurotransmitters that mediate interactions between stress and drugs of abuse are not well characterized, CRF has been shown to participate in stress-induced reinstatement of cocaine, heroin, and alcohol self-administration, insofar as CRF antagonists attenuate drug seeking induced by foot shock (Erb et al., 1998; Shaham et al., 1997, 1998). Significantly, cocaine seeking is reinstated by CRF infusions into VTA, whereas CRF antagonists injected into this region block cocaine seeking induced by foot shock (Wang et al., 2005). Here we provide evidence suggesting that CRF axon terminals contacting mostly dendrites in the VTA are the likely source of CRF that participates, at the level of the VTA, in cocaine seeking induced by stress (Wang et al., 2005).
In summary, we found that CRF in the VTA is present mostly in axon terminals making either asymmetric or symmetric synapses. The presence of VTA axon terminals coexpressing CRF and VGluT2 and others coexpressing CRF and GAD suggests that these CRF terminals come from at least two different types of neurons, CRF-glutamatergic and CRF-GABAergic. We observed that the majority of CRF axon terminals that contact DAergic neurons form asymmetric synapses (glutamatergic). Data shown here on the preferential excitatory connectivity between CRF axon terminals and DAergic neurons in the VTA suggest a mechanism for the well-known interaction of stress response— coordinated in the brain in part by the neuropeptide CRF—with the mesocorticolimbic DA system.
Acknowledgments
Grant sponsor: Intramural Research Program of the National Institute on Drug Abuse.
Footnotes
†
This article is a US Government work and, as such, is in the public domain in the United States of America.
LITERATURE CITED
- Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52:1655–1658. doi: 10.1111/j.1471-4159.1989.tb09224.x. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Chang YC, Gottlieb DI. Characterization of the proteins purified with monoclonal antibodies to glutamic acid decarboxylase. J Neurosci. 1988;8(6):2123–2130. doi: 10.1523/JNEUROSCI.08-06-02123.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutch AY, Roth RH. The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog Brain Res. 1990;85:367–402. doi: 10.1016/s0079-6123(08)62691-6. discussion 402–363. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Bean AJ, Bissette G, Nemeroff CB, Robbins RJ, Roth RH. Stress-induced alterations in neurotensin, somatostatin and corticotropin-releasing factor in mesotelencephalic dopamine system regions. Brain Res. 1987;417:350–354. doi: 10.1016/0006-8993(87)90462-8. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev. 1990;15:71–100. doi: 10.1016/0165-0173(90)90012-d. [DOI] [PubMed] [Google Scholar]
- Enrico P, Bouma M, de Vries JB, Westerink BH. The role of afferents to the ventral tegmental area in the handling stress-induced increase in the release of dopamine in the medial prefrontal cortex: a dual-probe microdialysis study in the rat brain. Brain Res. 1998;779:205–213. doi: 10.1016/s0006-8993(97)01132-3. [DOI] [PubMed] [Google Scholar]
- Erb S, Shaham Y, Stewart J. The role of corticotropin-releasing factor and corticosterone in stress- and cocaine-induced relapse to cocaine seeking in rats. J Neurosci. 1998;18:5529–5536. doi: 10.1523/JNEUROSCI.18-14-05529.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci U S A. 2002;99:14488–14493. doi: 10.1073/pnas.222546799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci. 2002;22:5442–5451. doi: 10.1523/JNEUROSCI.22-13-05442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis FM, Moghaddam B. Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem. 1999;72:1088–1094. doi: 10.1046/j.1471-4159.1999.0721088.x. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. Selective activation of dopamine transmission in the shell of the nucleus accumbens by stress. Brain Res. 1995;675:325–328. doi: 10.1016/0006-8993(95)00013-g. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P, Latimer LG. Neurochemical and behavioral effects of corticotropin-releasing factor in the ventral tegmental area of the rat. J Pharmacol Exp Ther. 1987;242:757–763. [PubMed] [Google Scholar]
- Kalivas PW, Duffy P, Barrow J. Regulation of the mesocorticolimbic dopamine system by glutamic acid receptor subtypes. J Pharmacol Exp Ther. 1989;251:378–387. [PubMed] [Google Scholar]
- Koob GF, Heinrichs SC. A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res. 1999;848:141–152. doi: 10.1016/s0006-8993(99)01991-5. [DOI] [PubMed] [Google Scholar]
- Korotkova TM, Brown RE, Sergeeva OA, Ponomarenko AA, Haas HL. Effects of arousal- and feeding-related neuropeptides on dopaminergic and GABAergic neurons in the ventral tegmental area of the rat. Eur J Neurosci. 2006;23:3407. doi: 10.1111/j.1460-9568.2006.04792.x. [DOI] [PubMed] [Google Scholar]
- Lavicky J, Dunn AJ. Corticotropin-releasing factor stimulates catecholamine release in hypothalamus and prefrontal cortex in freely moving rats as assessed by microdialysis. J Neurochem. 1993;60:602–612. doi: 10.1111/j.1471-4159.1993.tb03191.x. [DOI] [PubMed] [Google Scholar]
- Le AD, Harding S, Juzytsch W, Watchus J, Shalev U, Shaham Y. The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology. 2000;150:317–324. doi: 10.1007/s002130000411. [DOI] [PubMed] [Google Scholar]
- Morales M, Battenberg E, de Lecea L, Sanna PP, Bloom FE. Cellular and subcellular immunolocalization of the type 3 serotonin receptor in the rat central nervous system. Brain Res Mol Brain Res. 1996;36:251–260. doi: 10.1016/0169-328x(96)88406-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. xliii. Amsterdam; Boston: Elsevier Academic Press; 2005. p. 166. [Google Scholar]
- Peters A, Palay SL, Webster H. The fine structure of the nervous system: neurons and their supporting cells. xviii. New York: Oxford University Press; 1991. p. 494. [Google Scholar]
- Piazza PV, Le Moal M. The role of stress in drug self-administration. Trends Pharmacol Sci. 1998;19:67–74. doi: 10.1016/s0165-6147(97)01115-2. [DOI] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Sarabi A, Hoffer BJ, Olson L, Morales M. GFRalpha-1 mRNA in dopaminergic and nondopaminergic neurons in the substantia nigra and ventral tegmental area. J Comp Neurol. 2001;441:106–117. doi: 10.1002/cne.1400. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Imaki T, Potter E, Kovacs K, Imaki J, Vale W. The functional neuroanatomy of corticotropin-releasing factor. Ciba Found Symp. 1993;172:5–21. doi: 10.1002/9780470514368.ch2. discussion 21–29. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Funk D, Erb S, Brown TJ, Walker CD, Stewart J. Corticotropin-releasing factor, but not corticosterone, is involved in stress-induced relapse to heroin-seeking in rats. J Neurosci. 1997;17:2605–2614. doi: 10.1523/JNEUROSCI.17-07-02605.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Leung S, Buczek Y, Stewart J. CP-154,526, a selective, non-peptide antagonist of the corticotropin-releasing factor1 receptor attenuates stress-induced relapse to drug seeking in cocaine-and heroin-trained rats. Psychopharmacology. 1998;137:184–190. doi: 10.1007/s002130050608. [DOI] [PubMed] [Google Scholar]
- Shepard JD, Chuang DT, Shaham Y, Morales M. Effect of methamphetamine self-administration on tyrosine hydroxylase and dopamine transporter levels in mesolimbic and nigrostriatal dopamine pathways of the rat. Psychopharmacology. 2006;185:505–513. doi: 10.1007/s00213-006-0316-4. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Thierry AM, Tassin JP, Blanc G, Glowinski J. Selective activation of mesocortical DA system by stress. Nature. 1976;263:242–244. doi: 10.1038/263242a0. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A. Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron. 2003;39:401–407. doi: 10.1016/s0896-6273(03)00461-6. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Rudoy C, Saunders A, Liu XB, Van Bockstaele EJ. Corticotropin-releasing factor is preferentially colocalized with excitatory rather than inhibitory amino acids in axon terminals in the perilocus coeruleus region. Neuroscience. 2001;106:375–384. doi: 10.1016/s0306-4522(01)00279-2. [DOI] [PubMed] [Google Scholar]
- Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB. Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci. 2005;25:5389–5396. doi: 10.1523/JNEUROSCI.0955-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Brain reward circuitry: insights from unsensed incentives. Neuron. 2002;36:229–240. doi: 10.1016/s0896-6273(02)00965-0. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- Wolf ME, Kapatos G. Flow cytometric analysis and isolation of permeabilized dopamine nerve terminals from rat striatum. J Neurosci. 1989;9:106–114. doi: 10.1523/JNEUROSCI.09-01-00106.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]