Microenvironment Determines Lineage Fate in a Human Model of MLL-AF9 Leukemia (original) (raw)
. Author manuscript; available in PMC: 2009 Jun 1.
Published in final edited form as: Cancer Cell. 2008 Jun;13(6):483–495. doi: 10.1016/j.ccr.2008.04.020
Summary
Faithful modeling of mixed lineage leukemia in murine cells has been difficult to achieve. We show that expression of MLL-AF9 in human CD34+ cells induces acute myeloid, lymphoid or mixed lineage leukemia in immunodeficient mice. Some leukemia stem cells (LSC) were multipotent and could be lineage directed by altering either the growth factors or the recipient strain of mouse, highlighting the importance of microenviromental cues. Other LSC were strictly lineage committed, demonstrating the heterogeneity of the stem cell compartment in MLL disease. Targeting the Rac signaling pathway by pharmacologic or genetic means resulted in rapid and specific apoptosis of MLL-AF9 cells, suggesting that the Rac signaling pathway may be a valid therapeutic target in MLL-rearranged AML.
Significance
Mouse models of leukemia have elegantly defined the nature of the LSC implicated in AML-associated 11q23 leukemias but have proven more limited in modeling the lymphoid and biphenotypic aspects of the disease. We show that transduction of human CD34+ cells and transplantation into immunodeficient mice enables efficient modeling of mixed lineage leukemia associated with MLL-AF9 expression. Our data demonstrate that some MLL-AF9-expressing LSCs are multipotent while others are lineage restricted, demonstrating the heterogeneity of the LSC in mixed lineage leukemias. The role of microenvironment appears dominant over an instructive signal contributed by the fusion partner. This human-based system should prove useful in addressing the lineage promiscuity of MLL leukemias and allow testing of different therapeutic strategies.
Introduction
As its name implies, the Mixed Lineage Leukemia (MLL) gene on chromosome 11q23 is rearranged in both acute myeloid and acute lymphoid leukemia (AML and ALL) (De Braekeleer et al., 2005; Dimartino and Cleary, 1999). Reciprocal translocations that fuse the amino terminus of MLL to a diverse group of partner genes are found in 7% of AML and 10% of ALL. In AML, the most common partner gene for MLL is AF9 on chromosome 9p22. MLL-AF9 expressing AML cells have morphologic and immunophenotypic features consistent with myelomonocytic differentiation. Patients with an MLL-AF9 fusion have an intermediate to poor prognosis, suggesting that MLL-AF9 expression is associated with a more aggressive disease including resistance to chemotherapy (Schoch et al., 2003).
The propensity for MLL fusion proteins to transform primary hematopoietic progenitor cells (HPC) as independent, dominant oncogenes has been recapitulated in various mouse model systems. Knock-in of MLL-AF9 induces AML with a latency of 5 months (Corral et al., 1996). Retroviral gene transfer of MLL fusion genes into murine HPC has demonstrated that both MLL and fusion partner sequences are necessary and sufficient for transformation and that either HSC or more differentiated HPC can be transformed and function as leukemia stem cells (LSC) (Corral et al., 1996; Cozzio et al., 2003; DiMartino et al., 2002; Krivtsov et al., 2006; Lavau et al., 1997; So et al., 2003b; Somervaille and Cleary, 2006). Despite the elegance of these studies, the extent to which murine HPCs mirror the processes of the naturally occurring human disease is uncertain. In particular, the B cell leukemia associated with MLL fusion protein expression is not readily modeled in the mouse (Lavau et al., 1997).
In humans, MLL-ENL translocations are found equally represented in AML and ALL disease, yet the mouse model is highly biased towards myeloid leukemias (Lavau et al., 1997). It has recently been shown that MLL-ENL induces acute B-lymphocytic leukemia (B-ALL) when expressed in human HPC, with no development of AML (Barabe et al., 2007). Whether this disparity is due primarily to species-specific differences intrinsic to the hematopoietic cell or is an effect of microenvironment differences (the xenograft model versus the pure murine system) is an open question. This study went on to show that the MLL-AF9 oncogene, which is almost exclusively associated with AML in humans, induced AML in only 18% of recipient mice (2/11 mice) and B-ALL or MLL in 82% (9/11 mice). These data are used to argue for an instructive role of the fusion partner in MLL translocations, although neither MLL-ENL nor MLL-AF9 are primarily instructive for the B cell lineage in human clinical disease. The small sample size in this study does not permit definitive conclusions to be drawn; it is equally possible that factors other than the translocation fusion partner may be playing dominant roles in lineage decision, including microenvironment effects.
Here we report that retroviral mediated transfer of an MLL-AF9 fusion cDNA into normal human CD34+ cord blood (CB) cells leads to the establishment of cultures that produce a fatal leukemia in NOD/SCID mice. These cells proliferate indefinitely in vitro and in vivo, providing ample material for experimental manipulation and characterization. Gene expression profiling of MLL-AF9 expressing myeloid cultures reveals a signature that closely parallels that of patient AML samples containing the MLL-AF9 fusion. In addition, the mixed lineage nature of the leukemia is readily modeled in this system upon manipulation of environmental cues, implying that the lineage promiscuity of samples expressing the MLL-AF9 fusion protein may be primarily affected by the microenvironment in which the leukemia stem cell resides. This system provides a useful tool for the identification and validation of much needed therapeutic targets for MLL-rearranged leukemia. In particular, we identify the Rac signaling pathway as a critical regulator in MLL fusion leukemias and demonstrate that treatment of these cells with a Rac inhibitor or genetic ablation of Rac signaling induces cell cycle arrest and apoptosis.
Results
MLL-AF9 expression immortalizes human CD34+ cord blood cells
To determine whether MLL-AF9 would transform human CD34+ cord blood (CB) cells, we transduced cells with retroviral vectors expressing MLL-AF9 (MA9) as a bicistronic mRNA with the enhanced green fluorescent protein (EGFP) and independently with a second MA9 construct that did not co-express EGFP (Figure S1). Results using the two different constructs were similar, and most experiments were subsequently performed using the MA9-PuroR construct. Transduction efficiencies were 5–60% for MA9 vectors and 20–80% for control vectors (Figure 1A and data not shown). There was no significant skewing of the cell population two days after transduction as shown by surface expression of progenitor and myeloid specific surface antigens, with the majority of cells expressing CD34, CD117 (c-kit) and CD13 (Figure 1A). MA9 transduced cells became immortal and doubled in number every 2–3 days while control cells stopped proliferating between weeks 5 and 10 (Figure 1B). Expression of CD33, CD11b, CD13, CD14 and CD15 indicated a myelomonocytic lineage, presumably beyond the progenitor compartment given the lack of CD34 expression (Table S1). Long-term cultured MA9 cells (myeloid growth conditions) failed to differentiate towards the erythroid or B lymphoid lineages under conditions that support differentiation by control CB cells [data not shown and (Mulloy et al., 2003; Wunderlich et al., 2006)]. MA9 cells remained cytokine dependent for growth. In contrast to their murine counterparts, however, the human MA9 cells demonstrated a particular dependence on FLT3L that could not be readily overcome even with the combined use of SCF, GM-CSF and IL-3 (Figure 1C). An essential component in malignant transformation is the ability to maintain telomere length, a function attributed to the activity of telomerase in most human cancers (Flores et al., 2006). MA9 cells reproducibly demonstrated telomerase activity at early and late times in vitro (Figure 1D). To determine whether this might be an immediate effect of MA9 expression on the hTERT promoter, we analyzed cells for levels of telomerase mRNA expression shortly after transduction. Cord blood cells transduced with the GFP-expressing control and MA9 viruses were analyzed at day 7, when the majority of cells were positive for GFP expression. Transcript levels of MA9 were readily detected in two separate MA9-transduced cord blood cultures, while hTERT expression levels were unchanged (Figure 1E). To confirm these results in a different system, we used the U2OS osteosarcoma cell line, which is hTERT negative. Transduction followed by puromycin selection demonstrated that in this system as well, hTERT levels were unchanged while MA9 expression was easily detected by Q-PCR as early as day 4 after transduction (Figure 1E).
Figure 1. MLL-AF9 expression supports long-term proliferation and maturation arrest of CD34+ cord blood cells.
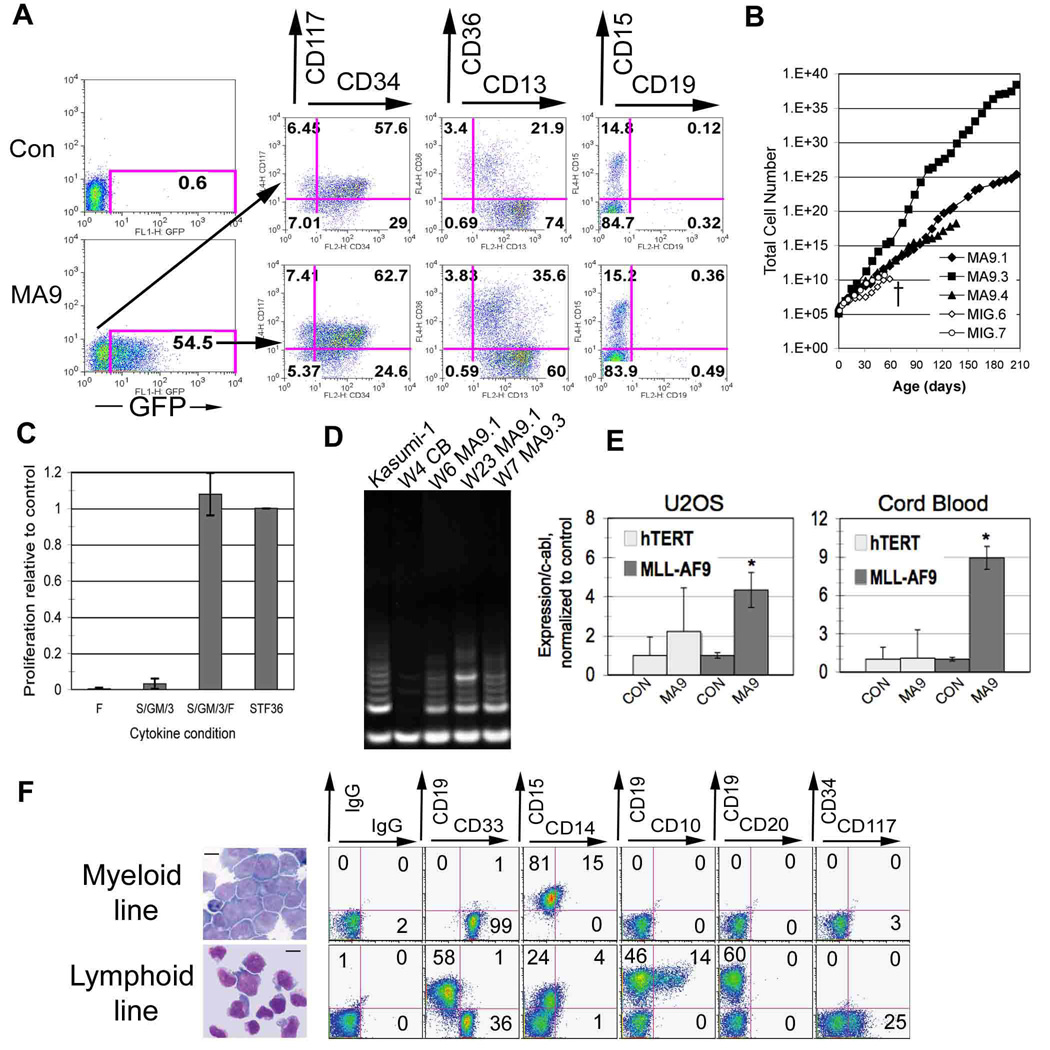
A) Immunophenotype of MLL-AF9 positive and negative cells on day 2 after transduction. B) MLL-AF9 expression results in immortalization of primary CB cells. A growth curve charts the differences in proliferative capacity between control (MIG) and MLL-AF9 positive (MA9) cells. C) MA9 expressing cells are particularly sensitive to FLT3L for proliferation. Cells cultured in the presence of FLT3L alone, SGM3, SGM3/F and (+) (S/T/F/3/6) respectively, were counted by trypan blue dye exclusion after one week. Results are expressed as mean ± standard deviation (SD). D) Telomerase activity was analyzed in MA9 cells grown for varying periods of time in vitro. Cord blood cells and the leukemia cell line Kasumi-1 were used as controls. W=weeks. E) hTERT and MLL-AF9 transcript levels were analyzed by Q-PCR using Sybr Green and the standard curve method, with two different primer sets for hTERT. The U2OS cells were analyzed four days after transduction and puromycin selection and the cord blood samples were tested at day 7 when the majority of cells in control and MA9 cultures were GFP+. Two independent cord blood samples were used and showed identical results. Results are shown as mean + SD with asterisks indicating significance of p≤ 0.05. hTERT transcript levels were essentially negative in U2OS, with signals detected at amplification cycles similar to no template control. F) Cytospins and flow cytometric analysis of long-term cultured cells grown under myeloid and lymphoid conditions. Scale bars represent 10 µm.
MLL-AF9 is occasionally associated with B-ALL (Secker-Walker, 1998). The clonal relatedness of the myeloid and B-lymphoid diseases is currently unclear, and presently there is no reproducible system for generating a B cell leukemia using the available mouse models for MLL-AF9. We transduced human CD34+ cells and cultured the cells under conditions that promote B cell outgrowth (Mulloy et al., 2003). MA9 cells became immortal and displayed a mixed lineage phenotype, with CD19+CD10(+/−)CD20- cells dominating the culture and variable levels of CD33+CD19- cells also present (Figure 1F). Thus, MA9 transduction of CD34+ cells results in immortalized myeloid and B-lymphoid cells, and the culture conditions play a critical role in determining the lineage outcome of the resulting culture.
MLL-AF9 expressing cells produce acute leukemia in NOD/SCID mice
MA9 cells were injected into non-obese diabetic/severe combined immunodeficient [NOD/SCID (NS)], NS-β2M−/− (NS-B2M) and NS mice that are transgenic for the human cytokine genes SCF, GM-CSF and IL-3 (NS-SGM3) (Feuring-Buske et al., 2003). In NS-SGM3 mice, cell lines as well as newly transduced cells (direct inject) induced AML with a median latency of 5 weeks and 7 weeks, respectively (Figure 2A). Cells from the myeloid and lymphoid cultures both resulted in AML in these mice. The disease resembled aggressive myelomonocytic leukemia with blast cells present in the peripheral blood, BM, spleen and liver (Figure 2A and Figure 3). Flow cytometry confirmed the myelomonocytic lineage of the leukemic cells (Figure 2A). Interestingly, most cells expressed the NK-associated surface molecule NCAM-1 (CD56), which is aberrantly expressed on up to 40% of MLL-fusion AMLs (Baer et al., 1998). Occasionally a leg tumor developed in those mice that were injected by the intrafemoral route, implying that the leukemic cells are able to form granulocytic sarcomas (Figure 3C). The median latency of secondary disease was 5 weeks, not significantly different than the latency of the primary AML (Figure 2A). However, when analyzed as paired samples, a significantly shorter latency was apparent in the secondary animals (Table S2).
Figure 2. MLL-AF9 expressing cells produce leukemia in immunodeficient mice.
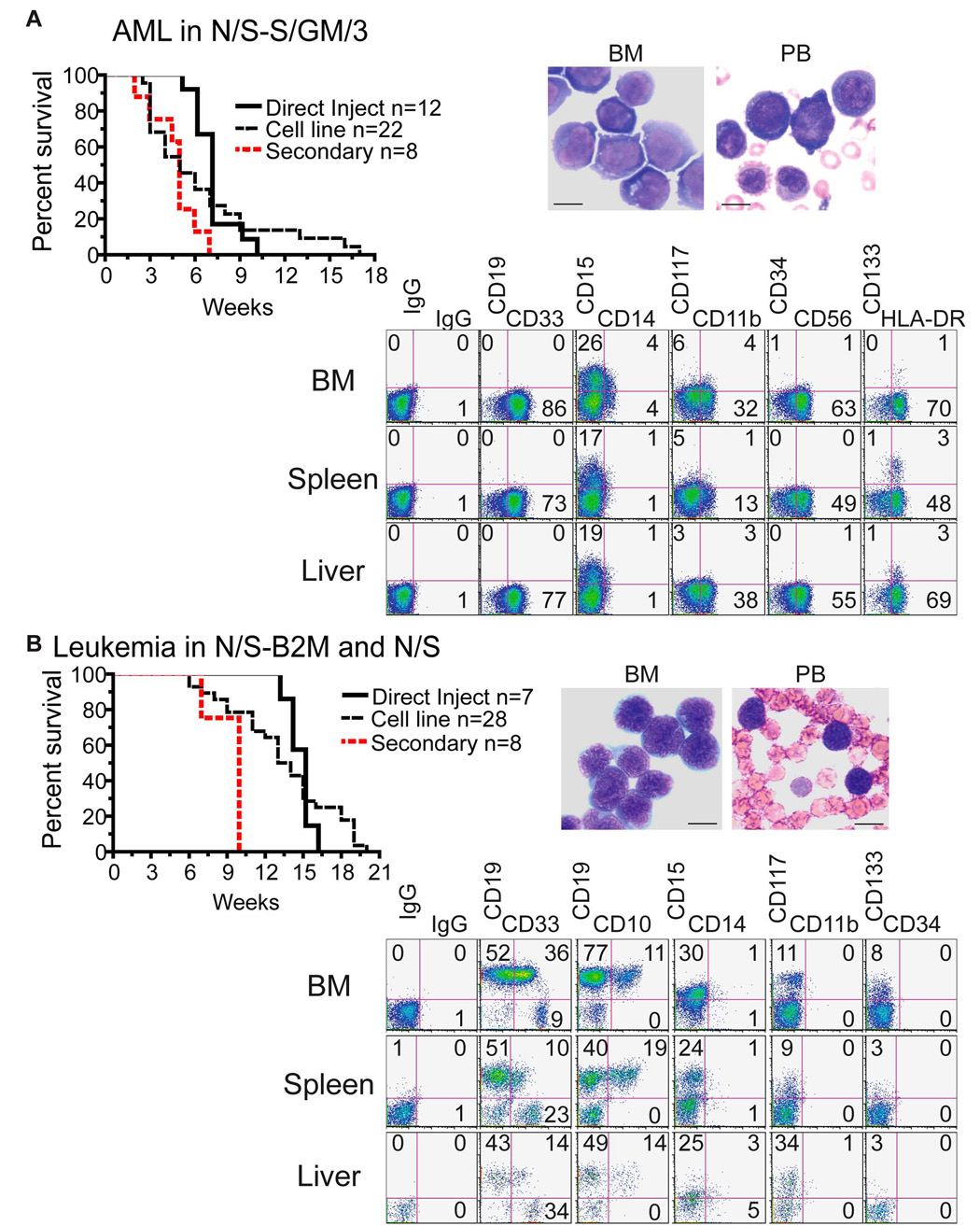
MA9 myeloid or lymphoid cultures (cell line), and freshly transduced CB cells (direct inject), were injected into NS-SGM3 mice (panel A) and NS or NS-B2M mice (panel B). Mice were sacrificed upon signs of illness. A Kaplan-Meier survival plot is shown for each cohort of animals. Wright-Giemsa stained BM cytospins and peripheral blood smears indicate that the circulatory system is full of immature myeloid or lymphoid blast cells. Scale bars represent 10 µm. Cells from mouse bone marrow, spleen and liver were stained for human lineage differentiation markers. Representative samples from an AML and B-ALL/ABL mouse are shown.
Figure 3. Haematoxylin and eosin stained paraffin sections demonstrate disruption of organ architecture due to tumor infiltration.
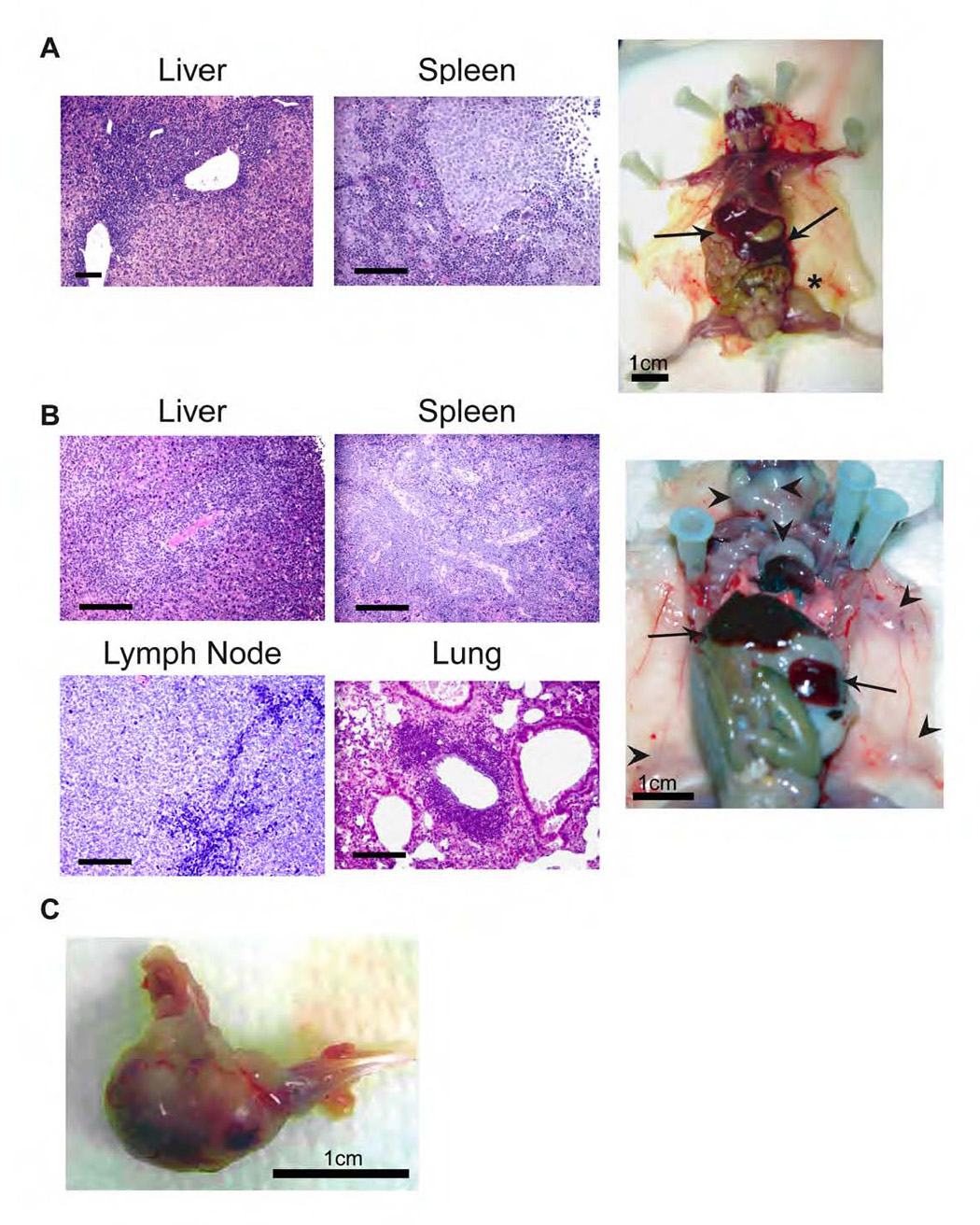
Organs and representative mice from animals with A) human AML disease and B) human B-ALL disease. Arrows indicate spleen and liver, and arrowheads show the enlarged lymph nodes in the B-ALL mouse. The asterisk highlights a leg tumor that is forming in the AML animal, which occasionally occurred after intrafemoral injection. A representative leg tumor is shown in panel C. Scale bars represent 100 µm unless indicated otherwise.
Cells injected into NS or NS-B2M mice resulted in a mix of AML, B-ALL and acute biphenotypic leukemia (ABL; defined as a leukemia with 20% or more of cells co-expressing myeloid and lymphoid markers). There was no significant difference in latency between disease in NS or NS-B2M mice (Figure S2A). Direct inject cells induced B-ALL or ABL (7/7 injections) in NS or NS-B2M mice with a median latency of 14.5 weeks (Figure 2B). This is similar to what has recently been published, with the exception that we did not observe any AML disease upon direct inject into NS mice (Barabe et al., 2007). However, these same cells uniformly induced AML in NS-SGM3, indicating the primacy of microenvironment on leukemia development (Figure 2A). In NS mice, the peripheral blood, spleen, liver, lung, kidney and lymph nodes were infiltrated with blast cells displaying a pro/pre-B phenotype (Figure 2B and Figure 3). In addition to B-ALL or ABL leukemia, there was often a separate population of cells that resembled AML disease, implying that independent LSCs were expanding in these mice or that the LSC was able to promote both AML and ALL in the same mouse. Cell lines cultured under myeloid conditions reproducibly induced only AML, with a latency of approximately 10 weeks (7/7 mice). In contrast, lymphoid cultures induced both AML (2/21 mice) and B-ALL or ABL (19/21 mice) (Figure 2B and Figure S2B). Median latency using lymphoid cell lines was 13.5 weeks, similar to that observed upon direct injection of freshly transduced CB cells (14.5 weeks) (Figure 2B). Injection of B-ALL samples into secondary recipients induced a significantly more rapid disease, suggesting that the B-ALL cells had acquired additional “hits” during the primary in vivo phase (Figure 2B). When separated into type of disease, AML occurred significantly faster than B-ALL/ABL in these mice, but was still slower than the AML that resulted upon injection into NS-SGM3 (Figures S2C and S2D). This strongly implies that the human cytokines supplied by the transgenes in NS-SGM3 are not only skewing the disease to a myeloid phenotype but are also shortening the latency of the leukemia. Results of all mouse experiments are summarized in Table 1.
Table 1.
Summary of mouse leukemia experiments
| Direct Inject | Myeloid Lines | Lymphoid Lines | |
|---|---|---|---|
| N/S | ND | AML(3);11 | ALL(8);15 |
| N/S-B2M | ALL(7);15 | AML(4);10 | AML(2);8ALL(11);15 |
| N/S-S/GM/3 | AML(12);7 | AML(18);4 | AML(4);12 |
Chromosome aneuploidy and oligoclonal expansion are evident in the cultured cells as well as in the leukemia samples
Chromosome changes were observed in some of the cell cultures as well as in some leukemia samples, but this was not uniformly seen (Table S3). Clonal evolution was evident in some samples, but gross karyotypic abnormalities were not required for disease induction (Table S3). All of the MA9 cell cultures displayed a polyclonal to oligoclonal pattern of retroviral integration at early time points in vitro, which became less complex over time (Figure 4A). Injection of week 3 MA9.6 cells into two mice (NS-SGM3) resulted in the induction of AML in each mouse after approximately 8 weeks, with clonal patterns present in each of the AML samples that were distinct from the in vitro long-term culture (9.6, 9.6#1 and 9.6#2 in Figure 4A). From a direct injection experiment, five separate mice displayed unique mono- and oligoclonal integration patterns in each of the resulting leukemias, again indicating that separate LSC populations were inducing these diseases (Figure 4B). This data would indicate that more than a single clone had acquired leukemogenic potential upon MA9 expression, and that transformation is a rapid event in human HPC upon expression of the MLL-AF9 fusion protein.
Figure 4. Oligoclonal outgrowth is detected in vivo as well as in vitro.
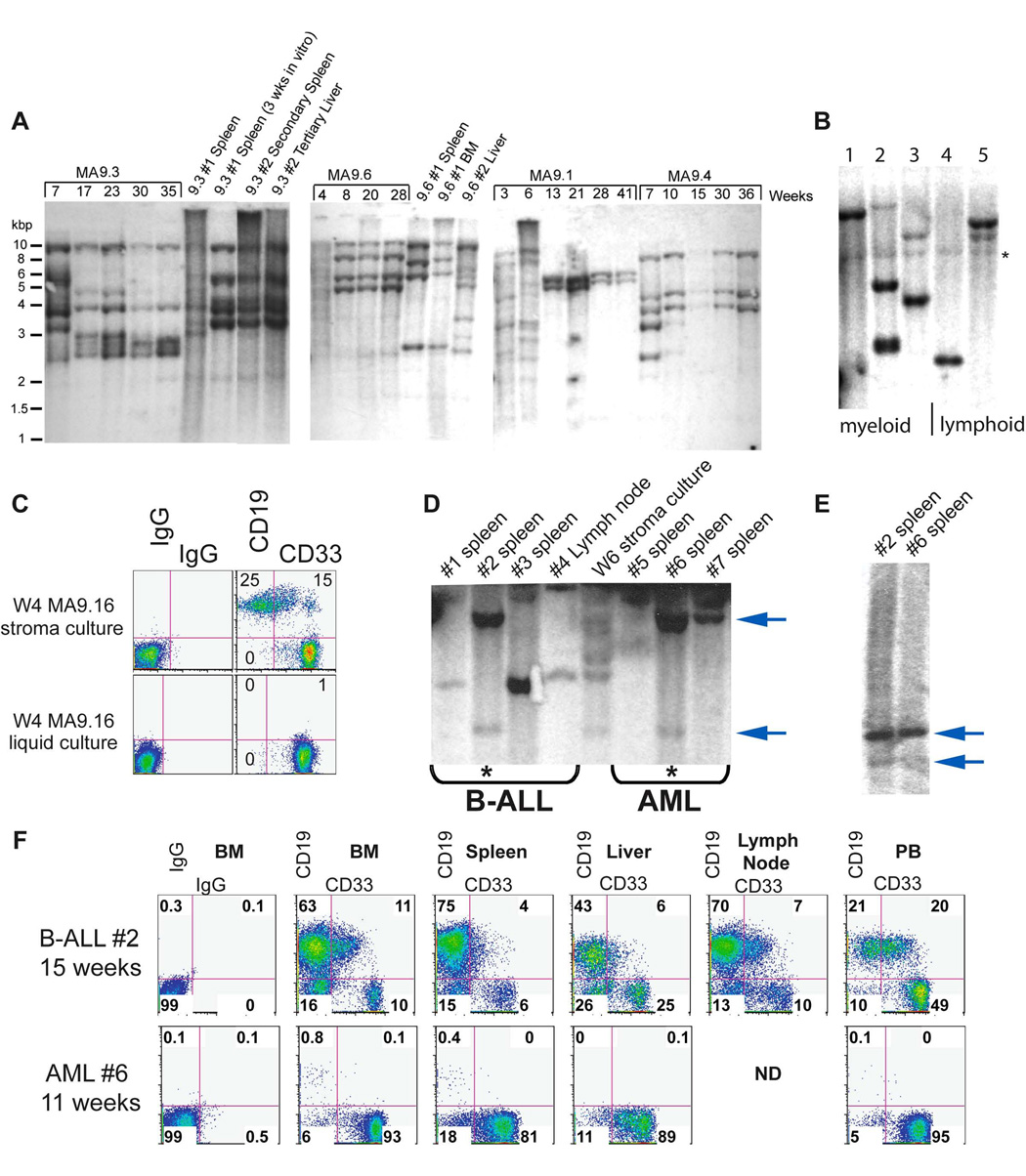
A) Samples were collected from culture and from affected mice at the indicated times and genomic DNA was digested with HinDIII or EcoR1 to detect clonal integration of provirus using a GFP probe. Polyclonal to oligoclonal patterns are present at early time-points in vitro, with oligoclonal to monoclonal integrations detected after extended growth. B) MA9-transduced CB cells were injected immediately after transduction and leukemic cells from sick mice were analyzed for integration site by Southern blotting. *background band. C–F) Cells from MA9.16 myeloid and lymphoid cultures were injected into NS-B2M mice. Upon illness, leukemic cells were recovered and analyzed by Southern blotting for clonal relatedness. Organs from the clonally related leukemias shown in panel D were analyzed for myeloid and lymphoid markers by flow cytometry.
The LSC in MLL-AF9 mixed lineage leukemias is heterogeneous
To determine whether cell culture conditions could influence disease phenotype, we injected a Week 4 myeloid culture and a Week 4 lymphoid culture (both resulting from the same cord blood transduction) into NS-B2M mice (Figure 4C). These injections resulted in AML from the myeloid cell culture (4/4 mice) and B-ALL from the lymphoid cell culture (4/4 mice) after 11–18 weeks. Southern blot analysis revealed that at least one B-ALL and one AML were clonally related, even though the predominant phenotype of each disease was clearly unique (Figure 4, panels D–F). The clonal identity was confirmed using a different restriction enzyme (Figure 4E). Thus, the same LSC can be influenced by the culture microenvironment to promote myeloid or lymphoid expansion and induce either AML or B-ALL/ABL, respectively.
The clonal relatedness of phenotypically unique leukemias implies that a leukemia stem cell expressing MA9 can be multipotent. Whether this is always the case and whether a multipotent cell is an obligate target for MLL fusion protein function in human cells is currently unknown. We separated the myeloid (CD19−CD33+) and lymphoid (CD19+CD33−) populations from a mixed culture by cell sorting and found that the CD19+CD33− cells were able to regenerate a CD19−CD33+ cell type, while the CD19−CD33+ cells were committed to the myeloid lineage and could not regenerate CD19+ cells even under lymphoid culture conditions (Figure 4A). Clonal analysis by Southern blotting showed that the original CD33+ LSC was a unique and independent leukemia population in this culture. However, the CD19−CD33+ population that was generated from the CD19+ sorted cells showed a clonal integration pattern identical to the CD19+ cells, demonstrating that this CD33+ population was in fact a progeny of the CD19+ LSC (Figure 4B). All populations of cells were able to proliferate robustly and also generated leukemia in vivo (data not shown). The morphology of the cells indicated that the surface phenotype was an accurate representation of the identity of the cells (Figure 4C). Interestingly, in contrast to the myeloid-restricted CD19−CD33+ LSC from this population, some CD19−CD33+ cells from other samples gave rise to both CD33+ AML and CD19+ ALL (Figure 4D). These data suggest that MLL-AF9-expressing LSCs can be lineage restricted as well as multipotent, demonstrating the heterogeneity of the LSC in mixed lineage leukemias.
Gene expression profile of MLL-AF9 expressing cells resembles human AML with 11q23 translocations
Subsets of human leukemia exhibit characteristic and robust gene expression signatures, defined on the basis of specific clonal chromosomal rearrangements (Armstrong et al., 2002; Bullinger et al., 2004; Lacayo et al., 2004; Ross et al., 2004; Valk et al., 2004). To compare the gene expression profile of MA9 cells to primary AML samples, we assessed global gene expression for several independently derived MA9 cultures using microarrays. As comparison samples, we used cell lines expressing one of the CBF fusion genes, AML1-ETO or CBFβ-MYH11 (AE and CM), cultured under the same conditions. Transduction of normal CB with AE or CM also results in cultures with long-term proliferative potential (Mulloy et al., 2003; Wunderlich et al., 2006). In total, 9 MA9 cultures and 9 CBF cultures were analyzed using the Affymetrix HGU133-Plus2.0 chip. The publicly available data from two large AML microarray studies were used for comparison (Ross et al., 2004; Valk et al., 2004). Samples from the two public datasets were combined and processed as described in the Methods section to generate a list of the top 100 most differentially expressed genes between CBF patient samples and MLL patient samples (Table S4). Both patient and culture samples were then analyzed using Principal Component Analysis. Two major groups were identified corresponding to the CBF and MLL patient samples (Figure 6A). All of the cell culture samples segregated according to fusion gene status, with the cell lines tending to cluster more closely with the samples from the pediatric AML dataset (Ross) than to the adult samples (Valk). This separation is also apparent in the two-dimensional dendrogram resulting from the hierarchical clustering of the samples shown in Figure 6B. For the MLL group, the cell culture samples behave as a subset within the Ross dataset, implying that these samples are even more closely related to the pediatric patient samples than are the adult AML samples. K-means clustering also demonstrated that the MLL culture samples were highly related to the primary pediatric samples (Figure S3). We used a simple Support Vector Machine [(SVM);linear kernel function and least-squares optimization] for classification of leukemia samples based on the top 10 or the top 7 most differentially expressed genes in CBF versus MLL patient samples and found that the classification accuracies were 100% for the cell culture samples. In addition, class assignment accuracies of randomly chosen training and test data sets from the pool of patient and cell culture samples were assessed by 10-fold cross validation. An average classification accuracy of 99.63% was observed when utilizing the same parameters for the SVM. We analyzed three genes by qPCR that have previously been identified as differentially expressed in MLL leukemias (HOXA9, FLT3 and SPARC) (Bullinger et al., 2004; DiMartino et al., 2006; Ross et al., 2004; Valk et al., 2004). HOXA9 and FLT3 were highly expressed in four MA9 samples compared to four AE samples, and SPARC was underexpressed in the MA9 samples (Figure S4). There was no difference in the expression of these three genes in the MA9 samples that were recovered from mice with leukemia (n=2) compared to the same samples prior to injection (Figure S4). Thus, the transcriptome of these experimentally created cell lines extensively parallels that of primary leukemia cells from AML patients with MLL fusions.
Figure 6. Gene expression profile of MLL-AF9 expressing cells resembles human AML.
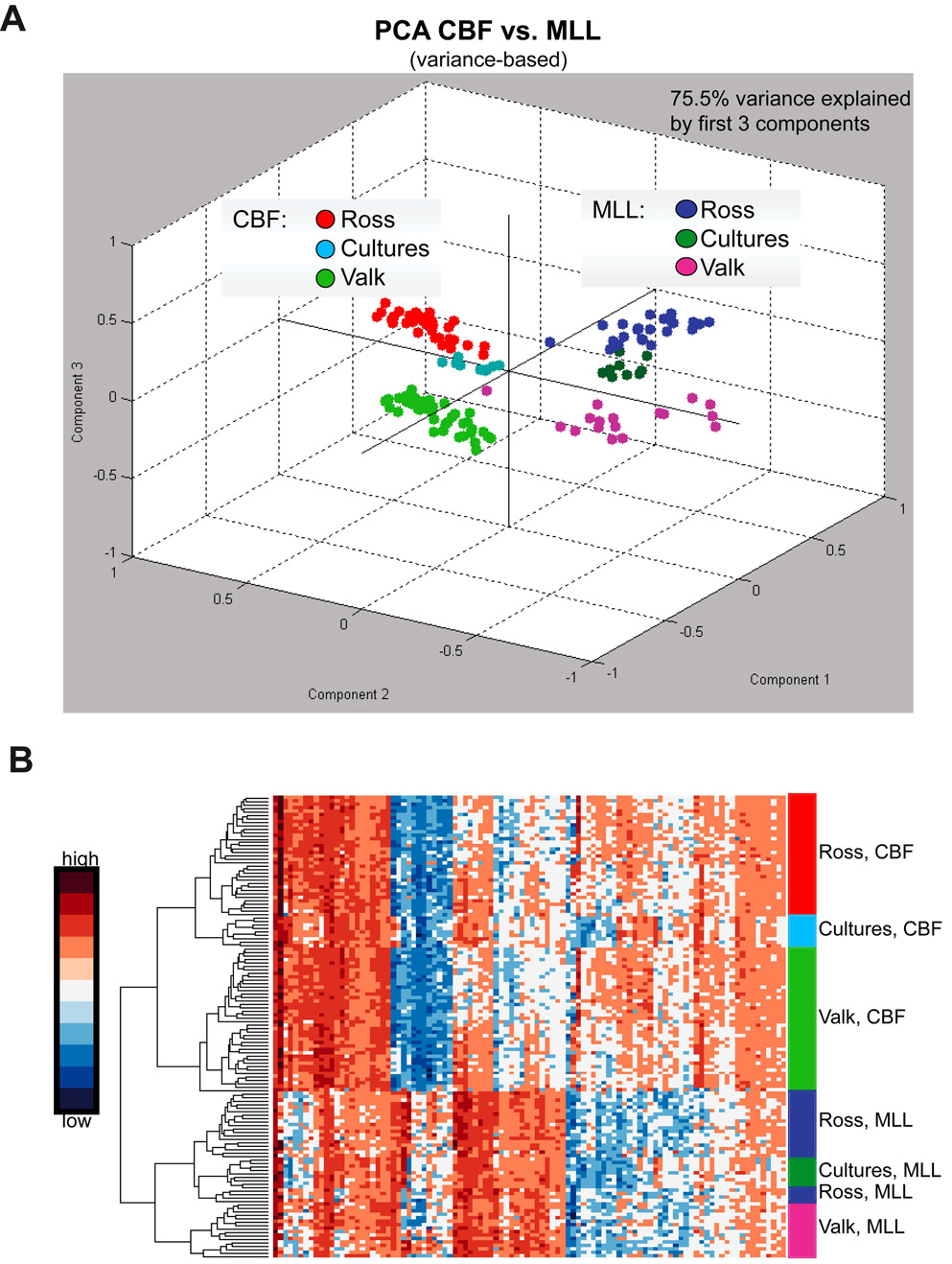
A) The plot depicts the principal component analysis of data for the 100 most differentially expressed genes in the CBF versus MLL leukemia samples from two published studies. Cell culture samples group closely with the respective patient samples from the 2 independent studies. Additionally, the plot suggests a closer relationship of cell culture samples to the pediatric AML data set (Ross). B) Hierarchical clustering of differentially expressed genes. The graph shows the heatmap of CBF and MLL leukemia patient samples as well as their respective cell culture samples (rows) based on the 100 genes (columns) most differentially expressed in CBF versus MLL leukemia samples.
Signaling through the Rac pathway is critical for MLL-AF9 induced AML
The specific signaling pathways downstream of MLL fusion proteins are only beginning to be understood. Recently, Somervaille et al. showed that the activity of the small GTPase proteins Rac1 and CDC42 are specifically increased in murine cells expressing MA9 (Somervaille and Cleary, 2006). We used the small molecule inhibitor of Rac, NSC23766, to determine the role of this signaling pathway in MLL leukemogenesis (Cancelas et al., 2005; Gao et al., 2004; Thomas et al., 2007). Total protein levels of Rac1 and CDC42 were not consistently different between MA9 cells, control cord blood cells and the preleukemia cell cultures expressing AML1-ETO (Figure S6A). We confirmed the published findings that NSC23766 specifically affects the activation of Rac and does not interfere with the activity of the closely related small GTPase CDC42 (Figure S6B). Interestingly, a dose dependent effect of NSC23766 on cell proliferation was realized which was specific for MA9 cells, with little to no effect on control cord blood cells or the AE cultures (Figure 7A). Inhibition correlated with a decrease in cycling cells (S/G2/M phase) and a significant increase in Annexin V+ cells, indicating that loss of Rac signaling in MA9 cells resulted in cell cycle arrest and apoptosis (Figure 7, panels B and C). It has previously been shown that Bcl-2 family members are downstream of Rac signaling (Yang et al., 2000). We analyzed cells 24 hours after drug treatment and found that the Bcl-xL protein was targeted for degradation specifically in the MA9 cells, with no effects detected in either CB or AE cells (Figure 7D). A slight effect on bcl-2 protein was also detected at 24 hours in MA9 cells. To determine whether these effects were specifically mediated by Rac inhibition, we used lentiviral constructs co-expressing the yellow fluorescent protein (YFP) to deliver shRNA targeting human Rac1 to the MA9 cells. Apoptosis was detected specifically in those MA9 cells expressing either of two independent shRNA targeting Rac, but not in scramble-control transduced cells or in AE cells targeted with the same lentiviral constructs (Figure 7E). The increase in apoptosis in the MA9 cells expressing Rac shRNA was statistically significant (Figure 7F). Protein levels of Rac1 were significantly decreased in the cultures expressing Rac shRNA (Figure 7G). Thus, the Rac signaling pathway is essential for the growth and survival of MA9 cells, likely via induction of cell cycle progression and Bcl-xL/Bcl-2 mediated survival.
Figure 7. Suppression of MA9 cell growth by a Rac-specific inhibitor, NSC23766 and by Rac1 knockdown.
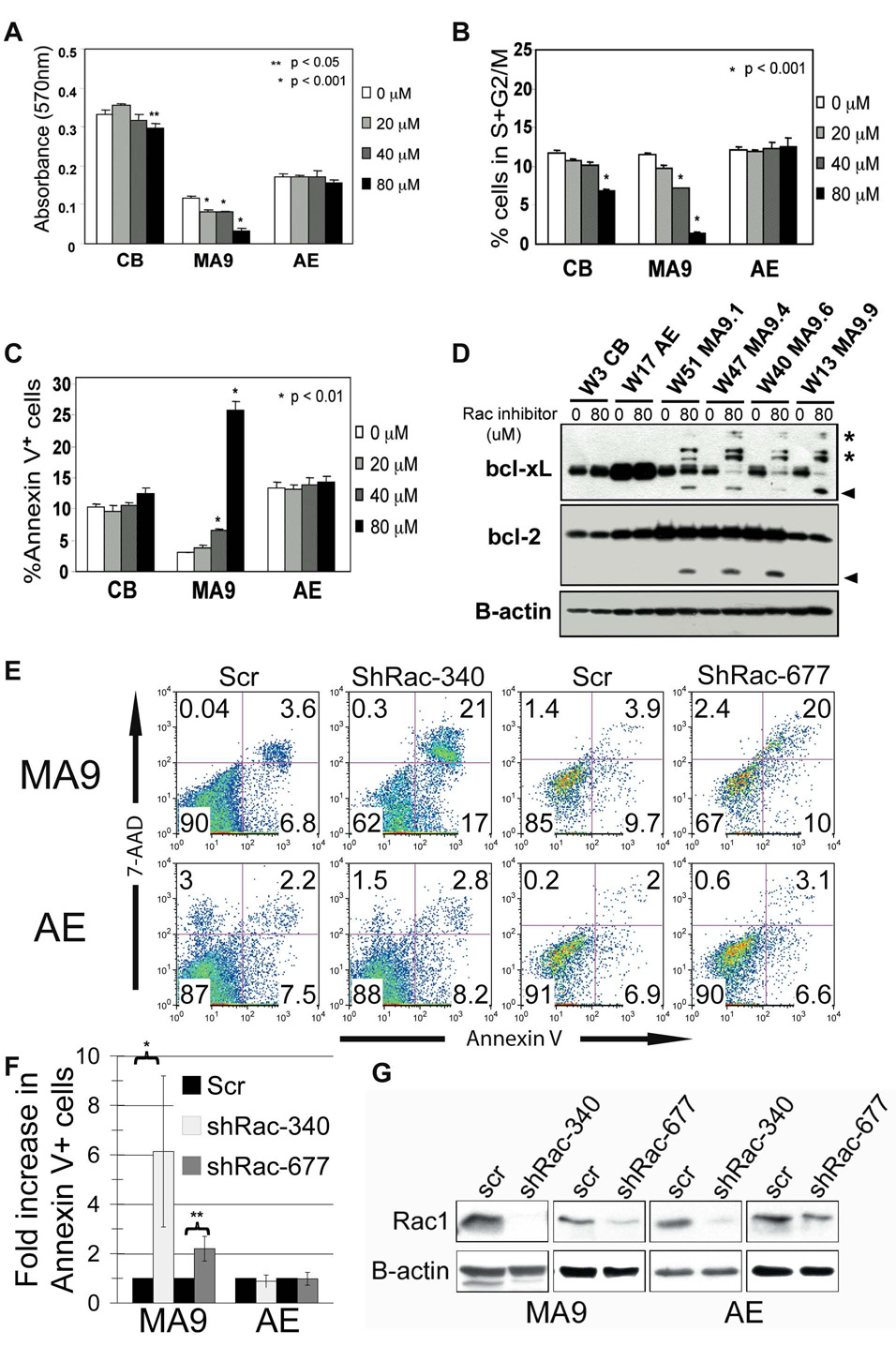
The transduced, long-term cultured MA9 and AE cord blood cells and two-week cultured cord blood cells were plated in the presence of various concentrations of NSC23766 as indicated. A) 48hr cell growth was measured by a MTT assay using absorbance at 570nm. B) 48hr cell cycle arrest was analyzed by propidium iodine staining and flow cytometry. C) 48hr apoptosis response was analyzed by Annexin V staining and flow cytometry. In A–C, data represent the average ±SD. At least three experiments were performed using three different MA9 cell cultures. *, P < 0.001, or **, P < 0.05, NSC23766-treated vs. non-treated cells. D) Bcl-2 and Bcl-xL protein levels after 24 hours of drug treatment. Asterisks indicate potentially ubiquitinated protein, and arrowheads designate possible degradation products. E) Lentivrial knockdown of Rac1 in MA9 and AE cells results in specific induction of apoptosis in the MA9 cells as shown by Annexin V/7-AAD staining. Two independent Rac-targeting shRNA were used. F) The mean ± SD of 3–5 experiments is shown. *p<.05, **p<.001 G) Western blot for Rac1 protein expression demonstrates the loss of expression in the Venus-sorted cells that received the shRac constructs.
Discussion
Mouse models have proven to be invaluable tools for the understanding of human cancer. Nevertheless, significant differences between the genetic makeup of mice and humans make it difficult to directly extrapolate observations made in the former to a clinical disease in the latter. Unlike outbred human populations, mouse strains are genetically homogeneous and homozygous across all or most loci. The relative importance of a particular mutation or gene expression pattern to oncogenesis may be over or under-estimated in this context. Normal primary human cells offer a potentially more relevant target for the study of oncogene function. However, these cells have historically proven to be resistant to neoplastic transformation by a single oncogene(Hahn et al., 1999). We show here that primary human HPC can, in fact, undergo leukemic transformation in response to the MLL-AF9 chimeric oncogene in a manner that faithfully recapitulates many features of the clinical disease. Like MLL-AF9 patient samples, normal human CB cells retrovirally transduced with MA9 display essentially unlimited replicative potential, myelomonocytic or pro-B cell features and are leukemogenic in mice. Moreover, the transcriptome of these experimentally created cell lines extensively parallels that of primary leukemia cells from AML patients with MLL-fusions. This model should prove useful for screening potential therapeutic compounds, as demonstrated by the exquisite sensitivity of the MA9 cells to inhibition of the Rac signaling pathway.
These data suggest that expression of MA9 is the primary molecular defect responsible for the defining characteristics of this disease. Our results support the hypothesis, based on clinical observations, that MLL fusion genes require fewer independent oncogenic events for leukemic transformation than other fusion oncoproteins. MLL fusions are associated with the shortest latency periods documented between the appearance of a karyotypic abnormality and the clinical manifestation of malignancy. MLL rearrangements arise in utero, are found in the majority of infants with acute leukemia and are unique in their ability to produce overt clinical disease after only a few months (Ford et al., 1993). In the present study, the primary leukemia in NS-SGM3 mice was extremely rapid, with a median latency of 7 weeks for direct injection of MLL-AF9 transduced cells (Figure 2A). It could be argued that the non-physiologic levels of human growth factors in the NS-SGM3 mice are contributing to the leukemogenesis, which is possible. However, even in the NS and NS-B2M mice, the rapidity of the AML (median latency of 9 weeks; Figure S2C) and the multi-clonal as well as multi-lineage nature of the disease argues that the need for additional cooperating events is rapidly met under these experimental conditions. Combined with the clinical observations, our data suggest that although MLL fusions alone are not sufficient to drive leukemogenesis, they somehow facilitate the rapid acquisition of the additional genetic or epigenetic events that are required.
The finding of telomerase activity in all MA9 clones suggests that the initiation of a self-renewal program by MA9, as demonstrated by others (Krivtsov et al., 2006), includes this critical enzyme. Although the hTERT promoter itself may not be a direct target of MLL fusion proteins, sustained hTERT activity is likely to be one of the essential components of transformation and is a valid therapeutic target for MLL leukemias. Numerous failed attempts have been made to transform the human HSPC using leukemia-associated oncogenes, and in all cases where telomerase activity has been analyzed it was shown to be low to absent (Mulloy et al., 2003; Warner et al., 2005; Wunderlich et al., 2006). It is likely not coincidental that the oncogene which successfully transforms the human HSPC also results in sustained activity of telomerase. Our demonstration that FLT3L is also critical for self-renewal of the MLL LSC is in keeping with previous experimental and clinical findings associating increased FLT3 expression with MLL leukemia(Armstrong et al., 2003; Armstrong et al., 2004; Ozeki et al., 2004; Stam et al., 2005; Stubbs and Armstrong, 2007; Stubbs et al., 2008). This could also explain our ability to expand the MLL LSC in vitro indefinitely, while Barabe et al. were unable to maintain the leukemic clone beyond 3 months; in their study, only the cytokines IL-3 and SCF were used (Barabe et al., 2007).
The clonal relatedness of phenotypically unique leukemias implies that a leukemia stem cell expressing MA9 can be multipotent, and our data demonstrates that this ability resides in both the CD19+CD33− and CD19−CD33+ cells. Although this has been previously described for murine cells expressing the MLL-GAS7 fusion, it is not found with the more common MLL-ENL or MLL-AF9 fusions, which almost exclusively result in AML in the mouse(So et al., 2003a). Whether this is a mouse/human difference remains to be determined but seems likely based on current data. Zeisig et al. showed that although MLL-ENL transduced murine BM cells appeared myeloid by morphology, even under lymphoid growth conditions, their gene expression profile and the presence of a rearranged immunoglobulin locus strongly favored a B lymphoid ancestry and a continued transcription factor promiscuity that belied a simple AML classification (Zeisig et al., 2003). Thus it may be that murine cells will not readily display the phenotypic and morphologic readout of the lymphoid disease associated with the common MLL fusions.
In our model, lineage restricted MLL LSC are immortal and leukemogenic, although they are not multipotent. This raises questions as to the true nature of the LSC in the human disease. Since MA9 expression is predominantly associated with AML in humans, our data could imply that the target cell in MA9-associated disease is frequently a committed myeloid progenitor cell. Alternatively, it is possible that the microenvironment in the human, or the fusion protein itself, strongly promotes a myeloid phenotype from a primitive LSC. It has been proposed that the fusion partner is instructive as to lineage (Barabe et al., 2007; Chen et al., 2006). However, given the ease with which the MA9 oncogene immortalizes human B cells and induces B-ALL, it seems unlikely that the fusion partner is the major determinant for lineage choice. Although Barabe et al. have argued that the xenograft model may skew the outcome towards over-representation of B-lineage cells, our data would instead support the hypothesis that environmental cues supplied by the microenvironment are playing a primary role in lineage determination. We clearly show that a B-cell outcome is readily attained in vitro upon expression of the MA9 fusion protein, a finding that is independent of xenograft effects (Figure 1F). In addition, the rapid occurrence of AML in NS-SGM3 would support the primary effect of microenvironment on lineage decision. It is obvious, given the definitive associations between fusion partner identity and lineage outcome, that additional factors are involved, including contributions from the individual fusion partner as well as possible differences in the cell that is targeted by the translocation. It will be interesting to analyze the effects of MLL-ENL (and MLL-AF4) in NS-SGM3 mice, and to determine whether MA9 is able to transform a committed myeloid or lymphoid human progenitor cell.
The progenitor cell in the MA9 lymphoid cultures is reminiscent of the bipotential B-macrophage progenitor that has been described in murine fetal liver and adult mouse bone marrow (Cumano et al., 1992; Montecino-Rodriguez et al., 2001). It is possible that the transcriptional program initiated and sustained by MA9 expression favors this particular progenitor cell. Our ability to manipulate these cells using specific growth factors, to induce either B lineage or myeloid/macrophage progeny, implies that the interplay between transcription factors downstream of these cytokine receptors are finalizing the fate of the cell and promoting lineage restriction. This effect must be instructive rather than simply promoting survival and outgrowth of a limited number of previously committed myeloid cells, based on our ability to promote myeloid differentiation from a pure CD19+CD33− population (Fig. 5). It is clear from a number of recent studies that the plasticity of the hematopoietic system is significantly greater than the simplified hierarchies that have been used in the past, and our data would support the conclusions of these studies (Iwasaki et al., 2006; Iwasaki et al., 2003; Xie et al., 2004). This human-based cell culture system should allow us to address which factors are involved in these decisions.
Figure 5. CD19+ cells generate myeloid and lymphoid progeny.
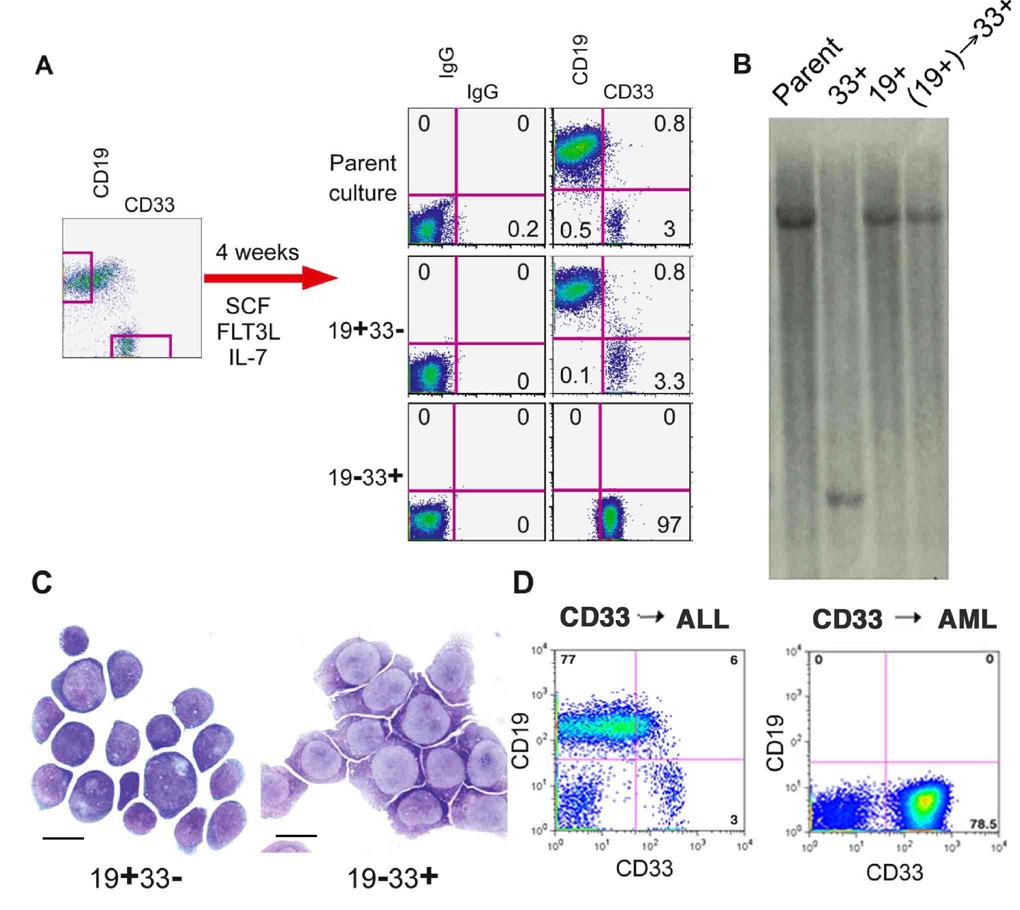
A) A mixed culture was separated by cell sorting into CD19+33− and CD19−33+ populations. Cells were grown for one month in the indicated cytokines, and stained for the indicated surface molecules. Only the 19+33− population of cells was able to regenerate the lineage potential of the parent population. B) Southern blot analysis was performed on the indicated populations of cells. The original CD33+ population is clonally unique and represents an AML LSC present in the parent population. The CD33+ population generated from the CD19+ sorted cells [(CD19+)→CD33+] shows clonal identity to the B-ALL LSC present in the parent culture. C) Cells from each population were analyzed by Wright-Giemsa staining one month after sorting. Scale bars represent 10 µm. D) Some CD33+ clones were able to produce B-ALL while others gave rise to pure AML in NOD/SCID mice.
The significant and specific effect of NSC23766 on MA9 cells could reveal a potential vulnerability of MLL fusion leukemias. Due to its relatively low affinity and pharmacodynamic properties, this particular version of the compound may not be clinically applicable. However, the results highlight the usefulness of this human-based system for identifying additional pathways for therapeutic targeting. When better Rac inhibitors become available, they may prove particularly useful in leukemia patients with 11q23 disease. Rac knockdown was highly effective in recapitulating the drug response, resulting in rapid induction of apoptosis that was specific for the MA9 cells and did not occur in the AML1-ETO-expressing cells. How the AML1-ETO cells are bypassing this particular pathway, and the specific signals upstream of Rac that are inducing this addiction, are interesting questions that can be addressed using this model system.
It is intriguing to speculate that MLL rearrangements, by simultaneously creating a state of haploinsufficiency and inducing expression of a chimeric form of MLL, may result in global epigenetic changes. These changes could provide opportunities for individual clones to rapidly achieve a selective advantage based on aberrant gene expression patterns. This could lead, for example, to failure to downregulate hTERT, allowing cells to continue replication beyond the normal limits imposed by telomere shortening. The striking overlap between the transcriptome of MA9 transduced human CB cells and their clinical counterparts suggests that whatever effects MLL-AF9 is having on its direct target genes and their downstream effectors can be modeled in the laboratory using primary human CD34+ cells. This system promises to yield valuable insights into the early events in MLL fusion driven leukemogenesis, some of which may be directly translated into clinical interventions.
Experimental Procedures
CD34+ cord blood cells
Human umbilical cord blood was obtained by the Translational Trials Support Laboratory at CCHMC under a protocol approved by the CCHMC Institutional Review Board. No identifying information related to the infant or mother was obtained with these collections. CD34+ cells were enriched to > 90% purity by immunomagnetic bead selection and cryopreserved. Cells were cultured in IMDM 20% bovine calf serum (or serum-free for some experiments) and supplemented with SCF, IL-3, IL-6, Flt-3L and TPO. Viable cell counts in MA9 and control cultures were assessed one to two times per week and cultures were split into fresh media as needed to maintain a cell density of between 5 × 105 and 2 × 106 cells/ml. For clonogenicity experiments, cells were plated in 96 well plates at 10,000 cells/well and serially diluted to 11 additional rows. After 5 weeks, plates were scored and Poisson statistics were used to determine clone frequencies.
Flow cytometry
Cells were analyzed on a FACSCalibur or FACSCanto flow cytometer (BD). Approximately 2×105 cells were stained with fluorochrome conjugated antibodies for 30 minutes at 4C and were washed with PBS/2%FBS prior to analysis. Animal tissues were processed according to standard procedures. Following red cell lysis, cells were incubated with an antibody to block non-specific binding (anti-mouse CD16/CD32 Fcγ receptor, BD). 7AAD was used to gate viable cells. Antibodies (all BD unless noted) used were the phycoerythrin (PE) conjugates anti-CD4, CD8, CD11b, CD13, CD14, CD16, CD19, CD20. CD33, CD34, CD38, CD41, CD56 (Caltag), CD117, CD123, CD135 and HLA-DR (Caltag) and allophycoerythrin (APC) conjugates anti-CD15, CD19, CD33, CD34, CD36, CD110, CD117, CD135 and CD133 (Miltenyi Biotech). Appropriate IgG isotype control antibodies (BD) were used. Cell cycle analysis (BrdU-APC kit, BD) and apoptosis analysis (Annexin V-PE kit, BD) were performed according to the manufacturer’s recommendations. All flow cytometry data was analyzed with FloJo software (TreeStar).
Retroviral vectors and viral transduction
The MIEG3 vector and the RD114 pseudotyping vector were obtained from Dr. David Williams. SFβ91 (REW) vector was from C. Baum. MSCV-MLL-AF9 plasmid was obtained from Eric So. An amino terminal FLAG-tagged cDNA encoding MLL-AF9 was a kind gift from J. Kersey and J. Hess. A 4.7 kbp fragment containing MLL-AF9 was cloned, in frame, downstream of the foot and mouth disease virus (FMDV) 2A peptide (a kind gift from C. Baum). The 2A-MLL-AF9 fragment was cloned downstream of an EGFP cassette in SFβ91. MIEG3, MA9 and AE vector constructs were transfected transiently into the Phoenix packaging cell line with either the RD114 or amphotropic env construct and the gag-pol expression plasmid. 40 ml of supernatant was concentrated ~8X for RD114-pseudotyped virus and snap frozen. For transduction, CD34+ cells were cultured in the presence of retroviral supernatant supplemented SCF, IL-3, IL-6 Flt-3L and TPO on retronectin coated plates for 2 days.
shRNA Constructs and lentiviral transduction
Lentiviral shRac1 vector MISSION pLKO.1-shRac1-puro constructs were obtained from Sigma. The insert encoding YFP was excised from the pLKO.1-scrambled-YFP vector (gift of Dr. Lee Grimes) with Kpn1/BamH1 and subcloned into the Kpn1/BamH1 site of pLKO.1–shRac1 vector #677 (CCGGGCTAAGGAGATTGGTGCTGTACTCGAGTACAGCACCAATCTCCTTAGCTTTTT) and #340 (CCGGCCCTACTGTCTTTGACAATTACTCGAGTAATTGTCAAAGACAGTAGGGTTTTT), replacing puromycin. 293T cells cultured in DMEM with 10% FBS were co-transfected with the pMD.2 VSV-G envelope plasmid, the pCDLN helper plasmid and the lentiviral vectors by calcium phosphate transfection. Virus was collected and filtered through a 0.45µm filter, concentrated and purified with 20% sucrose. For transduction, AE and MA9 cells were cultured in the presence of lentiviral supernatant supplemented with SCF, IL-3, IL-6, Flt-3L (and for AE cells, TPO was also included) on retronectin coated plates overnight. Two to three days after transduction, cells were sorted for YFP expression on a FACSVantage (Becton Dickinson, San Jose, CA). Two, five and seven days after sorting, cells were assessed for apoptosis using the annexin V-PE kit (Becton Dickinson) according to the manufacturer’s directions.
Telomerase assays
Cellular extracts were prepared from control and MA9 cells in 1x CHAPS lysis buffer and cleared by centrifugation for 20 min at 4°C. Telomerase activity was assayed with the kit from Intergen.
Mouse studies
Cultured MA9 cells were injected by tail vein or intrafemoral injection into sublethally irradiated (300–350 rad) 6–8 week old NOD/SCID, NOD/SCID-B2M or NOD/SCID-SGM3 mice(Feuring-Buske et al., 2003). Mouse experiments were performed in accordance with relevant institutional and national regulations and approved by the Institutional Animal Care and Use Committee of CCHMC. Mice were kept on chow supplemented with doxycycline for one week before and after injections. Mice were sacrificed when they showed signs of illness. Organs were homogenized and processed for flow cytometry after fixing a piece in 10% formalin for histopathologic analysis. Cells were grown in vitro for karyotype analysis. The four long bones of the hind legs were flushed to extract the bone marrow, and the majority of cells were frozen for injection into secondary recipients.
Histopathology and karyotype studies
Organ samples were preserved in 10% formalin before being embedded in paraffin, sectioned, and stained with Hematoxylin and Eosin by the Pathology department at CCHMC. Stained sections were visualized using a Nikon Optiphot-2 microscope and photographed with a Spot RT color camera (Diagnostic Instruments Inc). Cytospins were made using the Cytospin-4 centrifuge (Thermo Shandon). Cytospins were stained by Wright Giemsa (Fisher Scientific). Metaphase cells were prepared by standard cytogenetics methods. Karyotypes were described according to the International System for Human Cytogenetic Nomenclature (Mitelman, 1995).
NSC treatment studies, apoptosis and cell cycle analysis
NSC23766 was resuspended in PBS to obtain a 100uM stock solution. In vitro cultures were seeded at 106 cells/mL and incubated with the indicated doses. 24 to 48 hours later, cells were analyzed for Annexin V staining by flow cyometry according to the manufacturer’s recommendations (Annexin V-PE kit, BD). Cell cycle was assessed by BrdU incorporation for 1 hour followed by flow cytometry using the Anti-BrdU-PE kit (BD). Whole cell lysates were made with glycerol lysis buffer (10% Glycerol, 20mM Tris-HCl, 135mM NaCl, 1mM EGTA, 1.5mM MgCl2, 1% TritonX-100) supplemented with Complete Protease Inhibitor Cocktail (Roche). Protein concentrations were determined using the BCA protein kit (Pierce) and 25ug of protein were loaded onto 14% Tris-Glycine gels (Invitrogen). Western blot analysis was preformed with primary antibodies against Bcl-2 (BD transduction labs), Bcl-xL (Cell Signaling), and B-actin (Sigma) followed by incubation with HRP-conjugated secondary antibodies (Cell Signaling) and detection of signal with the SuperSignal ECL kit (Pierce) with BioMax L1 film (Kodak).
GTPase pulldown assay
To detect active GTP-bound Rac1 and Cdc42, we performed pull-down assays using Glutathione S-transferase (GST)-PAK1-PBD beads. 15×106 cells were plated with/without NSC23766 (40 µm) for 24 hours and lysed in 600µl lysis/binding/washing buffer. 1/10th of the lysate was used for total Rac1 and total cdc42 determination by Western blot with the remaining lysate incubated with GST-Pak1-PBD at 4°C for 1 hour. After incubation, the mixture was centrifuged at 8,000xg. The resins were washed three times with wash buffer and eluted by adding 50 µl of 1X SDS sample buffer and boiling at 100°C for 5 minutes. 20 µl of the sample volume was analyzed by SDS-PAGE (15% polyacrylamide gel) and transferred to nitrocellulose membrane. Active Rac1 and Cdc42 were detected by Western blotting. Detection was performed using SuperSignal West Pico Chemiluminescent Substrate followed by exposure to X-ray film. The band intensity was quantified and levels of Rac1-GTP and Cdc42-GTP were normalized to the amount of total protein.
Real time RT-PCR
RNA was isolated with the RNAeasy kit (Qiagen). RNA was reversed transcribed using MuLV RT and random hexamers (Applied Biosystems). The cDNA was then subject to QPCR using SYBR Green technology (Applied Biosystems). Reactions were run on a 7700 system and data were analyzed using SDS version 1.9 (Applied Biosystems). Expression levels of hTERT, HOXA9, FLT3 and SPARC were calculated by the standard curve method relative to THP-1 and K562 cells. Only samples with the appropriate dissociation curves were considered for analysis. Results were normalized to c-abl expression. Q-PCR primers are available upon request.
Southern blot analysis
Genomic DNA was prepared using the QIAamp DNA Blood Mini kit (Qiagen). 10ug DNA was cut with either HinDIII (REW-MA9 construct) or EcoR1 (MSCV-MA9-puro construct) ON, run on a 0.7% agarose gel for 16–20 hours at 20V and transferred to a positively charged nylon membrane (GE Magnacharge) by upward capillary transfer for 16–20h in 20x SSC. The DNA was fixed to the membrane in a UV Stratalinker 1800 (Stratagene). The membrane was pre-hybridized for 45 min in Hybrisol (Chemicon). The probe (NEBlot kit, New England Biolabs) was added to 10ml fresh Hybrisol and incubated ON. The membrane was then washed and exposed to Kodak BioMAX MS film (Fisher).
Microarray Data Analysis
We pooled Affymetrix GeneChip® microarray (U133A) data from MLL and CBF patients from 2 published leukemia studies(Ross et al., 2004; Valk et al., 2004). Within these data sets we identified a total of 40 MLL and 76 CBF leukemia samples (training data). Training data were combined with expression data for the probe sets of the U133A array from our leukemia culture model microarray (U133+2) data (test data). For further processing of this data matrix we utilized the statistical programming language R (www.R-project.org) with the Bioconductor package (www.bioconductor.org). The data were pre-processed using the MAS5 function (Affy package). A 3 parameter linear model was fitted to the training data. Using the empirical Bayes function (limma package) we identified probe sets differentially expressed between CBF and MLL patient samples. Probe sets were declared significantly differentially expressed if their Bonferroni-adjusted p-value < 0.01. We identified the 100 most significantly differentially expressed probe sets representing distinct genes excluding those probe sets specific for fusion gene partners. To visualize the relation of patient leukemia samples and leukemia model culture data we used dimensionality-reducing principal component analysis (PCA) (Matlab, Math Works Inc., version 7.1). Hierarchical clustering (squared Euclidean distance measure) of samples was performed using R/Bioconductor. Furthermore, k-means clustering with a correlation-based metric was conducted using Matlab.
Sample Classification using Support Vector Machines (SVM)
To investigate whether (a subset of) the 100 differentially expressed genes is able to discriminate MLL and CBF cultures we used classifiers generated by a linear support vector machine (SVM). We trained the SVM (Matlab) with expression data from the 10 most differentially expressed genes of the training data set. Our culture data (test data) were then classified according to the classification rule based on the leukemia data (training data). Also, we performed 10-fold cross-validation by repeatedly building classifiers based on 90% of randomly selected samples from the combined test and training data to classify the remaining 10% of samples.
Supplementary Material
01
Acknowledgements
We thank the mouse core at Cincinnati Children’s Hospital for help with animal experiments, Eric So for the MSCV-MLL-AF9 plasmid, Lee Grimes for the pLKO.1-venus plasmid, Kirin Brewery for the cytokine TPO and Amgen for FLT3L, SCF, and IL-6. This work was funded by National Institutes of Health grants CA118319 and CA90370 (JCM), University of Cincinnati Cancer Center grant (JCM), the American Society of Hematology (JFD and JP), the Ministerio de Sanidad Grant FIS04-0555 (JCC) and by U.S.P.H.S Grant Number MO1 RR 08084, General Clinical Research Centers Program, National Center for Research Resources, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession number
The microarray data is available at Gene Expression Omnibus, accession number GSE7011.
References
- Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, Pieters R, Kersey JH, Sallan SE, Fletcher JA, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3:173–183. doi: 10.1016/s1535-6108(03)00003-5. [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, Sallan SE, Korsmeyer SJ. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004;103:3544–3546. doi: 10.1182/blood-2003-07-2441. [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- Baer MR, Stewart CC, Lawrence D, Arthur DC, Mrozek K, Strout MP, Davey FR, Schiffer CA, Bloomfield CD. Acute myeloid leukemia with 11q23 translocations: myelomonocytic immunophenotype by multiparameter flow cytometry. Leukemia. 1998;12:317–325. doi: 10.1038/sj.leu.2400933. [DOI] [PubMed] [Google Scholar]
- Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science. 2007;316:600–604. doi: 10.1126/science.1139851. [DOI] [PubMed] [Google Scholar]
- Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, Dohner H, Pollack JR. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, Williams DA. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. 2005;11:886–891. doi: 10.1038/nm1274. [DOI] [PubMed] [Google Scholar]
- Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood. 2006;108:669–677. doi: 10.1182/blood-2005-08-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, Bell S, McKenzie AN, King G, Rabbitts TH. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell. 1996;85:853–861. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumano A, Paige CJ, Iscove NN, Brady G. Bipotential precursors of B cells and macrophages in murine fetal liver. Nature. 1992;356:612–615. doi: 10.1038/356612a0. [DOI] [PubMed] [Google Scholar]
- De Braekeleer M, Morel F, Le Bris MJ, Herry A, Douet-Guilbert N. The MLL gene and translocations involving chromosomal band 11q23 in acute leukemia. Anticancer Res. 2005;25:1931–1944. [PubMed] [Google Scholar]
- DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. The AF10 leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10. Blood. 2002;99:3780–3785. doi: 10.1182/blood.v99.10.3780. [DOI] [PubMed] [Google Scholar]
- Dimartino JF, Cleary ML. Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol. 1999;106:614–626. doi: 10.1046/j.1365-2141.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- DiMartino JF, Lacayo NJ, Varadi M, Li L, Saraiya C, Ravindranath Y, Yu R, Sikic BI, Raimondi SC, Dahl GV. Low or absent SPARC expression in acute myeloid leukemia with MLL rearrangements is associated with sensitivity to growth inhibition by exogenous SPARC protein. Leukemia. 2006;20:426–432. doi: 10.1038/sj.leu.2404102. [DOI] [PubMed] [Google Scholar]
- Feuring-Buske M, Gerhard B, Cashman J, Humphries RK, Eaves CJ, Hogge DE. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia. 2003;17:760–763. doi: 10.1038/sj.leu.2402882. [DOI] [PubMed] [Google Scholar]
- Flores I, Benetti R, Blasco MA. Telomerase regulation and stem cell behaviour. Curr Opin Cell Biol. 2006;18:254–260. doi: 10.1016/j.ceb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, Greaves M. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363:358–360. doi: 10.1038/363358a0. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Mizuno S, Arinobu Y, Ozawa H, Mori Y, Shigematsu H, Takatsu K, Tenen DG, Akashi K. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20:3010–3021. doi: 10.1101/gad.1493506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H, Mizuno S, Wells RA, Cantor AB, Watanabe S, Akashi K. GATA-1 converts lymphoid and myelomonocytic progenitors into the megakaryocyte/erythrocyte lineages. Immunity. 2003;19:451–462. doi: 10.1016/s1074-7613(03)00242-5. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Lacayo NJ, Meshinchi S, Kinnunen P, Yu R, Wang Y, Stuber CM, Douglas L, Wahab R, Becton DL, Weinstein H, et al. Gene expression profiles at diagnosis in de novo childhood AML patients identify FLT3 mutations with good clinical outcomes. Blood. 2004;104:2646–2654. doi: 10.1182/blood-2003-12-4449. [DOI] [PubMed] [Google Scholar]
- Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. Embo J. 1997;16:4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Leathers H, Dorshkind K. Bipotential B-macrophage progenitors are present in adult bone marrow. Nat Immunol. 2001;2:83–88. doi: 10.1038/83210. [DOI] [PubMed] [Google Scholar]
- Mulloy JC, Cammenga J, Berguido FJ, Wu K, Zhou P, Comenzo RL, Jhanwar S, Moore MA, Nimer SD. Maintaining the self-renewal and differentiation potential of human CD34+ hematopoietic cells using a single genetic element. Blood. 2003;102:4369–4376. doi: 10.1182/blood-2003-05-1762. [DOI] [PubMed] [Google Scholar]
- Ozeki K, Kiyoi H, Hirose Y, Iwai M, Ninomiya M, Kodera Y, Miyawaki S, Kuriyama K, Shimazaki C, Akiyama H, et al. Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood. 2004;103:1901–1908. doi: 10.1182/blood-2003-06-1845. [DOI] [PubMed] [Google Scholar]
- Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou X, Song G, Shurtleff SA, Pounds S, Cheng C, Ma J, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–3687. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- Schoch C, Schnittger S, Klaus M, Kern W, Hiddemann W, Haferlach T. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood. 2003;102:2395–2402. doi: 10.1182/blood-2003-02-0434. [DOI] [PubMed] [Google Scholar]
- Secker-Walker LM. General Report on the European Union Concerted Action Workshop on 11q23, London, UK, May 1997. Leukemia. 1998;12:776–778. doi: 10.1038/sj.leu.2401011. [DOI] [PubMed] [Google Scholar]
- So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, Cleary ML. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell. 2003a;3:161–171. doi: 10.1016/s1535-6108(03)00019-9. [DOI] [PubMed] [Google Scholar]
- So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer Cell. 2003b;4:99–110. doi: 10.1016/s1535-6108(03)00188-0. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–268. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, van der Voort E, Valsecchi MG, de Lorenzo P, Sallan SE, et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood. 2005;106:2484–2490. doi: 10.1182/blood-2004-09-3667. [DOI] [PubMed] [Google Scholar]
- Stubbs MC, Armstrong SA. FLT3 as a therapeutic target in childhood acute leukemia. Curr Drug Targets. 2007;8:703–714. doi: 10.2174/138945007780830782. [DOI] [PubMed] [Google Scholar]
- Stubbs MC, Kim YM, Krivtsov AV, Wright RD, Feng Z, Agarwal J, Kung AL, Armstrong SA. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia. 2008;22:66–77. doi: 10.1038/sj.leu.2404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EK, Cancelas JA, Chae HD, Cox AD, Keller PJ, Perrotti D, Neviani P, Druker BJ, Setchell KD, Zheng Y, et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell. 2007;12:467–478. doi: 10.1016/j.ccr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Warner JK, Wang JC, Takenaka K, Doulatov S, McKenzie JL, Harrington L, Dick JE. Direct evidence for cooperating genetic events in the leukemic transformation of normal human hematopoietic cells. Leukemia. 2005;19:1794–1805. doi: 10.1038/sj.leu.2403917. [DOI] [PubMed] [Google Scholar]
- Wunderlich M, Krejci O, Wei J, Mulloy JC. Human CD34+ cells expressing the inv(16) fusion protein exhibit a myelomonocytic phenotype with greatly enhanced proliferative ability. Blood. 2006;108:1690–1697. doi: 10.1182/blood-2005-12-012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- Yang FC, Kapur R, King AJ, Tao W, Kim C, Borneo J, Breese R, Marshall M, Dinauer MC, Williams DA. Rac2 stimulates Akt activation affecting BAD/Bcl-XL expression while mediating survival and actin function in primary mast cells. Immunity. 2000;12:557–568. doi: 10.1016/s1074-7613(00)80207-1. [DOI] [PubMed] [Google Scholar]
- Zeisig BB, Garcia-Cuellar MP, Winkler TH, Slany RK. The oncoprotein MLL-ENL disturbs hematopoietic lineage determination and transforms a biphenotypic lymphoid/myeloid cell. Oncogene. 2003;22:1629–1637. doi: 10.1038/sj.onc.1206104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01